

Abstract
Recent findings have demonstrated that the gut microbiome complements our human genome with at least 100-fold more genes. In contrast to our Homo sapiens–derived genes, the microbiome is much more plastic, and its composition changes with age and diet, among other factors. An altered gut microbiota has been associated with several diseases, including obesity and diabetes, but the mechanisms involved remain elusive. Here we discuss factors that affect the gut microbiome, how the gut microbiome may contribute to metabolic diseases, and how to study the gut microbiome. Next-generation sequencing and development of software packages have led to the development of large-scale sequencing efforts to catalog the human microbiome. Furthermore, the use of genetically engineered gnotobiotic mouse models may increase our understanding of mechanisms by which the gut microbiome modulates host metabolism. A combination of classical microbiology, sequencing, and animal experiments may provide further insights into how the gut microbiota affect host metabolism and physiology.
We have coevolved with microbes in the environment, and each body habitat has a unique set of microorganisms (microbiota) (1). The most abundant and well-studied microbiota are found in the gut, where the bacterial density reaches 1011–1012 cells/g in the distal human colon (2). The number of bacteria in the human gut has been estimated to exceed the number of somatic cells in the body by an order of magnitude and that the biomass of the gut microbiota may reach up to 1.5 kg (2). Thus, one may consider the gut microbiota as a multicellular organ similar in size to the liver (3). Furthermore, the combined genomes of the gut microbiota—the microbiome—contain >100-fold more genes than are encoded in the human genome (4), and these genes contribute significantly to our physiology and metabolism (5,6).
The introduction of high-throughput sequencing techniques has helped to reveal the complexity and composition of the gut microbiota. Most bacterial species in the human and mouse gut belong to the phyla Bacteroidetes and Firmicutes (7), but less abundant bacterial phyla, such as Actinobacteria, Proteobacteria, and Verrucomicrobia, as well as methanogenic archaea, mainly Methanobrevibacter smithii, are also present (4,8). The total number of bacterial species has been estimated to exceed 1,000, and at least 160 species are shared among individuals (4). Interestingly, a recent study demonstrated marked differences among populations in the U.S., rural Malawi, and Venezuela (9). Non-U.S. adult residents had higher levels of Prevotella, whereas the differences between the Malawi and Venezuela populations were more subtle, with differences in the abundance of several species in the Clostridials order. This finding highlights the necessity to perform metagenomic studies in different ethnic populations. Vast numbers of viruses are also present in the human gut (10), but our knowledge about their function is limited, and they will not be discussed here.
The gut microbiota have the capacity to affect host physiology within and outside the gut. For example, the gut microbiota are essential for normal development and homeostasis of the immune system in the gut, modulate epithelial cell proliferation, protect against pathogenic bacteria, and modulate villus architecture and angiogenesis within the intestine (5,6). Furthermore, the gut microbiota affect xenobiotic metabolism, bone mineral density, behavior, and several metabolic functions (5,6), and emerging data from humans and mouse models suggest that the gut microbiota play a role in the development of metabolic diseases. Here, we will review factors that affect the gut microbiota, how the gut microbiota may contribute to metabolic diseases, and how to assess the composition and function of the gut microbiota.
ESTABLISHMENT AND DEVELOPMENT OF THE MICROBIOTA
Age.
The fetal gut is sterile and is colonized at birth with microbes from the mother’s vaginal and fecal microbiota as well as with other environmental microbes encountered in the first days of life. Early colonization depends on the mode of delivery, diet (breast- vs. formula-feeding), hygiene, and antibiotic treatment (11). The first colonizers are facultative anaerobes, such as Escherichia coli and Streptococcus spp., and obligate anaerobic species colonize as the oxygen levels in the gut decrease. A large study involving three populations in different geographic locations found that a child’s microbiota stabilize and become adult-like at ∼3 years of age (9). The metagenome of the infant gut is characterized by enrichment of genes for simple sugar breakdown, such as lactose and galactose, whereas the weaned microbiota are enriched in genes for polysaccharide breakdown and vitamin production (9,12).
In the elderly, changes in the microbiota occur, resulting in reduced microbial diversity, which is accompanied by increased inflammation (13). Compared with elderly subjects in long-stay residential care units, elderly individuals living in the community have higher levels of fecal butyrate and other short-chain fatty acids (SCFAs), such as acetate propionate and valerate; the presence of SCFAs is generally believed to be associated with reduced inflammation.
Diet has profound effect on the gut microbiota.
Many of the nutrients in the diet are digested by human enzymes and absorbed in the small intestine. However, the gut microbiota have a central role for the metabolism of dietary fibers, which are not degraded by human enzymes. Comparative studies of the gut microbiota across mammals have shown that several bacterial species are shared and that their presence is influenced by host diet and phylogeny (14). Herbivores have a more diverse microbiota than carnivores, indicating that degradation of plant polysaccharides is more complex and demanding, which is also reflected in longer intestinal tracts and transit times. Metagenomic sequencing of the gut microbiota revealed that herbivores carry more genes for nitrogen assimilation into proteins compared with carnivores, which reflects that amino acids are less abundant in herbivore diets (15). Similarly, the gut microbiota of vegetarians and vegans are unable to metabolize carnitine, which is present in red meat (16).
In humans, microbes respond differently to dietary components, and long-term dietary habits have been linked to the abundance of microbial genera: Bacteroides correlates positively with a protein-rich diet, whereas Prevotella is associated with a diet rich in fiber (17). A short-term controlled-feeding study in which subjects were randomized to high-fat/low-fiber or low-fat/high-fiber diets and followed up for 10 days showed that although diet change had an initial rapid effect on the microbiota, the interindividual differences dominated (17). Furthermore, a study comparing the gut microbiota of children living in Italy with those living in Burkina Faso showed marked differences between the populations: the children from Burkina Faso, who consumed higher amounts of plant polysaccharides, had higher levels of Prevotella together with higher levels of SCFAs, which are likely linked to increased fermentation of indigestible plant polysaccharides (18). Thus, the gut microbiota are important for processing, but at the same time, diet may alter the gut microbial community (16).
MICROBIAL ALTERATIONS ASSOCIATED WITH METABOLIC DISEASES IN HUMANS
Obesity.
Studies in humans and mice have shown that obesity is associated with changes in the composition of the gut microbiota. Early studies reported an enrichment in Firmicutes and a corresponding decrease in Bacteroidetes levels in the microbiota of obese individuals; the Bacteroidetes-to-Firmicutes ratio normalized to the level observed in lean individuals after weight loss (19). An increased ratio of Firmicutes to Bacteroidetes has also been observed in mice genetically predisposed to obesity (ob/ob) (7). However, more recent studies have not observed this association (20) or observed an opposite association, with an increase in Bacteroidetes in obese individuals (21). When considering these results, it should be noted that the initial studies were limited in sample size and differed in study design and subjects (adults, adolescents, and some were compared before and after weight loss). Furthermore, the studies differed in methodology (16S rRNA gene sequencing, fluorescence in situ hybridization, and quantitative PCR). It should also be noted that the Bacteroidetes-to-Firmicutes ratio is a rough measure because these broad classifications of bacterial taxa that include pathogens such as Clostridium botulinum and Listeria monocytogenes as well as species such as Eubacterium rectale and Faecalibacterium prausnitzii that are known butyrate producers and generally regarded as beneficial to the host. Therefore, more standardized study protocols are needed to allow cross-comparisons between studies as well as a more taxonomically detailed description than phylum-level changes.
To determine whether the altered gut microbiota contribute to obesity or whether obesity alters the gut microbiota requires prospective studies. A prospective Finnish study of 49 infants sampled at 6 and 12 months of age showed that children who were overweight at 7 years of age had higher levels of Staphylococcus aureus and lower levels of Bifidobacteria during infancy (22).
Analysis of the metagenome of twins concordant for obesity showed that obese individuals harbor more genes for phosphotransferase systems involved in carbohydrate processing (23), suggesting an increased capacity to degrade polysaccharide-rich diets. Importantly, transfer of microbiota harvested from lean or obese individuals to germ-free mice demonstrated that the obese phenotype can be transferred by the microbiota, thus suggesting a causal relationship between the altered microbiota and obesity development (24,25).
Little is known about the mechanisms by which the gut microbiota modulate obesity apart from a potential role in energy harvest from the diet. Colonized mice consume less food than their germ-free counterparts and so increased food consumption cannot explain the obese phenotype in colonized mice (26). Activation of AMP-activated protein kinase and expression of angiopoietin-like protein 4 (also known as fasting-induced adipose factor), which are both associated with reduced energy expenditure (27), are suppressed by the gut microbiota and thus may be part of the potential mechanism through which the gut microbiota promote obesity. However, these studies need to be expanded, and the use of genetically modified germ-free mice may facilitate to delineate the molecular mechanisms by which specific microbes or consortia affect host metabolism.
Type 2 diabetes.
The incidence of type 2 diabetes (T2D) is increasing in parallel with obesity, and environmental factors that are associated with T2D risk include diet and the gut microbiota (28). Low-grade inflammation is observed in T2D patients, and diabetic mice and humans have increased plasma levels of lipopolysaccharide (LPS), a membrane component of Gram-negative bacteria, which has been shown to impair glucose metabolism in mice (29,30). Germ-free mice have fewer macrophages in their adipose tissue and improved glucose metabolism compared with colonized mice (31).
Recent metagenomics approaches have investigated whether the gut microbiota are altered in patients with T2D. Shotgun sequencing of the gut metagenome revealed that butyrate-producing bacteria, known to be anti-inflammatory (e.g., Roseburia spp. and Faecalibacterium spp.), are less abundant in T2D patients than in healthy control subjects (32,33). By comparing metagenomic data from Chinese and Swedish subjects, we showed that T2D-associated metagenomes encode similar functions, but the species involved are markedly different (33). Furthermore, we developed a model based on metagenomic data that could distinguish T2D subjects from control subjects with a predictive power that was far better than that of the body mass index (33). The predictive power of the metagenome was similar when trained on Chinese subjects (33), suggesting that it may be possible to develop novel diagnostic approaches based on analysis of the gut metagenome.
Gastric bypass surgery in obese patients not only promotes sustained weight reduction but also reduces the risks of T2D and cardiovascular mortality (34,35). Importantly, diabetes resolution occurs before weight reduction, suggesting that gastric bypass has direct antidiabetic effects. The mechanisms are not defined, but two independent studies in humans observed shifts in the composition of the fecal microbiota (36,37), thus suggesting that the gut microbiota may contribute to the improved metabolic phenotype associated with gastric bypass. In particular, abundance of the beneficial microbe F. prausnitzii was decreased in obese T2D patients and increased after surgery (36). The levels of F. prausnitzii negatively correlated with inflammatory markers, thus indicating that this microbe may contribute to the amelioration of T2D after gastric bypass by modulating systemic inflammation. Similar microbial changes are also seen in rats and mice undergoing bariatric surgery (38,39). Interestingly, mice that received microbiota from mice that underwent gastric bypass surgery exhibited improved metabolism compared with mice that received microbiota from mice that underwent sham surgery (39), providing direct evidence that an altered gut microbiota contribute to the beneficial effects of gastric bypass surgery.
A direct link between an altered gut microbiota and insulin resistance in humans was recently provided: insulin sensitivity and levels of butyrate-producing bacteria increased in patients with the metabolic syndrome after transplantation with intestinal microbiota from lean healthy donors (40). Transplanting unfractionated microbiota into humans is not without risk, and a more direct and targeted approach on single microbes or a community of microbes is desirable. In this context, more work is needed to identify individual species or groups of species that contribute to improvements in health and how these can be safely and effectively administered to patients.
Type 1 diabetes.
Even though the autoimmune disease type 1 diabetes (T1D) has a known genetic risk factor involving mutation in the human leukocyte antigen genes, the recent rise in incidence of this disease points to environmental factors playing an increasing role (41). A study of four children with newly developed T1D and four matched control children found differences in the composition of the gut metagenome between the groups and reduced diversity in T1D-associated metagenomes (42). Studies in nonobese diabetic (NOD) mice have shown that germ-free NOD mice or those housed in specific-pathogen free conditions are more likely to develop diabetes, suggesting that the gut microbiota are involved in the development of autoimmune diabetes. Specifically, a species of segmented filamentous bacteria was found to protect against autoimmune diabetes in NOD mice (43). Interestingly, children who progress to develop T1D have a markedly altered serum metabolome that could already be detected in the cord blood (44), and many of these metabolites are microbially regulated (unpublished observation).
Atherosclerosis.
Accumulation of cholesterol and recruitment of macrophages to the arterial wall promote the formation of atherosclerotic plaques, which may lead to myocardial infarction and stroke. Bacterial species from the genera Chryseomonas, Veillonella, and Streptococcus have been found in plaques and are also present in the oral cavity or the gut (45). We recently demonstrated that patients who had experienced an atherosclerotic event had higher levels of Collinsella and lower levels of Eubacterium and Roseburia in their gut microbiota than healthy control subjects (46). The health status in these patients correlated with several aspects of the functional metagenome, such as an increase in proinflammatory peptidoglycan genes and a decrease in genes involved in the synthesis of anti-inflammatory molecules (e.g., butyrate) (46). A particularly interesting finding was the increased prevalence of genes involved in biosynthesis of the antioxidant β-carotene, together with increased blood levels of β-carotene in healthy control subjects (46). These observations suggest that it may be possible to develop strategies to prevent atherosclerotic events based on the gut microbiota.
Recent findings have revealed that the microbial metabolism of dietary choline to betaine and trimethylamine, which can be further metabolized in the liver to trimethylamine N-oxide, strongly correlates with cardiovascular events (47,48). The authors also showed that feeding mice with choline promoted the formation of atherosclerotic plaques and that plaque formation could be prevented by antibiotic treatment (47). Dietary l-carnitine, which is abundant in red meat and has a similar polar head group to choline, was recently shown to be metabolized by the gut microbiota and to contribute to atherosclerosis and cardiovascular disease (16).
Studying the microbiota
16S rRNA–based profiling.
Most of the species in the human gut are strictly anaerobic and are difficult to culture. Molecular methods have thus been developed to analyze microbial composition in a given sample (Fig. 1, Table 1). For bacteria and Archaea, methods are primarily based on the 16S ribosomal gene (49,50). A number of fingerprinting analyses have been developed for studying the human gut microbiota. These include temperature gradient gel electrophoresis, denaturing gradient gel electrophoresis, and terminal restriction fragment length polymorphism (51,52). These methods are semiquantitative and provide a rapid profiling of the microbiota but normally do not provide detailed taxonomic information.
FIG. 1.
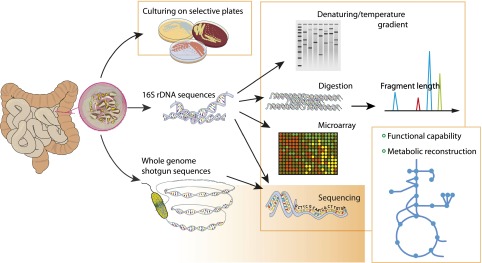
Methods for studying the microbiota. Traditionally, microbial communities have been characterized by culturing on specific plates, but this is only amenable to the culturable fraction of the members (20–50% [84,86]) and has limited resolution. Culture-independent methods based on characterization of the 16S rRNA genes have been developed and also provide information for organisms that cannot be cultured. Shotgun sequencing of the whole genome provides information about the functional and metabolic potential of the community.
TABLE 1.
Methods for the study of the gut microbiota
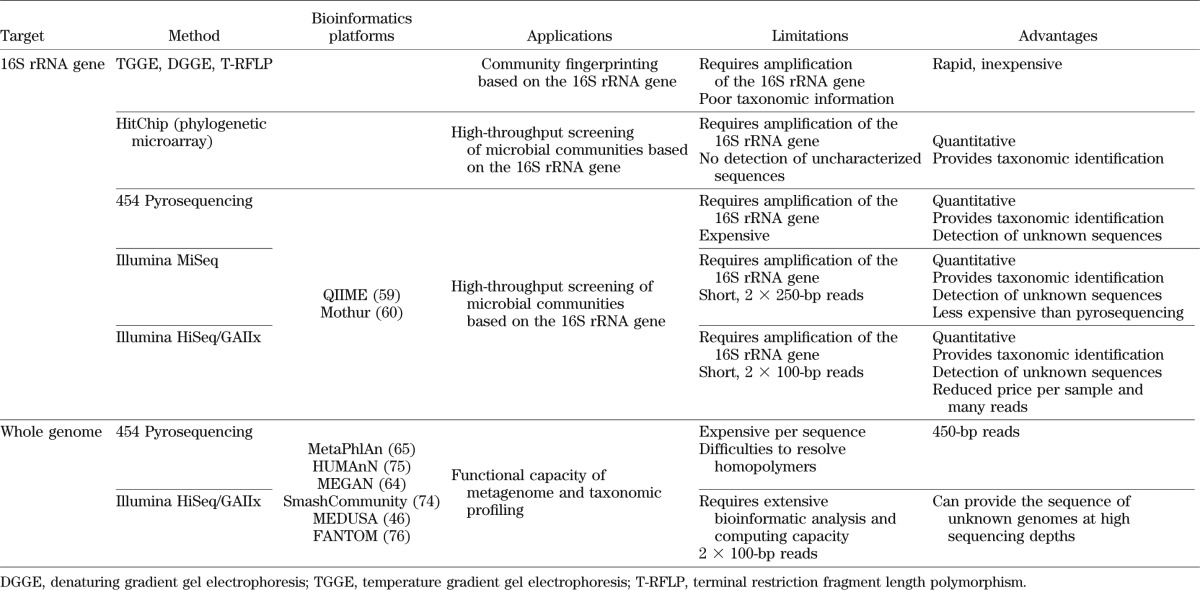
Microarrays with probes complementary to 16S rRNA sequences are high-throughput tools for characterizing abundance and diversity. The human intestinal tract chip (HITChip) probes were designed based on the hypervariable region of 1,140 unique rRNA (<98% identity) sequences that were clustered from a set of more than 16,000 human intestinal 16S rRNA sequences (53). The HITChip targets only known sequences and gives information on the relative abundance.
Direct sequencing of 16S rRNA genes has become increasingly used for assessing microbial diversity and abundance in the human gut because of reduced costs of sequencing, new bioinformatics algorithms and methods for data analysis, and better databases with sequences of known taxonomy. Initial studies used Sanger sequencing that produced nearly full-length sequences of the 16S rRNA gene (7,8). This procedure was time-consuming because it included the amplification of 16S rRNA gene sequences with universal primers, cloning into vectors, transformation into E. coli, and picking colonies for sequencing, purification of plasmid and bidirectional sequencing.
With the introduction of 454 sequencing technology, amplified sequences could be directly sequenced at a lower price, and more data could be generated (54,55). Read lengths obtained with 454 sequencing range from ∼100 to ∼450 bp and encompass regions of the 16S rRNA gene that are hypervariable but surrounded by conserved regions. The conserved regions allow for design of universal primers; however, the affinity is not universal across different species so there is still a bias in amplification efficiency between taxonomic groups.
A recent study showed that sequencing on the Illumina Genome AnalyzerIix (GAIIx) platform using paired reads of 100 bp could be used for 16S rRNA gene studies, providing consistent results with previous platforms and allowing a large number of samples to be analyzed simultaneously at a lower cost per sample (56). The Illumina MiSeq platform promises to deliver easy-to-use interface and sequence reads of 2 × 250 bp with the possibility to generate overlapping paired end reads with up to 30 million reads.
The choice of sequencing technology is coupled with the choice of primers for PCR amplification, and there is a large number to choose from. Primer choice will affect the taxonomic coverage, phylogenetic information of the generated fragments, and length of fragments. Common regions for amplification of the 16S rRNA gene are the V1, V2, V4, and V6 regions; V6 has been shown to perform poorly in taxonomic classification of sequences with a length of 250 bp, and the V1–V2 region underestimates Bifidobacteria (57). Longer read lengths are better for defining novel taxa but short reads, down to 100 bp, have been shown to resolve differences between communities (58). The choice of primer, 16S rRNA gene region, sequencing technology, number of sequences, cost, read length, and purpose are tightly coupled and need to be balanced to get the most out of each study.
Several tools for analysis of 16S rRNA gene sequences have been developed and are increasingly becoming more user-friendly and available to a large number of scientists. Quantitative Insights Into Microbial Ecology (QIIME) (59) and mothur (60) are highly used software packages that can be run on a laptop or computer cluster, depending on the size of the dataset, and can analyze millions of 16S rRNA gene sequences from microbial communities. The analysis is based on command line scripts that take raw sequences as input and cluster them into operational taxonomic units, producing phylogenetic trees and measurements of microbial diversity within and between samples. The analysis can be performed de novo when all operational taxonomic units are considered and compared across samples, but common sequences are compared with a reference database, such as Greengenes (61) and SILVA (62), and taxonomically annotated. A reference-based analysis is more straightforward to interpret and allows for comparison of different datasets sequencing different regions of the 16S rRNA gene.
Shotgun metagenome sequencing.
By sequencing the whole genomic content of a microbiota, the microbiome, and not only a marker gene, such as the 16S rRNA gene, a more detailed understanding of the functional potential of the community can be acquired. This is important because reference genomes are lacking for many organisms, and the gene content in different strains with an identical 16S rRNA gene sequence can differ in important aspects (e.g., in toxicity and pathogenicity genes). Sequencing of metagenomic reads enables taxonomic classification and diversity of community members as well as assessment of functional potential. The high density of microbial cells in the fecal content combined with the high diversity requires deep sequencing if low abundant species and genes should be studied. Because an individual carries at least 500,000 genes in a fecal sample, with a wide range of relative abundance, tens of million reads must be sequenced to cover the least abundant ones (4). The Illumina HiSeq and GAIIx systems can be used to obtain a quantitative measure of these genes, but the data amounts generated require that analyses are performed on a computer cluster with a large amount of storage and computing power.
Analysis of shotgun sequence data starts with removal of low-quality reads and trimming of reads with poor quality at the 3′ end. Removal of human reads by alignment to the human genome or other suspected contaminants is also required. After high-quality reads are generated, two approaches are typically performed: taxonomical classification of metagenomic reads and their functional classification. Several approaches for taxonomic classification of metagenomic reads exist and are commonly based on sequence alignment to a database, such as Basic Local Alignment Search Tool (BLAST), or short-reads sequence aligners or sequence composition methods that make use of short substring (k-mer) frequencies (63). The tool MEtaGenome ANalyzer (MEGAN) relies on a BLAST search to a reference catalog, such as National Center for Biotechnology Information nr, and analyzes and displays the results in a graphical user interphase (64). With large datasets, alignment with BLAST to a full reference catalog can be infeasible; alternative approaches include use of tools such as Metagenomic Phylogenetic Analysis (MetaPhlAn), which reduces the size by removing redundancy (65), or the use of accelerated but often less sensitive aligners (66).
For the functional classification of metagenomic reads, de novo assembly is often performed: single reads are assembled into contigs that are sequences of typical gene lengths or longer. The Metagenomics of the Human Intestinal Tract (MetaHIT) project used this approach to construct a gene catalog of 3.3 million genes found in 124 individuals, and each individual carried about half a million genes (4). Several different short read assemblers have been used for metagenomic data such as Short Oligonucleotide Analysis Package (SOAP) de novo (67) and Velvet (68) and, recently, MetaVelvet designed especially for metagenomic datasets (69). Dedicated pipelines for assembly of metagenomic data have also been developed: for example, MOCAT, which assembles metagenomic reads, predicts genes from contigs, and performs quality control of assembled contigs (70), and Metagenomic Data Utilization and Analysis (MEDUSA) (Fig. 2), which has been used for analysis of data from two metagenomics studies (33,46). De novo assembly is typically performed for each sample separately and then unassembled reads are used in a global assembly to maximize data use.
FIG. 2.

In the bioinformatic pipeline for analysis of whole metagenome shotgun sequences, sequences are subjected to quality control by removing uncertain base calls and contaminant sequences. Alignment of sequence reads to reference genomes is used for calculating species abundance. De novo assembly is used to identify genes not present in public databases. Genes can be functionally annotated and mapped onto metabolic networks such as in KEGG. Abundance of genes and species are compared among groups, and associations with disease can be tested.
Functional analysis and annotation can be done on predicted genes or directly on sequenced reads. Typically, sequences are annotated to genes and functions in the Kyoto Encyclopedia of Genes and Genomes (KEGG) (71), Clusters of Orthologous Groups (COG) (72), Pfam (73), or more specialized databases such as Carbohydrate-Active enZYmes (CAZy), which is important because of the extensive breakdown and fermentation of indigestible fibers in the gut. The KEGG database organizes genes into KEGG orthologs, enzymes, and pathways that are suitable for interpretation of metabolic capabilities of the community. Pipelines for automating some of these tasks include SmashCommunity (74) and the Human Microbiome Project Unified Metabolic Analysis Network (HUMAnN) (75), which are efficient but lack some of the flexibility of a custom pipeline. The recently developed pipeline Functional Annotation and Taxonomic Analysis off Metagenomes (FANTOM) is user-friendly and also enables visualization at different stages of the analysis (76). Several Web services exist for analysis of metagenomic data where the user uploads the data and can get a taxonomic and functional annotation as well as the possibility to compare with other published metagenomes (77–79).
Modeling of the microbiota.
Metagenomics provides a parts list of the gut microbiome, enabling the presence of genes and metabolic functions to be determined. Because of the extreme complexity of the gut ecosystem, it is, however, difficult to dissect specific metabolic functions solely from gene lists and statistical analysis. As for studies of other complex biological systems, mathematical modeling can assist to gain increased functional insight, particularly because setting up mathematical models may enable integration of different data types as well as evaluation of different hypotheses (80). Genome-scale metabolic models (GEMs) are particularly well suited for this kind of analysis because they are comprehensive collections of gene-protein reaction mappings that can take the parts list of genes in an organism and predict intracellular fluxes of metabolites during biomass production and growth. For example, GEMs have been used to model the metabolic interaction between Mycobacterium tuberculosis and the human alveolar cell (81). New improved drug targets could be predicted with the integrated bacteria and human model. Efforts to reconstruct GEMs for species of the gut microbiota have been initiated, and the prospects of simulating the production of SCFAs from carbohydrates will increase our understanding of the interactions between organisms in the human gut. GEMs provide excellent scaffolds for interpreting the metabolic implications of transcriptomic and other high-throughput data (82).
Personalized gnotobiotics.
Transplantation of human fecal microbiota into germ-free mice can be viewed as capturing an individual’s microbial community at a fixed moment in time (83). Importantly, the structure and composition of the transplanted human microbiota is well maintained in the mouse (84). Thus, humanized mice can be monitored over time and under highly controlled conditions. Therefore, potentially confounding variables can be constrained in ways that are not achievable in human studies to demonstrate whether specific phenotypes are transferred and mediated by the gut microbiota (Fig. 3). For example, the gut microbiota are altered during pregnancy in humans and at the third trimester resemble that of obese individuals (85). Interestingly, pregnant women develop a state that resembles insulin resistance to allow nutrients to be shuttled to the fetus. We investigated whether the altered microbiota conferred any of the metabolic effects by transferring the gut microbiota from women in the first and third trimester to germ-free female mice, and we observed that mice transplanted with microbes from the third trimester had increased body fat and also exhibited some impairment of glucose metabolism (85).
FIG. 3.
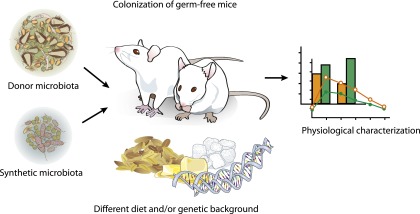
Germ-free mice can be used to study the effect of a gut microbiota on its host. Colonization of germ-free mice with human microbiota from different donors can test if there are functional differences between communities. Colonization of germ-free mice also allows investigation of the interaction between the microbiota and specific diets. Synthetic microbiota are defined communities with known species composition and provide a controlled environment for testing the interaction of microbes with diet and host.
Perspectives
Our knowledge of the gut microbiota and the microbiome has advanced at a rapid pace because of the improvements and cost reduction in DNA sequencing technology. Further development of sequencing technology promises longer read lengths and more data from a reduced amount of sample DNA at a lower cost, which will further enable deeper and more detailed studies of the gut microbiome. Advances in data analysis tools and statistical methods are needed and are being developed to speed up data analysis and make it available to a broader range of scientists. Cloud-based services, such as the Amazon Elastic Compute Cloud, can make data analysis available to researchers without an in-house computer cluster and eliminate the need for investment in hardware.
One key challenge is to perform prospective studies where serial fecal samples are obtained and the patients are carefully phenotyped to demonstrate if the altered gut microbiota is altered before metabolic disease or if the microbiota merely reflect the disease state. Because diet is an important modulator of the microbiota, it is essential to also record the composition and quantity of food intake. Care should be taken with regard to how samples are obtained and stored as well as which extraction protocol is used.
Metagenomic analyses provide information on the genomic content of microorganisms (dead or alive) whereas other methods such as metatranscriptomics and metaproteomics are needed to gain information on the active part of the microbiome. Metatranscriptomic studies are scarce and require more care when samples are taken to capture the actual in vivo activity of gene transcription. Yet another challenge is how to assess the composition of the microbiota in the small intestine, which requires invasive sampling in contrast to the fecal samples that are typically used today.
Despite the challenges in the field, the rapid advances made during the past decade suggest that the gut microbiota may constitute an important environmental factor that contributes to metabolic diseases. Further analysis of this “second” human genome is required to get additional insights into factors that determine its composition and function and how it interacts with key human cellular functions.
ACKNOWLEDGMENTS
Work in the authors’ laboratory is supported by Swedish Research Council, Swedish Diabetes Foundation, Swedish Foundation for Strategic Research, Knut and Alice Wallenberg Foundation, Ingbritt and Arne Lundberg’s Foundation, Swedish Heart Lung Foundation, Torsten Söderberg’s, Ragnar Söderberg’s, Novo Nordisk Foundation, AFA Insurances, and LUA-ALF grants from Västra Götalandsregionen and the Stockholm County Council.
J.N. and F.B. are founders and shareholders of Metabogen AB. No other potential conflicts of interest relevant to this article were reported.
F.K., V.T., J.N., and F.B. wrote the manuscript.
The authors thank Rosie Perkins, University of Gothenburg, for editing the manuscript and Anna Hallén, University of Gothenburg, for assisting in producing figures.
REFERENCES
- 1.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R. Bacterial community variation in human body habitats across space and time. Science 2009;326:1694–1697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bäckhed F, Ley RE, Sonnenburg JL, Peterson DA, Gordon JI. Host-bacterial mutualism in the human intestine. Science 2005;307:1915–1920 [DOI] [PubMed] [Google Scholar]
- 3.O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep 2006;7:688–693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Qin J, Li R, Raes J, et al. MetaHIT Consortium A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010;464:59–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sommer F, Bäckhed F. The gut microbiota—masters of host development and physiology. Nat Rev Microbiol 2013;11:227–238 [DOI] [PubMed] [Google Scholar]
- 6.Tremaroli V, Bäckhed F. Functional interactions between the gut microbiota and host metabolism. Nature 2012;489:242–249 [DOI] [PubMed] [Google Scholar]
- 7.Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI. Obesity alters gut microbial ecology. Proc Natl Acad Sci U S A 2005;102:11070–11075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Eckburg PB, Bik EM, Bernstein CN, et al. Diversity of the human intestinal microbial flora. Science 2005;308:1635–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yatsunenko T, Rey FE, Manary MJ, et al. Human gut microbiome viewed across age and geography. Nature 2012;486:222–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reyes A, Haynes M, Hanson N, et al. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature 2010;466:334–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wall R, Ross RP, Ryan CA, et al. Role of gut microbiota in early infant development. Clin Med Pediatr 2009;3:45–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koenig JE, Spor A, Scalfone N, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A 2011;108(Suppl. 1):4578–4585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Claesson MJ, Jeffery IB, Conde S, et al. Gut microbiota composition correlates with diet and health in the elderly. Nature 2012;488:178–184 [DOI] [PubMed] [Google Scholar]
- 14.Ley RE, Hamady M, Lozupone C, et al. Evolution of mammals and their gut microbes. Science 2008;320:1647–1651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Muegge BD, Kuczynski J, Knights D, et al. Diet drives convergence in gut microbiome functions across mammalian phylogeny and within humans. Science 2011;332:970–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koeth RA, Wang Z, Levison BS, et al. Intestinal microbiota metabolism of L-carnitine, a nutrient in red meat, promotes atherosclerosis. Nat Med 2013;19:576–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu GD, Chen J, Hoffmann C, et al. Linking long-term dietary patterns with gut microbial enterotypes. Science 2011;334:105–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.De Filippo C, Cavalieri D, Di Paola M, et al. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc Natl Acad Sci U S A 2010;107:14691–14696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ley RE, Turnbaugh PJ, Klein S, Gordon JI. Microbial ecology: human gut microbes associated with obesity. Nature 2006;444:1022–1023 [DOI] [PubMed] [Google Scholar]
- 20.Duncan SH, Lobley GE, Holtrop G, et al. Human colonic microbiota associated with diet, obesity and weight loss. Int J Obes (Lond) 2008;32:1720–1724 [DOI] [PubMed] [Google Scholar]
- 21.Schwiertz A, Taras D, Schäfer K, et al. Microbiota and SCFA in lean and overweight healthy subjects. Obesity (Silver Spring) 2010;18:190–195 [DOI] [PubMed] [Google Scholar]
- 22.Kalliomäki M, Collado MC, Salminen S, Isolauri E. Early differences in fecal microbiota composition in children may predict overweight. Am J Clin Nutr 2008;87:534–538 [DOI] [PubMed] [Google Scholar]
- 23.Turnbaugh PJ, Hamady M, Yatsunenko T, et al. A core gut microbiome in obese and lean twins. Nature 2009;457:480–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 2006;444:1027–1031 [DOI] [PubMed] [Google Scholar]
- 25.Turnbaugh PJ, Bäckhed F, Fulton L, Gordon JI. Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 2008;3:213–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bäckhed F, Ding H, Wang T, et al. The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 2004;101:15718–15723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bäckhed F, Manchester JK, Semenkovich CF, Gordon JI. Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A 2007;104:979–984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Larsen N, Vogensen FK, van den Berg FW, et al. Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS ONE 2010;5:e9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cani PD, Amar J, Iglesias MA, et al. Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 2007;56:1761–1772 [DOI] [PubMed] [Google Scholar]
- 30.Creely SJ, McTernan PG, Kusminski CM, et al. Lipopolysaccharide activates an innate immune system response in human adipose tissue in obesity and type 2 diabetes. Am J Physiol Endocrinol Metab 2007;292:E740–E747 [DOI] [PubMed] [Google Scholar]
- 31.Caesar R, Reigstad CS, Bäckhed HK, et al. Gut-derived lipopolysaccharide augments adipose macrophage accumulation but is not essential for impaired glucose or insulin tolerance in mice. Gut 2012;61:1701–1707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Qin J, Li Y, Cai Z, et al. A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 2012;490:55–60 [DOI] [PubMed] [Google Scholar]
- 33.Karlsson FH, Tremaroli V, Nookaew I, et al. Gut metagenome in European women with normal, impaired and diabeteic glucose control. Nature 2013;498:99–103 [DOI] [PubMed] [Google Scholar]
- 34.Sjöström L, Lindroos AK, Peltonen M, et al. Swedish Obese Subjects Study Scientific Group Lifestyle, diabetes, and cardiovascular risk factors 10 years after bariatric surgery. N Engl J Med 2004;351:2683–2693 [DOI] [PubMed] [Google Scholar]
- 35.Sjöström L, Narbro K, Sjöström CD, et al. Swedish Obese Subjects Study Effects of bariatric surgery on mortality in Swedish obese subjects. N Engl J Med 2007;357:741–752 [DOI] [PubMed] [Google Scholar]
- 36.Furet JP, Kong LC, Tap J, et al. Differential adaptation of human gut microbiota to bariatric surgery-induced weight loss: links with metabolic and low-grade inflammation markers. Diabetes 2010;59:3049–3057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhang H, DiBaise JK, Zuccolo A, et al. Human gut microbiota in obesity and after gastric bypass. Proc Natl Acad Sci U S A 2009;106:2365–2370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li JV, Ashrafian H, Bueter M, et al. Metabolic surgery profoundly influences gut microbial-host metabolic cross-talk. Gut 2011;60:1214–1223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liou AP, Paziuk M, Luevano JM Jr, Machineni S, Turnbaugh PJ, Kaplan LM. Conserved shifts in the gut microbiota due to gastric bypass reduce host weight and adiposity. Sci Transl Med 2013;5:178ra141 [DOI] [PMC free article] [PubMed]
- 40.Vrieze A, Van Nood E, Holleman F, et al. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology 2012;143:913–916.e7 [DOI] [PubMed]
- 41.Harrison LC, Honeyman MC, Morahan G, et al. Type 1 diabetes: lessons for other autoimmune diseases? J Autoimmun 2008;31:306–310 [DOI] [PubMed] [Google Scholar]
- 42.Brown CT, Davis-Richardson AG, Giongo A, et al. Gut microbiome metagenomics analysis suggests a functional model for the development of autoimmunity for type 1 diabetes. PLoS ONE 2011;6:e25792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kriegel MA, Sefik E, Hill JA, Wu HJ, Benoist C, Mathis D. Naturally transmitted segmented filamentous bacteria segregate with diabetes protection in nonobese diabetic mice. Proc Natl Acad Sci U S A 2011;108:11548–11553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Oresic M, Simell S, Sysi-Aho M, et al. Dysregulation of lipid and amino acid metabolism precedes islet autoimmunity in children who later progress to type 1 diabetes. J Exp Med 2008;205:2975–2984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koren O, Spor A, Felin J, et al. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A 2011;108(Suppl. 1):4592–4598 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Karlsson FH, Fåk F, Nookaew I, et al. Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun 2012;3:1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Z, Klipfell E, Bennett BJ, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011;472:57–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang WH, Wang Z, Levison BS, et al. Intestinal microbial metabolism of phosphatidylcholine and cardiovascular risk. N Engl J Med 2013;368:1575–1584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Woese CR. Bacterial evolution. Microbiol Rev 1987;51:221–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Clarridge JE., 3rd Impact of 16S rRNA gene sequence analysis for identification of bacteria on clinical microbiology and infectious diseases. Clin Microbiol Rev 2004;17:840–862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tzeneva VA, Heilig HG, van Vliet WA, Akkermans AD, de Vos WM, Smidt H. 16S rRNA targeted DGGE fingerprinting of microbial communities. Methods Mol Biol 2008;410:335–349 [DOI] [PubMed] [Google Scholar]
- 52.Li F, Hullar MA, Lampe JW. Optimization of terminal restriction fragment polymorphism (TRFLP) analysis of human gut microbiota. J Microbiol Methods 2007;68:303–311 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Rajilić-Stojanović M, Heilig HG, Molenaar D, et al. Development and application of the human intestinal tract chip, a phylogenetic microarray: analysis of universally conserved phylotypes in the abundant microbiota of young and elderly adults. Environ Microbiol 2009;11:1736–1751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sogin ML, Morrison HG, Huber JA, et al. Microbial diversity in the deep sea and the underexplored “rare biosphere.” Proc Natl Acad Sci U S A 2006;103:12115–12120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Andersson AF, Lindberg M, Jakobsson H, Bäckhed F, Nyrén P, Engstrand L. Comparative analysis of human gut microbiota by barcoded pyrosequencing. PLoS ONE 2008;3:e2836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Caporaso JG, Lauber CL, Walters WA, et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 2011;108(Suppl. 1):4516–4522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu Z, DeSantis TZ, Andersen GL, Knight R. Accurate taxonomy assignments from 16S rRNA sequences produced by highly parallel pyrosequencers. Nucleic Acids Res 2008;36:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu Z, Lozupone C, Hamady M, Bushman FD, Knight R. Short pyrosequencing reads suffice for accurate microbial community analysis. Nucleic Acids Res 2007;35:e120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Caporaso JG, Kuczynski J, Stombaugh J, et al. QIIME allows analysis of high-throughput community sequencing data. Nat Methods 2010;7:335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schloss PD, Westcott SL, Ryabin T, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009;75:7537–7541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.DeSantis TZ, Hugenholtz P, Larsen N, et al. Greengenes, a chimera-checked 16S rRNA gene database and workbench compatible with ARB. Appl Environ Microbiol 2006;72:5069–5072 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pruesse E, Quast C, Knittel K, et al. SILVA: a comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res 2007;35:7188–7196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Patil KR, Haider P, Pope PB, et al. Taxonomic metagenome sequence assignment with structured output models. Nat Methods 2011;8:191–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Huson DH, Auch AF, Qi J, Schuster SC. MEGAN analysis of metagenomic data. Genome Res 2007;17:377–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, Huttenhower C. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat Methods 2012;9:811–814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Martin J, Sykes S, Young S, et al. Optimizing read mapping to reference genomes to determine composition and species prevalence in microbial communities. PLoS ONE 2012;7:e36427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li R, Zhu H, Ruan J, et al. De novo assembly of human genomes with massively parallel short read sequencing. Genome Res 2010;20:265–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 2008;18:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Namiki T, Hachiya T, Tanaka H, Sakakibara Y. MetaVelvet: an extension of Velvet assembler to de novo metagenome assembly from short sequence reads. Nucleic Acids Res 2012;40:e155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kultima JR, Sunagawa S, Li J, et al. MOCAT: a metagenomics assembly and gene prediction toolkit. PLoS ONE 2012;7:e47656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kanehisa M, Goto S, Furumichi M, Tanabe M, Hirakawa M. KEGG for representation and analysis of molecular networks involving diseases and drugs. Nucleic Acids Res 2010;38(Database issue):D355–D360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Tatusov RL, Fedorova ND, Jackson JD, et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 2003;4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Punta M, Coggill PC, Eberhardt RY, et al. The Pfam protein families database. Nucleic Acids Res 2012;40(Database issue):D290–D301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Arumugam M, Harrington ED, Foerstner KU, Raes J, Bork P. SmashCommunity: a metagenomic annotation and analysis tool. Bioinformatics 2010;26:2977–2978 [DOI] [PubMed] [Google Scholar]
- 75.Abubucker S, Segata N, Goll J, et al. Metabolic reconstruction for metagenomic data and its application to the human microbiome. PLOS Comput Biol 2012;8:e1002358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sanli K, Karlsson FH, Nookaew I, Nielsen J. FANTOM: Functional and taxonomic analysis of metagenomes. BMC Bioinformatics 2013;14:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Seshadri R, Kravitz SA, Smarr L, Gilna P, Frazier M. CAMERA: a community resource for metagenomics. PLoS Biol 2007;5:e75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Markowitz VM, Ivanova NN, Szeto E, et al. IMG/M: a data management and analysis system for metagenomes. Nucleic Acids Res 2008;36:D534–D538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meyer F, Paarmann D, D’Souza M, et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 2008;9:386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Mardinoglu A, Gatto F, Nielsen J. Genome-scale modeling of human metabolism—a systems biology approach. Biotechnol J 24 April 2013 [Epub ahead of print] [DOI] [PubMed]
- 81.Bordbar A, Lewis NE, Schellenberger J, Palsson BO, Jamshidi N. Insight into human alveolar macrophage and M. tuberculosis interactions via metabolic reconstructions. Mol Syst Biol 2010;6:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Karlsson FH, Nookaew I, Petranovic D, Nielsen J. Prospects for systems biology and modeling of the gut microbiome. Trends Biotechnol 2011;29:251–258 [DOI] [PubMed] [Google Scholar]
- 83.Kau AL, Ahern PP, Griffin NW, Goodman AL, Gordon JI. Human nutrition, the gut microbiome and the immune system. Nature 2011;474:327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Goodman AL, Kallstrom G, Faith JJ, et al. Extensive personal human gut microbiota culture collections characterized and manipulated in gnotobiotic mice. Proc Natl Acad Sci U S A 2011;108:6252–6257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Koren O, Goodrich JK, Cullender TC, et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012;150:470–480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zoetendal EG, Vaughan EE, de Vos WM. A microbial world within us. Mol Microbiol 2006;59:1639–1650 [DOI] [PubMed] [Google Scholar]