

Abstract
The most highly abused prescription drugs are opioids used for the treatment of pain. Physician-reported drug-seeking behavior has resulted in a significant health concern among doctors trying to adequately treat pain while limiting the misuse or diversion of pain medications. In addition to abuse liability, opioid use is associated with unwanted side effects that complicate pain management, including opioid-induced emesis and constipation. This has resulted in restricting long-term doses of opioids and inadequate treatment of both acute and chronic debilitating pain, demonstrating a compelling need for novel analgesics. Recent reports indicate that adaptations in endogenous substance P/neurokinin-1 receptor (NK1) are induced by chronic pain and sustained opioid exposure, and these changes may contribute to processes responsible for opioid abuse liability, emesis, and analgesic tolerance. Here, we describe a multifunctional mu-/delta-opioid agonist/NK1 antagonist compound [Tyr-d-Ala-Gly-Phe-Met-Pro-Leu-Trp-NH-Bn(CF3)2 (TY027)] that has a preclinical profile of excellent antinociceptive efficacy, low abuse liability, and no opioid-related emesis or constipation. In rodent models of acute and neuropathic pain, TY027 demonstrates analgesic efficacy following central or systemic administration with a plasma half-life of more than 4 hours and central nervous system penetration. These data demonstrate that an innovative opioid designed to contest the pathology created by chronic pain and sustained opioids results in antinociceptive efficacy in rodent models, with significantly fewer side effects than morphine. Such rationally designed, multitargeted compounds are a promising therapeutic approach in treating patients who suffer from acute and chronic pain.
Introduction
Approximately 1.5 billion patients worldwide suffer from chronic pain of various etiologies—each with a unique pathology (Institute of Medicine, 2011). Opioids effectively alleviate many of these pains but are limited by the development of adverse events (e.g., nausea, constipation, dependence) and decreased efficacy at tolerable doses over time (Jensen and Finnerup, 2009). Despite a myriad of analgesic compounds on the market, nearly two-thirds of patients report inadequate pain relief (Jensen and Finnerup, 2009), whereas the risks associated with opioid addiction have enhanced concerns in both patients (36%) and physicians (68%) (Institute of Medicine, 2011). In the United States, prescribed opioids [e.g., μ-opioid receptor (MOP) agonists) have become some of the most highly abused drugs as measured by treatment center admission/cause of overdose (Hall et al., 2008; Centers for Disease Control and Prevention, 2011), with a National Institute on Drug Abuse survey reporting a 13% increase in prescription drug abuse in 2009. Efforts to develop abuse-deterrent formulations have done little to assuage the misuse of opioids and may be contributing to an increase in use of more potent formulations (Cicero et al., 2012). Although many studies in the last 15 years concerning these opioid abuses examine the idea that both injury and sustained MOP therapy can induce neuroplastic adaptations, no drug-development strategies have targeted such changes.
Adaptations that may contribute to the development of opioid adverse events include altered opioid receptor regulation, trafficking, activation, signaling, and interactions with nonopioid receptors (Vanderah et al., 2001; Szücs et al., 2004; Christie, 2008; Yu et al., 2009). It is noteworthy that adaptations in the δ-opioid receptor (DOP) may be of interest for drug discovery (Cahill et al., 2003; Gendron et al., 2007). DOP ligands lack side effects often associated with MOP activation in vivo, including antinociceptive tolerance, physical dependence, and self-administration (Negus et al., 1998). DOP knockout mice show enhanced behavioral responses to inflammation and neuropathic injury (Nadal et al., 2006; Gavériaux-Ruff et al., 2008). Interactions between MOP/DOP have been reported and indicate that both activation and blockade of the DOP can improve analgesia and attenuate morphine-induced tolerance (Gendron et al., 2007; Dietis et al., 2009; Schramm and Honda, 2010). Dual targeting of MOP and DOP with agonists may provide enhanced opioid-mediated antinociception (Vanderah, 2010).
Adaptations in pronociceptive systems are linked to “antiopioid” effects. In particular, expression and function of substance P (SP) and its primary receptor, the neurokinin-1 receptor (NK1), are altered following injury and sustained exposure to opioids. Anatomic (Aicher et al., 2000; Wan et al., 2006) and system-based interactions between opioids and SP-NK1 (Hylden and Wilcox, 1982; Cahill and Coderre, 2002; King et al., 2005) have been reported. SP content/release is altered in injury (Abbadie et al., 1996; Malcangio et al., 2000) and after sustained opioids (King et al., 2005; Lu et al., 2011) with enhanced activation of NK1 receptors throughout the nociceptive axis (Budai et al., 2007). The SP-NK1 system is implicated in the mechanisms underlying opioid antinociceptive tolerance, withdrawal, and reward (Commons, 2010). Chemical ablation of NK1-expressing neurons in rodents attenuated hyperalgesia (Mantyh et al., 1997; Vera-Portocarrero et al., 2007; Rivat et al., 2009), reward/anxiety (Gadd et al., 2003), and reduced symptoms of physical withdrawal (Maldonado et al., 1993). Coadministration of morphine with an NK1 antagonist attenuated the development of opioid antinociceptive tolerance in rats (Powell et al., 2003), with NK1 antagonists acting as antiemetics (Munoz and Covenas, 2010). Yet a pure NK1 antagonist failed in several clinical pain trials, likely due to actions in blocking a single neuromodulator associated with a specific sensory function at a selective postsynaptic target while alternative pronociceptive pathways simultaneously persist (Hill, 2000). Compounds that integrate blockade of antiopioid systems with activation of opioid receptors may better enhance pain relief with fewer side effects.
We previously reported on peptidomimetic compounds that targeted MOP/DOP/NK1 (Yamamoto et al., 2007) and characterized them in rodent models of pain (Largent-Milnes et al., 2010). The 3′,5′-bis(trifluoromethyl)-benzyl amide derivative of Tyr-d-Ala-Gly-Phe-Met-Pro-Leu-Trp-O-Bn(CF3)2 (TY005) produced Tyr-d-Ala-Gly-Phe-Met-Pro-Leu-Trp-NH-Bn(CF3)2 (TY027), which has high affinity for the MOP, DOP, and NK1 receptors; acts as an agonist at MOP and DOP and as an NK1 antagonist; and is biologically stable (Yamamoto et al., 2007, 2009). Here we demonstrate that TY027 is highly efficacious in rodent models of acute and neuropathic pain following multiple routes of administration while reducing adverse effects associated with opioid therapy, including antinociceptive tolerance, reward liability, gastrointestinal impairment, and physical dependence over a long duration. Such multifunctional compounds with a reduced side effect profile will address the urgent need for effective chronic pain therapy and minimize patient/physician concerns.
Materials and Methods
Animals
All animal procedures were performed in accordance with the policies and recommendations of the International Association for the Study of Pain, the National Institutes of Health, and with approval from the Animal Care and Use Committee of the University of Arizona for the handling and use of laboratory animals. Adult male Sprague-Dawley rats (175–300 g; Harlan, Indianapolis, IN), male ICR mice (20–30 g; Harlan), and male ferrets (1.1–1.5 kg; Marshall BioResources, North Rose, NY) were kept in a temperature-controlled environment with lights on 07:00–19:00 with food and water available ad libitum.
Drugs
Morphine sulfate (MS; 7,8-didehydro-4,5a-epoxy-17-methylmorphinan-3,6-a-diol sulfate) was obtained from the National Institute on Drug Abuse. The nonselective opioid antagonist naloxone [NLX; (5α)-4,5-epoxy-3,14-dihydro-17-(2-propenyl)morphinan-6-one hydrochloride), [14C]sucrose, and peptidergic compounds d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2 (CTAP), [d-Pen2,5]enkephalin, [d-Pen2, d-Pen5]enkephalin (DPDPE), and (Tyr-d-Ala-Gly-Phe-NH)2 (biphalin) were purchased from Tocris Biosciences (Bristol, UK) or synthesized in house; unless otherwise stated, MS and NLX were dissolved in 0.9% saline. SP (Arg-Pro-Lys-Pro-Gln-Gln-Phe-Phe-Gly-Leu-Met; American Peptide, Sunnyvale, CA) was prepared as a solution in saline or 5% acetic acid/saline. TY027 and 125I-TY027 were synthesized as described by Yamamoto et al. (2007). Central dosing of TY027 ranged between 0.38 and 100.0 μg in 5 μl and 1 and 30 mg/kg (1 ml/kg volume) for experiments with systemic dosing. TY027 was dissolved in dimethylsulfoxide (DMSO; Tocris Biosciences) or hydroxy-cyclo-β-dextran (Sigma-Aldrich, St. Louis, MO) and brought to final volume in either Millipore water (central doses; Millipore, Billerica, MA) or Tween 80/saline (systemic doses).
In Situ Brain Perfusion
Mice and rats were anesthetized and injected with heparin (10,000 U/kg i.p.), and the common carotid arteries were cannulated, perfused with erythrocyte-free modified mammalian Ringer’s solution [117 mM NaCl, 4.7 mM KCl, 0.8 mM MgSO4, 1.2 mM KH2PO4, 2.5 mM CaCl2, 10 d-glucose, 3.9% dextran (w/v; Mr 60,000)] and 1.0 g/l bovine serum albumin-type IV (pH 7.4), and then oxygenated with 95% O2,5% CO2 at a constant pressure (95–105 mm Hg) and rate (3.1 ml/min). Evans blue dye (55 mg/l) was added to the perfusate to visually mark blood-brain barrier (BBB) integrity. [14C]Sucrose (10 μCi of sucrose per 20 ml of perfusate) or 125I-TY027 (10 μCi per 20 ml of perfusate TY027) was added to the inflowing solution at a rate of 0.5 ml/min for 15 minutes. After perfusion, animals were decapitated, the meninges and choroid plexus were excised, and the cerebral hemispheres were sectioned. TS2 tissue solubilizer (Research Products International Corp., Mt. Prospect, IL) was added to the tissue samples and solubilized for 2 days at room temperature. To eliminate chemiluminescence, 100 μl of 30% glacial acetic acid was added with 2.0 ml of OptiPhase Supermix liquid scintillation cocktail (PerkinElmer Life and Analytical Sciences, Waltham, MA). Radioactivity was measured to determine the BBB penetration of TY027 using a model 1450 liquid scintillation and luminescence counter (PerkinElmer Life and Analytical Sciences). Results are reported as the percentage ratio of radioactivity in the brain to that in the perfusate (RBr) for rat perfusions, which is equal to the total amount of radiolabeled isotope in the whole brain [eq. 1: (Cbrain, in disintegrations/min/g) divided by amount of radiolabeled isotope in perfusate (Cperfusate, in disintegrations/min/ml)]. Mouse data are presented as unidirectional uptake constant Kin (RBr: µl/g brain tissue/time in minutes); CTAP, DPDPE, and biphalin (MOP/DOP selective agonists) served as positive controls and peptide controls.
| (1) |
Surgical Procedures
Intracerebroventricular.
Direct injection of solution into the lateral ventricles allowed study of any supraspinal effect in both mice and rats, eliminating pharmacokinetic alterations and giving direct proof of concept of TY027 activity at the site of action. Short-term injections in mice were performed by direct puncture into the ventricle. In brief, mice were anesthetized, and a small incision through the skin was made midline on top of the head to expose the skull. Compounds were injected (5 µl) 2 mm posterior, 2 mm lateral from the bregma, and 3 mm below the skull. For long-term studies in rats, indwelling cannulae were inserted to allow for multiple intracerebroventricular injections. Rats were fixed in a stereotaxic head holder (−3.5-mm nose bar; Stoelting, Wood Dale, IL), and the skull was exposed 2 cm. A guide cannula (Plastics One, Inc., Roanoke, VA) was inserted according to Paxinos and Watson (1997) from the bregma (antero/posterior: −1.5 mm; medio/lateral: −1 mm; and dorso/ventral: −3 mm from the skull). Cannulae were fixed to the skull with cyanoacrylate glue and application of dental cement around anchor screws (antero/posterior: −3 mm; medio/lateral: ±2 mm; Small Parts, Inc., Miami, FL). Injections (5 µl i.c.v.) of compound or vehicle were performed and the movement of fluid visually monitored. The injector was slowly removed after 30 seconds and replaced with a dummy cannula to prevent any backflow of drug.
Spinal, Intrathecal.
Rats were anesthetized (ketamine/xylazine anesthesia, 80/12 mg/kg i.p.; Sigma-Aldrich) and placed in a stereotaxic head holder. The cisterna magna was exposed and incised, and an 8-cm catheter (PE-10; Stoelting) was implanted as previously reported (Largent-Milnes et al., 2010), terminating in the lumbar region of the spinal cord. Catheters were sutured (3-0 silk suture) into the deep muscle and externalized at the back of the neck. Vehicle and compounds (5 µl) were injected while rats moved freely in their home cages.
Intravenous.
Short-term intravenous injections were performed in awake rats by placing the animals in an appropriate restrainer (VWR International, Tempe, AZ) and dilating the distal third of the tail in warm water for 5 seconds. Injections were performed over a 10-second period and were noted as positively placed by the presence of blood in the tip of the syringe before injection and the absence of an outpocketing of the tail skin at the site of injection. For repeated intravenous injections, a cannula was inserted into the right jugular vein of anesthetized rats. The vein was exposed, tied off using a 4-0 silk suture, and pierced with a 27-gauge needle to create an opening for an 8-cm length of PE-50 tubing filled with 10% heparinized saline inserted 2 cm into the vein. The closed cannula was then threaded subcutaneously and externalized at the back of the neck to prevent displacement by the animal.
Spinal Nerve Ligation.
A well-accepted nerve injury model is the rat spinal nerve L5/L6 ligation. In brief, the skin over the caudal lumbar region was incised, and the muscles were retracted. Under 2.5% isoflurane (Thermo Fisher Scientific, Waltham, MA) in O2 anesthesia delivered at 2 l/min, the L5 and L6 spinal nerves were exposed, carefully isolated, and tightly ligated with 4-0 silk distal to the dorsal root ganglion, as previously cited (Largent-Milnes et al., 2010). Any animals exhibiting signs of motor deficiency, infection, neurologic deficit, or >10% loss in total body weight were euthanized. Control animals underwent sham surgeries in which the nerves were exposed but not ligated.
Recovery.
Animals were housed individually and allowed to recover for 5–7 days between the time of surgery and the first manipulation. When multiple surgical procedures were performed on the same animal [e.g., intrathecal/spinal nerve ligation (SNL)], the same recovery period was allowed between procedures followed by another 7 days prior to any behavioral testing/drug manipulation.
Behavioral Assays
Antinociception/Thermal Hypersensitivity: Tail-Flick Latency.
The distal third of the mouse tail was immersed in a 55°C water bath, and the tail-flick latency (TFL) was recorded. Drug-naïve TFL values were between 1.5 and 3.0 seconds; animals with a baseline TFL >5 seconds were excluded. A cutoff latency of 10.0 seconds was established to prevent tissue injury. Paw-flick latency [as performed by Largent-Milnes et al. (2010)] was determined as follows. Rats were allowed to acclimate to the testing room for 30 minutes prior to testing. Basal paw withdrawal latencies (PWLs) to an infrared radiant heat source were measured and ranged between 20.0 and 25.0 seconds. A cutoff time of 33.0 seconds was used to prevent tissue damage.
Tactile Hypersensitivity.
Calibrated von Frey filaments (0.4–15.0 g) were used to probe the plantar surface of the left hind paw (ipsilateral to the SNL) of rats for 7 seconds, before and after SNL, and following drug administration utilizing the up-down method. Probes of greater force were not used because lifting of the paw with the filament confounded results; therefore, only mechanical allodynia, and not hyperalgesia, was assessed. Lifting or licking of the paw or vocalizing counted as positive responses to the mechanical probing (Largent-Milnes et al., 2010). Paw withdrawal thresholds (PWTs) were calculated in grams using the Dixon nonparametric test.
SP-Induced Nocifensive Behaviors.
Testing was performed as described by Seybold et al. (1982). SP administration (1 mM, 10 µl i.t.) was performed at t = 0 minutes with a total SP dose of 13.47 µg/rat. Some rats received either saline or NLX (2 mg/kg s.c.) at t = −25 minutes to block endogenous and TY027 opioid activity. TY027 (30 µg) or 10% DMSO was injected 15 minutes prior to SP, and the number of flinches, bites, or scratches per minute for the first 5 minutes was counted.
Conditioned Place Preference/Aversion.
Three chambered, rat-conditioned place preference/aversion (CPP/CPA) apparatus (San Diego Instruments, San Diego, CA) were customized as follows: 1) end chamber of horizontal black/white striped walls, smooth floor; 2) end chamber of black walls, rough floor; and 3) middle chamber of neutral gray walls and metal rod flooring. Rats were allowed to habituate to the testing room for 30–60 minutes prior to 20-minute recordings. Two baseline recordings were obtained on days 1 and 4; the average was used as the overall baseline for the putative conditioning chamber. Any rat with >80% preference to any chamber during baseline was excluded. At random, rats were separated into treatment groups and paired to associate one of the end chambers with drug and the other end chamber with vehicle on alternating days for a total of five drug exposures. After both central (intracerebroventricular) and systemic dosing (intravenous), CPP/CPA was assessed for control rats receiving vehicle-vehicle or vehicle-MS (10 µg or 3 mg/kg). Vehicle TY027 was administered at 20 µg i.c.v. or 6 mg/kg i.v. to provide equivalent dosing of the opioid component compared with MS. Evaluations of compound-induced CPP/CPA occurred in a drug-free state, 24 hours after drug exposure.
Naloxone-Precipitated Opioid Withdrawal.
Rats were given intravenous injections of vehicle [10% DMSO/10% Tween 80/80% saline (DTS) and saline], MS (3 mg/kg), or TY027 (10 mg/kg) twice daily for 3 days and then monitored for physical signs of spontaneous and NLX-precipitated opioid withdrawal on the fourth day, as previously described (Vera-Portocarrero et al., 2011). After a 30-minute habituation period, animals were evaluated for signs of spontaneous opioid withdrawal for 30 minutes. After this period of observations, rats were administered NLX (3 mg/kg s.c.) and observed for 30 minutes for signs of precipitated withdrawal. NLX-induced opioid withdrawal signs, including teeth chattering, grooming, wet-dog shakes, yawning, jumping, and writhing, were counted. Autonomic responses to NLX were recorded, including the development of diarrhea (yes/no) and weight loss (in grams; pre-NLX–post-NLX weight).
Opioid-Induced Emesis.
Ferrets, identified using metal ear tags and numbered collars, were handled before/after every experiment and at least three times a week. TY027 (3 mg/kg i.p.) and MS (0.6 mg/kg i.p.) were administered following a 30-minute acclimation period. Saline and DTS were used as vehicle controls for MS and TY027, respectively. The order of injections was randomized for each ferret in a blinded fashion, with 4 days allowed for washout between exposures. Animals were observed for the first 30 minutes after each dosing, and the number of retches/vomits was recorded in 5-minute intervals. Retching was defined as any rhythmic abdominal contraction without expulsion, whereas vomiting was any oral expulsion episode.
Gastrointestinal Transit.
Gastric emptying and geometric center were evaluated in rats receiving a nutritive meal of 1.5 ml of 2% milk marked with 51Cr by oral gavage immediately after intravenous tail vein administration of vehicle (20% DMSO), MS (1 mg/kg), or TY027 (3 mg/kg). Fifteen minutes later, animals were anesthetized with isoflurane and sacrificed by cervical dislocation, and the stomach and small bowel were excised. The gut was divided into 10 equal segments between the stomach and the cecum and placed in a glass culture tube. The amount of radioactivity per section was measured in a gamma counter with the percentage of gastric emptying determined by the proportion of the 51Cr marker in small intestine segments. Gastrointestinal propulsion was evaluated by determining movement of the marker along the small intestine using a geometric center calculation, where values 1–10 indicate total inhibition (1) or complete transit through the intestine (10), respectively.
Statistical Analysis
Data were analyzed by nonparametric two-way analysis of variance (ANOVA; post-hoc Student-Newman-Keuls), one-way ANOVA, and Student’s t-test when appropriate in FlashCalc (Dr. Michael H. Ossipov, University of Arizona, Tucson, AZ). Data were plotted in GraphPad Prism 4 (GraphPad Software, La Jolla, CA) and represent the mean ± S.E.M. where statistical significance was P ≤ 0.05 unless otherwise stated.
Results
TY027 Produces Antinociception in Noninjured Animals
Five doses of TY027 (0.38–38.0 µg in 5µl i.c.v.) were evaluated using the tail-flick assay (cutoff 10 seconds) in male ICR mice until the observed effect returned to baseline levels (Fig. 1A). Short-term supraspinal TY027 (0.38 µg) produced significant antinociception compared with baseline TFLs (21.5 ± 4.6%) 10 minutes after administration. Increasing the dose of TY027 yielded a maximal effect observed 20 minutes after injection, with TFLs calculated to be 68.1 ± 13.6 and 83.9 ± 6.2% above baseline for 1.21 and 3.80 µg, respectively. Maximal antinociception (100.0 ± 0.0%) was observed with 12.1 and 38.0 µg of TY027 as the TFLs reached cutoff values. Furthermore, the peptide’s duration of action increased dose-dependently (Fig. 1A), where TFL in mice treated with 1 bolus of the highest doses remained above baseline levels for 180 minutes. The calculated intracerebroventricular A50 was 0.93 µg [95% confidence interval (CI) = 0.62–1.39 µg] in mice, similar to the published data for morphine (A50 = 0.68 µg, 95% CI = 0.51–0.92 µg; Ananthan et al., 1999).
Fig. 1.

TY027 dose-dependently attenuated acute pain in noninjured mice and rats by either supraspinal or spinal administration. (A) Mice, intracerebroventricular dose- and time-response tail-flick latencies of TY027. (B) Rats, intrathecal dose- and time-response paw withdrawal latencies of TY027. (C) Pretreatment of mice with the nonselective antagonist NLX (10 mg/kg i.p., t = −20 minutes) blocked TY027 (intracerebroventricular)-mediated antinociception, indicating that the peptide was active at supraspinal opioid receptors in mice. (D) Pretreatment of rats with NLX (2 mg/kg i.p., t = −20 minutes) blocked TY027 (intrathecal)-mediated antinociception, indicating that the peptide was active at spinal opioid receptors in rats. Data represent the mean ± S.E.M. (n = 6–8/dose). *P ≤ 0.05; **P ≤ 0.001. BL, baseline.
Some species differences and sites of action in antinociceptive activity of novel compounds may exist given minor differences in receptor affinities and in vitro activities; therefore, rats were also used to assess antinociception by a spinal route of administration. All doses of TY027 (1–30 µg in 5 µl i.t.; Fig. 1B) resulted in a significant increase in PWL in rats in response to application of infrared heat within 15 minutes of the injection compared with control values (baseline PWL and vehicle-treated rats; P ≤ 0.05). The duration of TY027-mediated antinociception continued to be significant 30 minutes after dosing with 3 (67.4± 17.4%), 10 (100.0 ± 0.0%), and 30 µg (100.0 ± 0.0%) in 5 µl, but not with 1 µg, compared with baseline PWLs (Fig. 1B). Both minimal and maximal effects were observed at 30 minutes with the range of doses used; therefore, this time point was used to calculate the A50 value of TY027 in the acute pain model after spinal administration. TY027 (intrathecally) dose-dependently attenuated acute pain, with A50 = 1.8 µg (R2 = 0.77, 95% CI = 0.5–5.0 µg in 5 µl), a value lower than we have previously cited for morphine (5.3 µg; Largent-Milnes et al., 2010). For the higher doses, the duration of TY027 antinociception lasted 45 minutes after injection of 10 and 30 µg in 5 µl (60.9 ± 12.8 and 51.2 ± 19.6%, respectively; P ≤ 0.05, ANOVA).
To ensure that PWL results were not confounded by a drug-induced motor impairment, rotarod latencies were obtained every 15 minutes for the first hour and every 30 minutes for a maximum of 3 hours in naïve rats (n = 7–8 per group). Neither vehicle (10% DMSO) nor TY027 (30 µg) nor a 3-fold higher dose (100 µg) significantly changed rotarod latencies relative to baseline values at any time (P > 0.05).
On-Target Activity of TY027 In Vivo
To determine the in vivo opioid actions of TY027 at opioid receptors, experiments were performed such that a nonselective opioid antagonist was given as a pretreatment to TY027 in both mice (55°C tail flick; Fig. 1C) and rats (paw flick; Fig. 1D). The mean TFL in male ICR mice prior to compound exposure was determined to be 2.0 ± 0.1 seconds. In the absence of NLX, TY027 (3.8 µg i.c.v.) resulted in a significant increase in tail-flick latency (8.7 ± 0.6 seconds) compared with vehicle and baseline values (P ≤ 0.001). When NLX (10 mg/kg i.p.) was given 20 minutes prior to TY027 (3.8 µg i.c.v.), mice withdrew the tail at significantly lower latencies (P ≤ 0.05; Fig. 1C). In rats, the mean basal PWL was 22.0 ± 0.5 seconds. Neither pretreatment (20 minutes) with saline nor NLX altered PWLs of vehicle control rats compared with baseline. TY027 (30 µg) maximally increased the PWL 20 minutes postinjection (30.8 ± 1.4 seconds; P ≤ 0.001), and this increase was blocked by NLX (2 mg/kg i.p.) pretreatment (P ≤ 0.001; Fig. 1D).
We next evaluated the NK1 antagonist activity of TY027 against SP-induced nocifensive behaviors of flinching, biting, and scratching. Animals were pretreated with either saline or NLX (2 mg/kg s.c.) to block opioid receptor–mediated activity followed by 10% DMSO (5 µl) or TY027 (30 µg/5 µl). Spinal injection of SP (10 µl, 1 mM) induced flinching in rats pretreated with both saline and 10% DMSO (Table 1). Injection of TY027 (30 µg) significantly reduced flinching (P ≤ 0.05). NLX given before vehicle/SP did not significantly reduce flinching (P > 0.05), whereas rats exposed to TY027 (30 µg) in the presence of NLX flinched significantly less over 5 minutes for a total of 8.0 ± 3.0 flinches compared with the SP alone and SP + NLX control groups (P ≤ 0.01; Table 1).
TABLE 1.
TY027 acts as an NK1 antagonist in vivo: effects on SP-induced flinching
| Treatment | N | Mean Number of Flinches in 5 Min (± S.E.M.) |
|---|---|---|
| Sa-10% DMSO-SP | 5 | 37.6 (± 7.9) |
| Sa-TY02730-SP | 6 | 10.6 (± 3.4)* |
| NLXb- 10% DMSO-SP | 6 | 26.3 (± 5.5) |
| NLXb-TY02730-SP | 6 | 8.0 (± 3.0)** |
S, 0.9% saline (1 ml/kg s.c.) given 25 minutes before SP (10 µl, 1 mM i.t.), 10% DMSO, or TY027 (dose given in subscript = micrograms in 5 μl i.t.) 15 minutes prior to SP.
NLX (2 mg/kg s.c.) given 25 minutes before SP (10 µl, 1 mM i.t.), 10% DMSO, or TY027 (dose given in subscript = micrograms in 5 μl i.t.) 15 minutes prior to SP.
P ≤ 0.05 and **P ≤ 0.01, determined using one-way ANOVA (FlashCalc) with Newman-Kuels post-hoc analysis.
Blood-Brain Barrier Penetration of TY027
Before in vivo evaluation of systemic TY027, in situ perfusion was performed to assess the potential for TY027 to cross the BBB. Single time point analyses were used for mice (15 minutes) and rats (20 minutes). In both species, the amount of 125I-TY027 distributed in the brain was significantly higher than in the perfusate, as indicated by the uptake rate constant (Kin) calculated as 8.0 ± 0.6 µl/min/g for mice (P ≤ 0.05; Fig. 2A), the value determined for rats (2.1 ± 1.0 µl/min/g, P ≤ 0.05; data not shown). Despite these small species differences in Kin, the amount of TY027 in rats was significantly higher than sucrose (RBr percentage, P ≤ 0.05; Fig. 2B), consistent with the concept that TY027 crosses the BBB in vivo.
Fig. 2.
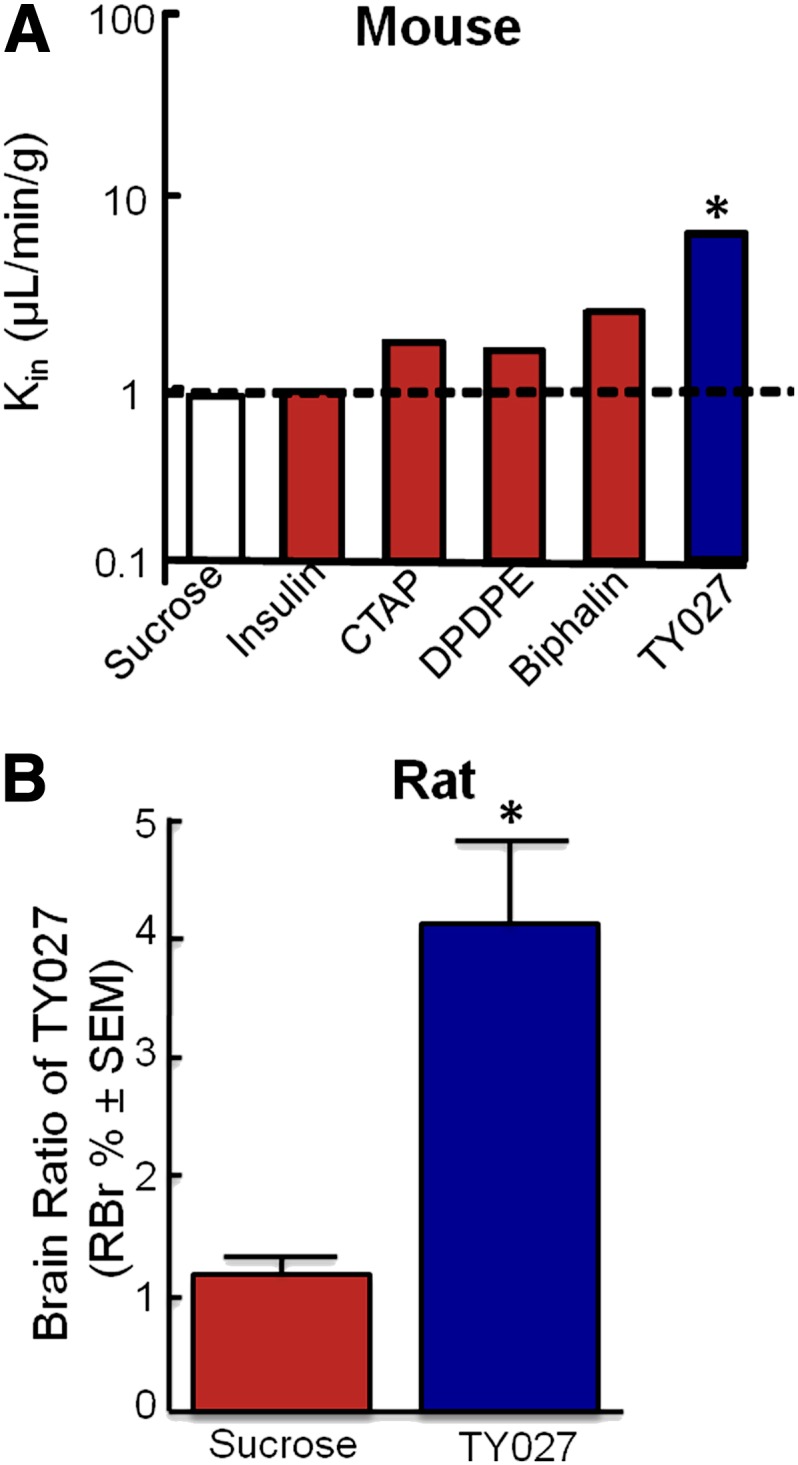
TY027 crosses the BBB. (A) The rate at which TY027 is taken into the brain, Kin, in mice relative to negative controls—sucrose and insulin—and known values of positive peptide controls (CTAP, DPDPE, biphalin) following a 15-minute in situ perfusion. Dotted line denotes the threshold for crossing the BBB; below the line indicates the vascular component, whereas above the dotted line represents compound BBB penetration. (B) The relative ratio of TY027 in the rat brain compared with sucrose following in situ perfusion of the compound for 20 minutes (n = 3–5/group). *P ≤ 0.05 compared with sucrose.
Nerve Injury–Induced Hypersensitivities Are Attenuated by Spinal TY027
All nerve injury studies were performed in rats to allow for easy identification of nerves to be ligated using a standard neuropathic pain model and successive testing of the injured hind paw for tactile and thermal hypersensitivities. Radiant heat was selected for the latter, as water immersion of the paw may sensitize the area to tactile stimulation, potentially confounding the results. Rats underwent surgery to implant an intrathecal catheter for spinal drug delivery followed by ligation of the L5/L6 spinal nerves (SNL) to create a model of neuropathic pain. Seven days post-SNL, rats were behaviorally assessed for injury-induced hypersensitivities, and either TY027 or vehicle was administered spinally followed by re-evaluation of hypersensitivities. SNL-induced thermal hyperalgesia prior to drug or vehicle administration was observed as a decrease in PWL from 22.8 ± 0.3 to 14.1 ± 0.4 seconds. Neither vehicle nor TY027 (1 µg/5 µl i.t.) resulted in a significant difference in paw withdrawal latency compared with post-SNL throughout the experiment. However, in the SNL neuropathic model, TY027 (3 µg/5 µl i.t.) significantly increased PWLs (P ≤ 0.05) with a peak time effect of 15 minutes after injection that lasted until 30 minutes after the spinal injection (Fig. 3A). Increasing the dose of TY027 to 10 or 30 µg resulted in a complete reversal of SNL-induced thermal hyperalgesia peaking 30 minutes after administration through 45 minutes (P ≤ 0.05; Fig. 3A). From data collected 30 minutes post-TY027 (the last time point where more than two doses induced significant antihyperalgesia), the A50 value was calculated to be 7.8 µg in 5 µl (95% CI = 3.0–19.8 µg; R2 = 0.70), similar to what we have cited for morphine (Largent-Milnes et al., 2010). Unlike morphine, a second phase of TY027-mediated antihyperalgesia was observed starting 2 hours after spinal administration and peaking at 3 hours (180 minutes). In this late phase, only the highest dose of TY027, 30 µg/5 µl (i.t.), significantly increased PWLs above post-SNL values (percentage antihyperalgesia180 minute: 64.3 ± 10.5%, n = 19; P ≤ 0.05); in 11 of the 19 rats, TY027 reversed SNL-induced thermal hyperalgesia completely.
Fig. 3.

Spinal TY027 attenuates SNL-induced hypersensitivities in rats. (A) Nerve injury induces a significant decrease in thermal PWLs that are reversed by spinal administration of TY027 in a dose-dependent and biphasic manner (*P ≤ 0.05; n = 10–19). (B) Nerve injury induces a significant decrease in mechanical PWTs that are reversed by spinal administration of TY027 in a dose-dependent and biphasic manner (*P ≤ 0.05; n = 10–19). Arrows indicate the time of compound or vehicle administration (in 5 µl).
Neuropathic pain often induces allodynia, an increased sensitivity to normally innocuous touch that is often insensitive to morphine therapy (10 µg i.t., 8.5% antiallodynic activity; Largent-Milnes et al., 2010). SNL decreased PWTs in response to probing with calibrated von Frey filaments from 14.6 ± 0.2 to 3.1 ± 0.2 g, indicating the development of allodynia in rats. TY027 (1 µg/5µl i.t.) significantly increased the PWT of SNL animals (P ≤ 0.05) compared with baseline and control rats (Fig. 3B) and reached a peak effect within 15 minutes. Higher doses of TY027 (3, 10, and 30 µg) reached maximal antiallodynic effect 30 minutes after spinal injection, and PWTs remained significantly elevated for 45 minutes (P ≤ 0.05; Fig. 3B). A dose-response curve was generated [30 minutes; R2 = 0.97; A50 = 4.5 µg (95% CI = 3.4–5.8 µg in 5 µl)]. Similar to our observations described earlier, spinal TY027 (30 µg/5µl i.t.) attenuated SNL-induced tactile allodynia in a biphasic manner with the second peak time of effect occurring 2 hours postintrathecal bolus (percentage antiallodynia120 minute: 63.2 ± 13.3%, n = 10; P ≤ 0.05). Likewise, in 5 of 10 of these rats, TY027 completely reversed SNL-induced allodynia.
Systemic Administration of TY027 Is Efficacious in Nerve-Injured Rats
The antihyperalgesic and antiallodynic effects of systemically administered TY027 (1, 3, 10, and 30 mg/kg i.v.) were evaluated in SNL-operated rats up to 5 hours postinjection (t1/2 = 4.83 hour; Supplemental Table 1; Yamamoto et al., 2009). SNL decreased PWL values to 12.8 ± 0.4 seconds, which were not significantly altered by either vehicle or low-dose TY027 (1 mg/kg i.v.; Fig. 4A). TY027 (3, 10, and 30 mg/kg i.v.) significantly reversed SNL-induced thermal hypersensitivity beginning 15 minutes after administration. At the highest doses (10 and 30 mg/kg), an initial duration of action of 90 minutes was observed (PWL: 17.2 ± 3.4 and 17.6 ± 1.7 seconds, respectively). Maximal effects were observed 30 minutes following the injection (Fig. 4A), and antihyperalgesia percentages ranged from 36.4 ± 11.3 to 90.2 ± 5.1% (P ≤ 0.05, P ≤ 0.001). The calculated A50 at 30 minutes was 1.6 mg/kg (95% CI = 0.9–2.7 mg/kg i.v.) with an R2 value of 0.85.
Fig. 4.

Systemic TY027 attenuates SNL-induced hypersensitivities in rats. (A) Nerve injury induces a significant decrease in thermal PWLs that are reversed by systemic administration of TY027 in a dose-dependent manner 30 minutes after intravenous administration (*P < 0.05; **P ≤ 0.001). (B) Nerve injury induces a significant decrease in mechanical PWTs that are reversed by TY027 in a dose-dependent manner 30 minutes after intravenous administration (*P ≤ 0.05; n = 8–10 in all experiments). BL, baseline; Veh, vehicle.
Peripheral nerve injury resulted in a significant decrease in PWT (Fig. 4B). Intravenous TY027 administration significantly increased PWTs within 30 minutes of 10 and 30 mg/kg compared with vehicle (P ≤ 0.05), with an onset of 15 minutes. TY027 (30 mg/kg i.v.) significantly reversed SNL-induced tactile allodynia with a peak response of 85.5 ± 10.1% (15 minutes) and 73.0 ± 13.6% (30 minutes); these values were not significantly different from each other. We observed that PWTs of rats treated with TY027 (30 mg/kg i.v.) returned to significantly higher values than post-SNL and vehicle values 150 minutes after intravenous administration (70.7 ± 14.7%) until the 420-minute time point (antiallodynia420 minute: 35.8 ± 10.9%, n = 10; P ≤ 0.05; data not shown), despite the initial observation of a dose-independent duration of action of TY027 against injury-induced allodynia (90 minutes). A dose-response curve was generated from data obtained 30 minutes after TY027 administration, and the calculated A50 for attenuating SNL-induced tactile allodynia was 13.8 mg/kg i.v. (R2 = 0.995; 95% CI = 9.7–20.4 mg/kg).
Evaluation of Opioid-Associated Side Effects
Characterization of the TY027 side-effect profile required dosing choices that would be efficacious in a chronic pain state. Appropriate doses used met the following criteria: ≥A90 dose of TY027 derived from nerve-injury and thermal data and contained an opioid component equipotent to the dose of MS that induced a given side effect for each route of administration evaluated.
Reward Liability of TY027.
To determine whether TY027 was potentially rewarding, rats were conditioned to associate each compound with the tactile-visual cues for a total of five supraspinal or systemic injections of vehicle, MS, or TY027. Doses were chosen using antihyperalgesia dose-response curves constructed after short-term spinal (central administration) and intravenous injection (systemic exposure). Twenty-four hours following the second and fifth drug exposures, absolute latencies in each chamber of a 3-chamber CPP/CPA system were assessed for each rat.
Supraspinal administration.
Rats with an intracerebroventricular cannula spent 549.0 ± 25.1 seconds in each putative conditioning chamber prior to drug pairing (baseline). To minimize potentially confounding effects of long-term central nervous system (CNS) administration of DMSO, both MS and TY027 were dissolved in 45% hydroxy-cyclo-β-dextran (final volume = 18%). Two injections of vehicle (5 µl), MS (10 µg), or TY027 (20 µg) did not significantly alter the total time spent in the drug-associated chamber when compared with baseline (Fig. 5A, day 2). After the fifth paired supraspinal injection of vehicle, the total time animals spent in each end chamber did not significantly change when compared with baseline values, whereas the fifth paired supraspinal injection of MS significantly increased the total time spent in the preconditioned chamber (P ≤ 0.001; Fig. 5A, day 5). Conversely, after the fifth paired supraspinal injection of TY027, there was no significant alteration of time spent in the drug-paired chamber compared with baseline or vehicle-treated controls (Fig. 5A, day 5).
Fig. 5.

Supraspinal or systemic TY027 does not induce CPP in rats. Baseline prior to drug administration resulted in animals spending approximately equal time on both sides of the CPP chambers. Twenty-four hours after two and/or five exposures to morphine (MS, red bars), rats demonstrated a significant increase in the total time spent in the putative conditioned chamber after intracerebroventricular (A) and intravenous (B) injections. Unlike morphine, TY027 (blue bars) administered either intracerebroventricularly (A) or intravenously (B) did not result in significantly increased total time spent in the putative conditioned chamber 24 hours post two and five exposures of TY027 (n = 8–10/treatment/route). Data represent the mean ± S.E.M. *P ≤ 0.05; **P ≤ 0.001.
Systemic administration.
The mean baseline latency in the putative chamber of conditioning was 462.7 ± 27.5 seconds prior to group randomization. To avoid masking any potential observations made using TY027 or MS following systemic administration, three different vehicles were used (1 ml/kg). Saline, 20% DMSO, and DTS vehicles did not differ significantly from each other after either two or five exposures (P > 0.05), nor did they affect the observed effects with MS or TY027 (Fig. 5B). Therefore, data were pooled according to compound, regardless of solution preparation as follows: vehicle, MS, or TY027, respectively. Vehicle did not alter the time spent in the preconditioned chamber compared with baseline after two or five injections. MS (3 mg/kg i.v.) significantly increased the total time spent in the drug-paired chamber 24 hours after the second and fifth systemic injection (600.8 ± 62.5 seconds, P ≤ 0.01 and 597.4 ± 75.7 seconds, P ≤ 0.05, respectively; Fig. 5B). However, TY027 (6 mg/kg i.v.) did not significantly increase the total amount of time spent in the preconditioned chamber after two exposures or after five intravenous injections of TY027; rats spent a mean total time of 322.2 ± 77.0 seconds in the paired chamber (P = 0.06 versus baseline; Fig. 5B).
Multiple Injections of TY027 Did Not Induce Paradoxical Allodynia or Antinociceptive Tolerance.
To assess the development of paradoxical allodynia using a more relevant dosing schedule, PWTs of rats used in CPP/CPA studies were collected following completion of conditioning/testing protocols. MS [10 μg i.c.v. (Supplemental Fig. 1A); 3 mg/kg i.v. (Supplemental Fig. 1B)] induced significant hind-paw mechanical allodynia in rats after two injections (P ≤ 0.05; P ≤ 0.01) that reached maximal levels after five exposures (P ≤ 0.05; P ≤ 0.01). Neither central (20 µg i.c.v.) nor systemic (6 mg/kg i.v.) TY027 significantly decreased PWTs (two injections) from pretreatment levels, unlike MS. Rats injected with TY027 (intravenously) did begin to withdraw the hind paw at lower thresholds than baseline values after five exposures (day 8, P ≤ 0.01; Supplemental Fig. 1B), with a return to basal levels within 24 hours (13.0 ± 0.9 g).
To determine antinociceptive tolerance, rats with spinal catheters and that underwent either sham or SNL surgery were assessed for responses to tactile and thermal stimuli after multiple drug injections with dose-response curves constructed on days 1 and 7 after long-term treatment with either MS or TY027 (Supplemental Fig. 2, A–C). Sham surgery did not significantly alter PWLs (P > 0.05; Supplemental Fig. 2A). On day 1, sham rats (noninjured) injected with TY027 (1, 3, 10, and 30 µg/5 µl i.t.) withdrew the injured paw with increasing latencies, as in previous studies demonstrating antinociception. Twice-daily administration of TY027 (20 µg/5 µl i.t.) did not induce a loss of antinociceptive activity at 30 minutes after each morning injection, and antinociceptive activity remained significant after reconstruction of the dose-response curve on day 7 (P = 0.02; Supplemental Fig. 2A). In SNL-operated rats with a significant decrease in PWL, a 7-day regimen of TY027 (i.t.) induced significant antihyperalgesia compared with postinjury baseline values and vehicle up to day 5 (55–80%, P ≤ 0.05; Supplemental Fig. 2B) ipsilateral to injury. We observed that the degree of antihypersensitivity (the attenuation of SNL-induced thermal hyperalgesia) was reduced on day 7 compared with day 1, suggesting some tolerance, but still paralleled levels observed in sham-operated animals. Spinal TY027 significantly attenuated injury-induced tactile allodynia dose-dependently on both days 1 and 7 of the experiment, and this attenuation was maintained on days 2–6 (Supplemental Fig. 2C), suggesting a lack of tolerance. The mean contralateral PWTs in sham and SNL rats were assessed for the development of paradoxical allodynia after TY027. Neither vehicle nor TY027 (intrathecal) induced significant tactile allodynia in the contralateral hind paw in SNL- or sham-operated rats (P > 0.05; Supplemental Fig. 2D).
Cessation of TY027 Did Not Induce Symptoms of Physical Dependence following Long-Term Administration.
Physical dependence is one side effect of opioids that can be evaluated both spontaneously and in an antagonist-precipitated state. Symptoms of opioid withdrawal include teeth chattering, grooming, “wet-dog” shakes, yawning, jumping, and writhing. Rats exposed to twice-daily vehicle [1 ml/kg, DTS (n = 7) or saline (n = 6)], MS (3 mg/kg i.v., n = 9), or TY027 (10 mg/kg i.v., n = 9) for 3 days did not show signs of significant spontaneous withdrawal prior to the final drug administration on day 4 (P > 0.05). Administration of NLX (3 mg/kg s.c.) 50 minutes after the final MS injection on the morning of the fourth day induced signs of precipitated withdrawal within 30 minutes (Table 2); however, animals treated with saline, DTS, or TY027 did not demonstrate signs of precipitated withdrawal (Table 2). NLX induced significantly fewer bouts of teeth chattering and wet-dog shakes (P ≤ 0.05) in TY027 compared with MS-pretreated rats. No differences were seen among vehicle control, MS, and TY027 rats in grooming behavior or yawning, and no rats exhibited jumping or writhing behavior, regardless of treatment. Autonomic responses of withdrawal, including the development of diarrhea and rapid weight loss after NLX, resulted in diarrhea in eight of nine MS rats and significant weight loss in all MS rats (n = 9/9), whereas no rat treated with TY027 developed diarrhea, and weight loss by two of nine rats was not statistically different than the vehicle rats (DTS) (P > 0.05; Table 2). Amounts of weight loss in TY027 rats were significantly lower than MS rats (P = 0.003).
TABLE 2.
NLX does not precipitate withdrawal in TY027-pretreated rats
All rats received twice-daily injections of the treatment for 3 days. On day 4, 30 minutes after the final injection, NLX (3 mg/kg s.c.) was injected to precipitate signs of physical withdrawal. Data represent the mean (± S.E.); n = 6–9/treatment.
| Treatment (Dose) |
Symptom |
||||
|---|---|---|---|---|---|
| Teeth Chattering |
Grooming Bouts |
Wet-Dog Shakes |
Weight Loss |
Diarrhea |
|
| g | N/Ntotal | ||||
| Saline | 0.00 (0.00) | 5.00 (1.26) | 0.00 (0.00) | 0.33 (0.49) | 0/6 |
| MS (3 mg/kg) | 24.33 (9.33)* | 4.22 (1.75) | 1.11 (0.39)* | 3.79 (0.32)** | 8/9 |
| DTS | 0.14 (0.14) | 4.71 (1.48) | 0.14 (0.14) | 2.86 (0.63) | 1/7 |
| TY027 (10 mg/kg) | 0.11 (0.11)‡ | 2.89 (0.79) | 0.00 (0.00)‡ | 1.89 (0.45)‡ | 0/9 |
P ≤ 0.05; **P ≤ 0.01 compared with vehicle; ‡P ≤ 0.05 compared with MS determined by ANOVA, post-hoc Student-Newman-Keuls.
Gastrointestinal Transit Is Not Impaired by TY027.
The effect of vehicle, MS, or TY027 (intravenous) on gastric transit was evaluated in rats 15 minutes after gavage administration of a nutritive meal supplement marked with 51chromium (51Cr). The total percentage of 51Cr in the cecum was determined as distribution of 51Cr throughout the cecum [(geometric center (GC)]. Vehicle (20% DMSO in saline) treatment resulted in transit of 56.2 ± 13.1% (Fig. 6A) of the total 51Cr through the small intestine, which correlated to a GC of 4.8 ± 0.8 (Fig. 6B). Gastric transit in rats treated with MS (1 mg/kg i.v.) was significantly slowed compared with vehicle, as proven by movement of 20.6 ± 9.3% of 51Cr from the stomach to the cecum (GC = 2.5 ± 0.7, P ≤ 0.001) (Fig. 6, A and B). However, TY027 (3 mg/kg i.v.) induced gastric emptying of 52.0 ± 11.8% of 51Cr (GC = 4.2 ± 0.8), which was not significantly different from vehicle (P > 0.05; Fig. 6, A and B).
Fig. 6.

TY027 does not impair gastrointestinal transit in mice. Gastrointestinal transit was evaluated in mice after intravenous administration of either vehicle, MS, or TY027 by measuring gastric emptying of stomach gavaged 51Cr into the intestine (A) and calculating the geometric center to determine the distance of propulsion (B). MS (1 mg/kg i.v.; n = 8) significantly slowed emptying and reduced the geometric center compared with vehicle, whereas TY027 (3 mg/kg i.v.; n = 8) did not significantly decrease gastric emptying nor reduce the distance of 51Cr propulsion. Data represent the mean ± S.E.M. **P ≤ 0.001.
TY027 Does Not Result in Opioid-Induced Emesis.
The ability of TY027 to induce retching and vomiting was compared with MS in ferrets (n = 12) following short-term systemic exposure (Fig. 7). Neither saline nor DTS vehicle administration produced retches or vomits in any animals (Fig. 7, A and C). A single exposure to MS (0.6 mg/kg) induced a significant number of retching episodes in 91% of animals (22 ± 6 retches/ferret, P ≤ 0.01; Fig. 7, A and B) and 5.0 ± 3.0 episodes of vomiting in 58% of animals (7 of 12; Fig. 7D). Across all ferrets (12 of 12), MS induced 3.0 ± 1.3 vomiting episodes overall (Fig. 7C). At a single dose, TY027 (3 mg/kg) induced 13 retches in 1 of 12 ferrets (8%). Neither the percentage of animals retching/vomiting nor the number of TY027-induced retches was significantly different from control values, and both were significantly lower than MS (P ≤ 0.05; P ≤ 0.01).
Fig. 7.

Opioid-induced emesis occurs with MS but not TY027. (A and B) MS (0.6 mg/kg i.p.), but not TY027 (3 mg/kg i.p.), induced retching in >90% of ferrets, each with an average of 22 retches per animal. (C and D) In the same animals, a higher percentage (58%) of MS-exposed ferrets experienced more vomiting episodes, whereas of those treated with TY027, only 1 of 12 animals experienced a vomiting episode. All ferrets were exposed to vehicle, MS, and TY027 in a randomized and blinded fashion by the observer. *P ≤ 0.05; **P ≤ 0.01 (n = 12 in all groups).
Discussion
Despite the 76 million people currently suffering from chronic pain, opioid misuse is one of the fastest growing drug problems in the United States. Nearly one-third of Americans assessed in the National Survey on Drug Use and Health >12 years of age began abusing drugs by misusing a prescription drug (Substance Abuse and Mental Health Services Administration, 2010). Therapies with reduced abuse liability are largely restricted to antidepressants and anticonvulsants (Dworkin et al., 2010), each with incomplete efficacy and significant side effects.
Novel drug design has not taken advantage of the neuroadaptations that occur after injury/prolonged opioid exposure. Neural plasticity throughout the pain axis (Abbadie et al., 1996; Malcangio et al., 2000; Vanderah et al., 2001; Szücs et al., 2004; King et al., 2005; Wan et al., 2006; Budai et al., 2007; Christie, 2008; Richebe et al., 2012), gastrointestinal (GI)/emesis axis (Bountra et al., 1993; Hargreaves et al., 2011), and reward pathways (Maldonado et al., 1993; Powell et al., 2003) includes enhanced signaling of substance P through NK1 receptors, resulting from chronic pain and opioids. These data suggest that targeting multiple molecular components may translate into safer and more efficacious pain therapeutics (Woodcock et al., 2007). We synthesized single compounds with distinct mechanisms of action: activate MOP/DOP while inhibiting endogenous SP at the NK1 receptor (Yamamoto et al., 2007).
Our recent MOP/DOP agonist–NK1 antagonist peptide, TY027, has improved in vitro stability and BBB access while retaining high affinity and functional activities over our prototype analog, TY005 (Supplemental Table 1; Yamamoto et al., 2007, 2009). In vivo, central and systemic administration of TY027 attenuated acute and neuropathic pain after single and long-term exposure and demonstrated a significant reduction in adverse events, including dependence, tolerance, and GI impairment in rodent models. Central TY027 dose-dependently induced antinociception in rodents that was blocked by the opioid antagonist NLX, suggesting an opioid receptor–mediated effect. Addition of NLX to block both endogenous opioid actions and the opioid component of TY027 did not significantly alter the degree to which TY027 reduced SP-induced behaviors, supporting on-target activity of TY027 at central NK1 receptors.
To determine potential BBB penetration, the ratio of 125I-TY027 in the brain 15–20 minutes following in situ perfusion was compared with levels of control compounds in naïve animals. A significantly higher ratio of 125I-TY027 in the brain relative to the vascular component was observed in both mouse and rat preparations. These data support compound access to the CNS following systemic administration, with a possible enhancement in the setting of inflammation and nerve injury (Beggs et al., 2010; Echeverry et al., 2011), increasing analgesic efficacy.
Reported compounds that act discriminately against tactile and thermal hypersensitivities often lack efficacy for neuropathic pain (Dworkin et al., 2010). In mice, activation of the MOP attenuates heat-induced acute pain without changing mechanical hypersensitivity, whereas DOP activation increases mechanical PWTs short-term during inflammation and in nerve injury (Scherrer et al., 2009) without altering thermal responses. DOP−/− mice do not have altered heat thresholds (Scherrer et al., 2009). In a model of peripheral neuropathy, TY027 was dose-dependently antiallodynic and antihyperalgesic following central and systemic administration. We observed a longer duration of action for TY027 against thermal hyperalgesia versus tactile allodynia. Differences in the modality-dependent duration and the observed biphasic activity of TY027 may be explained by unique mechanisms driving tactile versus thermal sensation, multiple sites of action, compound bioavailability, or potential metabolites; these hypotheses require further investigation.
The long-term effects of TY027 in control and SNL rats were evaluated. Prolonged TY027 reduced ipsilateral PWLs in sham rats after 7-day administration compared with basal values; significant antinociception was maintained over vehicle for the duration of the experiment. Injury-induced tactile and thermal hypersensitivities were attenuated after prolonged exposure to TY027. It is noteworthy that thermal PWLs on day 1 reached levels of antinociception (exceeding preinjury baseline), whereas on day 7, TY027-treated rats had PWLs consistent with antihyperalgesia (above postinjury and below preinjury baseline). This selective development of antinociceptive tolerance, but not antihypersensitivity, may result from differing mechanisms driving protective and pathologic pain. Furthermore, we observed that TY027 antiallodynic responses on days 1 and 7 were not different from each other. These data suggest that different molecular adaptations may underlie the maintenance of tactile and thermal hypersensitivities in chronic pain. Ablation of NK1-expressing neurons in rats using saporin-conjugated SP blocked opioid hypersensitivity/tolerance in naïve and injured rats (Vera-Portocarrero et al., 2007; Rivat et al., 2009). Results with TY027 may reflect accumulation of peptide in lipid membranes known to alter the bioactivity (Yamamoto et al., 2009). Significant TY027 antihyperalgesia to thermal stimulation was consistent with our previous observations (Tumati et al., 2012) and supports dual targeting of the opioid and NK1 systems for effective attenuation of nerve injury–induced hypersensitivities.
The search for analgesics with a decreased abuse liability is prodded by the increasing abuse of prescription opioids (Centers for Disease Control and Prevention, 2011). Opioids can produce euphoria (MOP) and dysphoria (kappa-opioid receptor; Lenard et al., 2007). Unlike the MOP and kappa-opioid receptor, the role of the DOP in reward/aversion is less understood. DOP may play a role in clinical comorbidities to pain (e.g., depression and anxiety) and has recently been implicated in the facilitation of CPP induced by morphine but not reinforcement, indicating a role in contextual cues (Lutz and Kieffer, 2013). The interdependency of opioid receptors with the neurokinin system in reward/motivation pathways (Lessard et al., 2009; Commons, 2010) is reported. SP administration induces CPP, whereas NK1 receptor antagonism decreases morphine reinforcement in self-administration studies (Holzhauer-Oitzl et al., 1988; Placenza et al., 2006). NK1−/− mice do not develop CPP to, or self-administer, morphine but still develop CPP when cocaine or food is used; opioid antinociception remains intact (Murtra et al., 2000; Ripley et al., 2002). These studies indicate that selective modulation of opioids by SP via NK1 receptors in pain pathways is distinct from their interactions in the reward axis (De Felipe et al., 1998). We demonstrate that the opioid agonist/NK1 antagonist TY027 did not produce significant CPP compared with morphine, suggesting that the introduction of an NK1 antagonist moiety prevents the opioid-induced reward effects in rats.
Long-term exposure to opioids often induces physical dependence in patients, and cessation of therapy can induce withdrawal symptoms including abdominal cramping, anorexia/weight loss, and diarrhea. In rats, stopping opioid therapy or administration of opioid antagonists can induce similar withdrawal symptoms (Vera-Portocarrero et al., 2011), as well as cultivate a state of opioid dependence by avoiding withdrawal. Injection of NLX in morphine-dependent rats can enhance the release of SP in the midbrain, hypothalamus, spinal cord, and in the guinea pig ileum (Lu et al., 2011). Administration of an NK1 antagonist to opioid-dependent rats reduced symptoms of physical dependence (Maldonado et al., 1993). Neither cessation of TY027 nor administration of NLX precipitated withdrawal symptoms in rats, suggesting that extended use of TY027 may limit opioid-induced physical dependence as measured by signs of physical withdrawal and autonomic responses.
Opioids induce nausea/vomiting and gastrointestinal complications. Blocking SP actions at NK1 is an effective approach against a number of emetogens, including morphine (Hargreaves et al., 2011). These antiemetic actions of NK1 antagonists may be the result of central actions in the chemoreceptor trigger zone, indicating the importance of BBB penetration for these compounds (Bountra et al., 1993). At a dose that was effective against SNL-induced thermal hyperalgesia in rats and twice that of morphine, systemic TY027 did not induce significant retching and vomiting in ferrets. This lack of an emetic effect with TY027 likely indicates NK1 antagonism at central sites. Our results are consistent with findings that pretreatment with an NK1 antagonist reduces opioid- and copper-induced emesis when injected into the area postrema (Ariumi et al., 2000). TY027 did not impair gastrointestinal propulsion or emptying compared with vehicle, which, unlike morphine, may result from a reduction in the ratio of MOP to DOP activation by TY027 as well as NK1 antagonist activity. Although additional investigation is required to elucidate the mechanisms underlying the effects of TY027 in the GI/emetic axis, these studies indicate that multifunctional compounds acting on both the opioid and neurokinin systems produce fewer gastrointestinal complications than traditional opioids, suggesting that such a compound has significantly reduced unwanted effects and will not be dose limited, a phenomenon of current opioids clinically used for chronic pain.
The use of a single compound such as TY027 as described by scientists at the U.S. Food and Drug Administration and National Institutes of Health (Woodcock et al., 2007), rather than coadministration of opioids with adjuvant therapies (Dworkin et al., 2010), introduces an alternate approach to clinical pain management. The use of multitarget, single molecules has advantages, including a simple absorption, distribution, metabolism, and excretion property; no drug-drug interactions; and a potential higher local concentration of active components. A multifunctional compound may counteract molecular adaptations induced by opioid agonists given the significant anatomic overlap of the systems leading to enhanced potency and efficacy as seen presently. We provide significant evidence that opioid/neurokinin compounds can target opioid-induced changes, introducing alternatives to available analgesics. These studies are the first to demonstrate the use of a multifunctional peptidergic opioid for acute and neuropathic pain with a reduction in gastrointestinal disturbances and reward liability that may help to diminish the growing epidemic of prescription drug misuse. Our findings support making a nonaddictive, nonconstipating opioid available to patients with chronic pain, reducing physicians’ rapidly rising concerns about prescribing opioids.
Supplementary Material
Acknowledgments
The authors thank Janice Oyarzo (University of Arizona) and Laine Greene for their assistance.
Abbreviations
- ANOVA
analysis of variance
- BBB
blood-brain barrier
- CNS
central nervous system
- CPP/CPA
conditioned place preference/aversion
- 51Cr
51chromium
- CTAP
d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2
- DOP
δ-opioid receptor
- DPDPE
[d-Pen2,5]enkephalin, [d-Pen2, d-Pen5]enkephalin
- DTS
10% dimethylsulfoxide/10% Tween 80/80% saline
- GC
geometric center
- GI
gastrointestinal
- MS
morphine sulfate
- MOP
μ-opioid receptor
- NLX
naloxone
- NK1
neurokinin-1 receptor
- PWL
paw withdrawal latencies
- PWT
paw withdrawal thresholds
- RBr
percentage ratio of radioactivity in the brain to that in the perfusate
- SNL
spinal nerve ligation
- SP
substance P
- TFL
tail-flick latency
- TY005
Tyr-d-Ala-Gly-Phe-Met-Pro-Leu-Trp-O-Bn(CF3)2
- TY027
Tyr-d-Ala-Gly-Phe-Met-Pro-Leu-Trp-NH-Bn(CF3)2
Authorship Contributions
Participated in research design: Largent-Milnes, Campos, Giuvelis, Bilsky, Hruby, Porreca, Vanderah.
Conducted experiments: Largent-Milnes, Brookshire, Hanlon, Skinner, Davis, Giuvelis, Campos.
Contributed new reagents or analytic tools: Yamamoto, Nair, Deekonda, Hruby.
Performed data analysis: Largent-Milnes, Hanlon, Davis, Brookshire, Giuvelis.
Wrote or contributed to the writing of the manuscript: Largent-Milnes, Vanderah, Bilsky, Giuvelis, Hruby, Porreca.
Footnotes
This work was supported by the National Institutes of Health National Institute on Drug Abuse [Grants DA-13449 and DA-06284].
Portions of data included in this article have been presented previously in abstract form:
Largent-Milnes TM, Yamamoto T, Campos CR, Corral-Frias NS, Jimenez-Andrade JM, Davis P, Ma S-W, Mantyh PW, French ED, Davis TP, et al. (2009) Dual acting opioid agonist/NK1 antagonist does not produce antinociceptive tolerance or reward in an animal model of neuropathic pain. Society for Neuroscience; 2009 Oct 11–17; Chicago, IL; and Largent-Milnes TM, Yamamoto T, Nair P, Navratilova E, Davis P, Hruby VJ, Yamamura HI, Lai J, Porreca F, and Vanderah TW (2008) Dual acting opioid agonist/NK1 antagonist peptide reverses neuropathic pain in an animal model without opioid side effects. International Association for the Study of Pain; 2008 Aug 17–22; Glasgow, Scotland, UK.
T.W.V. and T.M.L.-M. accept responsibility for the integrity of data analysis and interpretation.
 This article has supplemental material available at jpet.aspetjournals.org.
This article has supplemental material available at jpet.aspetjournals.org.
References
- Abbadie C, Brown JL, Mantyh PW, Basbaum AI. (1996) Spinal cord substance P receptor immunoreactivity increases in both inflammatory and nerve injury models of persistent pain. Neuroscience 70:201–209 [DOI] [PubMed] [Google Scholar]
- Aicher SA, Punnoose A, Goldberg A. (2000) mu-Opioid receptors often colocalize with the substance P receptor (NK1) in the trigeminal dorsal horn. J Neurosci 20:4345–4354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ananthan S, Kezar HS, 3rd, Carter RL, Saini SK, Rice KC, Wells JL, Davis P, Xu H, Dersch CM, Bilsky EJ, et al. (1999) Synthesis, opioid receptor binding, and biological activities of naltrexone-derived pyrido- and pyrimidomorphinans. J Med Chem 42:3527–3538 [DOI] [PubMed] [Google Scholar]
- Ariumi H, Saito R, Nago S, Hyakusoku M, Takano Y, Kamiya H. (2000) The role of tachykinin NK-1 receptors in the area postrema of ferrets in emesis. Neurosci Lett 286:123–126 [DOI] [PubMed] [Google Scholar]
- Beggs S, Liu XJ, Kwan C, Salter MW. (2010) Peripheral nerve injury and TRPV1-expressing primary afferent C-fibers cause opening of the blood-brain barrier. Mol Pain 6:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bountra C, Bunce K, Dale T, Gardner C, Jordan C, Twissell D, Ward P. (1993) Anti-emetic profile of a non-peptide neurokinin NK1 receptor antagonist, CP-99,994, in ferrets. Eur J Pharmacol 249:R3–R4 [DOI] [PubMed] [Google Scholar]
- Budai D, Khasabov SG, Mantyh PW, Simone DA. (2007) NK-1 receptors modulate the excitability of ON cells in the rostral ventromedial medulla. J Neurophysiol 97:1388–1395 [DOI] [PubMed] [Google Scholar]
- Cahill CM, Coderre TJ. (2002) Attenuation of hyperalgesia in a rat model of neuropathic pain after intrathecal pre- or post-treatment with a neurokinin-1 antagonist. Pain 95:277–285 [DOI] [PubMed] [Google Scholar]
- Cahill CM, Morinville A, Hoffert C, O’Donnell D, Beaudet A. (2003) Up-regulation and trafficking of delta opioid receptor in a model of chronic inflammation: implications for pain control. Pain 101:199–208 [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC) (2011) Vital signs: overdoses of prescription opioid pain relievers---United States, 1999--2008. MMWR Morb Mortal Wkly Rep 60:1487–1492 [PubMed] [Google Scholar]
- Christie MJ. (2008) Cellular neuroadaptations to chronic opioids: tolerance, withdrawal and addiction. Br J Pharmacol 154:384–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cicero TJ, Ellis MS, Surratt HL. (2012) Effect of abuse-deterrent formulation of OxyContin. N Engl J Med 367:187–189 [DOI] [PubMed] [Google Scholar]
- Commons KG. (2010) Neuronal pathways linking substance P to drug addiction and stress. Brain Res 1314:175–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Felipe C, Herrero JF, O’Brien JA, Palmer JA, Doyle CA, Smith AJ, Laird JM, Belmonte C, Cervero F, Hunt SP. (1998) Altered nociception, analgesia and aggression in mice lacking the receptor for substance P. Nature 392:394–397 [DOI] [PubMed] [Google Scholar]
- Dietis N, Guerrini R, Calo G, Salvadori S, Rowbotham DJ, Lambert DG. (2009) Simultaneous targeting of multiple opioid receptors: a strategy to improve side-effect profile. Br J Anaesth 103:38–49 [DOI] [PubMed] [Google Scholar]
- Dworkin RH, O’Connor AB, Audette J, Baron R, Gourlay GK, Haanpää ML, Kent JL, Krane EJ, Lebel AA, Levy RM, et al. (2010) Recommendations for the pharmacological management of neuropathic pain: an overview and literature update. Mayo Clin Proc 85(3, Suppl)S3–S14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Echeverry S, Shi XQ, Rivest S, Zhang J. (2011) Peripheral nerve injury alters blood-spinal cord barrier functional and molecular integrity through a selective inflammatory pathway. J Neurosci 31:10819–10828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadd CA, Murtra P, De Felipe C, Hunt SP. (2003) Neurokinin-1 receptor-expressing neurons in the amygdala modulate morphine reward and anxiety behaviors in the mouse. J Neurosci 23:8271–8280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavériaux-Ruff C, Karchewski LA, Hever X, Matifas A, Kieffer BL. (2008) Inflammatory pain is enhanced in delta opioid receptor-knockout mice. Eur J Neurosci 27:2558–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendron L, Pintar JE, Chavkin C. (2007) Essential role of mu opioid receptor in the regulation of delta opioid receptor-mediated antihyperalgesia. Neuroscience 150:807–817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall AJ, Logan JE, Toblin RL, Kaplan JA, Kraner JC, Bixler D, Crosby AE, Paulozzi LJ. (2008) Patterns of abuse among unintentional pharmaceutical overdose fatalities. JAMA 300:2613–2620 [DOI] [PubMed] [Google Scholar]
- Hargreaves R, Ferreira JC, Hughes D, Brands J, Hale J, Mattson B, Mills S. (2011) Development of aprepitant, the first neurokinin-1 receptor antagonist for the prevention of chemotherapy-induced nausea and vomiting. Ann N Y Acad Sci 1222:40–48 [DOI] [PubMed] [Google Scholar]
- Hill RG. (2000) NK1 (substance P) receptor antagonists—why are they not analgesic in humans? Trends Pharmacol Sci 21:244–246 [DOI] [PubMed] [Google Scholar]
- Holzhäuer-Oitzl MS, Hasenöhrl R, Huston JP. (1988) Reinforcing properties of substance P in the region of the nucleus basalis magnocellularis in rats. Neuropharmacology 27:749–756 [DOI] [PubMed] [Google Scholar]
- Hylden JL, Wilcox GL. (1982) Intrathecal opioids block a spinal action of substance P in mice: functional importance of both mu- and delta-receptors. Eur J Pharmacol 86:95–98 [DOI] [PubMed] [Google Scholar]
- Institute of Medicine (2011) Relieving Pain in America, A Blueprint for Transforming Prevention, Care, Education and Research, The National Academies Press, Washington, DC: [PubMed] [Google Scholar]
- Jensen TS, Finnerup NB. (2009) Neuropathic pain treatment: a further step forward. Lancet 374:1218–1219 [DOI] [PubMed] [Google Scholar]
- King T, Gardell LR, Wang R, Vardanyan A, Ossipov MH, Malan TP, Jr, Vanderah TW, Hunt SP, Hruby VJ, Lai J, et al. (2005) Role of NK-1 neurotransmission in opioid-induced hyperalgesia. Pain 116:276–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Largent-Milnes TM, Yamamoto T, Nair P, Moulton JW, Hruby VJ, Lai J, Porreca F, Vanderah TW. (2010) Spinal or systemic TY005, a peptidic opioid agonist/neurokinin 1 antagonist, attenuates pain with reduced tolerance. Br J Pharmacol 161:986–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenard NR, Daniels DJ, Portoghese PS, Roerig SC. (2007) Absence of conditioned place preference or reinstatement with bivalent ligands containing mu-opioid receptor agonist and delta-opioid receptor antagonist pharmacophores. Eur J Pharmacol 566:75–82 [DOI] [PubMed] [Google Scholar]
- Lessard A, Savard M, Gobeil F, Jr, Pierce JP, Pickel VM. (2009) The neurokinin-3 (NK3) and the neurokinin-1 (NK1) receptors are differentially targeted to mesocortical and mesolimbic projection neurons and to neuronal nuclei in the rat ventral tegmental area. Synapse 63:484–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu XF, Li YY, Wang CG, Wei JQ, Ye Y, Zhang LC, Cao JL. (2011) Substance P in the cerebrospinal fluid-contacting nucleus contributes to morphine physical dependence in rats. Neurosci Lett 488:188–192 [DOI] [PubMed] [Google Scholar]
- Lutz PE and Kieffer BL (2013) The multiple facets of opioid receptor function: implications for addiction. Curr Opin Neurobiol. pii S0959-4388(13)00053-6. 10.1016/j.conb.2013.02.005. [DOI] [PMC free article] [PubMed]
- Malcangio M, Ramer MS, Jones MG, McMahon SB. (2000) Abnormal substance P release from the spinal cord following injury to primary sensory neurons. Eur J Neurosci 12:397–399 [DOI] [PubMed] [Google Scholar]
- Maldonado R, Girdlestone D, Roques BP. (1993) RP 67580, a selective antagonist of neurokinin-1 receptors, modifies some of the naloxone-precipitated morphine withdrawal signs in rats. Neurosci Lett 156:135–140 [DOI] [PubMed] [Google Scholar]
- Mantyh PW, Rogers SD, Honore P, Allen BJ, Ghilardi JR, Li J, Daughters RS, Lappi DA, Wiley RG, Simone DA. (1997) Inhibition of hyperalgesia by ablation of lamina I spinal neurons expressing the substance P receptor. Science 278:275–279 [DOI] [PubMed] [Google Scholar]
- Muñoz M, Coveñas R. (2010) Neurokinin-1 receptor: a new promising target in the treatment of cancer. Discov Med 10:305–313 [PubMed] [Google Scholar]
- Murtra P, Sheasby AM, Hunt SP, De Felipe C. (2000) Rewarding effects of opiates are absent in mice lacking the receptor for substance P. Nature 405:180–183 [DOI] [PubMed] [Google Scholar]
- Nadal X, Baños JE, Kieffer BL, Maldonado R. (2006) Neuropathic pain is enhanced in delta-opioid receptor knockout mice. Eur J Neurosci 23:830–834 [DOI] [PubMed] [Google Scholar]
- Negus SS, Gatch MB, Mello NK, Zhang X, Rice K. (1998) Behavioral effects of the delta-selective opioid agonist SNC80 and related compounds in rhesus monkeys. J Pharmacol Exp Ther 286:362–375 [PubMed] [Google Scholar]
- Paxinos G, Watson WC, eds, ed (1997) The Rat Brain in Stereotaxic Coordinates, Academic Press, Sydney [Google Scholar]
- Placenza FM, Fletcher PJ, Vaccarino FJ, Erb S. (2006) Effects of central neurokinin-1 receptor antagonism on cocaine- and opiate-induced locomotor activity and self-administration behaviour in rats. Pharmacol Biochem Behav 84:94–101 [DOI] [PubMed] [Google Scholar]
- Powell KJ, Quirion R, Jhamandas K. (2003) Inhibition of neurokinin-1-substance P receptor and prostanoid activity prevents and reverses the development of morphine tolerance in vivo and the morphine-induced increase in CGRP expression in cultured dorsal root ganglion neurons. Eur J Neurosci 18:1572–1583 [DOI] [PubMed] [Google Scholar]
- Richebe P, Cahana A, Rivat C. (2012) Tolerance and opioid-induced hyperalgesia. Is a divorce imminent? Pain 153:1547–1548 [DOI] [PubMed] [Google Scholar]
- Ripley TL, Gadd CA, De Felipe C, Hunt SP, Stephens DN. (2002) Lack of self-administration and behavioural sensitisation to morphine, but not cocaine, in mice lacking NK1 receptors. Neuropharmacology 43:1258–1268 [DOI] [PubMed] [Google Scholar]
- Rivat C, Vera-Portocarrero LP, Ibrahim MM, Mata HP, Stagg NJ, De Felice M, Porreca F, Malan TP. (2009) Spinal NK-1 receptor-expressing neurons and descending pathways support fentanyl-induced pain hypersensitivity in a rat model of postoperative pain. Eur J Neurosci 29:727–737 [DOI] [PubMed] [Google Scholar]
- Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O’Donnell D, Kieffer BL, Basbaum AI. (2009) Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell 137:1148–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schramm CL, Honda CN. (2010) Co-administration of δ- and μ-opioid receptor agonists promotes peripheral opioid receptor function. Pain 151:763–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seybold VS, Hylden JL, Wilcox GL. (1982) Intrathecal substance P and somatostatin in rats: behaviors indicative of sensation. Peptides 3:49–54 [DOI] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration (2010) Results from the 2010 National Survey on Drug Use and Health: Summary of National Findings, NSDUH Series H-41, HHS Publication No. (SMA) 11-4658. http://www.samhsa.gov/data/NSDUH/2k10NSDUH/2k10Results.htm (accessed June 15, 2012).
- Szücs M, Boda K, Gintzler AR. (2004) Dual effects of DAMGO [d-Ala2,N-Me-Phe4,Gly5-ol]-enkephalin and CTAP (d-Phe-Cys-Tyr-d-Trp-Arg-Thr-Pen-Thr-NH2) on adenylyl cyclase activity: implications for mu-opioid receptor Gs coupling. J Pharmacol Exp Ther 310:256–262 [DOI] [PubMed] [Google Scholar]
- Tumati S, Largent-Milnes TM, Keresztes AI, Yamamoto T, Vanderah TW, Roeske WR, Hruby VJ, Varga EV. (2012) Tachykinin NK1 receptor antagonist co-administration attenuates opioid withdrawal-mediated spinal microglia and astrocyte activation. Eur J Pharmacol 684:64–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderah TW. (2010) Delta and kappa opioid receptors as suitable drug targets for pain. Clin J Pain 26 (Suppl 10):S10–S15 [DOI] [PubMed] [Google Scholar]
- Vanderah TW, Ossipov MH, Lai J, Malan TP, Jr, Porreca F. (2001) Mechanisms of opioid-induced pain and antinociceptive tolerance: descending facilitation and spinal dynorphin. Pain 92:5–9 [DOI] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Ossipov MH, Lai J, King T, Porreca F. (2011) Descending facilitatory pathways from the rostroventromedial medulla mediate naloxone-precipitated withdrawal in morphine-dependent rats. J Pain 12:667–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vera-Portocarrero LP, Zhang ET, King T, Ossipov MH, Vanderah TW, Lai J, Porreca F. (2007) Spinal NK-1 receptor expressing neurons mediate opioid-induced hyperalgesia and antinociceptive tolerance via activation of descending pathways. Pain 129:35–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Douglas SD, Wang X, Kolson DL, O’Donnell LA, Ho WZ. (2006) Morphine upregulates functional expression of neurokinin-1 receptor in neurons. J Neurosci Res 84:1588–1596 [DOI] [PubMed] [Google Scholar]
- Woodcock J, Witter J, Dionne RA. (2007) Stimulating the development of mechanism-based, individualized pain therapies. Nat Rev Drug Discov 6:703–710 [DOI] [PubMed] [Google Scholar]
- Yamamoto T, Nair P, Davis P, Ma S-W, Navratilova E, Moye S, Tumati S, Lai J, Vanderah TW, Yamamura HI, et al. (2007) Design, synthesis, and biological evaluation of novel bifunctional C-terminal-modified peptides for delta/mu opioid receptor agonists and neurokinin-1 receptor antagonists. J Med Chem 50:2779–2786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Nair P, Ma S-W, Davis P, Yamamura HI, Vanderah TW, Porreca F, Lai J, Hruby VJ. (2009) The biological activity and metabolic stability of peptidic bifunctional compounds that are opioid receptor agonists and neurokinin-1 receptor antagonists with a cystine moiety. Bioorg Med Chem 17:7337–7343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu YJ, Arttamangkul S, Evans CJ, Williams JT, von Zastrow M. (2009) Neurokinin 1 receptors regulate morphine-induced endocytosis and desensitization of mu-opioid receptors in CNS neurons. J Neurosci 29:222–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.