

Abstract
Anti-inflammatory and antifibrotic effects of the broad spectrum phosphodiesterase (PDE) inhibitor pentoxifylline have suggested an important role for cyclic nucleotides in the pathogenesis of hepatic fibrosis; however, studies examining the role of specific PDEs are lacking. Endotoxemia and Toll-like receptor 4 (TLR4)-mediated inflammatory and profibrotic signaling play a major role in the development of hepatic fibrosis. Because cAMP-specific PDE4 critically regulates lipopolysaccharide (LPS)-TLR4–induced inflammatory cytokine expression, its pathogenic role in bile duct ligation-induced hepatic injury and fibrogenesis in Sprague-Dawley rats was examined. Initiation of cholestatic liver injury and fibrosis was accompanied by a significant induction of PDE4A, B, and D expression and activity. Treatment with the PDE4-specific inhibitor rolipram significantly decreased liver PDE4 activity, hepatic inflammatory and profibrotic cytokine expression, injury, and fibrosis. At the cellular level, in relevance to endotoxemia and inflammatory cytokine production, PDE4B was observed to play a major regulatory role in the LPS-inducible tumor necrosis factor (TNF) production by isolated Kupffer cells. Moreover, PDE4 expression was also involved in the in vitro activation and transdifferentiation of isolated hepatic stellate cells (HSCs). Particularly, PDE4A, B, and D upregulation preceded induction of the HSC activation marker α-smooth muscle actin (α-SMA). In vitro treatment of HSCs with rolipram effectively attenuated α-SMA, collagen expression, and accompanying morphologic changes. Overall, these data strongly suggest that upregulation of PDE4 expression during cholestatic liver injury plays a potential pathogenic role in the development of inflammation, injury, and fibrosis.
Introduction
Hepatic fibrosis results from persistent liver injury and inflammation. Endotoxemia, a phenomenon that accompanies liver injury, plays a critical role in aggravation of inflammation via activation of Kupffer cells (KCs) through Toll-like receptor 4 (TLR4) signaling. Activated KCs produce a variety of proinflammatory cytokines, such as tumor necrosis factor (TNF)-α and monocyte chemoattractant protein (MCP-1), that promote the activation of hepatic stellate cells (HSCs) (Friedman, 2000). During activation, HSCs increase their expression of α-smooth muscle actin (α-SMA), become proliferative and migratory, and produce increasing amounts of extracellular matrix components such as collagen type I and fibronectin. Because transdifferentiation of HSCs plays a key role in the development of liver fibrosis, targeting their activation has become a focal point in treating liver fibrosis (Li et al., 2008). The critical role of TLR4 signaling in the development of fibrosis has been demonstrated by using TLR4 mutant mice as well as antibiotic treatment. Specifically, it has been shown that TLR4-mutant mice exhibit significantly reduced fibrosis in the bile duct ligation (BDL) model (Seki et al., 2007), and antibiotic treatment suppresses hepatic fibrosis (Seki et al., 2007).
Lipopolysaccharide (LPS)-inducible inflammatory cytokine expression is inhibited by cyclic adenosine monophosphate (cAMP) in human and murine monocytes/macrophages as well as rat Kupffer cells (Zidek, 1999); as a consequence, decreased cAMP levels can enhance LPS-inducible cytokine production (Gobejishvili et al., 2006). Work done by us and others has shown that regulation of cellular cAMP levels via degradation by phosphodiesterases (PDEs), particularly cAMP-specific PDE4B, plays an essential role in LPS-induced TLR4 signaling and inflammatory cytokine expression by monocytes/macrophages (Jin and Conti, 2002; Jin et al., 2005; Gobejishvili et al., 2011). In cholestatic liver injury, besides downregulating systemic and hepatic inflammatory cytokines, increases in cAMP have been shown to protect hepatocytes from apoptosis due to several related stimuli, including bile acids, LPS, Fas, and TNF-α (Fladmark et al., 1997; Li et al., 2000; Webster et al., 2002; Cullen et al., 2004; Reinehr and Haussinger, 2004).
Work done with PDE inhibitors has demonstrated their beneficial effect in experimental liver injury (Fischer et al., 1993; Gantner et al., 1997; Windmeier and Gressner, 1997; Taguchi et al., 1999; Xiang et al., 1999; Matsuhashi et al., 2005; Tukov et al., 2007), but there have been no studies examining the causal role of PDEs in the pathogenesis of liver fibrosis. cAMP-specific PDE4 isoforms have been shown to contribute to the pathogenesis of inflammation and fibrosis in lung tissue and fibroblast transdifferentiation; hence, in this study, we explored a potential pathogenic role of PDE4 subfamily members in an animal model of cholestatic liver injury/fibrosis. Additionally, the role of PDE4 in activation of primary hepatic Kupffer cells and HSCs was examined. The data obtained strongly suggest that PDE4 plays a significant pathogenic role in the development of hepatic inflammation, injury, and fibrosis during cholestatic liver injury.
Materials and Methods
Animals.
The bile duct ligation surgery was performed on 8-week-old Sprague-Dawley rats (Harlan, Indianapolis, IN), as described previously (Song et al., 2011), and assigned to three study groups (eight per group): 1) BDL, 2) BDL+rolipram (5 mg/kg body weight three times a week), and 3) BDL+dimethylsulfoxide (DMSO) serving as a vehicle control). Rolipram dose was chosen based on our preliminary studies and published work (Sanz et al., 2002; Odashima et al., 2005; Videla et al., 2006). Rolipram and DMSO were given intraperitoneally throughout the study period. Additional animals were assigned as sham-operated controls (n = 5). Rats were sacrificed after 1, 2, and 4 weeks. This study was approved by the Institutional Animal Care and Use Committee at the University of Louisville.
Materials.
PDE4-specific inhibitor rolipram (C16H21NO3) (Biomol, Enzo Life Sciences, Farmingdale, NY) was dissolved in sterile DMSO and diluted with sterile phosphate buffered saline just before injection. PDE4A, B, D, poly(ADP-ribose) polymerase 1 (PARP-1), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibodies were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX). Phospho-SMAD3 (pS423/425) antibody was purchased from Epitomics (Epitomics, Inc., Burlingame, CA); SMAD3 antibody was purchased from Cell Signaling Technology (Danvers, MA). Antisera specific for murine PDE4A and PDE4B for in vitro experiments were a generous gift from Dr. Marco Conti.
Cell Culture.
Cryopreserved purified Kupffer cells isolated from adult male Sprague-Dawley rats were obtained from Life Technologies (Grand Island, NY) and cultured according to the provided protocol. They were plated at 30,000 cells/well in 96-well plate in advanced Dulbecco’s modified Eagle’s medium (DMEM) supplemented by 10% heat-inactivated fetal bovine serum and stimulated with 1 μg/ml LPS with and without rolipram (10 μM) pretreatment. Cell-free supernatants were collected after 24 hours of LPS stimulation, and TNF protein levels were measured using enzyme-linked immunosorbent assay (ELISA) kit (Life Technologies). For evaluation of PDE4 mRNA and protein expression after LPS stimulation, Kupffer cells were plated in six-well plates at 0.5 million/well density. Primary hepatic stellate cells (HSCs) were isolated from livers of male Wistar rats and cultured on plastic dishes, as described previously (Wiercinska et al., 2006). For transdifferentiation experiments, cells were kept in medium containing 10% fetal calf serum.
Histopathology and Liver Enzymes.
Formalin-fixed, paraffin-embedded liver sections were stained with H&E for evaluation of histologic changes, and Sirius red staining was performed to detect collagen fibers (Song et al., 2011). Serum levels of alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels were measured using ALT and AST reagents per manufacturer’s instructions (Thermo Fisher Scientific, Pittsburgh, PA).
PDE4 Enzymatic Assay and Immunoblot Analysis.
PDE4-specific enzymatic activity was determined using PDE4 assay kit (FabGennix International, Inc., Frisco, TX) as described previously (Gobejishvili et al., 2011). In brief, liver tissue was lysed using SolO buffer (FabGennix International, Inc.), and 25 μg of protein was used per assay. The same lysates were analyzed by immunoblot, and bands were quantified, as described previously (Gobejishvili et al., 2011).
Terminal Deoxynucleotidyl Transferase dUTP Nick-End Labeling Assay for Apoptosis.
ApopTag in-situ apoptosis detection kit (Intergen Company, Purchase, NY) was used to detect the apoptotic cells.
RNA Isolation and Polymerase Chain Reaction Analysis.
Total RNA was isolated from 50 mg of liver tissue using TRIzol Reagent (Invitrogen, Carlsbad, CA). Real-time polymerase chain reaction (PCR) was performed with an ABI prism 7500 sequence detection system as described previously (Gobejishvili et al., 2011). The specific primers were purchased from SABiosciences (Qiagen, Valencia, CA). The relative gene expression was analyzed using 2–ΔΔCt method by normalizing 18S gene expression in all of the experiments and is presented as fold change over the values for sham-operated rats. For analysis of PDE4 isoform expression levels during in vitro activation of HSC, expression values of the target genes were normalized to corresponding GAPDH.
The following primers were used in regular PCR: ribosomal protein S6 (rS6)—forward 5′-GTG CCT CGT CGG TTG GGA C-3′; reverse 5′-GAC AGC CTA CGT CTC TTG GC-3′; fibronectin (FN)—forward 5′-GGT GTG GTC TAC TCT GTG G-3′; reverse 5′-CAC ACG TGC ACC TCA TCA TG-3′; and collagen I α2 (Col1α2)—forward 5′-GGC TTC CTG GTG AGA GAG G-3′; reverse 5′-CCT CTC TTT CCT TCT TCA CC-3′.
Statistical Analysis.
The results are presented as the means of five to eight animals per group or three to four HSC cell preparations per condition, respectively, ± S.D. Student’s t test was used for the determination of statistical significance. The differences between treatment groups were analyzed using analysis of variance followed by Tukey’s multiple comparison test. P < 0.05 was considered statistically significant.
Results
Upregulation of PDE4 Enzymes during Fibrogenesis.
Previously, we and others demonstrated that PDE4B is the predominant PDE that responds to endotoxin and is critically involved in LPS-signaling through TLR4 in macrophages. Because endotoxemia plays a critical role in the development of cholestatic liver injury, we examined the role of PDE4 enzymes in liver fibrogenesis following bile duct ligation (BDL). Sprague-Dawley rats were subjected to BDL surgery and sacrificed at 1, 2, and 4 weeks. Hepatic expression levels of PDE4A, B, C, and D were examined at mRNA, protein, and activity levels. PDE4A and B mRNA levels increased as early as 1week after BDL (Fig. 1A). At this time point, PDE4D mRNA levels were also slightly upregulated but did not reach significance (Fig. 1A). PDE4C did not change following BDL (data not shown). As expected, the most prominent increase was observed in the levels of PDE4B mRNA (>7-fold over sham controls) at an initiation stage of liver fibrosis (Fig. 1A). At 2 weeks post-BDL surgery, all three PDE4 isoforms, A, B, and D, were significantly increased and stayed elevated up to 4 weeks (>3-fold over sham controls). Examination of protein levels at 2 weeks confirmed that BDL caused an increase in PDE4A, B, and D isoforms (Fig. 1B). Moreover, liver-specific PDE4 enzymatic activity was significantly increased (Fig. 1C). These results indicate that cholestatic liver injury leads to upregulation of PDE4 expression and activity.
Fig. 1.
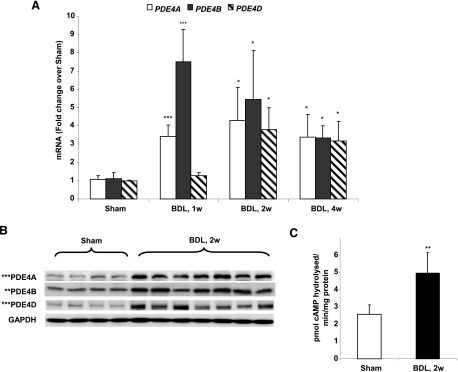
Upregulation of PDE4 enzymes during cholestatic liver injury. (A) Hepatic mRNA levels in rats subjected to BDL 1, 2,, and 4 weeks after surgery were quantified using real-time PCR represented as fold change over sham-operated rats. Data represent means ± S.D. (n = 5–8). (B) Protein levels of hepatic PDE4A, B, and D isoforms 2 weeks after BDL as measured by immunoblot. (C) Increase in hepatic PDE4 activity caused by BDL after 2 weeks. Data represent means ± S.D. (n = 5–8). Sham, sham-operated animals; 1w, 2w, and 4w, animals sacrificed 1, 2, and 4 weeks after BDL surgery. *P < 0.05; **P < 0.01; ***P < 0.001, compared with sham-operated animals.
Bile Duct Ligation–Induced Liver Injury Is Significantly Attenuated by PDE4 Inhibition.
To examine whether upregulation of PDE4 enzymes plays a causal role in the development of liver fibrosis, a group of bile duct–ligated animals was treated with the PDE4-specific inhibitor rolipram at 5 mg/kg body weight three times a week (BDL+Rol). To confirm the inhibitory effect of rolipram on liver PDE4 activity, we performed PDE4 assay with liver lysates purified from rats 2 weeks post-BDL. Rolipram treatment significantly attenuated the increase in PDE4 activity in the liver caused by BDL (Fig. 2A). The gross appearance of the livers from the rolipram-treated group was remarkably normal compared with untreated animals (Fig. 2B). We assessed hepatic injury by measuring liver enzyme levels and comparatively examining H&E staining of liver sections from BDL, BDL+Rol, and BDL+DMSO groups (DMSO was administered as a vehicle control in rats subjected to BDL surgery). H&E staining of the liver sections together with significant elevation of the liver enzymes ALT and AST confirmed the severe liver damage in BDL rats (Fig. 2, C and D). Overall, rolipram-treated animals (BDL+Rol) exhibited attenuated liver injury after bile duct ligation (Fig. 2), whereas the BDL+DMSO vehicle control group showed no difference as compared with the BDL group (Fig. 2, C and D).
Fig. 2.

PDE4 inhibitor treatment significantly attenuated liver injury induced by BDL. (A) Inhibition of hepatic PDE4 activity by rolipram treatment of 2 weeks after BDL. (B) Photographs of livers from sham-operated, BDL, and rolipram-treated rats (BDL+Rol) 4 weeks after BDL surgery. (C) Liver H&E staining, original magnification: 20×. (D) Liver enzymes ALT and AST. Data represent means ± S.D. (n = 5–8). *P < 0.05; **P < 0.01; ***P < 0.001.
PARP-1 cleavage represents a more stable hallmark of apoptosis and allows sustained signal detection even in late stages of this process (Lazebnik et al., 1994). Accordingly, PARP-1 status was evaluated by immunoblot analysis. Cholestatic liver injury induced by BDL at 2 weeks led to substantial PARP-1 cleavage, indicating significant hepatic apoptosis, which was attenuated by rolipram treatment (Fig. 3A). Furthermore, terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay showed that apoptotic cell numbers significantly increased at 1and 2 weeks after BDL treatment. Rolipram treatment also significantly reduced the level of TUNEL-positive cells (Fig. 3B), which correlates with PARP cleavage.
Fig. 3.

Rolipram treatment significantly decreased BDL-induced liver apoptosis. (A) PARP-1 protein levels 2 weeks after BDL. *P < 0.01 as compared with BDL+Rol. (B) TUNEL assay in livers with quantification. *P < 0.05; **P < 0.01 compared with sham; #P < 0.05 as compared with BDL. Original magnification: 20×.
PDE4 Inhibitor Significantly Decreased TNF-α.
As expected, BDL-induced hepatic inflammation, which was documented by a significant increase in TNF-α expression at both 2 and 4 weeks after surgery (only 2-week data are shown; Fig. 4A). Furthermore, the critical regulatory role of PDE4 in this process was strongly supported by the effect of rolipram treatment, which markedly suppressed TNF-α upregulation (Fig. 4A). Endotoxemia is a significant pathogenic feature of BDL-induced hepatic fibrosis since endotoxin is a key activator of hepatic Kupffer cells resulting in the production of inflammatory and fibrogenic cytokines (Aoyama et al., 2010); indeed, depletion of hepatic KCs decreases hepatic fibrosis. Accordingly, we examined the role of PDE4 in the activation and TNF production in isolated KCs in response to LPS. As seen in nonhepatic macrophages, LPS rapidly and robustly induced PDE4B mRNA expression in hepatic KCs, which peaked at 4 hours (Fig. 4B). In comparison, LPS did not induce PDE4A and 4D expression. Correspondent to the mRNA expression, significant protein expression of PDE4B was also evident by immunoblot analysis (Fig. 4C). Significantly, treatment of KCs with the PDE4-specific inhibitor rolipram markedly attenuated LPS-inducible TNF production (Fig. 4D).
Fig. 4.

Rolipram treatment significantly decreased TNF-α level. (A) Liver TNF-α mRNA levels without (BDL) and with rolipram treatment (BDL+Rol) 2 weeks after BDL surgery as compared with sham-operated rats. Data represent means ± S.D. (n = 5–8). *P < 0.05; **P < 0.01. (B) Kupffer cells were stimulated with 1 μg/ml LPS for 2, 4, and 8 hours and PDE4A, B, and D mRNA quantified by real-time PCR. Data obtained from three independent experiments is shown and presented as mean ± S.D. ***P < 0.001 compared with LPS-treated. (C) Western blot analysis of PDE4B protein levels in Kupffer cells after 4 and 6 hours of LPS stimulation. (D) KCs were pretreated with 10 μM rolipram for 30 minutes before LPS stimulation and TNF production was measured after 24 hours by TNF ELISA kit. Data obtained from three independent experiments is shown and presented as mean ± S.D. ***P < 0.001 compared with DMSO+LPS.
PDE4 Inhibition Attenuates Profibrotic Gene Expression and HSC Activation Induced by BDL.
Cholestatic liver injury results in an increase of liver profibrotic cytokines such as transforming growth factor β1 (TGF-β1), leading to activation of HSCs. Hence, we examined the effect of PDE4 inhibition on TGF-β1 and its type I receptor (TβRI, also called ALK5). Rolipram significantly inhibited the induction of TGF-β1 as well as TβRI mRNA expression (Fig. 5, A and B). Furthermore, rolipram prevented HSC activation as demonstrated by a marked reduction in immunohistochemical staining for hepatic α-SMA protein and α-SMA mRNA expression (Fig. 5, C and D).
Fig. 5.
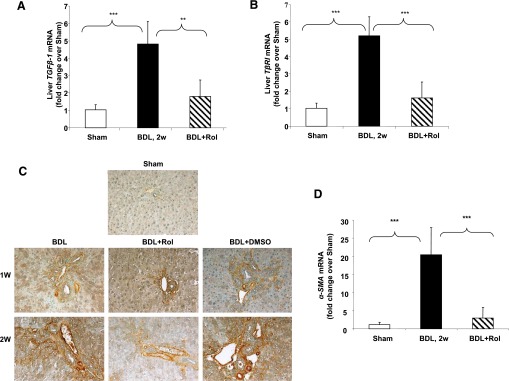
Significant attenuation of activation of hepatic stellate cells by rolipram treatment. Liver mRNA levels of (A) TGF-β1 and (B) TβRI 2 weeks after BDL surgery with and without rolipram administration. (C) Immunohistochemical staining of livers for α-SMA, original magnification: 20×. (D) Liver α-SMA mRNA expression 2 weeks after BDL surgery. Data represent means ± S.D. (n = 5–8). **P < 0.01; ***P < 0.001.
PDE4 Inhibition Markedly Attenuates Collagen Deposition.
HSCs play a major role in the development of liver fibrosis through their production of extracellular matrix (ECM) proteins, including collagen. Since Smad3 plays an important role in TGF-β1–mediated collagen production (Schnabl et al., 2001), the effect of rolipram on Smad3 activation was evaluated. As expected, BDL treatment led to an increase in Smad3 phosphorylation at Ser423/425, and this was significantly decreased by rolipram treatment, returning to near basal levels. DMSO vehicle control reduced Smad3 phosphorylation to some extent (Fig. 6A); however, it was still significantly greater than the basal level in the sham control group. Examination of collagen deposition by Sirius red staining of collagen fibers of the livers from the BDL and BDL+Rol groups showed significantly less staining in the group treated with rolipram (Fig. 6B). Liver Col1A1 mRNA levels were also significantly diminished by PDE4 inhibition, as compared with the BDL group (Fig. 6C).
Fig. 6.
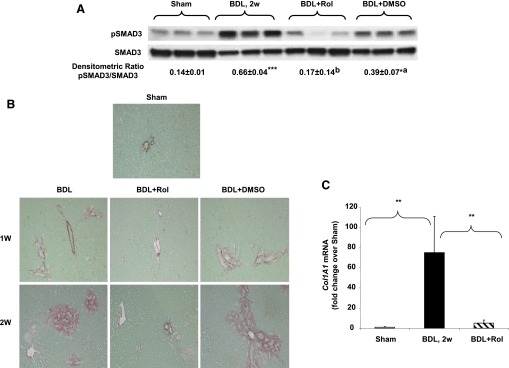
Decreased collagen deposition in rolipram-treated animals. (A) Liver pSMAD3 (pS423/425) protein levels. *P < 0.05; ***P < 0.001 compared with sham; aP < 0.05; bP < 0.001 as compared with BDL. (B) Sirius red staining, original magnification: 20×. (C) Liver ColA1 mRNA expression 2 weeks after BDL surgery. Data represent means ± S.D. (n = 5–8). **P < 0.01.
Expression of PDE4 in Primary Rat HSCs.
On the basis of the effects of PDE inhibitors, PDEs have been implicated in the activation and proliferative response of HSCs both in vivo and in vitro (Houglum et al., 1997; Desmouliere et al., 1999). Furthermore, PDE4 enzymes have been shown to play a significant role in the activation and proliferation of lung fibroblasts (Selige et al., 2011). Hence, the contribution of PDE4 isoforms in HSC transdifferentiation into myofibroblasts was examined in rat primary hepatic stellate cells. HSCs were isolated and cultured on tissue culture plastic as described previously (Xu et al., 1997; Desmouliere et al., 1999). At 1 hour, nonadherent cells were washed out and RNA was extracted from adhered cells starting from day 1 (1 hour of culture) up to day 5. We observed relatively higher mRNA levels of PDE4A, B, and D on day 1. On day 2, there was further increase in PDE4A and B mRNA levels (Fig. 7A). Examination of protein levels by Western blot demonstrated a significant induction of PDE4D in adhered cells (day 1) followed by A and B on day 2 (Fig. 7B). More importantly, we did not detect PDE4 protein in freshly isolated HSCs (day 0), indicating that quiescent HSCs do not express PDE4. Furthermore, there was no detectable α-SMA protein expression in the cells showing PDE4 expression on days 1 and 2 (Fig. 7C). These results indicate that PDE4 expression precedes α-SMA during the HSC activation process.
Fig. 7.
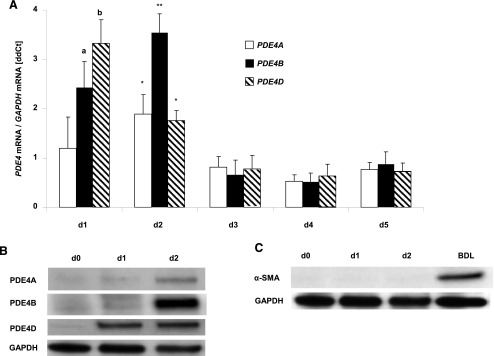
(A) PDE4 expression in isolated HSCs. Primary rat HSCs were isolated and plated to let them differentiate on plastic. PDE4A, B, and D mRNA levels were examined every day during culture-activation by real-time PCR. Data obtained from at least three independent preparations per time-point is shown and presented as mean ± S.D. *P < 0.05; **P < 0.01 compared with day 3. aP < 0.05; bP < 0.01 as compared with day 2. (B) PDE4A, B, and D protein levels in HSCs (d0, freshly isolated HSCs; d1, 1 hour after attachment of the cells; d2, 24 hours after plating). (C) α-SMA protein levels. BDL, liver lysate from BDL; 2w, as a positive control.
Inhibition of PDE4 by Rolipram Attenuates In Vitro Activation of HSC.
Having shown that HSCs express PDE4 isoforms very early during culture activation, we sought to determine whether their activity may play a role in the initiation of this process. To answer this question, we added rolipram, a specific inhibitor of PDE4 activity to the culture medium and plated freshly isolated rat HSCs in the presence of this inhibitor. In agreement with the in vivo results, expression of collagen type I mRNA and α-SMA protein, indicators of HSC activation, were attenuated in the presence of rolipram (Fig. 8, A–D). We also examined the effect of rolipram on morphologic changes in HSCs. In vitro activation of HSC needs several days to become morphologically obvious (Xu et al., 1997; Desmouliere et al., 1999). On day 5, morphologic analysis of HSCs demonstrated an elongated fibroblastic phenotype with a loss of fat droplets indicating their state of activation. In comparison, HSCs cultured in the presence of rolipram continued to be still round, with more fat droplets arranged in circles around the nuclei; DMSO (vehicle control) did not have any effect on the activated HSC morphology (Fig. 8E). These morphologic features together with attenuated expression of the activation markers collagen type I and α-SMA seen with rolipram treatment suggest that inhibition of PDE4 attenuates HSC activation in vitro.
Fig. 8.

Activation of HSCs is attenuated by rolipram treatment. HSCs were activated in culture for 5 days with and without rolipram. (A) Expression of Col1A2 and α-SMA mRNA was significantly decreased by rolipram. (B) Real-time PCR analysis of Col1A1. (C) α-SMA protein levels analyzed by Western blot. (D) Densitometric analysis of α-SMA normalized to GAPDH (n = 3). (E) Cell morphology of rat HSCs at day 5 in culture with or without rolipram. Original magnification: 100×.
Discussion
Clinical and experimental evidence indicates that endotoxemia and dysregulated inflammatory cytokine responses play a significant role in the activation of hepatic stellate cells and the resulting liver fibrosis. Work done by us and others has shown that endotoxin-stimulatable PDE4 expression plays a critical role in regulating the production of inflammatory cytokines (Jin et al., 2005; Gobejishvili et al., 2008, 2011). Hence, the pathogenic role of PDE4 in the BDL-induced hepatic inflammation, injury, and fibrogenesis was examined. The initial stage of BDL-induced cholestatic liver injury was characterized by hepatic upregulation of PDE4B and A isoforms followed by PDE4D, with PDE4B showing maximal expression; further, the PDE4 isoforms showed a sustained increase in expression up to 4 weeks (Fig. 1). Concomitant with the induction of PDE4 isoforms, a significant induction in critical inflammatory and fibrogenic gene expression (TNF, TGF-β1, α-SMA, and Col1A1) was also observed (Figs. 4–6). It is noteworthy that treatment with a specific PDE4 inhibitor, rolipram, considerably blunted these inductions (Figs. 4–6). Particularly with regard to the development of fibrosis, PDE4 inhibition almost completely inhibited not only the major fibrogenic cytokine, TGF-β1, but also its receptor, TβRI, leading to a marked attenuation of hepatic fibrosis (Fig. 5).
Endotoxemia and hepatic inflammation caused by gut-derived LPS play a major role in cholestatic liver injury and fibrosis. In this process, although other hepatic cells are also involved, Kupffer cells play a key role as principal inflammatory effectors in initiating the inflammatory cascade leading to tissue injury, remodeling, and fibrosis. Indeed, LPS-activated hepatic KCs and the production of inflammatory and fibrogenic cytokines play a significant contributory role in the development of fibrosis in the experimental cholestatic liver disease (Seki et al., 2007). In this regard, LPS-inducible PDE4, particularly PDE4B, was observed to play a major regulatory role in the production of TNF by KCs (Fig. 4). These data suggest that PDE4 expression plays a significant pathogenic role in the endotoxin-driven activation of hepatic Kupffer cells and inflammation in experimental cholestatic liver disease.
Besides endotoxemia, inflammatory triggers during cholestatic liver injury are also provided by hepatocytes undergoing bile acid–mediated apoptosis, which is an initiating step of this liver injury. Clearance of apoptotic bodies by phagocytosis leads to generation of TGF-β by Kupffer cells and HSCs, and dying hepatocytes also can release TGF-β (Canbay et al., 2004; Mehal and Imaeda, 2010). Our results show that BDL-mediated liver injury and apoptosis was significantly attenuated by PDE4 inhibition as assessed by reduced liver enzyme levels, decreased TUNEL staining, and PARP-1 cleavage (Fig. 3). Hence, in addition to its anti-inflammatory effects, PDE4 inhibition, which leads to increased cellular cAMP levels, could also attenuate hepatocyte death. Indeed, cAMP has been shown to protect hepatocytes from bile acid–induced death by affecting protein kinase A (PKA) and cAMP-guanine exchange factor–mediated signaling (Webster et al., 2002; Cullen et al., 2004; Reinehr and Haussinger, 2004; Johnston et al., 2011). Furthermore, along with inhibiting TGF-β and its receptor TβR1, PDE4 inhibition also reduced phosphorylation of Smad3 (Fig. 6A), an important downstream signaling component of fibrogenesis. With respect to the role of PDE4 in TGF-induced fibrogenic signaling, it has been shown that TGF-β1–induced activation of lung fibroblasts is critically regulated by PDE4B and D enzymes in normal human lung fibroblasts (Selige et al., 2011), whereas proliferation of normal human lung fibroblasts in response to cytokines is controlled by PDE4A and B. Overall, these data strongly support the notion that PDE4 not only regulates TGF-β/TβR1 expression but also their downstream signaling, which plays a major role in the development of hepatic fibrosis.
The key cellular process of hepatic fibrosis is activation and phenotypic change of HSCs into proliferative, contractile, and chemotactic myofibroblasts producing ECM (Friedman, 2000; Kisseleva and Brenner, 2008, 2011; Aoyama et al., 2010). Activation of HSCs by culturing freshly isolated cells on a stiff surface such as tissue culture plastic in the absence of a direct contact with matrix or other liver cell types represents a widely used model system to study fibrogenic responses in vitro. In this model, HSCs are activated and do not revert back to their quiescent phenotype. In relation to the role of PDE4 in HSC activation and fibrogenesis, its expression (PDE4A, B, and D), which was absent in freshly isolated HSCs, was significantly induced upon culturing preceded α-SMA expression. It is noteworthy that PDE4 inhibition significantly attenuated α-SMA and Col1a1 expression and the morphologic transformation that occurs in activated HSCs upon culturing. These data along with the earlier observations in cultured HSCs (Houglum et al., 1997; Desmouliere et al., 1999) signify that PDE4 expression plays a major role in the regulation of HSC activation.
Liver fibrosis occurs in multiple types of liver injury; unfortunately, there is no U.S. Food and Drug Administration-approved therapy for treatment. Thus, developing novel targets for antifibrotic therapy is an important undertaking, and antifibrotic agents are needed in many forms of liver disease. The present work identifies increased PDE4 expression as a critical pathogenic component in the development of hepatic fibrosis. Altered PDE4 expression and function drive the inflammatory changes during hepatic fibrosis and affect the pathologic inflammatory milieu within the fibrotic liver. It is noteworthy that the data also strongly implies that hepatic PDE4 expression is a potential clinically relevant target and that its inhibition can significantly attenuate cholestatic liver injury and fibrosis.
Abbreviations
- α-SMA
α-smooth muscle actin
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- BDL
bile duct ligation
- GAPDH
glyceraldehyde-3-phosphate dehydrogenase
- HSC
hepatic stellate cell
- KC
Kupffer cell
- LPS
lipopolysaccharide
- PARP-1
poly(ADP-ribose) polymerase 1
- PDE
phosphodiesterase
- TGF-β
transforming growth factor β
- TLR4
Toll-like receptor 4
- TNF-α
tumor necrosis factor-α
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick-end labeling
Authorship Contributions
Participated in research design: Gobejishvili, Barve, Breitkopf-Heinlein, McClain.
Conducted experiments: Gobejishvili, Breitkopf-Heinlein, Li, Zhang, Avila.
Contributed new reagents or analytic tools: Breitkopf-Heinlein, Dooley.
Performed data analysis: Gobejishvili, Barve, Breitkopf-Heinlein.
Wrote or contributed to the writing of the manuscript: Gobejishvili, Barve, Breitkopf-Heinlein, Dooley, McClain.
Footnotes
This work was supported by the National Institutes of Health National Institute on Alcohol Abuse and Alcoholism [Grants R01 AA018869, P01 AA017103, R01 AA018016, R37 AA010762, P30 AA019360, RC2AA019385, R01 AA015970] (to C.J.M.); Department of Veterans Affairs (to C.J.M.); and the Federal Ministry of Education and Research grants “The Virtual Liver” (to S.D.).
References
- Aoyama T, Paik YH, Seki E. (2010) Toll-like receptor signaling and liver fibrosis. Gastroenterol Res Pract 2010: DOI: 10.1155/2010/192543 [Epub]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canbay A, Friedman S, Gores GJ. (2004) Apoptosis: the nexus of liver injury and fibrosis. Hepatology 39:273–278 [DOI] [PubMed] [Google Scholar]
- Cullen KA, McCool J, Anwer MS, Webster CR. (2004) Activation of cAMP-guanine exchange factor confers PKA-independent protection from hepatocyte apoptosis. Am J Physiol Gastrointest Liver Physiol 287:G334–G343 [DOI] [PubMed] [Google Scholar]
- Desmoulière A, Xu G, Costa AM, Yousef IM, Gabbiani G, Tuchweber B. (1999) Effect of pentoxifylline on early proliferation and phenotypic modulation of fibrogenic cells in two rat models of liver fibrosis and on cultured hepatic stellate cells. J Hepatol 30:621–631 [DOI] [PubMed] [Google Scholar]
- Fischer W, Schudt C, Wendel A. (1993) Protection by phosphodiesterase inhibitors against endotoxin-induced liver injury in galactosamine-sensitized mice. Biochem Pharmacol 45:2399–2404 [DOI] [PubMed] [Google Scholar]
- Fladmark KE, Gjertsen BT, Døskeland SO, Vintermyr OK. (1997) Fas/APO-1(CD95)-induced apoptosis of primary hepatocytes is inhibited by cAMP. Biochem Biophys Res Commun 232:20–25 [DOI] [PubMed] [Google Scholar]
- Friedman SL. (2000) Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J Biol Chem 275:2247–2250 [DOI] [PubMed] [Google Scholar]
- Gantner F, Küsters S, Wendel A, Hatzelmann A, Schudt C, Tiegs G. (1997) Protection from T cell-mediated murine liver failure by phosphodiesterase inhibitors. J Pharmacol Exp Ther 280:53–60 [PubMed] [Google Scholar]
- Gobejishvili L, Barve S, Joshi-Barve S, Uriarte S, Song Z, McClain C. (2006) Chronic ethanol-mediated decrease in cAMP primes macrophages to enhanced LPS-inducible NF-kappaB activity and TNF expression: relevance to alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 291:G681–G688 [DOI] [PubMed] [Google Scholar]
- Gobejishvili L, Barve S, Joshi-Barve S, McClain C. (2008) Enhanced PDE4B expression augments LPS-inducible TNF expression in ethanol-primed monocytes: relevance to alcoholic liver disease. Am J Physiol Gastrointest Liver Physiol 295:G718–G724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gobejishvili L, Avila DV, Barker DF, Ghare S, Henderson D, Brock GN, Kirpich IA, Joshi-Barve S, Mokshagundam SP, McClain CJ, et al. (2011) S-Adenosylmethionine decreases lipopolysaccharide-induced phosphodiesterase 4B2 and attenuates tumor necrosis factor expression via cAMP/protein kinase A pathway. J Pharmacol Exp Ther 337:433–443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houglum K, Lee KS, Chojkier M. (1997) Proliferation of hepatic stellate cells is inhibited by phosphorylation of CREB on serine 133. J Clin Invest 99:1322–1328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SL, Conti M. (2002) Induction of the cyclic nucleotide phosphodiesterase PDE4B is essential for LPS-activated TNF-alpha responses. Proc Natl Acad Sci USA 99:7628–7633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SL, Lan L, Zoudilova M, Conti M. (2005) Specific role of phosphodiesterase 4B in lipopolysaccharide-induced signaling in mouse macrophages. J Immunol 175:1523–1531 [DOI] [PubMed] [Google Scholar]
- Johnston A, Ponzetti K, Anwer MS, Webster CR. (2011) cAMP-guanine exchange factor protection from bile acid-induced hepatocyte apoptosis involves glycogen synthase kinase regulation of c-Jun NH2-terminal kinase. Am J Physiol Gastrointest Liver Physiol 301:G385–G400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisseleva T, Brenner DA. (2008) Mechanisms of fibrogenesis. Exp Biol Med (Maywood) 233:109–122 [DOI] [PubMed] [Google Scholar]
- Kisseleva T, Brenner DA. (2011) Anti-fibrogenic strategies and the regression of fibrosis. Best Pract Res Clin Gastroenterol 25:305–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. (1994) Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature 371:346–347 [DOI] [PubMed] [Google Scholar]
- Li J, Yang S, Billiar TR. (2000) Cyclic nucleotides suppress tumor necrosis factor alpha-mediated apoptosis by inhibiting caspase activation and cytochrome c release in primary hepatocytes via a mechanism independent of Akt activation. J Biol Chem 275:13026–13034 [DOI] [PubMed] [Google Scholar]
- Li JT, Liao ZX, Ping J, Xu D, Wang H. (2008) Molecular mechanism of hepatic stellate cell activation and antifibrotic therapeutic strategies. J Gastroenterol 43:419–428 [DOI] [PubMed] [Google Scholar]
- Matsuhashi T, Otaka M, Odashima M, Jin M, Komatsu K, Konishi N, Wada I, Sato T, Horikawa Y, Ohba R, et al. (2005) Specific type IV phosphodiesterase inhibitor ameliorates thioacetamide-induced liver injury in rats. J Gastroenterol Hepatol 20:135–140 [DOI] [PubMed] [Google Scholar]
- Mehal W, Imaeda A. (2010) Cell death and fibrogenesis. Semin Liver Dis 30:226–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odashima M, Otaka M, Jin M, Komatsu K, Konishi N, Wada I, Horikawa Y, Matsuhashi T, Ohba R, Oyake J, et al. (2005) Rolipram, a specific type IV phosphodiesterase inhibitor, ameliorates aspirin-induced gastric mucosal injury in rats. Dig Dis Sci 50:1097–1102 [DOI] [PubMed] [Google Scholar]
- Reinehr R, Häussinger D. (2004) Inhibition of bile salt-induced apoptosis by cyclic AMP involves serine/threonine phosphorylation of CD95. Gastroenterology 126:249–262 [DOI] [PubMed] [Google Scholar]
- Sanz MJ, Alvarez A, Piqueras L, Cerdá M, Issekutz AC, Lobb RR, Cortijo J, Morcillo EJ. (2002) Rolipram inhibits leukocyte-endothelial cell interactions in vivo through P- and E-selectin downregulation. Br J Pharmacol 135:1872–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnabl B, Kweon YO, Frederick JP, Wang XF, Rippe RA, Brenner DA. (2001) The role of Smad3 in mediating mouse hepatic stellate cell activation. Hepatology 34:89–100 [DOI] [PubMed] [Google Scholar]
- Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. (2007) TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med 13:1324–1332 [DOI] [PubMed] [Google Scholar]
- Selige J, Hatzelmann A, Dunkern T. (2011) The differential impact of PDE4 subtypes in human lung fibroblasts on cytokine-induced proliferation and myofibroblast conversion. J Cell Physiol 226:1970–1980 [DOI] [PubMed] [Google Scholar]
- Song M, Zhou Z, Chen T, Zhang J, McClain CJ. (2011) Copper deficiency exacerbates bile duct ligation-induced liver injury and fibrosis in rats. J Pharmacol Exp Ther 339:298–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taguchi I, Oka K, Kitamura K, Sugiura M, Oku A and Matsumoto M (1999) Protection by a cyclic AMP-specific phosphodiesterase inhibitor, rolipram, and dibutyryl cyclic AMP against Propionibacterium acnes and lipopolysaccharide-induced mouse hepatitis. Inflamm Res 48:380–385. [DOI] [PubMed]
- Tukov FF, Luyendyk JP, Ganey PE and Roth RA (2007) The role of tumor necrosis factor alpha in lipopolysaccharide/ranitidine-induced inflammatory liver injury. Toxicol Sci 100:267–280. [DOI] [PubMed]
- Videla S, Vilaseca J, Medina C, Mourelle M, Guarner F, Salas A, Malagelada JR. (2006) Selective inhibition of phosphodiesterase-4 ameliorates chronic colitis and prevents intestinal fibrosis. J Pharmacol Exp Ther 316:940–945 [DOI] [PubMed] [Google Scholar]
- Webster CR, Usechak P, Anwer MS. (2002) cAMP inhibits bile acid-induced apoptosis by blocking caspase activation and cytochrome c release. Am J Physiol Gastrointest Liver Physiol 283:G727–G738 [DOI] [PubMed] [Google Scholar]
- Wiercinska E, Wickert L, Denecke B, Said HM, Hamzavi J, Gressner AM, Thorikay M, ten Dijke P, Mertens PR, Breitkopf K, et al. (2006) Id1 is a critical mediator in TGF-beta-induced transdifferentiation of rat hepatic stellate cells. Hepatology 43:1032–1041 [DOI] [PubMed] [Google Scholar]
- Windmeier C, Gressner AM. (1997) Pharmacological aspects of pentoxifylline with emphasis on its inhibitory actions on hepatic fibrogenesis. Gen Pharmacol 29:181–196 [DOI] [PubMed] [Google Scholar]
- Xiang M, Zaccone P, Di Marco R, Magro G, Di Mauro M, Beltrami B, Meroni PL, Nicoletti F. (1999) Prevention by rolipram of concanavalin A-induced T-cell-dependent hepatitis in mice. Eur J Pharmacol 367:399–404 [DOI] [PubMed] [Google Scholar]
- Xu G, Niki T, Virtanen I, Rogiers V, De Bleser P, Geerts A. (1997) Gene expression and synthesis of fibronectin isoforms in rat hepatic stellate cells. Comparison with liver parenchymal cells and skin fibroblasts. J Pathol 183:90–98 [DOI] [PubMed] [Google Scholar]
- Zidek Z. (1999) Adenosine-cyclic AMP pathways and cytokine expression. Eur Cytokine Netw 10:319–328 [PubMed] [Google Scholar]