

Abstract
G protein–coupled receptors (GPCRs) regulate virtually all aspects of human physiology and represent an important class of therapeutic drug targets. Many GPCR-targeted drugs resemble endogenous agonists, often resulting in poor selectivity among receptor subtypes and restricted pharmacologic profiles. The muscarinic acetylcholine receptor family exemplifies these problems; thousands of ligands are known, but few are receptor subtype–selective and nearly all are cationic in nature. Using structure-based docking against the M2 and M3 muscarinic receptors, we screened 3.1 million molecules for ligands with new physical properties, chemotypes, and receptor subtype selectivities. Of 19 docking-prioritized molecules tested against the M2 subtype, 11 had substantial activity and 8 represented new chemotypes. Intriguingly, two were uncharged ligands with low micromolar to high nanomolar Ki values, an observation with few precedents among aminergic GPCRs. To exploit a single amino-acid substitution among the binding pockets between the M2 and M3 receptors, we selected molecules predicted by docking to bind to the M3 and but not the M2 receptor. Of 16 molecules tested, 8 bound to the M3 receptor. Whereas selectivity remained modest for most of these, one was a partial agonist at the M3 receptor without measurable M2 agonism. Consistent with this activity, this compound stimulated insulin release from a mouse β-cell line. These results support the ability of structure-based discovery to identify new ligands with unexplored chemotypes and physical properties, leading to new biologic functions, even in an area as heavily explored as muscarinic pharmacology.
Introduction
G protein–coupled receptors (GPCRs) are integral transmembrane proteins that transduce extracellular signals from neurotransmitters, hormones, odorants, and many other signals across cellular membranes. The muscarinic acetylcholine receptors (M1–M5) are a subfamily of GPCRs recognizing the neurotransmitter acetylcholine and signaling through G proteins of the Gq/11 class (M1, M3, and M5 subtypes) and the Gi/o class (M2 and M4 subtypes). These receptors are targets for the treatment of many illnesses, including chronic obstructive pulmonary disease, urinary incontinence, and diabetes (Wess et al., 2007), and have been implicated in treatment of cognitive disorders such as Alzheimer’s disease (Messer, 2002) and schizophrenia (Chan et al., 2008).
Tool and drug development at muscarinic receptors has been complicated by difficulties in finding subtype-selective ligands. None of the muscarinic agonists and antagonists currently used in the clinic are selective for a particular muscarinic receptor subtype. This reflects the high sequence identities among the orthosteric sites of the M1–M5 receptors, differing, for instance, between the M2 and M3 subtypes by only a single residue. Muscarinic receptors can also mediate various side effects (e.g., adverse effects on heart rate, salivary secretion, and smooth muscle contractility). For example, whereas recent evidence suggests that an M3 agonist would promote insulin release in type 2 diabetes (Wess et al., 2007), M2 agonism would have substantial cardiac effects that would complicate clinical use. Similarly, M1 agonists have shown promise for treatment of Alzheimer’s disease, but dose-limiting side effects have precluded clinical use (Caccamo et al., 2009). Consequently, recent attempts to develop selective muscarinic drugs have focused on ligands targeting either an allosteric site (Conn et al., 2009b) or both orthosteric and allosteric sites simultaneously (Mohr et al., 2010).
The recent determination of the crystal structures for the M2 and M3 muscarinic receptor subtypes (Haga et al., 2012; Kruse et al., 2012) enables a structure-based discovery program for novel muscarinic ligands. To discover new chemotypes and compounds with novel physical and pharmacologic properties, we initially docked large compound libraries against the M2 muscarinic structure. A high discovery rate of new chemotypes and new physical properties inspired us to seek M3-selective molecules by exploiting the small region of sequence difference between the two receptor subtypes. Whereas most molecules displayed some selectivity for the M3 subtype, as designed in the docking screen, this selectivity was modest, illustrating the challenges of discovering subtype-selective orthosteric muscarinic ligands. However, the discovery of a partial M3 agonist that had no agonist activity at the M2 receptor, and its efficacy in a cell-based model to promote insulin release in β cells, also illustrates the potential of this approach.
Materials and Methods
Compounds were obtained from the vendors Molport (Riga, Latvia), Chembridge (San Diego, CA), Enamine (Kiev, Ukraine), Scientific Exchange (Center Ossipee, NH), Princeton Biomolecular Research (Princeton, NJ), and Asinex (Moscow, Russia), as well as from the Developmental Therapeutics Program at the National Institutes of Health National Cancer Institute. All compounds were sourced at 95% or greater purity as described by the vendors. All active compounds were further tested for purity by liquid chromatography–mass spectrometry at University of California at San Francisco, and were found to be at least 95% pure as judged by peak height and identity. For compounds 11 [(2S)-oxolan-2-ylmethyl 2-hydroxy-2,2-diphenylacetate] and 12 (benzyl2-hydroxy-2,2-diphenylacetate), liquid chromatography–mass spectrometry was inconclusive and purity was confirmed by 1H-NMR spectroscopy at the Stanford Magnetic Resonance Laboratory using a Varian Inova 600 mHz spectrometer (Varian, Palo Alto, CA). Compound 5 was not commercially available in sufficient purity, and details regarding its preparation are given below.
Chemistry.
Compound 5 (pyridin-3-ylmethyl 2-hydroxy-2,2-diphenylacetate) was not commercially available in sufficient purity, and was synthesized as follows. After stirring a suspension of 3-(hydroxymethyl)pyridine (30 µl, 0.31 mM) and K2CO3 (100 mg, 0.72 mM) in anhydrous dimethylformamide (12 ml) at room temperature for 1 hour, a solution of methyl benzilate (50 mg, 0.21 mM) in anhydrous dimethylformamide (3 ml) was added. The mixture was stirred at 65°C at 70–100 mbar for 6 hours and allowed to cool to room temperature. After addition of CH2Cl2 and water, the organic layer was washed with a saturated aqueous solution of NaCl, dried (Na2SO4), and evaporated. The residue was purified by flash chromatography (CH2Cl2–MeOH 60:1) to yield pure pyridin-3-ylmethyl 2-hydroxy-2,2-diphenylacetate (24.3 mg, 36%) as a white solid (melting point: 98–101°C). 1H-NMR (CDCl3, 600 MHz) δ from tetramethylsilane (ppm): 8.58 [brs (broad singlet), 1H], 8.51 (brs, 1H), 7.52 (broad doublet, J = 7.6 Hz, 1H), 7.37–7.41 (m, 4H), 7.30–7.34 (m, 6H), 7.26–7.27 (m, 1H), 5.31 (s, 2H), 4.15 (brs, 1H); 13C-NMR (CDCl3, 150 MHz) d from tetramethylsilane (ppm): 174.3, 149.3, 148.8, 141.7, 136.6, 131.1, 128.4, 128.3, 127.5, 123.9, 81.4, 65.7; IR (NaCl), n (cm−1): 3150, 3060, 1740, 1600, 1580, 1450, 1220, 1060, 700; high-performance liquid chromatography: tR = 18.55 minutes (eluent 1), tR = 16.41 minutes (eluent 2), purity >95%; high-resolution mass spectra (m/z): [M]+ calculated for C20H17NO3 (M + Na+) 342.1101, found 342.1111.
Infrared spectra were recorded on a JASCO model FTIR 410 instrument as a film on NaCl. 1H-NMR (600 MHz) and 13C-NMR (150 MHz) spectra were determined on a Bruker AVANCE 600 spectrometer. Electrospray ionization–time-of-flight high mass accuracy and resolution experiments were performed on a Bruker maXis MS (Bruker Avance, Karlsruhe, Germany) in the laboratory of the Chair of Bioinorganic Chemistry, Friedrich Alexander University. High-performance liquid chromatography analysis was performed on an analytical system [Agilent 1100 analytical series (Agilent Technologies, Waldbronn, Germany), VWD detector (Agilent Technologies, Tokyo, Japan), Zorbax Eclipse XDB-C8 analytical column (Agilent Technologies, Santa Clara, CA), 4.6 × 150 mm, 5 µm, flow rate: 0.5 ml/min]. Eluent 1: CH3OH in H2O + 0.1% HCO2H (0–3 minutes 10%, 3–18 minutes 10–100%, 18–24 minutes 100%); eluent 2: CH3CN in H2O + 0.1% HCO2H (0–3 minutes 5%, 3–18 minutes 5–85%, 18–24 minutes 85%). Flash chromatography was done using silica gel (40–63 µm) as the stationary phase. The purity of the test compound was determined to be >95%.
Molecular Docking.
To predict new muscarinic ligands, we used DOCK 3.6 (Lorber and Shoichet, 2005; Irwin et al., 2009; Mysinger and Shoichet, 2010) to virtually screen the approximately 3.1 million lead-like and fragment-like subsets of ZINC (Irwin and Shoichet, 2005; Irwin et al., 2012) against the M2 or M3 muscarinic receptor structure. Compounds were docked in multiple orientations and multiple conformations. Each geometry was scored for electrostatic and van der Waals complementarity, and corrected for desolvation using the solvent-excluded volume method, and the complex with the lowest energy was picked. Compounds were manually selected for experimental testing from the top-ranking 500 molecules based both on their physical complementarity and chemical novelty, using criteria previously described (Mysinger et al., 2012).
To identify compounds that selectively bind to the M3 receptor, a similar method was first employed to score lead-like and fragment-like subsets of ZINC against both receptors. The top 5000 ranked molecules against the M3 receptor were selected for further consideration. Each of these molecules was then ranked according to the difference in energy score between docking at M3 and M2. The 500 molecules with the largest energy score difference in favor of the M3 receptor were then inspected, and 16 were chosen for experimental testing on the basis of high physical and chemical complementarity to M3, poor complementarity to M2, and novelty.
Receptor Expression and Membrane Preparation.
Human M2 and rat M3 muscarinic receptors were expressed with an amino-terminal FLAG epitope tag in Sf9 insect cells using the BestBac system (Expression Systems, Davis, CA). Membranes were prepared using a glass dounce tissue grinder to homogenize cells in 20 mM Tris pH 7.5 and 1 mM EDTA. Homogenized cell material was then centrifuged at low speed (100g) for 5 minutes to remove debris. The supernatant was then centrifuged at 18,000g in an SA-800 rotor for 15 minutes to pellet membranes. Membranes were resuspended in binding buffer (75 mM Tris, pH 7.4, 12.5 mM MgCl2, 1 mM EDTA), aliquoted, and flash frozen in liquid nitrogen.
Radioligand Binding Assays.
Ligand affinities were measured by radioligand displacement binding assays. Binding assays were performed using 3H-N-methyl scopolamine (NMS; PerkinElmer, Waltham, MA) at 0.61 nM in all samples. Following mixing of membranes, cold ligand and NMS samples were shaken at 20°C for 2 hours. Samples were then filtered on a glass fiber filter with a 48-well harvester (Brandel, Gaithersburg, MD). Radioactivity was measured by liquid scintillation. Binding data are summarized in Tables 1 and 2, and representative binding curves are shown in Supplemental Figs. 1–3. Binding data analysis was performed using GraphPad Prism 4.0 software (GraphPad Software, La Jolla, CA).
TABLE 1.
Compounds identified by docking to M2 receptor
| Compound ID (ZINC ID)a | Structure | Docking Rankb | M2 Ki ± S.E.M.c | M3 Ki ± S.E.M. | Tcd | Closest Analoge |
|---|---|---|---|---|---|---|
| 1 (C30009023) | 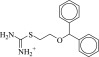 |
241f | 390 ± 32 nM | 130 ± 3 nM | 0.47 | 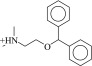 |
| 2 (C01571130) | 337g | 17 ± 2 μM | N.D. | 0.25 |  |
|
| 3 (C02293082) | 379g | 38 ± 6 μM | N.D. | 0.23 | ||
| 4 (C04202452) |  |
89g | 39 ± 3 μM | N.D. | 0.30 |  |
| 5 (C13283175) | 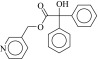 |
198f | 1.2 ± 0.2 μM | 360 ± 65 nM | 0.42 | 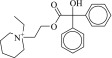 |
| 6 (C32628700) | 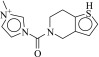 |
100g | 33 ± 8 μM | N.D. | 0.23 | 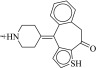 |
| 7 (C32810363) | 299g | 21 ± 5 μM | N.D. | 0.24 | 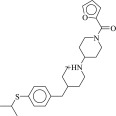 |
|
| 8 (C48231657) |  |
58f | 6.6 ± 1.4 μM | 1.8 ± 1.3 μM | 0.26 | 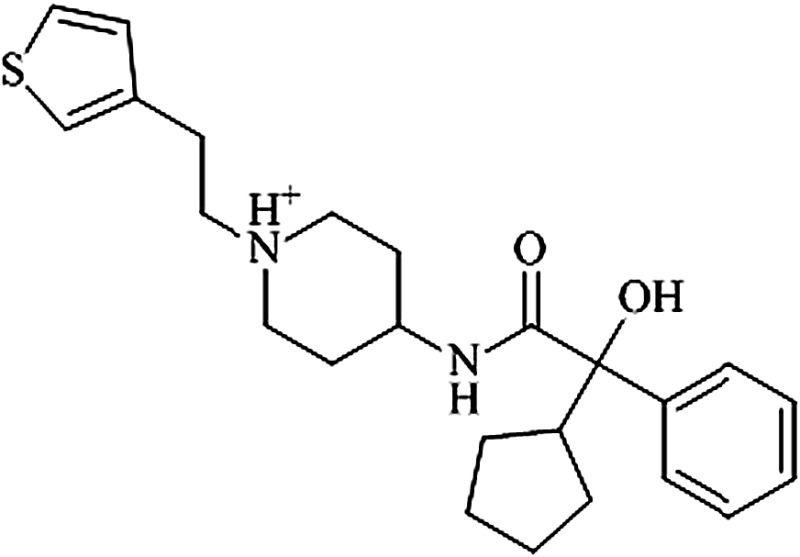 |
| 9 (C58162941) | 186g | 22 ± 4 μM | N.D. | 0.24 | 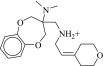 |
|
| 10 (C58406123) |  |
46f | 4.7 ± 0.8 μM | 5.8 ± 0.5 μM | 0.32 |  |
| 11 (C58857984) |  |
370f | 2.0 ± 0.1 μM | 1.2 ± 0.4 μM | 0.46 | 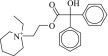 |
| 12 (C04547851) |  |
Analog of Compound 5 | 1.6 ± 0.1 μM | 290 ± 48 nM | 0.49 |  |
N.D., not determined.
From http://zinc.docking.org.
Out of 3.1 million fragments and “lead-like” molecules docked to the M2 receptor.
Values are from a minimum of two independent measurements performed in triplicate.
ECFP4-based Tanimoto coefficient to the most similar muscarinic ligand in ChEMBL.
Most similar molecule in ChEMBL.
Rank among to 2,662,342 lead-like compounds.
Rank among 357594 fragments.
TABLE 2.
Compounds docking well to M3 receptor and poorly to M2 receptor
| Compound ID (ZINC ID) | Structure | M2 Ki± S.E.M.a | M3 Ki ± S.E.M.a | M3/M2 | M3 Rank/ M2 Rank | Tc | Closest Analog |
|---|---|---|---|---|---|---|---|
| 13 (C18061786) | 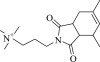 |
8.2 ± 0.8 μM | 1.3 ± 0.1 μM | 6.3-fold | 2496 / 1,238,745 | 0.21 | |
| 14 (C00181425) |  |
16 ± 1.6 μM | 10 ± 2.9 μM | 1.1-fold | 3278 / 1,018,801 | 0.24 | 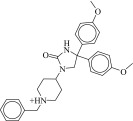 |
| 15 (C06850766) | 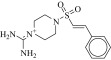 |
89 ± 7.7 μM | 64 ± 14 μM | 1.4-fold | 3728 / 1,157,022 | 0.29 | 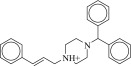 |
| 16 (C21270353) | >100 μM | >100 μM (Ki) 5.4 ± 2.8 μM (EC50)b | N/A | 4528 / 984,037 | 0.30 |  |
|
| 17 (C19866069) | 24 ± 2.7 μM | 5.1 ± 0.7 μM | 4.8-fold | 828/ 67,487 | 0.48 |  |
|
| 18 (C01694229) | 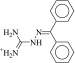 |
18 ± 1.8 μM | 8.8 ± 1.3 μM | 2.0-fold | 294 / 14,466 | 0.23 |  |
| 19 (C48433680) |  |
740 ± 37 nM | 780 ± 390 nM | 1.0-fold | 14 / 722 | 0.25 |  |
| 20 (C49524426) |  |
1.9 ± 0.5 μM | 1.4 ± 0.4 μM | 1.4-fold | 369 / 471,031 | 0.33 |  |
N/A, not applicable.
Values are from a minimum of two independent measurements performed in triplicate.
EC50 in a cell-based agonism assay (see Materials and Methods).
Calcium Mobilization Assay.
Chinese hamster ovary (CHO) cells stably expressing the human M3 receptor or CHO cells stably co-expressing the human M2 receptor and a hybrid G protein Gqi5 (Marlo et al., 2009) (a Gaq subunit in which the last five amino acids were replaced with the corresponding Gai sequence) were incubated with increasing concentrations of ligands, and changes in intracellular calcium levels were determined using fluorometric imaging plate reader technology (Molecular Devices, Sunnyvale, CA). All measurements were performed in 96-well plates, as described previously (Li et al., 2007; McMillin et al., 2011). Agonist concentration-response curves were analyzed using GraphPad Prism 4.0 software.
cAMP Assay.
CHO cells stably expressing the human M2 receptor were trypsinized, collected by centrifugation, and resuspended in phosphate-buffered saline containing glucose (1 mg/ml) and EDTA-free complete protease inhibitor (Roche Applied Science, Basel, Switzerland) at a density of 1 × 106 cells/ml. Subsequently, 20-μl aliquots were added to 200-μl polymerase chain reaction tubes and incubated with the same volume (20 μl) of increasing concentrations of ligands in the presence of 50 μM forskolin for 25 minutes at 37°C. The incubation mixtures were then transferred into white-bottom 384-well plates (∼5000 cells/well), and cells were lysed to determine drug-dependent changes in cAMP levels using a fluorescence resonance energy transfer–based cAMP detection technique (cAMP dynamic 2 kit; Cisbio Bioassays, Bedford, MA) according to the manufacturer's protocol. An elevated 665/620 nm ratio indicates decreased cAMP levels in this assay.
Insulin Release Assays (MIN6 Cells).
MIN6 cells (a kind gift from Dr. Abner Notkins, National Institute of Dental and Craniofacial Research, National Institutes of Health) were cultured as described previously (Ishihara et al., 1993). 60,000 cells were seeded into 96-well plates and cultured for 48 hours at 37°C in 5% CO2. After this time, MIN6 cells were washed with 3.3 mM glucose buffer (in Krebs-Ringer bicarbonate/HEPES buffer) and then incubated for 1 hour at 37°C in 5% CO2. After this step, MIN6 cells were incubated for another hour at 37°C in 5% CO2 with increasing concentrations of oxotremorine-M (OXO-M) or compound 16 [1-(3,5-dichlorobenzenesulfonyl)-4-methylpiperazine] in 16.7 mM glucose Krebs-Ringer buffer. Insulin release was determined by measuring insulin concentrations in the incubation medium using an insulin enzyme-linked immunosorbent assay kit (Crystal Chem, Inc., Downers Grove, IL). To confirm that the observed responses were mediated by muscarinic receptors, some assays were carried out in the presence of atropine (10 µM). Emax and EC50 values were obtained from OXO-M and compound 16 concentration-response curves using GraphPad Prism 4.0 software.
Antagonism Assay.
To examine whether compounds 12, 13 [3-(4,6-dimethyl-1,3-dioxo-1,3,3a,4,7,7a-hexahydro-2H-isoindol-2-yl)-N,N,N-trimethyl-1-propanaminium iodide], and 20 [1-(2-{[4-(4-methoxyphenyl)oxan-4-yl]formamido}ethyl)-2-methyl-1H-imidazol-3-ium] were able to block M3 receptor–mediated responses, we determined their ability to inhibit OXO-M–induced increases in intracellular calcium levels via activation of M3 receptors endogenously expressed by MIN6 cells. 50,000 cells were seeded into 96-well plates, and fluorometric imaging plate reader assays were carried out as described above (calcium mobilization assay). On the day of the assay, cells were preincubated with the calcium-chelating dye and the various compounds (atropine and compounds 12, 13, and 20) for 45 minutes, followed by the addition of the muscarinic receptor agonist OXO-M (1 μM). Compounds 12, 13, and 20 were used at a concentration of 10 μM (∼10 times their Ki). Atropine was employed at a concentration of 10 nM.
Results
Identification of New Muscarinic Ligands.
To identify new muscarinic ligands and to assess the suitability of muscarinic receptor structures as templates for ligand discovery, we pursued a docking campaign against the M2 muscarinic receptor structure. Like most GPCR structures available to date, the M2 receptor was solved in an inactive conformation bound to a small molecule antagonist. It presents a deep, almost completely buried ligand-binding site (Fig. 1A), covered by a layer of tyrosines long known to be critical for ligand binding. Such deeply buried cavities are well-suited to computational ligand discovery, and previous GPCR docking work has met with remarkable success (Sabio et al., 2008; Kolb et al., 2009; Carlsson et al., 2010; Katritch et al., 2010; de Graaf et al., 2011; Costanzi et al., 2012; Mysinger et al., 2012). Within the binding pocket, the crystallographic ligand quinuclidinyl benzilate (QNB) engages largely in hydrophobic interactions, while Asn4046.52 forms a pair of hydrogen bonds, and Asp1033.32 serves as a counter ion to the positive charge of the ligand (Fig. 1B; superscript numerals refer to the Ballesteros-Weinstein numbering system for GPCRs) (Ballesteros and Weinstein, 1995).
Fig. 1.

Docking poses for selected M2 muscarinic receptor hits. (A) The overall structure of the M2 receptor (Haga et al., 2012) with the orthosteric site outlined. (B) The chemical structure of the cocrystallized antagonist QNB, its crystallographic geometry, and key interactions (dashed lines). (C) Docking-discovered ligands (carbons in cyan) are superimposed in their docked poses on the crystallographic structure of QNB (carbons in yellow).
We screened 3.1 million fragments or “lead-like” molecules (Materials and Methods) from the ZINC database (Irwin and Shoichet, 2005; Irwin et al., 2012) against the structure of the M2 receptor. Each fragment and “lead-like” molecule was sampled in an average of 222 and 274 orientations and 437 and 700 conformations, respectively, in the orthosteric site; overall, over 547 billion configurations of the 3.1 million molecules were sampled. Molecules were ranked based on van der Waals and electrostatic complementarity and corrected for ligand desolvation using a receptor volume-based implementation of the Generalized-Born equation (Mysinger and Shoichet, 2010). From among the top 500-ranked molecules, we selected 18 that interacted with key residues such as Asp1033.32, Asn4046.52, and Trp4006.48, preferring molecules topologically or physically dissimilar to known muscarinic ligands. These 18 molecules were tested by single point competition binding against the high affinity antagonist 3H-NMS (Supplemental Table 1), and those with substantial inhibition at 20 μM were further tested in a competition binding assay. Of the 18 compounds tested, 11 had Ki values lower than 50 μM (Supplemental Fig. 1; Supplemental Table 2; Table 1). The compound with the highest affinity (compound 1 [2-(2-benzhydryloxyethyl)isothiourea]) displayed a Ki of 390 nM. Six of these compounds were fragments, with ligand efficiencies ranging from 0.36 to 0.44 kcal/heavy-atom. Most of the 11 molecules were topologically dissimilar to known muscarinic agents. Using two-dimensional ECFP4 fingerprints and Tanimoto coefficients (Tc) (Hert et al., 2004) to all known muscarinic ligands in ChEMBL11 (Gaulton et al., 2012), 8 of the 11 compounds were found to have a Tc < 0.33 to the closest muscarinic ligand of any class, a difference large enough to be typically considered a “scaffold hop” (Muchmore et al., 2008). Correspondingly, their binding poses differ substantially from that of the cocrystallized ligand (Fig. 1C).
Intriguingly, two of the higher affinity ligands, compounds 5 and 11 (Table 1), lack the defining cationic amine that is ubiquitous among muscarinic ligands and other aminergic GPCRs (e.g., histaminergic, adrenergic, dopaminergic, or serotonergic). Indeed, they were chosen for testing because of this unexpected physical property. Whereas in compound 5 the pyridine nitrogen might conceivably be cationic—although it would be expected to be neutral at physiologic pH, and is docked in this form—compound 11 is constitutively neutral at all accessible pH values. Correspondingly, the phenyl analog of 5 and 11, compound 12, is also a ligand with low micromolar affinity. The loss of the Asp1033.32 ion-pair with the ligand cation is a substantial insult, amounting to about 4 kcal/mol if one compares the affinity of compounds 11 and 12 to that of the analogous QNB, which binds with an affinity of 180 pM to the M2 receptor (Heitz et al., 1999). However, the fact that such ligands can even bind to muscarinic receptors at meaningful, reasonable concentrations has few precedents in the field (Barlow and Tubby, 1974). Indeed, no uncharged ligands of the M2 or M3 receptors are reported in the ChEMBL database (i.e., all are expected to be ionized at physiologic pH values) of the over 5000 ligands annotated, and while four neutral analogs of acetylcholine and other acetic-acid esters are reported to be active at acetylcholine receptors of the guinea-pig ileum (Barlow and Tubby, 1974), no further uncharged ligands have been reported subsequently, to the best of our knowledge.
Docking for Subtype Selectivity.
Though the docking against the M2 receptor had no selectivity goal—compounds were simply chosen based on complementarity to the M2 receptor—we were interested to learn whether the unusual chemotypes and physical properties of the new ligands conferred selectivity. We thus tested those M2 ligands with Ki values lower than 10 μM for binding to the M3 receptor (Supplemental Fig. 2; Table 1) (those molecules with weaker affinity were not pursued). Intriguingly, all three uncharged ligands (5, 11, and 12) bear some selectivity for the M3 over the M2 subtype. For example, compound 12 shows a 5-fold higher affinity for the M3 subtype (Ki = 290 nM) as compared with the M2 subtype. Prompted by this observation, we explicitly set out to exploit the few differences that do exist between the M2 and M3 orthosteric sites in docking screens for subtype-selective ligands, treating the M2 subtype as a docking “antitarget.” In the M3 receptor, M2 Phe181 is replaced by a leucine, creating an enlarged pocket that might be exploited to achieve binding selectivity (Fig. 2, A and B). We again docked the fragment and “lead-like” subsets of the ZINC database against both the M2 and M3 receptors, this time selecting the top-ranked 5000 molecules against the M3 receptor. From these compounds, we chose 500 molecules with the largest rank difference between subtypes (Fig. 2C; Supplemental Fig. 3; Supplemental Table 3; Table 2). For instance, compound 13 ranks 2496 out of 3.1 million (top 0.1%) docked against the M3 receptor, but ranks only 1,238,745 out of 3.1 million (top 40%) against the M2 receptor, suggesting much better complementarity to the M3 subtype. From these 500 molecules, 16 candidates were selected for testing, again weighing key interactions and chemical novelty (Table 2). Of these candidates, eight compounds showed detectable binding to the M3 receptor. We then tested each of these molecules for affinity against both receptor subtypes. Although most compounds showed detectably higher affinity for the M3 receptor, the selectivity ratios were typically modest, reaching at best 6-fold (Table 2). The one exception was compound 16, a ligand with an unprecedented sulfonamide core and a ECFP4-based Tc value of only 0.3 to the closest known muscarinic ligand in ChEMBL (Gaulton et al., 2012). This molecule proved to be a partial agonist at the M3 receptor in a cell-based functional assay (5 μM EC50 value) without detectable activity at the M2 receptor (see below).
Fig. 2.

Docking for selective M3 receptor ligands. (A) The M3 (green) and M2 receptor (orange) binding pockets are superimposed and rendered as solvent-accessible surfaces, highlighting the enlarged binding pocket in the M3 subtype (Kruse et al., 2012). (B) Specific interactions with the cocrystallized M3 antagonist tiotropium are shown. (C) Docking poses for select new ligands.
Efficacy of New Ligands.
Most docking screens against inactive GPCR structures have discovered only antagonists (Kolb et al., 2009; Carlsson et al., 2010, 2011; Katritch et al., 2010; de Graaf et al., 2011), while a docking screening against the activated state of the β2-adrenergic receptor discovered only agonists (Weiss et al., 2013). Thus far, the only exception to this pattern is the κ-opioid receptor, where an inactive state was used as a template for the docking-based discovery of specific agonists (Negri et al., 2013). We therefore investigated the efficacy of the new ligands against both M2 and M3 receptors, using a calcium mobilization assay to test for G protein activation. The M2 receptor couples primarily to the Gi class of G proteins, which mediate inhibition of adenylyl cyclase, while the M3 receptor preferentially couples to Gq/, mediating hydrolysis of phospoinositide lipids and consequent elevation of intracellular calcium. For these assays, we used CHO cells stably expressing the human M3 receptor or CHO cells stably coexpressing the human M2 receptor and a hybrid G protein Gqi5, which consists of a Gαq subunit in which the last five amino acids were replaced with the corresponding Gαi sequence, allowing coupling to the M2 receptor (Wess et al., 1997). Almost all compounds tested were devoid of agonist activity on either receptor. Additional functional studies with representative compounds showed that the uncharged compound 12 antagonized oxotremorine-M–induced activation of M3 receptors in cultured MIN6 cells, as did compounds 13 and 20 (Supplemental Fig. 4).
The only agent that showed agonist activity at the M3 receptor was compound 16. This molecule was a partial agonist at the M3 receptor, with an EC50 of 5.2 μM an Emax of 65%, but lacked detectable efficacy at the M2 subtype (Fig. 3). The lack of agonist activity of 16 at the M2 receptor was confirmed in both calcium mobilization (Fig. 3A) and adenylate cyclase inhibition (Fig. 3B) assays. To our knowledge, compound 16 represents the first pharmacological agent that can activate M3 but not M2 receptors. This novel activity profile mirrors its unusual chemotype: unlike most muscarinic ligands, compound 16 cannot form a paired hydrogen bond with Asn6.52, as seen in the M3 cocrystal structure with tiotropium, and instead may hydrogen bond through its unique sulfonamide to Tyr3.33 (Fig. 3C). Whether this configuration is conserved in the activated M3 structure to which it must bind is uncertain at this time; we cannot now rule out the possibility that compound 16 may even bind in a completely unexpected manner, including even to allosteric pockets that may initiate activation in their own right (Bluml et al., 1994; Avlani et al., 2010; Gregory et al., 2010). Further studies will be required to definitively establish the binding site for compound 16. For now, it is the novelty of this chemotype to which we attribute its unexpected activity and selectivity.
Fig. 3.
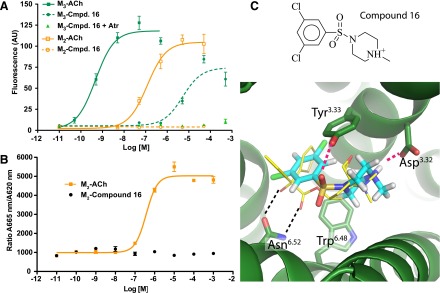
Compound 16 activates M3 but not M2 receptors. (A) Compound 16 showed partial agonism at the M3 subtype, but not at the M2 receptor in a calcium mobilization assay using CHO cells stably expressing M2 or M3 receptors (see Materials and Methods for details). This effect was blocked by the muscarinic antagonist atropine (Atr), consistent with direct activity at the M3 receptor. (B) In a fluorescence resonance energy transfer–based cAMP assay (see Materials and Methods for details), compound 16 did not lead to changes in intracellular cAMP levels in CHO-M2 cells, confirming that this agent lacks efficacy at M2 receptors. In this assay, an elevated 665 nm/620 nm ratio corresponds to decreased cAMP levels. The curves shown in A and B are representative of three independent experiments. (C) The unique structure and predicted binding mode of compound 16 may account for its novel activity profile. ACh, acetylcholine; Cmpd, compound.
Compound 16 Stimulates Insulin Release in Pancreatic β Cells.
The M3 receptor is a critical regulator of acetylcholine-mediated glucose-dependent insulin release from pancreatic β cells, and recent studies indicate that increasing M3 receptor signaling would be useful in the treatment of type 2 diabetes (Gautam et al., 2010; Ruiz de Azua et al., 2010). However, further study of this concept has been stymied by the lack of selective M3 agonists. We therefore tested the ability of compound 16, a selective M3 agonist, to stimulate insulin release from pancreatic β cells. Specifically, we incubated MIN6 insulinoma cells, a mouse β-cell line expressing endogenous M3 receptors, with increasing amounts of OXO-M, a potent muscarinic agonist, or compound 16. Both compounds evoked a dose-dependent increase in insulin secretion, with pEC50 values of −4.21 and −5.75, respectively, for compound 16 and OXO-M. Compound 16 induced insulin secretion with an Emax 58% that of OXO-M (Fig. 4). Insulin release could be blocked by 10 μM atropine (Fig. 4B), confirming the involvement of M3 receptors.
Fig. 4.
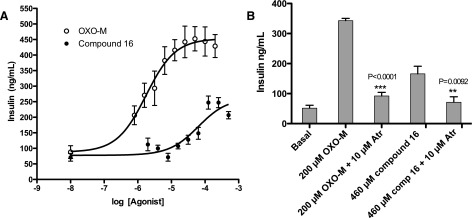
Ligand-stimulated insulin release in MIN6 cells. (A) MIN6 cells, which express endogenous M3 receptors, were incubated with increasing concentrations of OXO-M and compound 16, and ligand-induced insulin release was measured. (B) The responses to both agonists were sensitive to blockade by atropine, indicating that the observed effects result from direct M3 receptor activation. Data (mean ± S.E.) are from three independent experiments: OXO-M pEC50 = 5.75 ± 0.17; Emax = 453 ± 21; compound 16 pEC50 = 4.21 ± 0.18; Emax = 261 ± 21. Atr, atropine; comp, compound. **P < 0.0092; ***P < 0.0001.
Discussion
Four major observations emerge from this study. First, docking to the M2 and M3 muscarinic receptors led to the identification of multiple compounds with new physical properties and new chemical scaffolds. Second, as observed for other GPCRs, the docking hit-rates were high, between 50 and 60% of the compounds tested were active, with lead-like molecules often having affinities in the 0.1–1.0 μM range and with fragments with ligand efficiencies often above 0.4 kcal/heavy-atom (de Graaf et al., 2011). Third, an effort to explicitly dock for molecules specific for the M3 over the M2 subtype largely failed to successfully exploit the admittedly small difference between the two orthosteric sites, likely reflecting weaknesses in our current rigid-receptor docking models. Fourth, whereas it is not clear that the discovery of compound 16 reflects on our ability to select against binding to the M2 subtype—it may simply reflect the unexplored functionality of this compound—compound 16 represents an important novel pharmacologic tool in that it can activate M3 but not M2 receptors. These findings hint at the potential of a structure-based program to discover compounds with new chemistry and correspondingly new pharmacology.
A promise of structure-based discovery is the identification of molecules that physically complement a binding site but escape from trends emerging from classic structure-activity relationships. The muscarinic ligands are a good example of how a few key chemotypes and physical properties have come to dominate an area of pharmacology. Of over 5000 M2 or M3 receptor ligands annotated in ChEMBL, all bear at least a single cationic nitrogen. The discovery of ligands that are constitutively uncharged demonstrates that orthosteric site binding in muscarinic receptors is not contingent on the presence of such a cationic group. Since both cationic and uncharged ligands were found in our screen, and ranked about equally in the docking screen, this discovery also attests to the ability of a physics-based docking scoring function to balance high-magnitude ionic interactions (favoring charged ligands) and desolvation (favoring uncharged ligands) to arrive at a list of uncharged and cationic candidates. The uncharged ligands may balance the loss of the energy contributed by the Asp3.32 ion pair by hydrogen bonds with Asn6.52 and quadrupolar stacking with Tyr6.51 and Trp6.48, as observed in the docked poses (Fig. 1). These interactions are less common among cationic docking hits, which tend to be dominated by the Asp3.32 interaction (Fig. 1). Whereas the uncharged ligands bind as well as the new cationic ligands discovered here, they do lose about 4.5 kcal/mol in affinity compared with a structurally similar cationic ligand such as QNB, attesting to the importance of the ion-pair in contributing to high-affinity ligand binding. Still, as uncharged ligands will typically exhibit much greater membrane permeability than charged counterparts, such agents may show unique properties in vivo and may merit further exploration.
While the promise of discovering ligands with new chemotypes and new physical properties was realized in the docking screens, that of targeting particular differences between the M2 and M3 receptors to identify subtype-selective ligands was not. Although docking found molecules that fit much better against the rigid M3 than the M2 receptor structure owing to clashes with the larger Phe181 of the M2 site, these apparent structural specificities largely disappeared on pharmacologic testing. Despite much more favorable M3 docking ranks and scores (Table 2), experimental preference for the M3 subtype never rose above 6-fold in binding affinity. Thus, the steric clashes with Phe181 in the M2 site were not realized, or only to a small degree, presumably reflecting conformational flexibility in the site. This has largely been true of other recent efforts to find molecules selective among different GPCR subtypes: where selective molecules have been found directly from docking, they may reflect more on the chemical novelty of the compounds than on specific interactions captured by the modeling (Carlsson et al., 2011; de Graaf et al., 2011; Kolb et al., 2012). The exception to this is where chemical synthesis of multiple analogs, guided by structure, has followed initial hit-discovery by virtual screening (Langmead et al., 2012). Whereas there are now several methods that allow one to model local receptor flexibility in docking (Durrant and McCammon, 2010; Henzler and Rarey, 2011), implementing these prospectively in a way that does not lead to the appearance or even dominance of nonbinding decoys remains an ongoing challenge (Wei et al., 2004; Totrov and Abagyan, 2008). As the structures of more receptor subtypes are being solved or become amenable to homology modeling, the call for reliable methods that can exploit small differences in receptor structure among closely related subtypes will become increasingly pressing. Correspondingly, the call for strategies that exploit differences among allosteric sites, which are often substantially greater than those between the orthosteric sites of receptor subtypes (May et al., 2007; Conn et al., 2009a) is also supported by this study. In all such efforts, a close collaboration with medicinal chemistry will be crucial, as molecules that are at once new to a receptor and optimal for it are unlikely to be present in any library of available molecules.
Although we were unable to reliably exploit the subtle differences between the M2 and M3 orthosteric sites to identify M3 selective antagonists, the discovery of a selective M3 receptor agonist (compound 16) hints at the promise of a structure-based discovery program. Whereas the unusual pharmacology of compound 16 may owe as much to its chemical novelty as to the differential docking, the exploration of new chemotypes is something that has been often realized in docking campaigns against GPCRs (Evers and Klebe, 2004; de Graaf et al., 2011; Langmead et al., 2012) and that can be relied on. The observation that this agent can induce insulin release from pancreatic β cells in culture supports its status as a lead compound for chemical tool development, and this finding may have important therapeutic implications for the treatment of type 2 diabetes if selective M3 receptor agonists endowed with higher affinity can be developed. More broadly, a structure-based program of ligand discovery against the M3 receptor and related GPCRs holds out the promise of identifying new chemotypes with new physical properties and correspondingly new specificities and pharmacologic properties, with important implications for the discovery of new probes and therapeutic leads.
Supplementary Material
Acknowledgments
The authors thank Corey Liu and Aashish Manglik for technical assistance with NMR spectroscopy, and Dr. Allan I. Levey (Emory University) for kindly providing the two stable CHO cell lines.
Abbreviations
- brs
broad singlet
- CHO
Chinese hamster ovary
- compound 1
2-(2-benzhydryloxyethyl)isothiourea
- compound 5
pyridin-3-ylmethyl 2-hydroxy-2,2-diphenylacetate
- compound 11
(2S)-oxolan-2-ylmethyl 2-hydroxy-2,2-diphenylacetate
- compound 12
benzyl 2-hydroxy-2,2-diphenylacetate
- compound 13
3-(4,6-dimethyl-1,3-dioxo-1,3,3a,4,7,7a-hexahydro-2H-isoindol-2-yl)-N,N,N-trimethyl-1-propanaminium iodide
- compound 16
1-(3,5-dichlorobenzenesulfonyl)-4-methylpiperazine
- compound 20
1-(2-{[4-(4-methoxyphenyl)oxan-4-yl]formamido}ethyl)-2-methyl-1H-imidazol-3-ium
- GPCR
G protein–coupled receptors
- NMS
N-methyl scopolamine
- OXO-M
oxotremorine-M
- QNB
quinuclidinyl benzilate
- Tc
Tanimoto coefficient
Authorship Contributions
Participated in research design: Kruse, Weiss, Wess, Kobilka, Shoichet.
Conducted experiments: Kruse, Weiss, K. Hu, J. Hu, Rossi.
Contributed new reagents or analytic tools: Eitel, Gmeiner.
Performed data analysis: Kruse, Weiss, Wess, Kobilka, Shoichet.
Wrote or contributed to the writing of the manuscript: Kruse, Weiss, Wess, Kobilka, Shoichet.
Footnotes
This work was supported by the National Science Foundation [Grant CHE-1223785]; and the National Institutes of Health [Grant P01 GM106990] and [Grants R01 GM71896 (to B.K.S.), R01 NS028471 (to B.K.K.), and F32 GM093580 (to D.R.W.)]. Support was also provided by the Intramural Research Program of the National Institutes of Health [National Institute of Diabetes and Digestive and Kidney Diseases] (to J.W.); the Mathers Charitable Foundation (to BKK); Bavaria California Technology Center (to M.R., J.H., K.H., P.G., and K.E.); and the National Science Foundation (Graduate Research Fellowship to A.C.K.).
 This article has supplemental material available at molpharm.aspetjournals.org.
This article has supplemental material available at molpharm.aspetjournals.org.
References
- Avlani VA, Langmead CJ, Guida E, Wood MD, Tehan BG, Herdon HJ, Watson JM, Sexton PM, Christopoulos A. (2010) Orthosteric and allosteric modes of interaction of novel selective agonists of the M1 muscarinic acetylcholine receptor. Mol Pharmacol 78:94–104 [DOI] [PubMed] [Google Scholar]
- Barlow RB, Tubby JH. (1974) Actions of some esters of 3,3-dimethylbutan-1-ol (the carbon analogue of choline) on the guinea-pig ileum. Br J Pharmacol 51:95–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballesteros J, Weinstein H. (1995) Integrated methods for modeling G-protein coupled receptors. Methods Neurosci 25:366–428 [Google Scholar]
- Blüml K, Mutschler E, Wess J. (1994) Functional role in ligand binding and receptor activation of an asparagine residue present in the sixth transmembrane domain of all muscarinic acetylcholine receptors. J Biol Chem 269:18870–18876 [PubMed] [Google Scholar]
- Caccamo A, Fisher A, LaFerla FM. (2009) M1 agonists as a potential disease-modifying therapy for Alzheimer’s disease. Curr Alzheimer Res 6:112–117 [DOI] [PubMed] [Google Scholar]
- Carlsson J, Coleman RG, Setola V, Irwin JJ, Fan H, Schlessinger A, Sali A, Roth BL, Shoichet BK. (2011) Ligand discovery from a dopamine D3 receptor homology model and crystal structure. Nat Chem Biol 7:769–778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson J, Yoo L, Gao ZG, Irwin JJ, Shoichet BK, Jacobson KA. (2010) Structure-based discovery of A2A adenosine receptor ligands. J Med Chem 53:3748–3755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WY, McKinzie DL, Bose S, Mitchell SN, Witkin JM, Thompson RC, Christopoulos A, Lazareno S, Birdsall NJM, Bymaster FP, et al. (2008) Allosteric modulation of the muscarinic M4 receptor as an approach to treating schizophrenia. Proc Natl Acad Sci USA 105:10978–10983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Christopoulos A, Lindsley CW. (2009a) Allosteric modulators of GPCRs: a novel approach for the treatment of CNS disorders. Nat Rev Drug Discov 8:41–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conn PJ, Jones CK, Lindsley CW. (2009b) Subtype-selective allosteric modulators of muscarinic receptors for the treatment of CNS disorders. Trends Pharmacol Sci 30:148–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costanzi S, Santhosh Kumar T, Balasubramanian R, Kendall Harden T, Jacobson KA. (2012) Virtual screening leads to the discovery of novel non-nucleotide P2Y₁ receptor antagonists. Bioorg Med Chem 20:5254–5261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Graaf C, Kooistra AJ, Vischer HF, Katritch V, Kuijer M, Shiroishi M, Iwata S, Shimamura T, Stevens RC, de Esch IJ, et al. (2011) Crystal structure-based virtual screening for fragment-like ligands of the human histamine H(1) receptor. J Med Chem 54:8195–8206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durrant JD, McCammon JA. (2010) Computer-aided drug-discovery techniques that account for receptor flexibility. Curr Opin Pharmacol 10:770–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evers A, Klebe G. (2004) Successful virtual screening for a submicromolar antagonist of the neurokinin-1 receptor based on a ligand-supported homology model. J Med Chem 47:5381–5392 [DOI] [PubMed] [Google Scholar]
- Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, et al. (2012) ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res 40 (Database issue):D1100–D1107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gautam D, Ruiz de Azua I, Li JH, Guettier JM, Heard T, Cui Y, Lu H, Jou W, Gavrilova O, Zawalich WS, et al. (2010) Beneficial metabolic effects caused by persistent activation of beta-cell M3 muscarinic acetylcholine receptors in transgenic mice. Endocrinology 151:5185–5194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory KJ, Hall NE, Tobin AB, Sexton PM, Christopoulos A. (2010) Identification of orthosteric and allosteric site mutations in M2 muscarinic acetylcholine receptors that contribute to ligand-selective signaling bias. J Biol Chem 285:7459–7474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haga K, Kruse AC, Asada H, Yurugi-Kobayashi T, Shiroishi M, Zhang C, Weis WI, Okada T, Kobilka BK, Haga T, et al. (2012) Structure of the human M2 muscarinic acetylcholine receptor bound to an antagonist. Nature 482:547–551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitz F, Holzwarth JA, Gies JP, Pruss RM, Trumpp-Kallmeyer S, Hibert MF, Guenet C. (1999) Site-directed mutagenesis of the putative human muscarinic M2 receptor binding site. Eur J Pharmacol 380:183–195 [DOI] [PubMed] [Google Scholar]
- Henzler AM and Rarey M(2011) Protein flexibility in structure-based virtual screening: from models to algorithms, in Virtual Screening (Sotriffer C ed) pp 223–244, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, Germany. [Google Scholar]
- Hert J, Willett P, Wilton DJ, Acklin P, Azzaoui K, Jacoby E, Schuffenhauer A. (2004) Comparison of topological descriptors for similarity-based virtual screening using multiple bioactive reference structures. Org Biomol Chem 2:3256–3266 [DOI] [PubMed] [Google Scholar]
- Irwin JJ, Shoichet BK. (2005) ZINC—a free database of commercially available compounds for virtual screening. J Chem Inf Model 45:177–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Shoichet BK, Mysinger MM, Huang N, Colizzi F, Wassam P, Cao Y. (2009) Automated docking screens: a feasibility study. J Med Chem 52:5712–5720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin JJ, Sterling T, Mysinger MM, Bolstad ES, Coleman RG. (2012) ZINC: A Free Tool to Discover Chemistry for Biology. J Chem Inf Model 52:1757–1768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishihara H, Asano T, Tsukuda K, Katagiri H, Inukai K, Anai M, Kikuchi M, Yazaki Y, Miyazaki JI, Oka Y. (1993) Pancreatic beta cell line MIN6 exhibits characteristics of glucose metabolism and glucose-stimulated insulin secretion similar to those of normal islets. Diabetologia 36:1139–1145 [DOI] [PubMed] [Google Scholar]
- Katritch V, Jaakola VP, Lane JR, Lin J, Ijzerman AP, Yeager M, Kufareva I, Stevens RC, Abagyan R. (2010) Structure-based discovery of novel chemotypes for adenosine A(2A) receptor antagonists. J Med Chem 53:1799–1809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb P, Rosenbaum DM, Irwin JJ, Fung JJ, Kobilka BK, Shoichet BK. (2009) Structure-based discovery of beta2-adrenergic receptor ligands. Proc Natl Acad Sci USA 106:6843–6848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb P, Phan K, Gao ZG, Marko AC, Sali A, Jacobson KA. (2012) Limits of ligand selectivity from docking to models: in silico screening for A(1) adenosine receptor antagonists. PLoS ONE 7:e49910 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kruse AC, Hu J, Pan AC, Arlow DH, Rosenbaum DM, Rosemond E, Green HF, Liu T, Chae PS, Dror RO, et al. (2012) Structure and dynamics of the M3 muscarinic acetylcholine receptor. Nature 482:552–556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorber DM, Shoichet BK. (2005) Hierarchical docking of databases of multiple ligand conformations. Curr Top Med Chem 5:739–749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead CJ, Andrews SP, Congreve M, Errey JC, Hurrell E, Marshall FH, Mason JS, Richardson CM, Robertson N, Zhukov A, et al. (2012) Identification of novel adenosine A(2A) receptor antagonists by virtual screening. J Med Chem 55:1904–1909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Scarselli M, Knudsen CD, Kim S-K, Jacobson KA, McMillin SM, Wess J. (2007) Rapid identification of functionally critical amino acids in a G protein-coupled receptor. Nat Methods 4:169–174 [DOI] [PubMed] [Google Scholar]
- Marlo JE, Niswender CM, Days EL, Bridges TM, Xiang Y, Rodriguez AL, Shirey JK, Brady AE, Nalywajko T, Luo Q, et al. (2009) Discovery and characterization of novel allosteric potentiators of M1 muscarinic receptors reveals multiple modes of activity. Mol Pharmacol 75:577–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- May LT, Leach K, Sexton PM, Christopoulos A. (2007) Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 47:1–51 [DOI] [PubMed] [Google Scholar]
- McMillin SM, Heusel M, Liu T, Costanzi S, Wess J. (2011) Structural basis of M3 muscarinic receptor dimer/oligomer formation. J Biol Chem 286:28584–28598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer WS., Jr (2002) The utility of muscarinic agonists in the treatment of Alzheimer’s disease. J Mol Neurosci 19:187–193 [DOI] [PubMed] [Google Scholar]
- Mohr K, Tränkle C, Kostenis E, Barocelli E, De Amici M, Holzgrabe U. (2010) Rational design of dualsteric GPCR ligands: quests and promise. Br J Pharmacol 159:997–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muchmore SW, Debe DA, Metz JT, Brown SP, Martin YC, Hajduk PJ. (2008) Application of belief theory to similarity data fusion for use in analog searching and lead hopping. J Chem Inf Model 48:941–948 [DOI] [PubMed] [Google Scholar]
- Mysinger MM, Shoichet BK. (2010) Rapid context-dependent ligand desolvation in molecular docking. J Chem Inf Model 50:1561–1573 [DOI] [PubMed] [Google Scholar]
- Mysinger MM, Weiss DR, Ziarek JJ, Gravel S, Doak AK, Karpiak J, Heveker N, Shoichet BK, Volkman BF. (2012) Structure-based ligand discovery for the protein-protein interface of chemokine receptor CXCR4. Proc Natl Acad Sci USA 109:5517–5522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negri A, Rives ML, Caspers MJ, Prisinzano TE, Javitch JA, Filizola M. (2013) Discovery of a Novel Selective Kappa-Opioid Receptor Agonist Using Crystal Structure-Based Virtual Screening. J Chem Inf Model 53:521–526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz de Azua I, Scarselli M, Rosemond E, Gautam D, Jou W, Gavrilova O, Ebert PJ, Levitt P, Wess J. (2010) RGS4 is a negative regulator of insulin release from pancreatic β-cells in vitro and in vivo. Proc Natl Acad Sci USA 107:7999–8004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabio M, Jones K, Topiol S. (2008) Use of the X-ray structure of the beta2-adrenergic receptor for drug discovery. Part 2: Identification of active compounds. Bioorg Med Chem Lett 18:5391–5395 [DOI] [PubMed] [Google Scholar]
- Totrov M, Abagyan R. (2008) Flexible ligand docking to multiple receptor conformations: a practical alternative. Curr Opin Struct Biol 18:178–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei BQ, Weaver LH, Ferrari AM, Matthews BW, Shoichet BK. (2004) Testing a flexible-receptor docking algorithm in a model binding site. J Mol Biol 337:1161–1182 [DOI] [PubMed] [Google Scholar]
- Weiss DR, Ahn S, Sassano MF, Kleist A, Zhu X, Strachan R, Roth BL, Lefkowitz RJ, Shoichet BK. (2013) Conformation guides molecular efficacy in docking screens of activated β-2 adrenergic G protein coupled receptor. ACS Chem Biol 8:1018–1026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wess J, Liu J, Blin N, Yun J, Lerche C, Kostenis E. (1997) Structural basis of receptor/G protein coupling selectivity studied with muscarinic receptors as model systems. Life Sci 60:1007–1014 [DOI] [PubMed] [Google Scholar]
- Wess J, Eglen RM, Gautam D. (2007) Muscarinic acetylcholine receptors: mutant mice provide new insights for drug development. Nat Rev Drug Discov 6:721–733 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.