

Abstract
The orphan receptor GPR17 has been reported to be activated by UDP, UDP-sugars, and cysteinyl leukotrienes, and coupled to intracellular Ca2+ mobilization and inhibition of cAMP accumulation, but other studies have reported either a different agonist profile or lack of agonist activity altogether. To determine if GPR17 is activated by uracil nucleotides and leukotrienes, the hemagglutinin-tagged receptor was expressed in five different cell lines and the signaling properties of the receptor were investigated. In C6, 1321N1, or Chinese hamster ovary (CHO) cells stably expressing GPR17, UDP, UDP-glucose, UDP-galactose, and cysteinyl leukotriene C4 (LTC4) all failed to promote inhibition of forskolin-stimulated cAMP accumulation, whereas both UDP and UDP-glucose promoted marked inhibition (>80%) of forskolin-stimulated cAMP accumulation in C6 and CHO cells expressing the P2Y14 receptor. Likewise, none of these compounds promoted accumulation of inositol phosphates in COS-7 or human embryonic kidney 293 cells transiently transfected with GPR17 alone or cotransfected with Gαq/i5, which links Gi-coupled receptors to the Gq-regulated phospholipase C (PLC) signaling pathway, or PLCε, which is activated by the Gα12/13 signaling pathway. Moreover, none of these compounds promoted internalization of GPR17 in 1321N1-GPR17 cells. Consistent with previous reports, coexpression experiments of GPR17 with cysteinyl leukotriene receptor 1 (CysLTR1) suggested that GPR17 acts as a negative regulator of CysLTR1. Taken together, these data suggest that UDP, UDP-glucose, UDP-galactose, and LTC4 are not the cognate ligands of GPR17.
Introduction
Extracellular adenine and uracil nucleotides regulate a broad range of physiologic and pathophysiological responses by activating two main receptor families: P2X receptors, which are ATP-gated cation-selective ion channels; and P2Y receptors, which are G protein–coupled receptors (GPCRs) activated by adenine and uracil nucleotides. To date, molecular cloning and pharmacological characterization have identified seven mammalian P2X receptors (P2X1–7) and eight functional mammalian P2Y receptor subtypes (P2Y1, P2Y2, P2Y4, P2Y6, P2Y11, P2Y12, P2Y13, and P2Y14) (Costanzi et al., 2004; Abbracchio et al., 2006). Based on their sequence identity and signaling properties, P2Y receptors are commonly grouped into two subfamilies. The P2Y1 receptor subfamily, which encompasses P2Y1, P2Y2, P2Y4, P2Y6, and P2Y11 receptors, couples to Gq and activates phospholipase C (PLC). In addition to coupling to Gq, the P2Y11 receptor also couples to Gs and activates adenylyl cyclase, albeit with lower efficiency than Gq/PLC (Qi et al., 2001a; Torres et al., 2002). The P2Y12 receptor subfamily members, which include P2Y12, P2Y13, and P2Y14, are all located in a gene cluster on chromosome 3, share ∼43–47% sequence identity, and couple to Gi/o. An additional receptor in this cluster, GPR87, which shares ∼34–42% identity with the other P2Y receptors, apparently is not activated by nucleotides but has been reported to be activated by lysophosphatidic acid (Tabata et al., 2007).
Erroneous designation of new or orphan receptors as P2Y receptors has caused considerable confusion and controversy. Seven receptors (p2y3, p2y5, p2y7, p2y8, p2y9, p2y10, and p2y15) were originally classified as P2Y receptors (Akbar et al., 1996; Webb et al., 1996a,b; Bogdanov et al., 1997; Inbe et al., 2004), but were later shown either to not respond to nucleotides (p2y5, p2y7, p2y9, p2y10, and p2y15) (Herold et al., 1997; Janssens et al., 1997; Li et al., 1997; Qi et al., 2004) or likely to be species homologs of mammalian P2Y receptors (i.e., the avian P2Y3 receptor and mammalian P2Y6 receptors; the Xenopus P2Y8 and mammalian P2Y4 receptors) (Li et al., 1998). Several of the aforementioned receptors were deorphaned and shown to respond to either leukotriene B4 (p2y7) (Yokomizo et al., 1997), lysophosphatidic acid (p2y5, p2y9, and p2y10) (Noguchi et al., 2003; Murakami et al., 2008; Pasternack et al., 2008), sphingosine 1-phosphate (p2y10) (Murakami et al., 2008), or the citric acid cycle metabolite α-ketoglutarate (p2y15) (He et al., 2004; Qi et al., 2004).
An orphan GPCR, GPR17, which is located phylogenetically at an intermediate position between P2Y and cysteinyl leukotriene receptors (CysLTRs), was reported to be activated by cysteinyl leukotrienes, uracil nucleotides, and nucleotide sugars (UDP, UDP-glucose, and UDP-galactose) and promote mobilization of intracellular Ca2+ and inhibition of cAMP accumulation (Ciana et al., 2006). Although multiple follow-up studies consistent with this original report have been published by the same group (Parravicini et al., 2008; Pugliese et al., 2009; Buccioni et al., 2011; Daniele et al., 2011; Coppi et al., 2013), Benned-Jensen and Rosenkilde (2010) reported that UDP and UDP-glucose, but not cysteinyl leukotrienes, promoted GPR17-dependent guanosine 5′-O-(3-[35S]thio)triphosphate binding, and Maekawa et al. (2009) concluded that GPR17 was not activated by any of these presumed agonists but instead was a negative regulator of CysLTR1. Given these disparate results, we have reexamined the potential activation of GPR17 in a broad range of cell types that either stably or transiently express this GPCR. In addition, four independent downstream signaling responses were quantified in response to uracil nucleotides, nucleotide sugars, or cysteinyl leukotrienes. Our data indicate that GPR17 is neither activated nor internalized by these ligands, whereas receptors that are established targets of these ligands demonstrate robust activation and internalization. Moreover, our data are consistent with GPR17 acting as a negative regulator of CysLTR1 as reported previously (Maekawa et al., 2009).
Materials and Methods
All cell culture reagents were supplied by the Lineberger Comprehensive Cancer Center tissue culture facility (University of North Carolina at Chapel Hill). UDP, UDP-glucose, UDP-galactose, cysteinyl leukotriene C4 (LTC4), 3-isobutyl-1-methyl-xanthine, and carbachol were from Sigma-Aldrich (St. Louis, MO). ATP and FuGENE6 transfection reagent were from Roche (Indianapolis, IN). Forskolin was purchased from Calbiochem (La Jolla, CA). CysLTR1 [3× hemagglutinin (HA)-tagged CysLTR1 in pcDNA3.1(+)] was obtained from the cDNA Resource Center at the Missouri University of Science and Technology (Rolla, MO).
Construction of HA-Tagged GPR17 cDNAs.
The coding sequence of the short form of GPR17 was amplified from human genomic DNA by polymerase chain reaction with Pfu polymerase (Stratagene, Carlsbad, CA). The upstream primer sequence contained an MluI site at its 5′ end (5′-TGACGCGTCCAATGGCCTTGAAGTGGCTCC-3′), while the downstream primer contained an XhoI site following the stop codon (5′-TCCTCGAGCTACAGCTCTGACTTGGCA-3′). The amplified fragment was digested with MluI and XhoI and ligated in-frame into a similarly digested pLXSN retroviral expression vector containing an EcoRI site, Kozak initiation sequence, initiating methionine residue, and the HA epitope tag (YPYDVPDY), followed by an MluI site. To construct pcDNA3-HA-GPR17, pLXSN-HA-GPR17 was digested with EcoRI and XhoI and ligated in-frame into similarly digested pcDNA3 expression vector. The sequence of GPR17 was identical to accession #NP_001154888.1.
Stable Expression of GPR17 and Other GPCRs in 1321N1, Chinese Hamster Ovary, and C6 Cells.
Stable expression of GPR17 in 1321N1, Chinese hamster ovary (CHO), and C6 cells was accomplished by retroviral infection as described previously (Qi et al., 2001a). Briefly, recombinant retrovirus particles were produced by calcium phosphate–mediated transfection of PA317 cells with pLXSN alone or with pLXSN containing HA-tagged versions of GPR17, P2Y6, P2Y14, cysteinyl leukotriene receptor-1 (CysLTR1), or lysophosphatidic acid receptor-3 (LPA3) receptors. Recombinant retroviruses were harvested 2 days later and incubated with 1321N1, CHO, and C6 cells in the presence of 8 μg/ml hexadimethrine bromide (Polybrene). After 2 days, G418-resistant cells were selected with 1 mg/ml G418-containing medium.
Transient Expression of GPR17 and Other GPCRs in COS-7 and Human Embryonic Kidney 293 Cells.
Transient transfections were performed with FuGENE6 according to the manufacturer’s protocol. Briefly, 1 × 106 COS-7 or human embryonic kidney (HEK) 293 cells per 60-mm dish were incubated with 8 μl of FuGENE6 and 2.8 μg pcDNA3-based plasmid construct in 0.4 ml of serum-free Dulbecco’s modified Eagle’s medium (DMEM). For cotransfection experiments with Gαq/i5, 2 μg of receptor DNA and 0.8 μg of pcDNA1-Gαq/i5 DNA (Conklin et al., 1993) per dish were used. Following overnight incubation, the cells were treated with trypsin and plated in 24-well dishes for activity assays carried out 2 days later.
Expression of HA-Tagged CysLTR1.
The pcDNA3.1 expression plasmid for human CysLTR1 obtained from the cDNA Resource Center has three consecutive HA tags fused to the amino terminus of the receptor. This vector was used to transiently transfect COS-7 and HEK293 cells, but cell surface expression was not detected by radioimmunoassay (RIA) in two independent transfections. A previous study demonstrated that surface expression of mouse CysLTR1 could be improved by fusing a cleavable signal sequence to the amino terminus of the receptor (Martin et al., 2001). Therefore, we made a new construct by fusing the 16-amino-acid prolactin signal sequence (Guan et al., 1992) to the start of the 3× HA tag of the receptor, and significant cell surface expression was demonstrated when this plasmid was transfected into COS-7 and HEK293 cells. For stable expression in 1321N1 and CHO cells, the fragment encoding the receptor fused to the prolactin signal sequence was subcloned into pLXSN and the receptor was expressed by retroviral infection as described above.
Assay of Inositol Phosphate and cAMP Accumulation.
1321N1 and CHO cells were seeded in 24-well plates at 105 and 5 × 104 cells/well, respectively, and assayed 3 days later when confluent. Transiently transfected COS-7 and HEK293 cells were seeded in 24-well plates at 2 × 105 cells/well and assayed 2 days later. Inositol lipids were radiolabeled by overnight incubation of cells with 200 μl serum-free, inositol-free DMEM containing 0.4 μCi myo-[3H]inositol. Agonists or antagonists were added at 5× concentration in 50 μl of 50 mM LiCl and 250 mM HEPES, pH 7.25. Following a 30-minute incubation (10 minutes in Fig. 3) at 37°C, the medium was aspirated and the reaction was terminated by adding 0.75 ml boiling EDTA, pH 8.0. [3H]Inositol phosphates were resolved on Dowex AG1-X8 (Sigma-Aldrich) columns as described previously (Qi et al., 2001a).
Fig. 3.

Lack of uracil nucleotide– and cysteinyl leukotriene–promoted accumulation of inositol phosphates in 1321N1 or CHO cells stably expressing GPR17. UDP, UDP-glucose (UDP-Glu), UDP-galactose (UDP-Gal) (all at 100 μM), or LTC4 (100 nM) failed to promote significant accumulation of inositol phosphates in (A) 1321N1 or (B) CHO cells stably expressing GPR17 compared with cells expressing the empty vector. (C) Cell surface expression of HA-tagged GPR17 was verified by an intact cell RIA. Data shown are from a representative assay performed three times with similar results. Ab, antibody; cpm, counts per minute; WT, wild type.
To quantify cAMP accumulation, cells were labeled with 200 μl serum-free DMEM containing 0.8 μCi of [3H]adenine for 2 hours. Ten minutes prior to the assay, cells were supplemented with 50 μl of 40 mM HEPES, pH 7.4, and 200 μM isobutyl methylxanthine (final concentrations in the assay). Drugs were added at 6× concentration in 50 μl of Hanks’ balanced salt solution (HBSS) for 30 minutes at 37°C. Reactions were terminated by aspiration of the medium and addition of 1 ml of 5% trichloroacetic acid. [3H]cAMP was purified on Dowex and alumina columns as described previously (Qi et al., 2001a). For assays measuring inhibition of cAMP accumulation, cells were labeled and treated as above, except that forskolin (either 10 or 30 μM, final concentration) was included in the drug mixture to increase cAMP levels. Reactions were terminated and [3H]cAMP levels were quantified as described above.
Intracellular Calcium Measurements.
1321N1 cells expressing GPR17 were grown on glass coverslips for 1–2 days to ∼20% confluency. Prior to calcium measurements, cells were incubated with 1 μM Fura-2/acetoxymethyl ester (Fura-2/AM) for 30–40 minutes. Coverslips were then sealed in a Plexiglass chamber (200 μl total volume) and mounted on the stage of Nikon Diaphot inverted microscope (Nikon Instruments, Melville, NY). Cells were continuously superfused (1.4 ml/min) with HBSS with or without agonist. Solution changes were accomplished by means of a manual valve attached to a gravity-driven six-well reservoir. Cells were superfused with agonist for 1 minute. Calcium responses were recorded with a digital imaging system from Intracellular Imaging (Cincinnati, OH) as previously described (Qi et al., 2001b).
RIA of Cell Surface Receptors.
Cells were seeded in 24-well plates at densities between 5 × 104 cells/well and 1 × 105 cells/well and assayed 3 days later when confluent as described previously (Qi et al., 2001b). Briefly, cells were fixed in 4% paraformaldehyde, washed twice with HBSS containing Ca2+ and Mg2+ (HBSS++), and blocked with medium containing 10% fetal bovine serum (FBS). Cells were then incubated with a 1:1000 dilution of mouse anti-HA monoclonal antibody (clone HA.11; Covance, Denver, PA) in medium containing 5% FBS for 1 hour, washed twice with HBSS++, and then incubated for 2 hours with 125I-labeled rabbit anti-mouse antibody in medium containing 5% FBS. Cells were washed twice with HBSS++, solubilized overnight with 1 M NaOH, and transferred to glass tubes for quantification of radioactivity by γ-counting.
Receptor Internalization.
1321N1 cells expressing HA-GPR17, HA-P2Y2, or 3× HA-tagged CysLTR1 receptors were plated on glass coverslips at 20% confluence and allowed to grow for 48 hours. The cells were cooled to 4°C, labeled with mouse anti-HA antibody, and washed to remove excess antibody. The temperature was then increased to 37°C and the cells were treated with either phosphate-buffered saline (all cells) or with 300 nM LTC4 (GPR17), 100 μM UTP (P2Y2), 100 μM UDP (GPR17), or 100 μM UDP-glucose (GPR17) for 30 minutes. The cells were then fixed, permeabilized, and incubated with AlexaFluor 488 goat anti-mouse antibody (Invitrogen/Molecular Probes, Eugene, OR). Cells were imaged by confocal microscopy on an Olympus Fluoview 300 laser scanning confocal imaging system (Olympus Imaging, Center Valley, PA) configured with an IX70 fluorescence microscope fitted with a PlanApo 60× oil objective.
Functional Regulation of CysLTR1 by GPR17.
1321N1 cells were serially infected with retroviruses directing expression of 3× HA-tagged CysLTR1 and myc-tagged GPR17. Retroviruses directing CysLTR1 expression conferred resistance to G418, while those directing GPR17 expression conferred resistance to hygromycin. Cells expressing either CysLTR1 alone or CysLTR1 and GPR17 were submitted to RIA to determine the levels of CysLTR1 and challenged with LTC4 to quantify receptor signaling as described above.
Results
Inhibition of cAMP Accumulation in C6, CHO, and 1321N1 Cells Stably Expressing GPR17.
The orphan GPCR GPR17 was reported to be activated by uracil nucleotides (UDP), nucleotide sugars (UDP-glucose and UDP-galactose), and cysteinyl leukotrienes (LTC4 and leukotriene D4), and coupled to both inhibition of cAMP accumulation and intracellular Ca2+ mobilization (Ciana et al., 2006; Pugliese et al., 2009), although other studies have reported different agonist selectivity (Benned-Jensen and Rosenkilde, 2010) or no activation of GPR17 by these ligands (Heise et al., 2000; Maekawa et al., 2009). To investigate whether these compounds were agonists at GPR17, HA-tagged GPR17 was stably expressed in C6 and CHO cells by retroviral infection, and its capacity to promote inhibition of cAMP accumulation was assessed. UDP, UDP-glucose, and UDP-galactose (all at 100 μM) failed to promote inhibition of cAMP in either C6 or CHO cells expressing GPR17, regardless of whether cAMP levels were raised by 10 or 30 μM forskolin (Fig. 1, A and C). The lack of activity of these ligands was not due to the absence of GPR17 at the plasma membrane, because GPR17 was well expressed at the cell surface in both cell lines (Fig. 1E). In contrast, both UDP and UDP-glucose decreased cAMP accumulation by 65–75% in C6 and CHO cells expressing the P2Y14 receptor, a Gi-coupled P2Y receptor activated by UDP and UDP-sugars (Fig. 1, B and D) (Carter et al., 2009). C6 cells natively express the P2Y12 receptor (Boyer et al., 1993), and therefore 2-methylthio-ADP (2MeSADP) also significantly decreased cAMP accumulation in C6 cells (Fig. 1B). Thus, activation of bona fide Gi-coupled P2Y receptors, whether endogenously or recombinantly expressed, in two different cell lines markedly decreased cAMP accumulation, whereas addition of the same ligands to C6 and CHO cells expressing GPR17 had no effect on cAMP levels.
Fig. 1.

UDP and UDP-glucose (UDP-Glu) do not promote inhibition of cAMP accumulation in C6 or CHO cells stably expressing GPR17. UDP-Glu or UDP was added simultaneously with either 10 or 30 μM forskolin (Forsk), and the levels of cAMP accumulation were determined as described in Materials and Methods. (A) C6 cells stably expressing GPR17; (B) C6 cells stably expressing the P2Y14 receptor; (C) CHO cells stably expressing GPR17; (D) CHO cells stably expressing the P2Y14 receptor. (E) Cell surface levels of stably expressed HA-tagged GPR17 or P2Y14 receptors in C6 and CHO cells were determined by an intact cell RIA. Data shown are representative assays performed three times with similar results. Ab, antibody; cpm, counts per minute.
Activation of GPR17 also was reported to inhibit accumulation of cAMP when expressed in 1321N1 cells (Ciana et al., 2006; Daniele et al., 2011). Therefore, we also examined the capacity of uracil nucleotides, UDP-sugars, and cysteinyl leukotrienes to inhibit adenylyl cyclase in 1321N1 cells expressing GPR17. As in C6 and CHO cells, these compounds had no effect on cAMP levels in cells preincubated with 10 or 30 μM forskolin, even though a cell surface RIA confirmed robust expression of the receptor (data not shown).
Although HA-tagged receptors have been used routinely for receptor studies without any effect on signaling, we considered whether the presence of an HA tag at the N terminus interfered with receptor activity. Therefore, we stably expressed wild-type GPR17 lacking the HA tag in CHO cells and examined its capacity to inhibit cAMP accumulation. Similar to the HA-tagged receptor, neither UDP nor UDP-glucose (both at 100 μM) promoted inhibition of cAMP accumulation in CHO cells expressing the nontagged GPR17 (data not shown).
Inositol Phosphate Accumulation in HEK293 and COS-7 Cells Transiently Expressing GPR17 Alone or with Gαq/i5.
We also examined the capacity of GPR17 to couple to Gi in HEK293 and COS-7 cells. For these experiments, we used Gαq/i5, a chimeric Gα protein that couples Gi-linked receptors to activation of PLC (Conklin et al., 1993). HEK293 and COS-7 cells were transfected with GPR17 alone or with Gαq/i5, and surface expression was verified by an intact cell RIA (Fig. 2C). Neither UDP, UDP-glucose, UDP-galactose (all at 100 μM), nor LTC4 (100 nM) promoted inositol phosphate accumulation in cells transfected with GPR17 alone or with Gαq/i5 (Fig. 2, A and B). In contrast, these compounds promoted robust accumulation of [3H]inositol phosphates in cells expressing their respective cognate receptors, i.e., UDP with P2Y6 receptors, UDP-glucose and UDP-galactose with P2Y14 receptors (when coexpressed with Gαq/i5), and LTC4 with CysLTR1 receptors (Fig. 2, A and B).
Fig. 2.

UDP, UDP-sugars, and LTC4 do not promote accumulation of inositol phosphates in COS-7 or HEK293 cells transiently coexpressing GPR17 and Gαq/i5. Application of UDP, UDP-glucose (UDP-Glu), UDP-galactose (UDP-Gal) (all at 100 μM), and LTC4 (100 nM) all failed to promote significant inositol phosphate accumulation in (A) COS-7 or (B) HEK293 cells transfected with GPR17 alone or cotransfected with GPR17 and Gαq/i5, which links Gi/o-coupled receptors to the PLC signaling pathway. In contrast, UDP, UDP-Glu, UDP-Gal, and LTC4 promoted robust accumulation of inositol phosphates in cells expressing their cognate receptors (UDP at P2Y6 receptors, UDP-Glu and UDP-Gal at P2Y14 receptors, and LTC4 at CysLTR1 receptors). (C) Cell surface expression of HA-tagged GPR17 was verified by an intact cell RIA. Data shown are representative assays performed three times with similar results. cpm, counts per minute.
As reported in other studies (Lazarowski et al., 1995, 2003), basal levels of inositol phosphates were significantly increased in COS-7 cells expressing the P2Y6 or P2Y14 (cotransfected with Gαq/i5) (Fig. 2A) and in HEK293 cells coexpressing the P2Y14 receptor with Gαq/i5 (Fig. 2B). This increase is most likely the result of cellular release of nucleotides and nucleotide sugars, which activate their cognate receptors in the absence of added agonists. In contrast, expression of GPR17 alone or with Gαq/i5 in either COS-7 or HEK293 cells had no effect on basal levels of inositol phosphates.
Analysis of GPR17 Coupling to Other Gα Signaling Pathways.
Because no indication of GPR17 coupling to Gi was observed, we examined whether GPR17 coupled to other signaling pathways, i.e., Gq or G12/13. UDP, UDP-sugars, and LTC4 had no effect on [3H]inositol phosphate accumulation following transient expression of GPR17 in HEK293 and COS-7 cells (Fig. 2A). To examine the capacity of stably expressed GPR17 to couple to Gq, 1321N1 and CHO cells expressing GPR17 were challenged with UDP, UDP-sugars, or LTC4, and inositol phosphate accumulation was measured. None of the ligands tested stimulated inositol lipid hydrolysis above levels observed in wild-type cells (Fig. 3, A and B). This was not due to a lack of cell surface–expressed GPR17, as an intact cell RIA showed that the receptor was well expressed in both cell lines (Fig. 3C). In contrast, both carbachol, an agonist for endogenous M3-muscarinic receptors in 1321N1 cells, and ATP, an agonist for endogenous P2Y2 receptors in CHO cells, increased inositol phosphate accumulation 2- to 3-fold over basal in the respective cell lines (Fig. 3, A and B).
We also examined the possibility that GPR17 couples to G12/13 by cotransfecting COS-7 cells with pcDNA3-HA-GPR17 and pcDNA3-PLC-ε. In this system, activated Gα12/13 interacts with p115-RhoGEF, which in turn catalyzes the formation of GTP-bound RhoA and stimulation of PLC-ε activity (Hains et al., 2006). Neither UDP, UDP-glucose, nor LTC4 increased [3H]inositol phosphate accumulation in transfected cells, whereas lysophosphatidic acid increased [3H]inositol phosphate accumulation in COS-7 cells expressing the LPA1 receptor, which couples to Gα12/13, together with PLC-ε (Fig. 4). Moreover, the increase in [3H]inositol phosphate accumulation promoted by LPA was markedly decreased in the presence of the regulator of G protein signaling (RGS) domain from p115RhoGEF, indicating that the increase was the result of Gα12/13 and not from Gβγ or Gαq.
Fig. 4.

GPR17 does not couple to G12/13 signaling pathways. Cotransfection of the LPA1 receptor with PLC-ε (activated by Gα12/13) in COS-7 cells markedly increased LPA-promoted inositol phosphate accumulation, which was blocked by cotransfection of the RGS domain from p115RhoGEF. In contrast, LTC4 (100 nM) failed to promote significant inositol phosphate accumulation in COS-7 cells transfected with GPR17 or cotransfected with GPR17 and PLC-ε. GPR17-expressing cells were stimulated with 300 nM LTC4 (agonist, for 30 minutes), and all others were challenged with 10 μM LPA. Data shown are from a representative assay performed three times with similar results. cpm, counts per minute.
Intracellular Ca2+ Mobilization in 1321N1 Cells Expressing GPR17.
An additional observation in the study by Ciana et al. (2006) was that UDP, UDP-glucose, and LTC4 resulted in intracellular Ca2+ mobilization in a subset (30%) of 1321N1 cells transfected with GPR17 but not with empty vector. 1321N1 cells stably expressing GPR17 were seeded to coverslips and loaded with Fura-2/AM to measure intracellular Ca2+ mobilization promoted by the presumed agonists of the receptor. When UDP, UDP-glucose, UDP-galactose (all at 100 μM), or LTC4 (100 nM)) was superfused over 1321N1-GPR17 cells, no Ca2+ responses were observed in any of the >50 cells examined on four coverslips (Fig. 5). In contrast, 1 mM carbachol (acting through an endogenous M3-muscarinic receptor) consistently promoted robust intracellular Ca2+ mobilization in 100% of 1321N1 cells examined (Fig. 5).
Fig. 5.

Lack of uracil nucleotide– and leukotriene-promoted intracellular Ca2+ mobilization in 1321N1-GPR17 cells. GPR17-expressing 1321N1 cells were grown on glass coverslips to a density of 20% of confluence. Cells were placed in a sealed chamber and continuously superfused with HBSS with or without (A) 100 μM UDP, (B) 100 μM UDP-glucose (UDP-Glu), (C) 100 μM UDP-galactose (UDP-Gal), (D) 100 nM LTC4, or (A–D) 1 mM carbachol (Carb). Each compound was superfused for 1 minute. Traces of intracellular Ca2+ concentrations were averaged from 6–9 individual cells/coverslip. Each compound was tested on a minimum of three coverslips.
Internalization of Receptors in 1321N1 Cells.
With few exceptions, GPCRs internalize when challenged with their cognate agonists. Therefore, as an additional measure of agonist activity, we also examined whether any of the reported agonists of GPR17 were capable of promoting internalization of GPR17 in 1321N1 cells. 1321N1-GPR17, 1321N1-P2Y2, and 1321N1-CysLTR1 cells were incubated with anti-HA antibody at 4°C, the temperature was increased to 37°C, and ligands were added to promote internalization of the receptors. Addition of LTC4, UDP, or UDP-glucose to 1321N1-GPR17 cells for 30 minutes did not promote internalization of the receptor, whereas LTC4 and UTP promoted marked internalization of their cognate receptors in 1321N1-CysLTR1 and 1321N1-P2Y2 cells, respectively (Fig. 6).
Fig. 6.

Neither UDP, UDP-glucose (UDP-G), nor LTC4 promotes internalization of GPR17 stably expressed in 1321N1 cells. 1321N1 cells stably expressing CysLTR1, P2Y2, or GPR17 receptors were seeded on glass coverslips for 2 days, then cooled down to 4°C and labeled with mouse monoclonal anti-HA.11 antibody for 1 hour. Cells were washed, warmed to 37°C, and treated with either phosphate-buffered saline (PBS) or receptor agonists (100 μM UTP, UDP, or UDP-G or 100 nM LTC4) for 30 minutes. The cells were then fixed, permeabilized, and stained with goat anti-mouse A-488 secondary antibody. Localization of receptors was determined by confocal microscopy.
GPR17 As a Negative Regulator of CysLTR1-Promoted Signaling.
Maekawa et al. (2009) reported that GPR17 was not activated by UDP, UDP-sugars, or LTC4 in a number of cell lines examined; instead, GPR17 was shown to inhibit CysLTR1 signaling, presumably through receptor dimer formation, without any effect on cell surface expression of CysLTR1. Therefore, we investigated the capacity of GPR17 to inhibit CysLTR1-dependent signaling activity by infecting 1321N1 cells already expressing HA-tagged CysLTR1 with retroviruses harboring myc-tagged GPR17. LTC4 (300 nM) stimulated inositol phosphate accumulation was then measured in those cells expressing either CysLTR1 alone or CysLTR1 coexpressed with GPR17. LTC promoted a 3- to 4-fold increase in inositol phosphate accumulation in CysLTR1-expressing cells, but agonist-stimulated response in cells coexpressing CysLTR1 with GPR17 was reduced by ∼85%. Importantly, the surface expression of LTR1 was reduced by 50% in cells coexpressing this GPCR with GPR17 (Fig. 7). Transient transfection experiments with CysLTR1 and GPR17 gave similar results (data not shown). These data are consistent with those reported by Maekawa et al. (2009) showing that GPR17 is a negative regulator of CysLTR1.
Fig. 7.
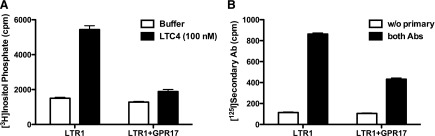
GPR17 negatively regulates CysLTR1 function. 1321N1 cells stably expressing 3× HA-tagged CysLTR1 were infected with retroviruses directing expression of myc-tagged GPR17, and the resulting cells were assessed for LTC4-promoted inositol phosphate accumulation (A) and CysLTR1 expression by a cell surface RIA (B). Data shown are from a representative assay performed three times with similar results. Ab, antibody; cpm, counts per minute.
Discussion
Recent studies highlight the importance of GPR17 in inflammation, neuronal damage sensing, neurorepair, food intake, and myelination (Lecca et al., 2008; Chen et al., 2009; Maekawa et al., 2010; Ren et al., 2012; Franke et al., 2013), but the true identity of the endogenous agonists of the receptor remains controversial. Ciana et al. (2006) reported that GPR17 is activated by UDP, UDP-sugars, and cysteinyl leukotrienes, whereas Benned-Jensen and Rosenkilde (2010) reported activation of GPR17 with nucleotides but not with leukotrienes. Heise et al. (2000) injected GPR17 cRNA into Xenopus oocytes and also observed no response to LTC4 or leukotriene D4, while these two compounds elicited strong calcium-dependent chloride current in CysLTR2 cRNA–injected oocytes. Maekawa et al. (2009) demonstrated that LTC4, leukotriene D4, and leukotriene E4 were unable to promote Ca2+ mobilization in a variety of cell lines (1321N1, CHO, and HEK293T) stably expressing the human or mouse GPR17.
Data presented in this study show that neither UDP, UDP-sugars, nor cysteinyl leukotrienes activate GPR17 when the receptor is expressed stably in C6, 1321N1, and CHO cells or transiently in COS-7 and HEK293 cells (with or without Gαq/i5). In all cases, expression of GPR17 at the cell surface was verified by an intact cell RIA (with the exception of experiments with a nontagged receptor). Our studies extended this question further by examining the potential coupling of GPR17 to each of the G protein signaling pathways. Although receptors known to couple to each G protein pathway mediated predictable positive responses, no GPCR signaling activity was observed in a variety of cells in response to nucleotides or leukotrienes. Receptor activation was measured by inhibition of cAMP accumulation, inositol phosphate production, and by monitoring Ca2+ mobilization with Fura-2/AM. None of the purported ligands of GPR17 activated the receptor in any of these test systems, whereas the cognate receptors for these ligands showed robust signaling activity. We also demonstrated that these ligands failed to promote internalization of GPR17 in 1321N1 cells, whereas receptors that are activated by these compounds were markedly internalized. Based on these data, we conclude that UDP, UDP-sugars, and cysteinyl leukotrienes are not activators of GPR17.
Maekawa et al. (2009) also reported that coexpression of GPR17 with CysLTR1 in 1321N1 cells significantly inhibited LTC4- and LTD4-promoted calcium influx and extracellular signal–regulated kinase phosphorylation compared with the response in cells expressing the CysLTR1 receptor alone. Because GPR17 coexpression did not affect surface expression of the CysLTR1 receptor, the authors concluded that GPR17 functions as a negative regulator of the CysLTR1 receptor. A more recent study has shown that knockout of GPR17 in mice increased the immune responses to dust mites, including the recruitment of inflammatory cells, increased levels of IgE, and increased Th2 and Th17 cytokine expression, which was due to the absence of inhibition of CysLTR1 signaling activity by GPR17 (Maekawa et al., 2010). We also observed a marked decrease (>80%) in CysLTR1 receptor activity when GPR17 was coexpressed, although we also saw a decrease in the level of CysLTR1 at the cell surface (∼50%). Given that CysLTR1 was expressed recombinantly, it is likely that the levels of receptors, although decreased, are more than sufficient for full activity and that the decrease in receptor signaling is a direct consequence of GPR17 coexpression. Thus, our data are fully consistent with those of Maekawa et al. (2009, 2010).
We are unsure why some laboratories have shown activity of UDP, UDP-sugars, and LTC4 at GPR17 whereas others (including ours) have not observed activity of some or all these ligands. Early characterization of GPR17 mostly focused on guanosine 5′-3-O-(thio)triphosphate binding studies instead of second messenger assays, but a recent study of GPR17 by the authors who originally reported activity of these ligands at the receptor showed inhibition of cAMP accumulation and receptor internalization in 1321N1 cells (Daniele et al., 2011). However, using the same conditions (cAMP levels elevated by 10 μM forskolin) and the same cells (1321N1), we failed to observe inhibition of cAMP accumulation in over a dozen independent experiments, even though the receptor was expressed at the cell surface based on an intact cell RIA.
We also examined the sequence of GPR17 in our clones to confirm that its sequence matched the sequence in GenBank. Surprisingly, of the six sequences in GenBank, two of them (accession numbers AAH31653.1 and BAG54380.1) harbored amino acid differences from the other four. AAH31653.1 contains an L55F mutation, while BAG54380.1 has three mutations: L44P, V225A, and S287R. Our sequence was identical to the other four sequences and matched the sequence reported in Ciana et al. (2006).
A recent study by a different group analyzed the pharmacological selectivity of GPR17 expressed in HEK293 cells using the GloSensor cAMP assay (Promega, Madison, WI) and observed activation by UDP, UDP-galactose, and UDP-glucose (Buccioni et al., 2011). However, the authors were unable to demonstrate inhibition of cAMP accumulation by two different immunocompetitive cAMP assays and required the increased sensitivity of the GloSensor assay to observe inhibition of adenylyl cyclase. In contrast, Daniele et al. (2011) reported robust inhibition of cAMP accumulation in 1321N1 cell using a competitive protein-binding assay. Thus, there are important discrepancies even among laboratories that observe activation of GPR17 with nucleotides and nucleotide sugars. We tested four different cell lines for inhibition of cAMP accumulation, including transfection experiments with Gαq/i5, but observed no agonist-dependent activity.
Identification and characterization of new P2Y receptors has been fraught with controversy and misidentification. At least part of the difficulty in identifying new P2Y receptors is the lack of sequence identity between known P2Y receptors; the two P2Y receptor subfamilies, P2Y1 and P2Y12, share minimal sequence identity (∼20%), and thus low homology of an orphan receptor to known P2Y receptors is not a criterion for exclusion. P2Y receptors are found in nearly all cell lines (with the exception of 1321N1 cells), and contribution of endogenous receptors to the signal supposedly emanating from an expressed recombinant one is often difficult to exclude. Finally, the possibility of hetero-oligomerization of an orphan receptor with endogenous P2Y receptors (including receptors that are not normally expressed or are expressed at low levels but are “recruited” to the cell surface by expression of the orphan receptor) also could lead to potentially incorrect identification (see, e.g., Bush et al., 2007).
Because of nucleotide release and/or cell lysis, basal levels of inositol phosphates in COS-7 cells transfected with P2Y receptors are consistently observed to be elevated over vector-transfected controls (Lazarowski et al., 1995, 2003). For example, both the P2Y6 receptor alone and the P2Y14 receptor cotransfected with Gαq/i5 result in elevated basal levels of inositol phosphates in COS-7 cells compared with cells transfected with empty vector (Fig. 2). In contrast, basal levels of [3H]inositol phosphates in COS-7 (or HEK293) cells expressing GPR17 either alone or with Gαq/i5 were not significantly higher than vector-transfected cells (Fig. 2, A and B). These data suggest that neither UDP nor UDP-sugars released into the medium of cells are capable of activating GPR17, although they do increase the activation of their cognate receptors, P2Y6 and P2Y14. These results are consistent with the lack of activity of nucleotides and nucleotide sugars at GPR17.
Even though nucleotides are unlikely to activate CysLTR1, there does appear to be a function interaction between CysLTR1 and P2Y receptors. For example, activation of endogenous P2Y receptors in U937 cells with ATP or UDP strongly cross-desensitized CysLTR1 receptors, but not vice versa, and a similar cross-desensitization was observed in COS-7 cells exogenously expressing CysLTR1 (Capra et al., 2005). This heterologous desensitization was dependent on protein kinase C, because GF109203X (bisindoylmaleimide I) blocked nucleotide-dependent desensitization of CysLTR1. A separate study demonstrated that antagonists of leukotriene receptors, montelukast and pranlukast, inhibit nucleotide-promoted activation of P2Y receptors both in differentiated U937 cells and in 1321N1 cells stably expressing P2Y1, P2Y2, P2Y4, or P2Y6 receptors (Mamedova et al., 2005). This antagonism was not competitive, however, as high concentrations of nucleotides were unable to surmount the inhibition, and these antagonists did not block binding of the P2Y1 receptor–selective antagonist [3H]MRS2279 [2-chloro-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate] to the P2Y1 receptor.
In conclusion, our data demonstrate that nucleotides, nucleotide sugars, and cysteinyl leukotrienes do not promote activation of GPR17 when assessed by standard and well established signaling assays. Although the reasons for the discrepancies between different laboratories are unclear, our data are consistent with an earlier study showing that GPR17 is a negative regulator of CysLTR1 function.
Abbreviations
- CHO
Chinese hamster ovary
- CysLTR1
cysteinyl leukotriene receptor 1
- DMEM
Dulbecco’s modified Eagle’s medium
- FBS
fetal bovine serum
- Fura-2/AM
Fura-2/acetoxymethyl ester
- GPCR
G protein–coupled receptor
- HA
hemagglutinin
- HBSS
Hanks' balanced salt solution
- LTC4
cysteinyl leukotriene C4
- MRS2279
2-chloro-N6-methyl-(N)-methanocarba-2′-deoxyadenosine-3′,5′-bisphosphate
- PLC
phospholipase C
- RIA
radioimmunoassay
Authorship Contributions
Participated in research design: Qi, Harden, Nicholas.
Conducted experiments: Qi.
Performed data analysis: Qi, Harden, Nicholas.
Wrote or contributed to the writing of the manuscript: Qi, Harden, Nicholas.
Footnotes
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grant R01HL071131 (to R.A.N.)].
References
- Abbracchio MP, Burnstock G, Boeynaems JM, Barnard EA, Boyer JL, Kennedy C, Knight GE, Fumagalli M, Gachet C, Jacobson KA, et al. (2006) International Union of Pharmacology LVIII: update on the P2Y G protein-coupled nucleotide receptors: from molecular mechanisms and pathophysiology to therapy. Pharmacol Rev 58:281–341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akbar GKM, Dasari VR, Webb TE, Ayyanathan K, Pillarisetti K, Sandhu AK, Athwal RS, Daniel JL, Ashby B, Barnard EA, et al. (1996) Molecular cloning of a novel P2 purinoceptor from human erythroleukemia cells. J Biol Chem 271:18363–18367 [DOI] [PubMed] [Google Scholar]
- Benned-Jensen T, Rosenkilde MM. (2010) Distinct expression and ligand-binding profiles of two constitutively active GPR17 splice variants. Br J Pharmacol 159:1092–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanov YD, Dale L, King BF, Whittock N, Burnstock G. (1997) Early expression of a novel nucleotide receptor in the neural plate of Xenopus embryos. J Biol Chem 272:12583–12590 [DOI] [PubMed] [Google Scholar]
- Boyer JL, Lazarowski ER, Chen XH, Harden TK. (1993) Identification of a P2Y-purinergic receptor that inhibits adenylyl cyclase. J Pharmacol Exp Ther 267:1140–1146 [PubMed] [Google Scholar]
- Buccioni M, Marucci G, Dal Ben D, Giacobbe D, Lambertucci C, Soverchia L, Thomas A, Volpini R, Cristalli G. (2011) Innovative functional cAMP assay for studying G protein-coupled receptors: application to the pharmacological characterization of GPR17. Purinergic Signal 7:463–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush CF, Jones SV, Lyle AN, Minneman KP, Ressler KJ, Hall RA. (2007) Specificity of olfactory receptor interactions with other G protein-coupled receptors. J Biol Chem 282:19042–19051 [DOI] [PubMed] [Google Scholar]
- Capra V, Ravasi S, Accomazzo MR, Citro S, Grimoldi M, Abbracchio MP, Rovati GE. (2005) CysLT1 receptor is a target for extracellular nucleotide-induced heterologous desensitization: a possible feedback mechanism in inflammation. J Cell Sci 118:5625–5636 [DOI] [PubMed] [Google Scholar]
- Carter RL, Fricks IP, Barrett MO, Burianek LE, Zhou Y, Ko H, Das A, Jacobson KA, Lazarowski ER, Harden TK. (2009) Quantification of Gi-mediated inhibition of adenylyl cyclase activity reveals that UDP is a potent agonist of the human P2Y14 receptor. Mol Pharmacol 76:1341–1348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Wu H, Wang S, Koito H, Li J, Ye F, Hoang J, Escobar SS, Gow A, Arnett HA, et al. (2009) The oligodendrocyte-specific G protein-coupled receptor GPR17 is a cell-intrinsic timer of myelination. Nat Neurosci 12:1398–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciana P, Fumagalli M, Trincavelli ML, Verderio C, Rosa P, Lecca D, Ferrario S, Parravicini C, Capra V, Gelosa P, et al. (2006) The orphan receptor GPR17 identified as a new dual uracil nucleotides/cysteinyl-leukotrienes receptor. EMBO J 25:4615–4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conklin BR, Farfel Z, Lustig KD, Julius D, Bourne HR. (1993) Substitution of three amino acids switches receptor specificity of Gq α to that of Gi α. Nature 363:274–276 [DOI] [PubMed] [Google Scholar]
- Coppi E, Maraula G, Fumagalli M, Failli P, Cellai L, Bonfanti E, Mazzoni L, Coppini R, Abbracchio MP, Pedata F, et al. (2013) UDP-glucose enhances outward K(+) currents necessary for cell differentiation and stimulates cell migration by activating the GPR17 receptor in oligodendrocyte precursors. Glia 61:1155–1171 [DOI] [PubMed] [Google Scholar]
- Costanzi S, Mamedova L, Gao ZG, Jacobson KA. (2004) Architecture of P2Y nucleotide receptors: structural comparison based on sequence analysis, mutagenesis, and homology modeling. J Med Chem 47:5393–5404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniele S, Trincavelli ML, Gabelloni P, Lecca D, Rosa P, Abbracchio MP, Martini C. (2011) Agonist-induced desensitization/resensitization of human G protein-coupled receptor 17: a functional cross-talk between purinergic and cysteinyl-leukotriene ligands. J Pharmacol Exp Ther 338:559–567 [DOI] [PubMed] [Google Scholar]
- Franke H, Parravicini C, Lecca D, Zanier ER, Heine C, Bremicker K, Fumagalli M, Rosa P, Longhi L, Stocchetti N, et al. (2013) Changes of the GPR17 receptor, a new target for neurorepair, in neurons and glial cells in patients with traumatic brain injury. Purinergic Signal DOI:10.1007/s11302-013-9366-3 [published ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan X-M, Kobilka TS, Kobilka BK. (1992) Enhancement of membrane insertion and function in a type IIIb membrane protein following introduction of a cleavable signal peptide. J Biol Chem 267:21995–21998 [PubMed] [Google Scholar]
- Hains MD, Wing MR, Maddileti S, Siderovski DP, Harden TK. (2006) Galpha12/13- and rho-dependent activation of phospholipase C-epsilon by lysophosphatidic acid and thrombin receptors. Mol Pharmacol 69:2068–2075 [DOI] [PubMed] [Google Scholar]
- He W, Miao FJ, Lin DC, Schwandner RT, Wang Z, Gao J, Chen JL, Tian H, Ling L. (2004) Citric acid cycle intermediates as ligands for orphan G-protein-coupled receptors. Nature 429:188–193 [DOI] [PubMed] [Google Scholar]
- Heise CE, O’Dowd BF, Figueroa DJ, Sawyer N, Nguyen T, Im DS, Stocco R, Bellefeuille JN, Abramovitz M, Cheng R, et al. (2000) Characterization of the human cysteinyl leukotriene 2 receptor. J Biol Chem 275:30531–30536 [DOI] [PubMed] [Google Scholar]
- Herold CL, Li Q, Schachter JB, Harden TK, Nicholas RA. (1997) Lack of nucleotide-promoted second messenger signaling responses in 1321N1 cells expressing the proposed P2Y receptor, p2y7. Biochem Biophys Res Commun 235:717–721 [DOI] [PubMed] [Google Scholar]
- Inbe H, Watanabe S, Miyawaki M, Tanabe E, Encinas JA. (2004) Identification and characterization of a cell-surface receptor, P2Y15, for AMP and adenosine. J Biol Chem 279:19790–19799 [DOI] [PubMed] [Google Scholar]
- Janssens R, Boeynaems JM, Godart M, Communi D. (1997) Cloning of a human heptahelical receptor closely related to the P2Y5 receptor. Biochem Biophys Res Commun 236:106–112 [DOI] [PubMed] [Google Scholar]
- Lazarowski ER, Shea DA, Boucher RC, Harden TK. (2003) Release of cellular UDP-glucose as a potential extracellular signaling molecule. Mol Pharmacol 63:1190–1197 [DOI] [PubMed] [Google Scholar]
- Lazarowski ER, Watt WC, Stutts MJ, Boucher RC, Harden TK. (1995) Pharmacological selectivity of the cloned human P2U-purinoceptor: potent activation by diadenosine tetraphosphate. Br J Pharmacol 116:1619–1627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lecca D, Trincavelli ML, Gelosa P, Sironi L, Ciana P, Fumagalli M, Villa G, Verderio C, Grumelli C, Guerrini U, et al. (2008) The recently identified P2Y-like receptor GPR17 is a sensor of brain damage and a new target for brain repair. PLoS ONE 3:e3579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Olesky M, Palmer RK, Harden TK, Nicholas RA. (1998) Evidence that the p2y3 receptor is the avian homologue of the mammalian P2Y6 receptor. Mol Pharmacol 54:541–546 [DOI] [PubMed] [Google Scholar]
- Li Q, Schachter JB, Harden TK, Nicholas RA. (1997) The 6H1 orphan receptor, claimed to be the p2y5 receptor, does not mediate nucleotide-promoted second messenger responses. Biochem Biophys Res Commun 236:455–460 [DOI] [PubMed] [Google Scholar]
- Maekawa A, Balestrieri B, Austen KF, Kanaoka Y. (2009) GPR17 is a negative regulator of the cysteinyl leukotriene 1 receptor response to leukotriene D4. Proc Natl Acad Sci USA 106:11685–11690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa A, Xing W, Austen KF, Kanaoka Y. (2010) GPR17 regulates immune pulmonary inflammation induced by house dust mites. J Immunol 185:1846–1854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mamedova L, Capra V, Accomazzo MR, Gao ZG, Ferrario S, Fumagalli M, Abbracchio MP, Rovati GE, Jacobson KA. (2005) CysLT1 leukotriene receptor antagonists inhibit the effects of nucleotides acting at P2Y receptors. Biochem Pharmacol 71:115–125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin V, Sawyer N, Stocco R, Unett D, Lerner MR, Abramovitz M, Funk CD. (2001) Molecular cloning and functional characterization of murine cysteinyl-leukotriene 1 (CysLT(1)) receptors. Biochem Pharmacol 62:1193–1200 [DOI] [PubMed] [Google Scholar]
- Murakami M, Shiraishi A, Tabata K, Fujita N. (2008) Identification of the orphan GPCR, P2Y(10) receptor as the sphingosine-1-phosphate and lysophosphatidic acid receptor. Biochem Biophys Res Commun 371:707–712 [DOI] [PubMed] [Google Scholar]
- Noguchi K, Ishii S, Shimizu T. (2003) Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J Biol Chem 278:25600–25606 [DOI] [PubMed] [Google Scholar]
- Parravicini C, Ranghino G, Abbracchio MP, Fantucci P. (2008) GPR17: molecular modeling and dynamics studies of the 3-D structure and purinergic ligand binding features in comparison with P2Y receptors. BMC Bioinformatics 9:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasternack SM, von Kügelgen I, Al Aboud K, Lee YA, Rüschendorf F, Voss K, Hillmer AM, Molderings GJ, Franz T, Ramirez A, et al. (2008) G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat Genet 40:329–334 [DOI] [PubMed] [Google Scholar]
- Pugliese AM, Trincavelli ML, Lecca D, Coppi E, Fumagalli M, Ferrario S, Failli P, Daniele S, Martini C, Pedata F, et al. (2009) Functional characterization of two isoforms of the P2Y-like receptor GPR17: [35S]GTPgammaS binding and electrophysiological studies in 1321N1 cells. Am J Physiol Cell Physiol 297:C1028–C1040 [DOI] [PubMed] [Google Scholar]
- Qi AD, Harden TK, Nicholas RA. (2004) GPR80/99, proposed to be the P2Y(15) receptor activated by adenosine and AMP, is not a P2Y receptor. Purinergic Signal 1:67–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi AD, Kennedy C, Harden TK, Nicholas RA. (2001a) Differential coupling of the human P2Y(11) receptor to phospholipase C and adenylyl cyclase. Br J Pharmacol 132:318–326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi AD, Zambon AC, Insel PA, Nicholas RA. (2001b) An arginine/glutamine difference at the juxtaposition of transmembrane domain 6 and the third extracellular loop contributes to the markedly different nucleotide selectivities of human and canine P2Y11 receptors. Mol Pharmacol 60:1375–1382 [DOI] [PubMed] [Google Scholar]
- Ren H, Orozco IJ, Su Y, Suyama S, Gutiérrez-Juárez R, Horvath TL, Wardlaw SL, Plum L, Arancio O, Accili D. (2012) FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell 149:1314–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabata K, Baba K, Shiraishi A, Ito M, Fujita N. (2007) The orphan GPCR GPR87 was deorphanized and shown to be a lysophosphatidic acid receptor. Biochem Biophys Res Commun 363:861–866 [DOI] [PubMed] [Google Scholar]
- Torres B, Zambon AC, Insel PA. (2002) P2Y11 receptors activate adenylyl cyclase and contribute to nucleotide-promoted cAMP formation in MDCK-D(1) cells. A mechanism for nucleotide-mediated autocrine-paracrine regulation. J Biol Chem 277:7761–7765 [DOI] [PubMed] [Google Scholar]
- Webb TE, Henderson D, King BF, Wang S, Simon J, Bateson AN, Burnstock G, Barnard EA. (1996a) A novel G protein-coupled P2 purinoceptor (P2Y3) activated preferentially by nucleoside diphosphates. Mol Pharmacol 50:258–265 [PubMed] [Google Scholar]
- Webb TE, Kaplan MG, Barnard EA. (1996b) Identification of 6H1 as a P2Y purinoceptor: P2Y5. Biochem Biophys Res Commun 219:105–110 [DOI] [PubMed] [Google Scholar]
- Yokomizo T, Izumi T, Chang K, Takuwa Y, Shimizu T. (1997) A G-protein-coupled receptor for leukotriene B4 that mediates chemotaxis. Nature 387:620–624 [DOI] [PubMed] [Google Scholar]