

INTRODUCTION
Aspirin-exacerbated respiratory disease (AERD) is characterized by the quatrad of asthma, eosinophilic rhinosinusitis, nasal polyposis, and the onset of respiratory reactions after ingestion of aspirin or any other inhibitor of the cyclooxygenase 1 (COX-1) enzyme. Although precipitation of acute reactions by ingestion of COX-1 inhibitors is the defining feature of the syndrome, the underlying inflammatory respiratory disease process begins and continues independently of exposure to aspirin or nonsteroidal antiinflammatory drugs (NSAIDs). The clinical course of the respiratory disease is often stereotypical, which suggests a common underlying cause and mechanism. Typically, the syndrome begins with severe nasal congestion, often after an apparent viral upper respiratory infection in young adulthood, which progresses to chronic eosinophilic rhinosinusitis and recurrent nasal polyposis. During the evolution of the sinus disease, symptoms of lower respiratory tract disease also begin and persistent asthma is diagnosed. The asthma tends to be severe, and individuals with AERD have significantly lower baseline lung function measurements than do those with aspirin-tolerant asthma, suggesting the effects of airway remodeling.1 If patients with AERD ingest aspirin or an NSAID, an acute reaction develops, characterized by both upper and lower respiratory symptoms. Neither the pathophysiology of the underlying disease nor the mechanisms of the reactions to NSAIDs are entirely understood. Both the baseline respiratory pathology of AERD and the clinical reactions to NSAIDs are accompanied by aberrant metabolism of arachidonic acid, the precursor of both leukotrienes (LTs) and prostaglandins (PGs). This article reviews potential casual mechanisms responsible for the underlying disease, the confirmatory reactions to COX-1-active drugs, and the disturbances and pathogenetic roles of the lipid mediator systems.
PATHOGENESIS OF THE UNDERLYING RESPIRATORY DISEASE
Histopathology, Cytokines, and Microbes
Histologically, AERD is characterized by intense inflammation in the upper and lower airways. The numbers of eosinophils identified in the mucosal and submucosal compartments of bronchial and nasal biopsies exceed the numbers found in biopsies from aspirin-tolerant asthmatic individuals by 2 to 3-fold.2,3 Degranulated mast cells are frequent. Lavage fluids obtained from the nasal and bronchial tissues show both higher percentages of eosinophils and higher levels of eosinophil cationic protein, when compared with lavage fluids from aspirin-tolerant controls.3–5 Thus, even without aspirin provocation, there is evidence for an active, ongoing inflammatory response dominated by eosinophils and mast cells in the respiratory tissue in AERD. This persistent inflammation likely contributes substantially to the recurrent development of severe nasal polyps,6 with the soluble products of eosinophils and mast cells causing edema and fibroproliferation.7
The mechanism by which granulocytes are drawn into the respiratory tissues is not fully understood. In individuals with AERD, aspirin challenges are associated with a significant increase in serum levels of eotaxin-2, a potent and selective chemoattractant for eosinophils.8 A recent study9 showed a markedly increased frequency of leukocytes associated with platelets in the nasal polyp specimens of patients with AERD compared with polyps from aspirin-tolerant controls (Fig. 1). Moreover, the percentages of eosinophils, neutrophils, and monocytes with adherent platelets were several-fold higher in the blood of individuals with AERD than in the blood of aspirin-tolerant controls. The platelet-adherent subsets of each leukocyte type expressed higher expression of both β1-integrins and β2-integrins than did the corresponding nonadherent subset. These findings suggest that a platelet-dependent pathway may facilitate inflammation of the respiratory mucosa in AERD by priming leukocytes for entry into the inflamed tissues. Resident eosinophils in the nasal polyps store interleukin 5 (IL-5) and granulocyte-macrophage colony-stimulating factor (GM-CSF), both of which are viability sustaining factors for eosinophils.10 Another study reported that both mast cells and eosinophils staining positively for IL-5, as well as mast cells staining for GM-CSF, were several-fold higher in bronchial biopsies from patients with AERD than from aspirin-tolerant individuals.11 These studies suggest that resident cells of the innate immune system contribute substantially to generating the cytokines that drive eosinophilic inflammation in a self-perpetuating fashion.
Fig. 1.
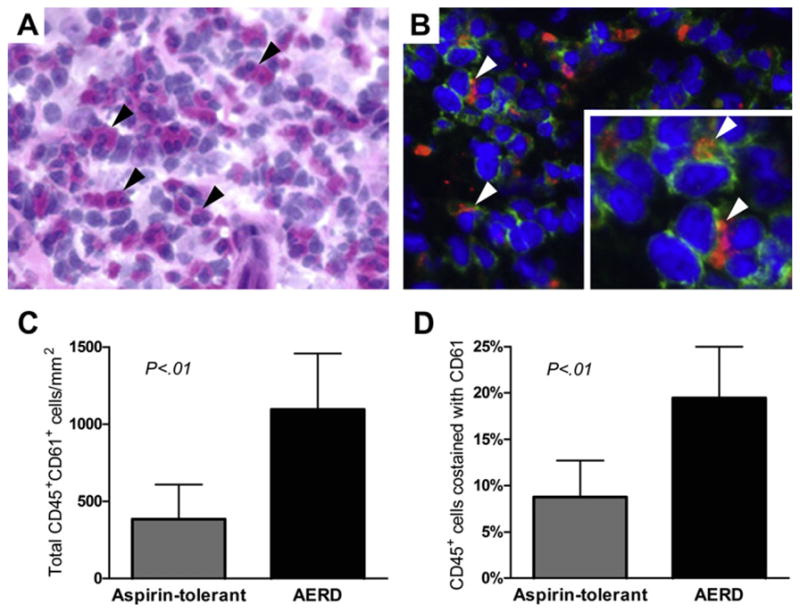
Detection of platelet-leukocyte aggregates in nasal polyp tissue. (A) Hematoxylin-eosin staining of nasal polyp tissue from a patient with AERD shows many eosinophils (black arrowheads) and (B) immunofluorescent staining of the same tissue shows leukocytes (green, CD45+) with adherent platelets (red, CD61+) (white arrowheads). Photographs are shown at 400× magnification. (C) Total numbers of CD45+cells that colocalized with CD61 and (D) percentages of CD45+ cells that colocalized with CD61 in the nasal polyp tissue from aspirin-tolerant controls with sinusitis (n = 4) and patients with AERD (n = 6). Data are expressed as mean + standard deviation. (Data from Laidlaw TM, Kidder MS, Bhattacharyya N, et al. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood 2012;119:3790–98.)
Although the histopathology of AERD supports the importance of effector cells of the allergic response, individuals with AERD frequently lack skin test reactivity to allergens commonly associated with allergic asthma and rhinosinusitis.1,12 On average, total IgE levels tend to be slightly increased in the serum of patients with AERD compared with nonasthmatic controls, but lower than the levels found in the serum of atopic, aspirin-tolerant individuals1,13 Some studies have identified higher levels of specific IgE against staphylococcal enterotoxin B in serum13 and nasal polyps14 from patients with AERD than from aspirin-tolerant controls. It is unclear whether these findings represent a direct causal relationship to staphylococcal colonization, or whether IgE responses to Staphylococcus aureus or its toxins reflect an upstream perturbation of an aberrant immune response to other microbes. Although a microbial pathogenesis has long been suspected in AERD, no consistent microbial isolates (including Staphylococcus) have been reported, nor are there geographic patterns suggestive of outbreaks.
LIPID MEDIATORS IN THE PATHOGENESIS OF AERD
Cysteinyl LTs
A fundamental characteristic of AERD is striking overproduction of cysteinyl LTs (cysLTs), a class of potent lipid inflammatory mediators derived from arachidonic acid.15,16 Mast cells, eosinophils, and platelet-adherent leukocytes, all of which abound in the respiratory tissue of patients with AERD, each have the capacity to synthesize LTC4, the parent cysLT, although it is unclear if the overproduction of cysLTs represents a fundamental abnormality or is a consequence of greater numbers and activation of these inflammatory cells. After liberation of arachidonic acid by cytosolic phospholipase A2, arachidonic acid is oxidized by 5-lipoxygenase (LO) to form the unstable intermediate LTA4, which is then conjugated to reduced glutathione by the terminal enzyme LTC4 synthase (LTC4S) to form LTC4. LTC4 is exported from the cell and enzymatically converted into LTD4 and then into the stable end-metabolite LTE4. Baseline urinary LTE4 levels, a marker of systemic cysLT production, are 3 to 5 times higher in patients with AERD than in their aspirin-tolerant counterparts.15 These levels increase further from the increased baseline (by as much as 10-fold) after challenges with COX-1-active drugs (see later discussion). Baseline levels of LTE4 predict the magnitude of bronchoconstriction occurring during provocative aspirin challenges.17 The short-lived LT mediators, LTC4 and LTD4, and their stable metabolite LTE4 can all induce edema, bronchoconstriction, and airway mucous secretion.18–20 In addition, LTE4, the weakest bronchoconstrictor of the 3 cysLTs, induces marked recruitment of eosinophils to the airways in asthmatic patients.21 Animal models also support a role for cysLTs in driving tissue fibrosis and airway remodeling.22,23 Thus, cysLTs almost certainly contribute to the chronic inflammation and airway remodeling characteristic of AERD. The pathogenetic importance of the cysLTs in AERD is validated by the efficacy of drugs that interfere with their synthesis or block 1 of their receptors,24,25 both of which improve disease control and sinonasal function.
There are several potential mechanistic explanations for the overproduction of cysLTs in AERD. Platelets, which express LTC4S, can convert leukocyte-derived LTA4 to LTC4 when they adhere to 5-LO-expressing leukocytes.26,27 Leukocyte-adherent platelets accounted for most LTC4S activity among peripheral blood granulocytes isolated from the blood of individuals with AERD, and the levels of urinary LTE4 correlated strongly with the frequencies of platelet-adherent eosinophils, neutrophils, and monocytes.9 Immunohistochemical studies indicated that eosinophils in bronchial and sinonasal biopsies from individuals with AERD show selective overexpression of LTC4S protein, but not 5-LO or 5-LO-activating protein.3 Accordingly, peripheral blood eosinophils from individuals with AERD express more mRNA for LTC4S than do eosinophils from aspirin-tolerant controls.28 A common polymorphic variant of the LTC4S allele shows a higher degree of promoter activity than the wild-type allele and was associated with an increased risk of AERD in a Polish population.28 However, this association was not replicated in a cohort from the United States.29 The percentages of mast cells and eosinophils in bronchial biopsies from patients with AERD staining positively for 5-LO were several-fold higher than in biopsies from aspirin-tolerant asthmatic controls.30 Impaired functions of PGE2, a COX pathway product that suppresses the activity of 5-LO (see later discussion), could also contribute to cysLT overproduction in AERD. Thus, overproduction of cysLTs likely relates to factors that both increase the activity of 5-LO and the availability of the substrate LTA4, and increase the availability of the terminal enzyme, LTC4S, including increased granulocyte-adherent platelets and increased LTC4S expression by eosinophils.
CysLT Receptors
In addition to overproduction of cysLTs, individuals with AERD also show enhanced end-organ reactivity to the cysLTs. The functions of cysLTs are mediated by at least 2 G protein-coupled receptors (GPCRs), termed CysLT1R and CysLT2R. The existence of yet-to-be-discovered cysLT receptors (CysLTRs) is likely, based on studies performed in receptor-null mice.31,32 The high-affinity receptor for LTD4, CysLT1R, is overexpressed in nasal tissue inflammatory cells of patients with AERD compared with aspirin-tolerant controls.33 The cause of this increased receptor expression is not known, although there are 3 single nucleotide polymorphisms (SNPs) in the promoter region of CYSLTR1 in which mutant variants have been found to be associated with AERD. The mutant variants showed higher promoter activity, suggesting that these polymorphisms could modulate CysLT1R expression and lead to increased susceptibility to AERD.34 Although CysLT1R has trivial affinity for LTE4, patients with AERD show a selective hyperresponsiveness to bronchoconstriction induced by inhaled LTE4 compared with aspirin-tolerant asthmatic controls,35 suggesting that AERD involves additional upregulation of an unidentified LTE4-specific receptor. Polymorphisms in the CYSLTR2 gene encoding CysLT2R, which negatively regulates the actions of CysLT1R,36,37 are also associated with AERD, and certain alleles were associated with the magnitude in the decrement in FEV1 (forced expiratory volume in first second of expiration) measured during aspirin provocation.38 Collectively, these observations suggest that the end-organ reactivity to cysLTs that characterizes AERD may be partly caused by genetically determined patterns of CysLTR expression or function. Moreover, the selective LTE4 hyperresponsiveness in the face of high systemic levels is consistent with a potentially unique role for this stable end-metabolite of cysLTs in driving the disease.
COX PATHWAY PRODUCTS IN AERD
Proinflammatory PGs
PGs derive from COX-dependent metabolism of arachidonic acid. The COX-derived precursor, PGH2, is converted to 1 of 5 bioactive terminal products (PGE2, PGF2, PGI2, PGD2, and thromboxane A2 [TXA2]) by corresponding isoform-specific synthases (Fig. 2). These terminal PG synthases show cell-selective expression and in some instances preferential coupling to COX-1, the more aspirin-sensitive COX isoform, or to COX-2, which is more resistant to inhibition by aspirin.39 Based on their pharmacologic properties, some PGs are candidate pathogenic effectors in AERD. TXA2, the dominant COX product of platelets, which can also be generated by monocytes, mast cells, and granulocytes, is a potent bronchoconstrictor40 and an inducer of endothelial adhesion molecule expression (intercellular adhesion molecule 1 [ICAM-1] and vascular cell adhesion molecule).41 Metabolites of TXA2 are present in higher levels in bronchoalveolar lavage fluid from patients with AERD compared with aspirin-tolerant asthmatics at baseline,5 potentially reflecting the presence of platelets in the respiratory tissue (see Fig. 1). Furthermore, TXA2 may be involved in the bronchoconstrictive effects of cysLTs, because the increase in airway resistance induced by either LTC4 or LTD4 in guinea pigs is prevented by a selective TXA2 synthase inhibitor.42 Moreover, the potentiation of histamine reactivity of human and guinea pig bronchi by LTE4 could be blocked by an antagonist of the T prostanoid (TP) receptor, the only high-affinity receptor for TXA2, or by pretreatment of the tissues with indo-methacin to block COX function.43 Thus, some of the clinical pharmacology of LTE4 may relate to an incompletely explained capacity for this mediator to use TXA2 or TP receptors as effectors. Although immunohistochemical studies of the TP receptor are lacking because of the lack of specific antibodies, there are 2 SNPs of the TBXA2R that are associated with AERD in a Korean population.44
Fig. 2.

Metabolism of arachidonic acid. Pathways of arachidonic acid metabolism involved in the pathogenesis of AERD. Enzymes are in italics, relevant receptors are in dashed boxes, and consequences of signaling through each receptor are in bulleted lists. Thick gray arrows show whether expression and function of each enzyme or product are increased or decreased in patients with AERD.
PGD2 is the dominant COX pathway product of mast cells.45 Base line levels of PGD2 metabolites are higher in the serum and nasal polyps from patients with AERD than in samples from aspirin-tolerant controls,46 possibly reflecting the ongoing activation of mast cells in AERD. PGD2 is a bronchoconstrictor,47 presumably through the effects of its stable metabolite, 9α,11β-PGF2, acting at TP receptors.48 PGD2, acting through the CRTH2 receptor, is also a potent chemoattractant for eosinophils.49 The persistent production of PGD2 in the inflamed respiratory tissues of patients with AERD may therefore contribute to the ongoing airway dysfunction and eosinophilic inflammation characteristic of the phenotype. Whereas metabolites of TXA2 usually decrease in response to aspirin challenge in AERD,50 PGD2 metabolites in the bronchoalveolar lavage fluid or urine do not (and even increase in some studies),46,51 suggesting that these COX-derived effectors either derive principally from separate cell types (platelets and mast cells, respectively), or are generated through different COX isoforms in AERD. In that regard, it is noteworthy that COX-2 protein is expressed more strongly by mast cells in bronchial biopsies from patients with AERD than by mast cells in biopsies from aspirin-tolerant controls,52 suggesting a potential explanation for aspirin-resistant PGD2 production in this disease.
Antiinflammatory PGs
PGE2 may be the most essential PG for maintaining homeostasis of inflammatory responses in the airway in general and in AERD in particular. Of all COX products, PGE2 is unique for its bronchoprotective and antiinflammatory effects in the airway. PGE2 blocks allergen-induced early-phase and late-phase responses in atopic asthmatics53 and blocks mast cell activation through a pathway dependent on 1 of its 4 GPCRs, termed the E prostanoid 2 (EP2) receptor, and its downstream effectors, adenylate cyclase and cyclic adenosine monophosphate.54,55 PGE2 also uses the same signaling mechanism to induce phosphorylation and prevent translocation of 5-LO to the nuclear envelope56 and can thus control the generation of cysLTs, a function that may be especially relevant to AERD. The importance of this potential mechanism is supported by the fact that inhaled PGE2 prevents both the airway obstruction and the increase in urinary LTE4 occurring in response to aspirin challenge in patients with AERD.57 PGE2 also potently inhibits eosinophil migration,58 and both endogenous and exogenous PGE2 suppress allergen-induced pulmonary eosinophil accumulation in humans53 and rodents.59 PGE2 signals through EP2 receptors to dampen the proliferation of fibroblasts and their production of collagen.60 Collectively, these observations suggest that deficient PGE2 generation or EP2 receptor function on both hematopoietic and structural cells could also be relevant to the pathogenesis of AERD.
PGE2 is generated primarily by COX-2 and a partner terminal synthetic enzyme, microsomal PGE2 synthase 1 (mPGES-1). These 2 enzymes are coexpressed by epithelial cells, fibroblasts, and macrophages and are frequently upregulated in tandem during inflammatory responses.61 There is substantial evidence that PGE2 production and the functions of COX-2/mPGES-1 are impaired in the respiratory tissues in AERD. Although urinary levels of PGE2 metabolites in AERD are similar to those in aspirin-tolerant controls, nasal polyps from patients with AERD contain markedly lower levels of PGE2 than do sinonasal tissues from aspirin-tolerant individuals.62 These levels are inversely related to the levels of cysLTs recovered from the same polyps. Peripheral blood leukocytes isolated from patients with AERD produce less PGE2 than aspirin-tolerant controls,63 and both nasal epithelial cells and cultured fibroblasts from nasal polyps of patients with AERD generate less PGE2 in vitro than do cells from aspirin-tolerant asthmatic controls.64,65 The expression of COX-2 mRNA is diminished in nasal polyps from patients with AERD compared with that in normal nasal mucosa and in polyps from aspirin-tolerant patients,66 and the IL-1β-induced upregulation of COX-2 mRNA and protein expression in cultured fibroblasts from nasal polyps of patients with AERD is markedly blunted compared with their upregulation in cells from aspirin-tolerant controls.65 Although there are no studies reporting the expression of mPGES-1 in AERD, the PTGES gene encoding this enzyme is hypermethylated (a mechanism for gene silencing) in nasal polyps from patients with AERD.67 Collectively, these studies support defective function of the major system responsible for maintaining local PGE2 in inflammation, potentially mediated by epigenetic mechanisms. A predicted outcome of the failure to appropriately upregulate COX-2 and mPGES-1 expression in the respiratory tissue is a dependency on COX-1 to provide the residual PGE2 needed to maintain homeostasis of eosinophilic inflammation, mast cell activation, and 5-LO pathway activity. As noted later, this hypothetical dependency on COX-1 could contribute to a state of aspirin sensitivity.
Alterations in EP2 Receptor Expression and Function in AERD
Given the EP2 receptor-dependent actions of PGE2 noted earlier, it is noteworthy that the percentages of neutrophils, mast cells, eosinophils, and T cells expressing the EP2 receptor are lower in nasal mucosa biopsies from patients with AERD compared with those in biopsies from aspirin-tolerant asthmatic controls.68 Similar findings were recently reported for bronchial biopsies.69 In addition, cultured fibroblasts from the nasal polyps of patients with AERD display decreased induction of the EP2 receptor protein by IL-1β compared with fibroblasts from aspirin-tolerant patients.65 Several SNPs in the promoter region of the EP2 gene have been found to be associated with AERD, and the most significantly associated SNP, uS5, is located in the regulatory region of the EP2 gene in a signal transducer and activator of transcription-binding consensus sequence and it confers reduced transcription activity.70 However, this regulatory SNP was identified in only ~30% of patients with AERD versus ~20% of controls. The expression of EP2 receptors (as well as that of COX-2) can become dysregulated by epigenetic mechanisms in cancer71,72 and in pulmonary fibrosis.73,74 It seems plausible that similar epigenetic mechanisms may promote deficiencies in these same targets in AERD, even in individuals who do not possess SNPs that dysregulate their expression.
Other Arachidonic Acid Metabolites in AERD
Lipoxins (LXs) are unstable antiinflammatory derivatives of arachidonic acid, synthesized through transcellular mechanisms involving cooperation between 5-LO and platelet 12-LO75 or 5-LO and epithelial cell-derived 15-LO.76 In the presence of aspirin, COX-2 can function as a 15-LO and convert leukocyte-derived LTA4 to 15-epi-LXA4, also known as aspirin-triggered LX.77 LXs inhibit cysLT production and eosinophilic infiltration of pulmonary tissue in rodent models of pulmonary inflammation,78 and in vitro studies suggest that they are high-affinity antagonists of CysLT1R.79 Calcium ionophore stimulation of peripheral blood leukocytes from patients with AERD results in the release of less LXA4 than does stimulation of leukocytes from the blood of aspirin-tolerant asthmatic control individuals.80 Given the predicted antiinflammatory function of LXA4, a deficient capacity to generate this mediator could contribute to the dysregulated inflammatory responses and cysLT-driven pathobiology of AERD, although this has not been validated in vivo.
CROSS-TALK BETWEEN ARACHIDONIC ACID PATHWAYS IN THE PATHOBIOLOGY OF AERD AND GENESIS OF ASPIRIN-INDUCED REACTIONS
Although the events that initiate the development of AERD are unknown, the complex set of disturbances in the homeostasis of inflammation and lipid mediator production that characterize the basal state of the disease may hinge on a deficient synthesis of PGE2 or on a diminished ability for it to signal through EP2 receptors (Fig. 3). First, lowered levels of PGE2 in the tissues combined with diminished expression of EP2 receptors on the respiratory tissue granulocytes would allow for the dramatically elevated levels of cysLTs found in patients with AERD: it would permit enhanced LTC4 synthesis by eosinophils and mast cells and would increase the neutrophil-derived LTA4 available for conversion to LTC4 by adherent platelets. Second, deficiencies in available PGE2 and EP2 receptor function would promote ongoing activation of mast cells and would remove a check on the chemotaxis of eosinophils. Third, EP2 receptor signaling maintains TP receptors in a relative state of phosphorylation/desensitization via protein kinase A, and a recent study59 revealed that EP2 receptor signaling and mPGES-1-derived PGE2 were essential to prevent exaggerated TP receptor-driven upregulation of endothelial cell ICAM-1 and consequent allergen-induced pulmonary eosinophilia and airway hyperreactivity. Last, deficient EP2 receptor function on fibroblasts would impair the capacity of local PGE2 to maintain a check on proliferation and collagen production. Thus, baseline impairment of local PGE2 generation and EP2 receptor signaling would likely promote persistent inflammation and ongoing tissue remodeling by several complementary mechanisms in AERD.
Fig. 3.

Summary of the pathogenesis of AERD at baseline and after COX-1 inhibition. Normal mechanisms for the maintenance of homeostasis at baseline and after COX-1 inhibition in aspirin-tolerant control patients are presented on the left. The consequences of abnormalities that lead to both the underlying respiratory disease and the development of reactions on ingestion of COX-1 inhibitors in patients with AERD are presented on the right.
The defining respiratory reactions of AERD occur exclusively with drugs that inhibit COX isoforms nonselectively. At the low doses required to cause reactions in AERD, aspirin selectively inhibits the COX-1 isoenzyme81 and it is inhibition of COX-1, but not of COX-2, which precipitates reactions.82–85 All COX-1 inhibitors induce stereotypically similar respiratory reactions in patients with AERD, indicating that the ability of these structurally diverse drugs to inhibit COX-1 per se is via their pharmacologic actions. Although the basal state of AERD does not require exposure to COX inhibitors, the remarkable ability for low doses of aspirin (or any COX-1-active NSAID) to induce reactions implies that the aspirin sensitivity of AERD is caused by a tenuous dependency on COX-1 products to maintain homeostasis. Based on several lines of evidence, COX-1-derived PGE2 is likely relevant to the pathophysiology of reactions to COX inhibitors. Although challenges with aspirin do not deplete PGE2 metabolites in the urine of patients with AERD,86 this measurement may be more reflective of ongoing renal tubular generation of PGE2 than of that from the respiratory tract. The markedly reduced expression of COX-2 by nasal polyp tissues from patients with AERD implies that the small, residual amounts of local PGE2 are generated primarily through COX-1. Indeed, local concentrations of PGE2 do decrease in nasal lavage fluid after oral aspirin challenges in patients with AERD.87 It seems likely that the additional low expression of EP2 receptors would magnify the physiologic effects of small changes in local PGE2 levels. In this scenario, depletion of residual PGE2 from the respiratory tract would permit increased activity of 5-LO and a surge of cysLT production and mast cell activation (as shown, respectively, by the characteristic increase in urinary LTE4 and increases in both serum tryptase and PGD2 metabolites occurring after challenge). The observation that inhalation of PGE2 can block both airway obstruction and the increase in urinary LTE4 occurring in response to aspirin challenge further supports a key causative function for depletion of PGE2 in the pathogenesis of reactions to COX-1 inhibitors.57
Although COX-1-derived PGE2 may be the major PG responsible for controlling the activity of 5-LO, other COX-1-derived PGs may also play a role in regulating cysLT production during reactions in AERD. TXA2 is largely COX-1-dependent, and urinary levels of its stable metabolite (11-dihidro TXB2) decrease during reactions to aspirin in patients with AERD.5 As noted earlier, TXA2 may mediate certain LTE4-dependent physiologic events, and this property may account for the rapid loss of selective LTE4 hyperresponsiveness observed after desensitization to aspirin.88 However, TXA2 in platelets also suppresses the activity of LTC4S, presumably through an auto-crine, TP receptor-dependent pathway that maintains phosphorylation of LTC4S by protein kinase C.89 Treatment of platelets ex vivo with aspirin rapidly removes this suppression and potentiates the activity of LTC4S. It is thus possible that simultaneous depletion of PGE2/EP2 signaling on leukocytes and TXA2/TP signaling on platelets by aspirin could further dysregulate cysLT generation, especially from platelet-leukocyte complexes.
Given the ability of the 5-LO inhibitor zileuton and antagonists of CysLT1R to blunt the clinical and physiologic features of reactions to COX-1 inhibitors, 5-LO pathway products in general and cysLTs in particular are unequivocal effectors of reactions. Nevertheless, neither the cellular sources of these mediators during reactions nor the targets of their effects in reactions are completely understood. Both mast cells and eosinophils possess the complete repertoire of enzymes needed to generate cysLTs from endogenous arachidonic acid. As noted earlier, soluble markers of both eosinophil and mast cell activation increase in the lavage fluids, serum, or urine of patients with AERD with aspirin or lysine aspirin challenges. Thus, both cell types are likely to contribute to the surge in cysLTs that typify reactions. Inhalation of the mast cell-stabilizing drugs cromolyn or nedocromyl block aspirin-induced bronchoconstriction, further supporting the role of mast cell activation (and likely mast cell-derived cysLTs) in the reactions to aspirin.90,91 Receptors for cysLTs are broadly expressed by both structural and hematopoietic cells, and immunohistochemical studies indicate that the expression of CysLT1R by respiratory tract eosinophils and mast cells is upregulated in AERD.33 The administration of zileuton to subjects with AERD not only dramatically reduces the nasal symptoms of reactions to lysine aspirin challenge, but markedly reduces the concentrations of mast cell tryptase recovered from the nasal lavage fluid compared with the levels found in fluids from placebo-treated challenged controls.24 It is thus likely that cysLTs induce a diverse array of physiologic effects during reactions beyond CysLT1R-dependent bronchoconstriction. The fact that LTE4, the most abundant mediator detected during reactions, acts by receptors other than CysLT1R and CysLT2R makes this hypothesis a virtual certainty Fig. 2.
SUMMARY
Physiologic and pharmacologic studies in humans strongly support the hypothesis that AERD involves fundamental disturbances in both the production of and endorgan responsiveness to eicosanoids that are regulators of homeostasis (PGE2) and those that are effectors (cysLTs). The likelihood that these disturbances are acquired in adulthood implies potential epigenetic modifications of the relevant mediator systems as a result of only partially clarified environmental factors (eg, potentially viral or bacterial infections, inhaled pollutants). The rapid development of technology permitting analyses of the epigenetic landscape and the microbiome may permit finer detailing of the evasive causal agents and lesions. Meanwhile, the putative pathobiological mechanisms of AERD introduce substantial opportunities for targeted therapeutic intervention. The advent of anti-IL-5,92 tyrosine kinase inhibitors,93 and antiplatelet drugs94 for other indications may permit novel therapeutic approaches to AERD by targeting some of the key cellular effectors (eosinophils, mast cells, platelets). Existing or new drugs that act by targeting of candidate mediators and receptors, such as PGD2/DP295 and TXA2/TP,96 should permit finer detail of their specific roles in disease pathogenesis, as well as providing additional potential efficacy of treatment. The anticipated identification of LTE4-selective receptors and the development of EP2 receptor-selective agonists have substantial therapeutic potential in AERD.
KEY POINTS.
Pathogenesis of aspirin-exacerbated respiratory disease (AERD) involves a consistent pattern of aberrant leukotriene generation.
AERD is associated with deregulated expression and function of enzymes and receptors responsible for the production and function of lipid mediators that are protective against reactions to cyclooxygenase inhibitors.
AERD is acquired rather than hereditary.
References
- 1.Szczeklik A, Nizankowska E, Duplaga M. Natural history of aspirin-induced asthma. AIANE Investigators. European Network on Aspirin-Induced Asthma. Eur Respir J. 2000;16:432. doi: 10.1034/j.1399-3003.2000.016003432.x. [DOI] [PubMed] [Google Scholar]
- 2.Adamjee J, Suh YJ, Park HS, et al. Expression of 5-lipoxygenase and cyclooxygenase pathway enzymes in nasal polyps of patients with aspirin-intolerant asthma. J Pathol. 2006;209:392. doi: 10.1002/path.1979. [DOI] [PubMed] [Google Scholar]
- 3.Cowburn AS, Sladek K, Soja J, et al. Overexpression of leukotriene C4 synthase in bronchial biopsies from patients with aspirin-intolerant asthma. J Clin Invest. 1998;101:834. doi: 10.1172/JCI620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yamashita T, Tsuji H, Maeda N, et al. Etiology of nasal polyps associated with aspirin-sensitive asthma. Rhinol Suppl. 1989;8:15. [PubMed] [Google Scholar]
- 5.Sladek K, Dworski R, Soja J, et al. Eicosanoids in bronchoalveolar lavage fluid of aspirin-intolerant patients with asthma after aspirin challenge. Am J Respir Crit Care Med. 1994;149:940. doi: 10.1164/ajrccm.149.4.8143059. [DOI] [PubMed] [Google Scholar]
- 6.Hosemann W. Surgical treatment of nasal polyposis in patients with aspirin intolerance. Thorax. 2000;55(Suppl 2):S87. doi: 10.1136/thorax.55.suppl_2.S87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kakoi H, Hiraide F. A histological study of formation and growth of nasal polyps. Acta Otolaryngol. 1987;103:137. doi: 10.3109/00016488709134709. [DOI] [PubMed] [Google Scholar]
- 8.Makowska JS, Grzegorczyk J, Bienkiewicz B, et al. Systemic responses after bronchial aspirin challenge in sensitive patients with asthma. J Allergy Clin Immunol. 2008;121:348. doi: 10.1016/j.jaci.2007.09.039. [DOI] [PubMed] [Google Scholar]
- 9.Laidlaw TM, Kidder MS, Bhattacharyya N, et al. Cysteinyl leukotriene overproduction in aspirin-exacerbated respiratory disease is driven by platelet-adherent leukocytes. Blood. 2012;119:3790. doi: 10.1182/blood-2011-10-384826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamilos DL, Leung DY, Wood R, et al. Evidence for distinct cytokine expression in allergic versus nonallergic chronic sinusitis. J Allergy Clin Immunol. 1995;96:537. doi: 10.1016/s0091-6749(95)70298-9. [DOI] [PubMed] [Google Scholar]
- 11.Sousa AR, Lams BE, Pfister R, et al. Expression of interleukin-5 and granulocyte-macrophage colony-stimulating factor in aspirin-sensitive and non-aspirin-sensitive asthmatic airways. Am J Respir Crit Care Med. 1997;156:1384. doi: 10.1164/ajrccm.156.5.9702072. [DOI] [PubMed] [Google Scholar]
- 12.Pearce N, Pekkanen J, Beasley R. How much asthma is really attributable to atopy? Thorax. 1999;54:268. doi: 10.1136/thx.54.3.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee JY, Kim HM, Ye YM, et al. Role of staphylococcal superantigen-specific IgE antibodies in aspirin-intolerant asthma. Allergy Asthma Proc. 2006;27:341. doi: 10.2500/aap.2006.27.2908. [DOI] [PubMed] [Google Scholar]
- 14.Suh YJ, Yoon SH, Sampson AP, et al. Specific immunoglobulin E for staphylococcal enterotoxins in nasal polyps from patients with aspirin-intolerant asthma. Clin Exp Allergy. 2004;34:1270. doi: 10.1111/j.1365-2222.2004.02051.x. [DOI] [PubMed] [Google Scholar]
- 15.Christie PE, Tagari P, Ford-Hutchinson AW, et al. Urinary leukotriene E4 concentrations increase after aspirin challenge in aspirin-sensitive asthmatic subjects. Am Rev Respir Dis. 1991;143:1025. doi: 10.1164/ajrccm/143.5_Pt_1.1025. [DOI] [PubMed] [Google Scholar]
- 16.Smith CM, Hawksworth RJ, Thien FC, et al. Urinary leukotriene E4 in bronchial asthma. Eur Respir J. 1992;5:693. [PubMed] [Google Scholar]
- 17.Daffern PJ, Muilenburg D, Hugli TE, et al. Association of urinary leukotriene E4 excretion during aspirin challenges with severity of respiratory responses. J Allergy Clin Immunol. 1999;104:559. doi: 10.1016/s0091-6749(99)70324-6. [DOI] [PubMed] [Google Scholar]
- 18.Dahlen SE, Hedqvist P, Hammarstrom S, et al. Leukotrienes are potent constrictors of human bronchi. Nature. 1980;288:484. doi: 10.1038/288484a0. [DOI] [PubMed] [Google Scholar]
- 19.Dahlen SE, Hansson G, Hedqvist P, et al. Allergen challenge of lung tissue from asthmatics elicits bronchial contraction that correlates with the release of leukotrienes C4, D4, and E4. Proc Natl Acad Sci U S A. 1983;80:1712. doi: 10.1073/pnas.80.6.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahlen SE, Bjork J, Hedqvist P, et al. Leukotrienes promote plasma leakage and leukocyte adhesion in postcapillary venules: in vivo effects with relevance to the acute inflammatory response. Proc Natl Acad Sci U S A. 1981;78:3887. doi: 10.1073/pnas.78.6.3887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gauvreau GM, Parameswaran KN, Watson RM, et al. Inhaled leukotriene E(4), but not leukotriene D(4), increased airway inflammatory cells in subjects with atopic asthma. Am J Respir Crit Care Med. 2001;164:1495. doi: 10.1164/ajrccm.164.8.2102033. [DOI] [PubMed] [Google Scholar]
- 22.Beller TC, Maekawa A, Friend DS, et al. Targeted gene disruption reveals the role of the cysteinyl leukotriene 2 receptor in increased vascular permeability and in bleomycin-induced pulmonary fibrosis in mice. J Biol Chem. 2004;279:46129. doi: 10.1074/jbc.M407057200. [DOI] [PubMed] [Google Scholar]
- 23.Henderson WR, Jr, Tang LO, Chu SJ, et al. A role for cysteinyl leukotrienes in airway remodeling in a mouse asthma model. Am J Respir Crit Care Med. 2002;165:108. doi: 10.1164/ajrccm.165.1.2105051. [DOI] [PubMed] [Google Scholar]
- 24.Fischer AR, Rosenberg MA, Lilly CM, et al. Direct evidence for a role of the mast cell in the nasal response to aspirin in aspirin-sensitive asthma. J Allergy Clin Immunol. 1994;94:1046. doi: 10.1016/0091-6749(94)90123-6. [DOI] [PubMed] [Google Scholar]
- 25.Dahlen SE, Malmstrom K, Nizankowska E, et al. Improvement of aspirin-intolerant asthma by montelukast, a leukotriene antagonist: a randomized, double-blind, placebo-controlled trial. Am J Respir Crit Care Med. 2002;165:9. doi: 10.1164/ajrccm.165.1.2010080. [DOI] [PubMed] [Google Scholar]
- 26.Maclouf JA, Murphy RC. Transcellular metabolism of neutrophil-derived leukotriene A4 by human platelets. A potential cellular source of leukotriene C4. J Biol Chem. 1988;263:174. [PubMed] [Google Scholar]
- 27.Maugeri N, Evangelista V, Celardo A, et al. Polymorphonuclear leukocyte-platelet interaction: role of P-selectin in thromboxane B2 and leukotriene C4 cooperative synthesis. Thromb Haemost. 1994;72:450. [PubMed] [Google Scholar]
- 28.Sanak M, Pierzchalska M, Bazan-Socha S, et al. Enhanced expression of the leukotriene C(4) synthase due to overactive transcription of an allelic variant associated with aspirin-intolerant asthma. Am J Respir Cell Mol Biol. 2000;23:290. doi: 10.1165/ajrcmb.23.3.4051. [DOI] [PubMed] [Google Scholar]
- 29.Van Sambeek R, Stevenson DD, Baldasaro M, et al. 5′ flanking region polymorphism of the gene encoding leukotriene C4 synthase does not correlate with the aspirin-intolerant asthma phenotype in the United States. J Allergy Clin Immunol. 2000;106:72. doi: 10.1067/mai.2000.107603. [DOI] [PubMed] [Google Scholar]
- 30.Nasser SM, Pfister R, Christie PE, et al. Inflammatory cell populations in bronchial biopsies from aspirin-sensitive asthmatic subjects. Am J Respir Crit Care Med. 1996;153:90. doi: 10.1164/ajrccm.153.1.8542168. [DOI] [PubMed] [Google Scholar]
- 31.Maekawa A, Kanaoka Y, Xing W, et al. Functional recognition of a distinct receptor preferential for leukotriene E4 in mice lacking the cysteinyl leukotriene 1 and 2 receptors. Proc Natl Acad Sci U S A. 2008;105:16695. doi: 10.1073/pnas.0808993105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Paruchuri S, Tashimo H, Feng C, et al. Leukotriene E4-induced pulmonary inflammation is mediated by the P2Y12 receptor. J Exp Med. 2009;206:2543. doi: 10.1084/jem.20091240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sousa AR, Parikh A, Scadding G, et al. Leukotriene-receptor expression on nasal mucosal inflammatory cells in aspirin-sensitive rhinosinusitis. N Engl J Med. 2002;347:1493. doi: 10.1056/NEJMoa013508. [DOI] [PubMed] [Google Scholar]
- 34.Kim SH, Oh JM, Kim YS, et al. Cysteinyl leukotriene receptor 1 promoter polymorphism is associated with aspirin-intolerant asthma in males. Clin Exp Allergy. 2006;36:433. doi: 10.1111/j.1365-2222.2006.02457.x. [DOI] [PubMed] [Google Scholar]
- 35.Christie PE, Schmitz-Schumann M, Spur BW, et al. Airway responsiveness to leukotriene C4 (LTC4), leukotriene E4 (LTE4) and histamine in aspirin-sensitive asthmatic subjects. Eur Respir J. 1993;6:1468. [PubMed] [Google Scholar]
- 36.Jiang Y, Borrelli LA, Kanaoka Y, et al. CysLT2 receptors interact with CysLT1 receptors and down-modulate cysteinyl leukotriene dependent mitogenic responses of mast cells. Blood. 2007;110:3263. doi: 10.1182/blood-2007-07-100453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barrett NA, Fernandez JM, Maekawa A, et al. Cysteinyl leukotriene 2 receptor on dendritic cells negatively regulates ligand-dependent allergic pulmonary inflammation. J Immunol. 2012;189:4556. doi: 10.4049/jimmunol.1201865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park JS, Chang HS, Park CS, et al. Association analysis of cysteinyl-leukotriene receptor 2 (CYSLTR2) polymorphisms with aspirin intolerance in asthmatics. Pharmacogenet Genomics. 2005;15:483. doi: 10.1097/01.fpc.0000166456.84905.a0. [DOI] [PubMed] [Google Scholar]
- 39.DeWitt DL, el-Harith EA, Kraemer SA, et al. The aspirinandheme-binding sites of ovine and murine prostaglandin endoperoxide synthases. J Biol Chem. 1990;265:5192. [PubMed] [Google Scholar]
- 40.Bureau MF, De Clerck F, Lefort J, et al. Thromboxane A2 accounts for broncho-constriction but not for platelet sequestration and microvascular albumin exchanges induced by fMLP in the guinea pig lung. J Pharmacol Exp Ther. 1992;260:832. [PubMed] [Google Scholar]
- 41.Ishizuka T, Kawakami M, Hidaka T, et al. Stimulation with thromboxane A2 (TXA2) receptor agonist enhances ICAM-1, VCAM-1 or ELAM-1 expression by human vascular endothelial cells. Clin Exp Immunol. 1998;112:464. doi: 10.1046/j.1365-2249.1998.00614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ueno A, Tanaka K, Katori M. Possible involvement of thromboxane in bronchoconstrictive and hypertensive effects of LTC4 and LTD4 in guinea pigs. Prostaglandins. 1982;23:865. doi: 10.1016/0090-6980(82)90130-7. [DOI] [PubMed] [Google Scholar]
- 43.Jacques CA, Spur BW, Johnson M, et al. The mechanism of LTE4-induced histamine hyperresponsiveness in guinea-pig tracheal and human bronchial smooth muscle, in vitro. Br J Pharmacol. 1991;104:859. doi: 10.1111/j.1476-5381.1991.tb12518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kim SH, Choi JH, Park HS, et al. Association of thromboxane A2 receptor gene polymorphism with the phenotype of acetyl salicylic acid-intolerant asthma. Clin Exp Allergy. 2005;35:585. doi: 10.1111/j.1365-2222.2005.02220.x. [DOI] [PubMed] [Google Scholar]
- 45.Mita H, Endoh S, Kudoh M, et al. Possible involvement of mast-cell activation in aspirin provocation of aspirin-induced asthma. Allergy. 2001;56:1061. doi: 10.1111/j.1398-9995.2001.00913.x. [DOI] [PubMed] [Google Scholar]
- 46.Bochenek G, Nagraba K, Nizankowska E, et al. A controlled study of 9alpha,11beta-PGF2 (a prostaglandin D2 metabolite) in plasma and urine of patients with bronchial asthma and healthy controls after aspirin challenge. J Allergy Clin Immunol. 2003;111:743. doi: 10.1067/mai.2003.1387. [DOI] [PubMed] [Google Scholar]
- 47.Johnston SL, Freezer NJ, Ritter W, et al. Prostaglandin D2-induced bronchoconstriction is mediated only in part by the thromboxane prostanoid receptor. Eur Respir J. 1995;8:411. doi: 10.1183/09031936.95.08030411. [DOI] [PubMed] [Google Scholar]
- 48.Larsson AK, Hagfjard A, Dahlen SE, et al. Prostaglandin D(2) induces contractions through activation of TP receptors in peripheral lung tissue from the guinea pig. Eur J Pharmacol. 2011;669:136. doi: 10.1016/j.ejphar.2011.07.046. [DOI] [PubMed] [Google Scholar]
- 49.Hirai H, Tanaka K, Yoshie O, et al. Prostaglandin D2 selectively induces chemotaxis in T helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J Exp Med. 2001;193:255. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sladek K, Szczeklik A. Cysteinyl leukotrienes overproduction and mast cell activation in aspirin-provoked bronchospasm in asthma. Eur Respir J. 1993;6:391. [PubMed] [Google Scholar]
- 51.Szczeklik A, Sladek K, Dworski R, et al. Bronchial aspirin challenge causes specific eicosanoid response in aspirin-sensitive asthmatics. Am J Respir Crit Care Med. 1996;154:1608. doi: 10.1164/ajrccm.154.6.8970343. [DOI] [PubMed] [Google Scholar]
- 52.Sousa A, Pfister R, Christie PE, et al. Enhanced expression of cyclooxygenase isoenzyme 2 (COX-2) in asthmatic airways and its cellular distribution in aspirin-sensitive asthma. Thorax. 1997;52:940. doi: 10.1136/thx.52.11.940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gauvreau GM, Watson RM, O’Byrne PM. Protective effects of inhaled PGE2 on allergen-induced airway responses and airway inflammation. Am J Respir Crit Care Med. 1999;159:31. doi: 10.1164/ajrccm.159.1.9804030. [DOI] [PubMed] [Google Scholar]
- 54.Peachell PT, MacGlashan DW, Jr, Lichtenstein LM, et al. Regulation of human basophil and lung mast cell function by cyclic adenosine monophosphate. J Immunol. 1988;140:571. [PubMed] [Google Scholar]
- 55.Feng C, Beller EM, Bagga S, et al. Human mast cells express multiple EP receptors for prostaglandin E2 that differentially modulate activation responses. Blood. 2006;107:3243. doi: 10.1182/blood-2005-07-2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Luo M, Jones SM, Flamand N, et al. Phosphorylation by protein kinase a inhibits nuclear import of 5-lipoxygenase. J Biol Chem. 2005;280:40609. doi: 10.1074/jbc.M507045200. [DOI] [PubMed] [Google Scholar]
- 57.Sestini P, Armetti L, Gambaro G, et al. Inhaled PGE2 prevents aspirin-induced bronchoconstriction and urinary LTE4 excretion in aspirin-sensitive asthma. Am J Respir Crit Care Med. 1996;153:572. doi: 10.1164/ajrccm.153.2.8564100. [DOI] [PubMed] [Google Scholar]
- 58.Sturm EM, Schratl P, Schuligoi R, et al. Prostaglandin E2 inhibits eosinophil trafficking through E-prostanoid 2 receptors. J Immunol. 2008;181:7273. doi: 10.4049/jimmunol.181.10.7273. [DOI] [PubMed] [Google Scholar]
- 59.Liu T, Laidlaw TM, Feng C, et al. Prostaglandin E2 deficiency uncovers a dominant role for thromboxane A2 in house dust mite-induced allergic pulmonary inflammation. Proc Natl Acad Sci U S A. 2012;109:12692. doi: 10.1073/pnas.1207816109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Huang SK, Wettlaufer SH, Hogaboam CM, et al. Variable prostaglandin E2 resistance in fibroblasts from patients with usual interstitial pneumonia. Am J Respir Crit Care Med. 2008;177:66. doi: 10.1164/rccm.200706-963OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Uematsu S, Matsumoto M, Takeda K, et al. Lipopolysaccharide-dependent prostaglandin E(2) production is regulated by the glutathione-dependent prostaglandin E(2) synthase gene induced by the Toll-like receptor 4/MyD88/NF-IL6 pathway. J Immunol. 2002;168:5811. doi: 10.4049/jimmunol.168.11.5811. [DOI] [PubMed] [Google Scholar]
- 62.Yoshimura T, Yoshikawa M, Otori N, et al. Correlation between the prostaglandin D(2)/E(2) ratio in nasal polyps and the recalcitrant pathophysiology of chronic rhinosinusitis associated with bronchial asthma. Allergol Int. 2008;57:429. doi: 10.2332/allergolint.o-08-545. [DOI] [PubMed] [Google Scholar]
- 63.Schafer D, Schmid M, Gode UC, et al. Dynamics of eicosanoids in peripheral blood cells during bronchial provocation in aspirin-intolerant asthmatics. Eur Respir J. 1999;13:638. doi: 10.1183/09031936.99.13363899. [DOI] [PubMed] [Google Scholar]
- 64.Kowalski ML, Pawliczak R, Wozniak J, et al. Differential metabolism of arachidonic acid in nasal polyp epithelial cells cultured from aspirin-sensitive and aspirin-tolerant patients. Am J Respir Crit Care Med. 2000;161:391. doi: 10.1164/ajrccm.161.2.9902034. [DOI] [PubMed] [Google Scholar]
- 65.Roca-Ferrer J, Garcia-Garcia FJ, Pereda J, et al. Reduced expression of COXs and production of prostaglandin E(2) in patients with nasal polyps with or without aspirin-intolerant asthma. J Allergy Clin Immunol. 2011;128:66. doi: 10.1016/j.jaci.2011.01.065. [DOI] [PubMed] [Google Scholar]
- 66.Picado C, Fernandez-Morata JC, Juan M, et al. Cyclooxygenase-2 mRNA is downexpressed in nasal polyps from aspirin-sensitive asthmatics. Am J Respir Crit Care Med. 1999;160:291. doi: 10.1164/ajrccm.160.1.9808048. [DOI] [PubMed] [Google Scholar]
- 67.Cheong HS, Park SM, Kim MO, et al. Genome-wide methylation profile of nasal polyps: relation to aspirin hypersensitivity in asthmatics. Allergy. 2011;66:637. doi: 10.1111/j.1398-9995.2010.02514.x. [DOI] [PubMed] [Google Scholar]
- 68.Ying S, Meng Q, Scadding G, et al. Aspirin-sensitive rhinosinusitis is associated with reduced E-prostanoid 2 receptor expression on nasal mucosal inflammatory cells. J Allergy Clin Immunol. 2006;117:312. doi: 10.1016/j.jaci.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 69.Corrigan CJ, Napoli RL, Meng Q, et al. Reduced expression of the prostaglandin E2 receptor E-prostanoid 2 on bronchial mucosal leukocytes in patients with aspirin-sensitive asthma. J Allergy Clin Immunol. 2012;129:1636. doi: 10.1016/j.jaci.2012.02.007. [DOI] [PubMed] [Google Scholar]
- 70.Jinnai N, Sakagami T, Sekigawa T, et al. Polymorphisms in the prostaglandin E2 receptor subtype 2 gene confer susceptibility to aspirin-intolerant asthma: a candidate gene approach. Hum Mol Genet. 2004;13:3203. doi: 10.1093/hmg/ddh332. [DOI] [PubMed] [Google Scholar]
- 71.Gray SG, Al-Sarraf N, Baird AM, et al. Regulation of EP receptors in non-small cell lung cancer by epigenetic modifications. Eur J Cancer. 2009;45:3087. doi: 10.1016/j.ejca.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 72.de Maat MF, van de Velde CJ, Umetani N, et al. Epigenetic silencing of cyclooxygenase-2 affects clinical outcome in gastric cancer. J Clin Oncol. 2007;25:4887. doi: 10.1200/JCO.2006.09.8921. [DOI] [PubMed] [Google Scholar]
- 73.Huang SK, Fisher AS, Scruggs AM, et al. Hypermethylation of PTGER2 confers prostaglandin E2 resistance in fibrotic fibroblasts from humans and mice. Am J Pathol. 2010;177:2245. doi: 10.2353/ajpath.2010.100446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Coward WR, Watts K, Feghali-Bostwick CA, et al. Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol Cell Biol. 2009;29:4325. doi: 10.1128/MCB.01776-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Serhan CN, Sheppard KA. Lipoxin formation during human neutrophil-platelet interactions. Evidence for the transformation of leukotriene A4 by platelet 12-lipoxygenase in vitro. J Clin Invest. 1990;85:772. doi: 10.1172/JCI114503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Edenius C, Kumlin M, Bjork T, et al. Lipoxin formation in human nasal polyps and bronchial tissue. FEBS Lett. 1990;272:25. doi: 10.1016/0014-5793(90)80440-t. [DOI] [PubMed] [Google Scholar]
- 77.Takano T, Fiore S, Maddox JF, et al. Aspirin-triggered 15-epilipoxin A4 (LXA4) and LXA4 stable analogues are potent inhibitors of acute inflammation: evidence for anti-inflammatory receptors. J Exp Med. 1997;185:1693. doi: 10.1084/jem.185.9.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Levy BD, De Sanctis GT, Devchand PR, et al. Multi-pronged inhibition of airway hyper-responsiveness and inflammation by lipoxin A(4) Nat Med. 2002;8:1018. doi: 10.1038/nm748. [DOI] [PubMed] [Google Scholar]
- 79.Gronert K, Martinsson-Niskanen T, Ravasi S, et al. Selectivity of recombinant human leukotriene D(4), leukotriene B(4), and lipoxin A(4) receptors with aspirin-triggered 15-epi-LXA(4) and regulation of vascular and inflammatory responses. Am J Pathol. 2001;158:3. doi: 10.1016/S0002-9440(10)63937-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sanak M, Levy BD, Clish CB, et al. Aspirin-tolerant asthmatics generate more lipoxins than aspirin-intolerant asthmatics. Eur Respir J. 2000;16:44. doi: 10.1034/j.1399-3003.2000.16a08.x. [DOI] [PubMed] [Google Scholar]
- 81.Meade EA, Smith WL, DeWitt DL. Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isozymes by aspirin and other non-steroidal anti-inflammatory drugs. J Biol Chem. 1993;268:6610. [PubMed] [Google Scholar]
- 82.Yoshida S, Ishizaki Y, Onuma K, et al. Selective cyclooxygenase 2 inhibitor in patients with aspirin-induced asthma. J Allergy Clin Immunol. 2000;106:1201. doi: 10.1067/mai.2000.110926. [DOI] [PubMed] [Google Scholar]
- 83.Szczeklik A, Nizankowska E, Bochenek G, et al. Safety of a specific COX-2 inhibitor in aspirin-induced asthma. Clin Exp Allergy. 2001;31:219. doi: 10.1046/j.1365-2222.2001.01075.x. [DOI] [PubMed] [Google Scholar]
- 84.Dahlen B, Szczeklik A, Murray JJ. Celecoxib in patients with asthma and aspirin intolerance. The Celecoxib in Aspirin-Intolerant Asthma Study Group. N Engl J Med. 2001;344:142. doi: 10.1056/NEJM200101113440215. [DOI] [PubMed] [Google Scholar]
- 85.Stevenson DD, Simon RA. Lack of cross-reactivity between rofecoxib and aspirin in aspirin-sensitive patients with asthma. J Allergy Clin Immunol. 2001;108:47. doi: 10.1067/mai.2001.116290. [DOI] [PubMed] [Google Scholar]
- 86.Mastalerz L, Sanak M, Gawlewicz-Mroczka A, et al. Prostaglandin E2 systemic production in patients with asthma with and without aspirin hypersensitivity. Thorax. 2008;63:27. doi: 10.1136/thx.2007.080903. [DOI] [PubMed] [Google Scholar]
- 87.Ferreri NR, Howland WC, Stevenson DD, et al. Release of leukotrienes, prostaglandins, and histamine into nasal secretions of aspirin-sensitive asthmatics during reaction to aspirin. Am Rev Respir Dis. 1988;137:847. doi: 10.1164/ajrccm/137.4.847. [DOI] [PubMed] [Google Scholar]
- 88.Arm JP, O’Hickey SP, Spur BW, et al. Airway responsiveness to histamine and leukotriene E4 in subjects with aspirin-induced asthma. Am Rev Respir Dis. 1989;140:148. doi: 10.1164/ajrccm/140.1.148. [DOI] [PubMed] [Google Scholar]
- 89.Tornhamre S, Ehnhage A, Kolbeck KG, et al. Uncoupled regulation of leukotriene C4 synthase in platelets from aspirin-intolerant asthmatics and healthy volunteers after aspirin treatment. Clin Exp Allergy. 2002;32:1566. doi: 10.1046/j.1365-2222.2002.01531.x. [DOI] [PubMed] [Google Scholar]
- 90.Yoshida S, Amayasu H, Sakamoto H, et al. Cromolyn sodium prevents bronchoconstriction and urinary LTE4 excretion in aspirin-induced asthma. Ann Allergy Asthma Immunol. 1998;80:171. doi: 10.1016/S1081-1206(10)62951-1. [DOI] [PubMed] [Google Scholar]
- 91.Robuschi M, Gambaro G, Sestini P, et al. Attenuation of aspirin-induced bronchoconstriction by sodium cromoglycate and nedocromil sodium. Am J Respir Crit Care Med. 1997;155:1461. doi: 10.1164/ajrccm.155.4.9105094. [DOI] [PubMed] [Google Scholar]
- 92.O’Byrne PM. The demise of anti IL-5 for asthma, or not. Am J Respir Crit Care Med. 2007;176:1059. doi: 10.1164/rccm.200708-1264ED. [DOI] [PubMed] [Google Scholar]
- 93.Pardanani A, Elliott M, Reeder T, et al. Imatinib for systemic mast-cell disease. Lancet. 2003;362:535. doi: 10.1016/s0140-6736(03)14115-3. [DOI] [PubMed] [Google Scholar]
- 94.Wiviott SD, Braunwald E, McCabe CH, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357:2001. doi: 10.1056/NEJMoa0706482. [DOI] [PubMed] [Google Scholar]
- 95.Uller L, Mathiesen JM, Alenmyr L, et al. Antagonism of the prostaglandin D2 receptor CRTH2 attenuates asthma pathology in mouse eosinophilic airway inflammation. Respir Res. 2007;8:16. doi: 10.1186/1465-9921-8-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zuccollo A, Shi C, Mastroianni R, et al. The thromboxane A2 receptor antagonist S18886 prevents enhanced atherogenesis caused by diabetes mellitus. Circulation. 2005;112:3001. doi: 10.1161/CIRCULATIONAHA.105.581892. [DOI] [PubMed] [Google Scholar]