

Abstract
In patients with diabetes, impaired ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats, member 13) proteolysis of highly thrombogenic von Willebrand factor (VWF) multimers may accelerate renal and cardiovascular complications. Restoring physiological VWF handling might contribute to ACE inhibitors’ (ACEi) reno- and cardioprotective effects. To assess how Pro618Ala ADAMTS13 variants and related proteolytic activity interact with ACEi therapy in predicting renal and cardiovascular complications, we genotyped 1,163 normoalbuminuric type 2 diabetic patients from BErgamo NEphrologic DIabetes Complications Trial (BENEDICT). Interaction between Pro618Ala and ACEi was significant in predicting both renal and combined renal and cardiovascular events. The risk for renal or combined events versus reference Ala carriers on ACEi progressively increased from Pro/Pro homozygotes on ACEi (hazard ratio 2.80 [95% CI 0.849–9.216] and 1.58 [0.737–3.379], respectively) to Pro/Pro homozygotes on non-ACEi (4.77 [1.484–15.357] and 1.99 [0.944–4.187]) to Ala carriers on non-ACEi (8.50 [2.416–29.962] and 4.00 [1.739–9.207]). In a substudy, serum ADAMTS13 activity was significantly lower in Ala carriers than in Pro/Pro homozygotes and in case subjects with renal, cardiovascular, or combined events than in diabetic control subjects without events. ADAMTS13 activity significantly and negatively correlated with all outcomes. In patients with diabetes, ADAMTS13 618Ala variant associated with less proteolytic activity, higher risk of chronic complications, and better response to ACEi therapy. Screening for Pro618Ala polymorphism may help identify patients with diabetes at highest risk who may benefit the most from early reno- and cardioprotective therapy.
In patients with type 2 diabetes, the incidence of coronary events and ischemic strokes is twice as high as in subjects without diabetes (1). This excess risk is even higher in diabetic patients with evidence of renal involvement as manifested by urinary albumin excretion (UAE) in the micro- or macroalbuminuric range (2). Overall, cardiovascular disease and microvascular complications such as nephropathy, retinopathy, and neuropathy are major causes of illness in this population and impose an enormous economic burden. Optimized blood pressure (BP) and metabolic control and inhibition of the renin-angiotensin system by ACE inhibitors (ACEi) or angiotensin receptor blockers may effectively prevent or delay the onset and progression of all chronic complications of diabetes (3–6). Despite treatment, however, most patients are at high risk of events. Thus, better understanding the pathogenic mechanisms underlying chronic complications of diabetes and identification of predictors of outcome and response to treatment are instrumental in optimizing the use of available or novel therapeutic tools.
Among the several factors associated with the abnormal metabolic state that accompanies diabetes, endothelial cell dysfunction and uncontrolled platelet activation have consistently been found to play a central role in the pathogenesis of vascular damage (7–10). Altered vascular handling of von Willebrand factor (VWF) has been suggested to be a key determinant of the excess platelet activation frequently observed in this population (7). VWF is a multimeric glycoprotein stored in endothelial Weibel-Palade bodies as highly thrombogenic ultralarge multimers (ULVWF). Upon endothelial injury, these multimers are secreted to mediate platelet adhesion to injured endothelium—the first step in thrombus formation (11). ULVWF multimers, however, are only transiently bound to the endothelial surface, since they are promptly cleaved by the plasma metalloprotease ADAMTS13 (a disintegrin and metalloprotease with thrombospondin type 1 repeats, member 13), produced by liver stellate cells and endothelial cells (12), and are released in the circulation as smaller multimers with less thrombogenic potential (13). This sequence of events is crucial in modulating the thrombotic process (11), as demonstrated by evidence that acquired or genetically determined ADAMTS13 deficiency associates with uncontrolled intravascular thrombosis of thrombotic thrombocytopenic purpura (14).
Chronic endothelial dysfunction induced by hyperglycemia, oxidative stress, and other factors associated with diabetes may sustain continued VWF multimer formation and release into the circulation (9,15,16), in particular in type 2 diabetic patients with microalbuminuria (8,17) or renal lesions (18). VWF levels independently predicted risk of progression to macroalbuminuria (19) or of cardiovascular events (20). Another consequence of endothelial dysfunction is impaired ADAMTS13 synthesis and secretion (21). Thus, in addition to enhanced ULVWF release, concomitant reduction of ADAMTS13 cleaving potential may also contribute to increase circulating ULVWF and consequent excess thrombotic risk. Indeed, in experimental post–ischemic brain stroke (22) and myocardial infarction (23,24), mice that genetically lack ADAMTS13 develop larger infarcts in the brain (22) and the heart (23,24) than wild-type mice. Exacerbated injury in ADAMTS13−/− mice was VWF dependent, since it was not observed in ADAMTS13−/−/VWF−/− mice (22,23). Thus, defective ADAMTS13 bioavailability may result in uncontrolled VWF-mediated thrombosis. This could explain why low ADAMTS13 levels are associated with renal and cardiovascular events in subjects with diabetes and even in the general population (25–27). ADAMTS13 activity and levels can be genetically determined (14). Actually, the ADAMTS13 gene is highly polymorphic (28,29), and several ADAMTS13 single nucleotide polymorphisms (SNPs) associate with altered protein secretion and activity in vitro (28,29). Among them, only the Pro457Ser, a SNP common in the Japanese population but extremely rare in Caucasians, has been investigated in vivo and was found to associate with decreased plasmatic ADAMTS13 activity (30) so far. In preliminary studies in human embryonic kidney (HEK293T) cells expressing recombinant ADAMTS13 proteins carrying the four nonsynonymous SNPs with a >0.05 minor allele frequency in the European population (rs34024143 [Arg7Trp], rs2301612 [Glu448Gln], rs28647808 [Pro618Ala], and rs685523 [Ala900Val] [http://www.ncbi.nlm.nih.gov/snp, CEU population]), we found that protease secretion and activity were reduced only with the 618Ala with respect to wild-type protein (Supplementary Figs. 1 and 2). The residual protease activity of the 618Ala variant was 27% (74% secretion × 37% activity). Consistently, analysis in 102 healthy Caucasians showed that serum ADAMTS13 activity was significantly lower in 618Ala carriers compared with homozygous carriers of the wild-type Pro618 allele but was not affected by the other variants, with the exception of the 7Trp variant that was in linkage disequilibrium with the 618Ala (Supplementary Figs. 2 and 3). Altogether, the above findings converged to indicate that the Pro618Ala is the only known common SNP that substantially affects ADAMTS13 activity levels, at least in Caucasians, and suggested that, among the several inborn factors associated with renal and cardiovascular complications of diabetes (31–34), a genetically determined reduced ADAMTS13 activity due to the 618Ala variant could play a role. Thus, we hypothesized that impaired ADAMTS13 production secondary to diabetes-associated endothelial dysfunction could result in a clinically relevant reduction of ADAMTS13 activity leading to excess risk of renal and cardiovascular disease, in particular in those patients who may have an intrinsic ADAMTS13 defect because of their specific Pro618Ala genotype.
To formally test this hypothesis, we investigated whether and to what extent Pro618Ala ADAMTS13 polymorphism predicted the risk of renal involvement (defined as new-onset of microalbuminuria) and cardiovascular events in the large cohort of normoalbuminuric type 2 diabetic patients included in the BErgamo NEphrologic DIabetes Complications Trial (BENEDICT) and randomly allocated to the ACEi trandolapril or to non-ACEi therapy (4). Since the trial had already demonstrated that trandolapril—alone or combined with verapamil—significantly affected the risk of progression to microalbuminuria, we first evaluated the interactive role of Pro618Ala and treatment in event prediction. We then tested the impact of the polymorphism on risk of developing events in patients on either ACEi or non-ACEi therapy. Finally, in a subgroup of patients we ran a case-control study to investigate whether the association of the Pro618Ala polymorphism with outcomes could be explained by the effect of the polymorphic variants on ADAMTS13 plasmatic activity.
RESEARCH DESIGN AND METHODS
The main study and the case-control substudy were both approved by the local ethics committee, and all study participants provided written informed consent according to the Declaration of Helsinki guidelines. All data were handled in respect of patient confidentiality and anonymity.
Main study.
We primarily evaluated the interactive role of Pro618Ala ADAMTS13 polymorphism and ACEi therapy in the prediction of renal events (new-onset microalbuminuria) in 1,163 of the 1,204 type 2 diabetic patients included in the BENEDICT phase A study who consented to genetic analyses (Fig. 1). We secondarily evaluated the role of the above interaction in predicting major cardiovascular events (fatal or nonfatal stroke; acute myocardial infarction, unstable angina; coronary, carotid, or peripheral artery revascularization; or surgical amputation for critical ischemic limbs) and a combined end point of renal and/or cardiovascular events.
FIG. 1.
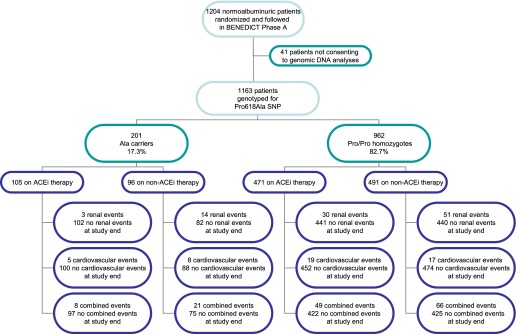
Schematic diagram of BENEDICT phase A type 2 diabetic patients screened for the Pro618Ala ADAMTS13. ACEi allocation: Ala carriers vs. Pro/Pro homozygotes, χ2 = 0.715, P = 0.398. Renal event development: Ala carriers, ACEi vs. non-ACEi, χ2 = 8.907, P = 0.003; Pro/Pro homozygotes, ACEi vs. non-ACEi, χ2 = 5.032, P = 0.025; ACEi, Ala carriers vs. Pro/Pro homozygotes, χ2 = 1.961, P = 0.161; non-ACEi, Ala carriers vs. Pro/Pro homozygotes, χ2 = 1.436, P = 0.231. Cardiovascular event development: Ala carriers, ACEi vs. non-ACEi, χ2 = 1.057, P = 0.304; Pro/Pro homozygotes, ACEi vs. non-ACEi, χ2 = 0.218, P = 0.641; ACEi, Ala carriers vs. Pro/Pro homozygotes, χ2 = 0.114, P = 0.736; non-ACEi, Ala carriers vs. Pro/Pro homozygotes, χ2 = 4.673, P = 0.031. Combined event development: Ala carriers, ACEi vs. non-ACEi, χ2=8.255, P = 0.004; Pro/Pro homozygotes, ACEi vs. non-ACEi, χ2 = 2.109, P = 0.146; ACEi, Ala carriers vs. Pro/Pro homozygotes, χ2 = 0.747, P = 0.388; non-ACEi, Ala carriers vs. Pro/Pro homozygotes, χ2 = 4.523, P = 0.033.
The new-onset of microalbuminuria was defined as a UAE ≥20 μg and <200 μg/min in at least two of three consecutive overnight urine collections at two consecutive visits 2 months apart (4,35). All cardiovascular events were adjudicated by two cardiologists (Brigitte Kalsh and Piero Ruggenenti) blinded to patient treatment allocation and genetic results.
Detailed information about the BENEDICT design, treatment arms, and biochemical measurement has previously been provided (4,35).
Case-control substudy.
We evaluated the associations among Pro618Ala, serum ADAMTS13 activity, and progression to different outcomes in a subset of the 487 BENEDICT patients enrolled at the Bergamo Center. Within this subgroup, we identified all cases that during BENEDICT had progressed to persistent microalbuminuria, major cardiovascular events, or both, respectively. For each case subject, one diabetic control subject who had not progressed to any considered event, who was matched by sex, age (±10 years vs. case), and ACEi therapy allocation in the core study, was identified. For each diabetic case and diabetic control subject, one healthy control subject matched by sex and age (±10 years) was also identified from a population of 250 blood donors.
Genotyping.
Genomic DNA was extracted from peripheral blood leukocytes by a Nucleon BACC2 kit (Amersham). Genotyping for Arg7Trp (exon 1), Gln448Glu (exon 12), and Ala900Val (exon 21) was performed with the Sanger direct sequencing method as previously described (36). Genotyping for Pro618Ala SNP was performed using a TaqMan Pre-Designed SNP Allelic Discrimination assay (assay C_3183368_20) from Applied Biosystems following the manufacturer’s instructions. Genotyping success rate was 99%. As a quality-control procedure, we double genotyped a subset of individuals (n = 169) by Sanger direct sequencing of ADAMTS13 exon 16 on the ABI-3730 Sequencer analyzer. The concordance rate between the two genotyping methods was 100%.
ADAMTS13 activity evaluation.
Serum ADAMTS13 activity was measured at completion of the BENEDICT core study when all diabetes case and control subjects were on the same ACEi therapy and in healthy control subjects by using the residual collagen-binding assay (37). The detection limit of the assay is 6%.
Statistical analyses.
Times to renal, major cardiovascular, and combined end points were the outcomes of interest. For patients who did not reach the end point, we censored time at the last follow-up visit with available data for renal end point and at the last follow-up visit for major cardiovascular and combined end points. The Kaplan-Meier method was used to plot the probability of achieving the end points according to Pro618Ala polymorphism and the ACEi treatment. All time-to-event end points were analyzed using Cox proportional hazard regression models, and results were expressed as hazard ratio (HR) and 95% CI. Multivariable models for the end points included Pro618Ala genotype, ACEi treatment, and all the baseline covariates that at the univariable Cox analysis significantly (P < 0.05) associated with the outcome, without exceeding the limit of one independent variable included in the model for every at least 10 outcome events available for the analyses (38). The above variables were also tested in Cox models with genotype × ACEi treatment interaction terms (39). Blood glucose was not considered because of its high collinearity with HbA1c levels.
To investigate the potentially relevant predictive value of clinical covariates not significantly associated with the outcome at univariable analysis, we performed sensitivity analyses by testing in the multivariable models with genotype × ACEi treatment interaction the addition of each of the following covariates considered separately: sex, age, smoking, BMI, duration of diabetes, HbA1c, triglycerides, creatinine, total cholesterol, LDL cholesterol, HDL cholesterol, systolic (SBP) and diastolic (DBP) BP, and mean arterial pressure (MAP), as appropriate. The same approach was used to consider in the above multivariable model the predictive value of mean SBP and DBP or median HbA1c calculated on the basis of follow-up values without considering the baseline.
Tests of the proportional hazards assumption were based on Schoenfeld residuals. If the global test was significant, we used a detailed variable-by-variable test based on scaled Schoenfeld residuals and we specified those variables that varied continuously with respect to time, introducing a time-varying covariate (variables × time interaction) in the models (40).
To test possible follow-up differences among the four groups derived by genotype × ACEi treatment interaction, we used linear mixed-effect models for DBP, SBP, and HbA1c. Not normally distributed covariates (UAE, HbA1c, triglycerides, and HDL cholesterol) were log transformed before analysis. Normality for continuous variables was assessed by means of the Q-Q plot.
The data of baseline characteristics were presented as n (%), means and SDs, or medians and interquartile ranges (IQRs) as appropriate. Comparisons between groups were made using one-way ANOVA, Kruskal-Wallis test, χ2 test, or paired t test as appropriate. Comparison between groups of UAE at the final visit was carried out by ANOVA and ANCOVA and by analysis for trend. Correlation analysis between continuous variables and dichotomous variables was carried out using the point-biserial correlation coefficient (rpb). All P values were two-sided. Analyses were carried out using SAS (version 9.1) and Stata (version 12).
RESULTS
Eight (0.7%) and 193 (16.6%) of 1,163 patients (all Caucasians from north Italy) consenting to genetic analyses carried two or one copy of the 618Ala allele, respectively, and 962 (82.7%) were Pro/Pro homozygotes (Fig. 1 and Supplementary Table 1). Genotype distribution was comparable with that of a healthy subject population from the same geographical region and did not deviate from Hardy-Weinberg equilibrium (Supplementary Table 1). Data from carriers of two or one Ala allele were pooled for comparative analysis versus Pro/Pro homozygotes.
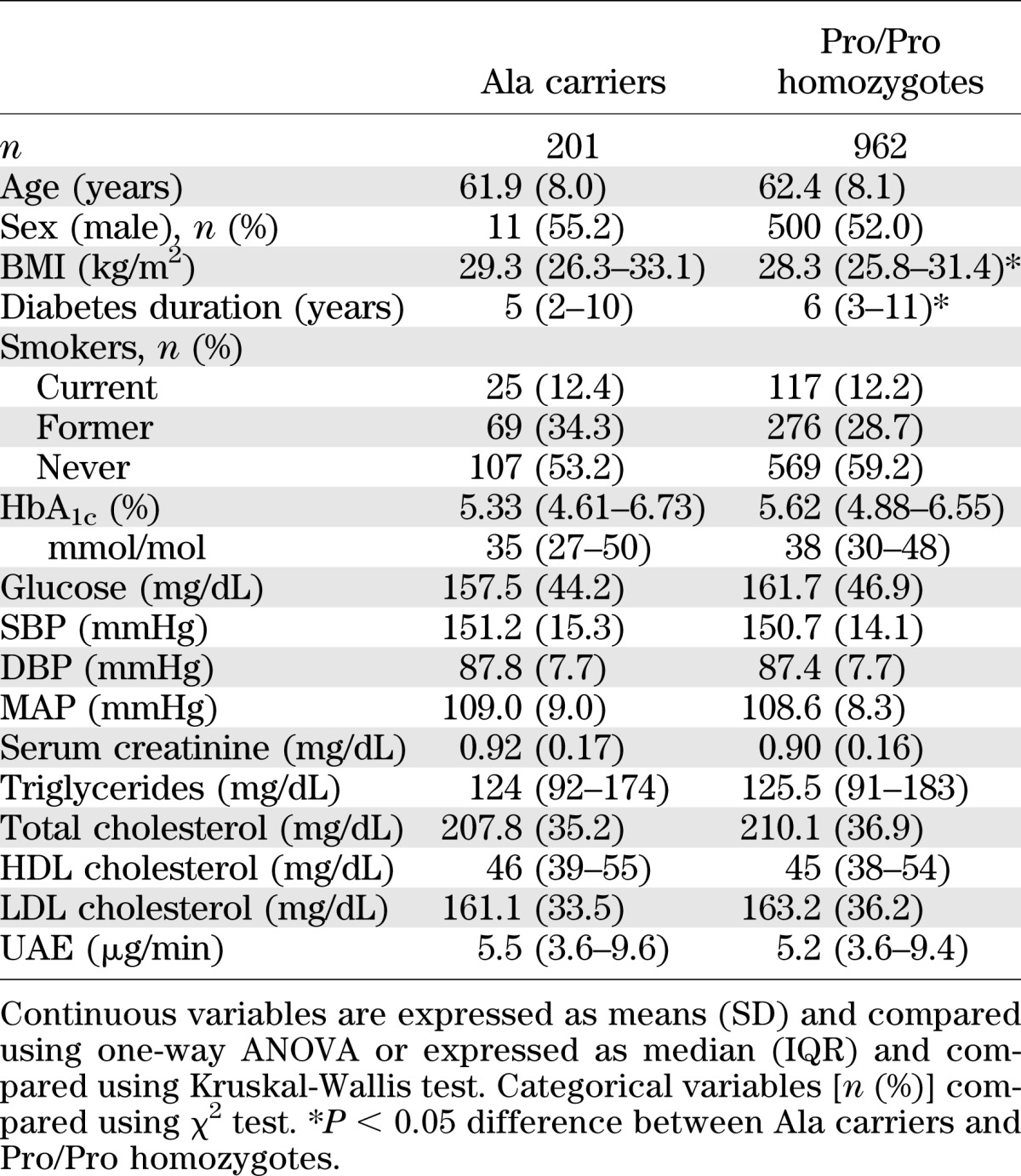
In the two genotype groups, the baseline characteristics (Table 1), the proportion of patients allocated to ACEi or non-ACEi therapy (Fig. 1), and the distribution of concomitant medications (Supplementary Table 2) were similar with the exception of a higher BMI and a lower duration of diabetes in Ala carriers.
TABLE 1.
Baseline clinical characteristics of patients with type 2 diabetes of BENEDICT phase A
Predictors of outcome and genotype-treatment interaction.
Over a median (interquartile range) follow-up of 43.6 (16.4–51.8) months, 98 of the 1,163 participants (8.4%) progressed to renal events (new-onset microalbuminuria), 49 (4.2%) to major cardiovascular events, and 144 (12.4%) to combined events, including 3 patients who progressed to both end points (Fig. 1). At univariable Cox analyses, male sex, smoking, and higher baseline UAE and HbA1c significantly associated with increased risk, and ACEi therapy significantly associated with reduced risk of renal events (Supplementary Table 3). Of the above variables, baseline UAE and HbA1c and ACEi therapy retained their independent predictive value also at multivariable analysis (Supplementary Table 4). Cardiovascular events were predicted by age and baseline UAE and HDL cholesterol both at univariable and multivariable analyses (Supplementary Tables 3 and 4). Univariable and multivariable models showed that older age and higher baseline UAE and HbA1c predicted an increased risk and ACEi therapy predicted a reduced risk of progression to combined events, while sex, smoking habits, and HDL cholesterol predicted the outcome only at univariable analysis (Supplementary Tables 3 and 4).
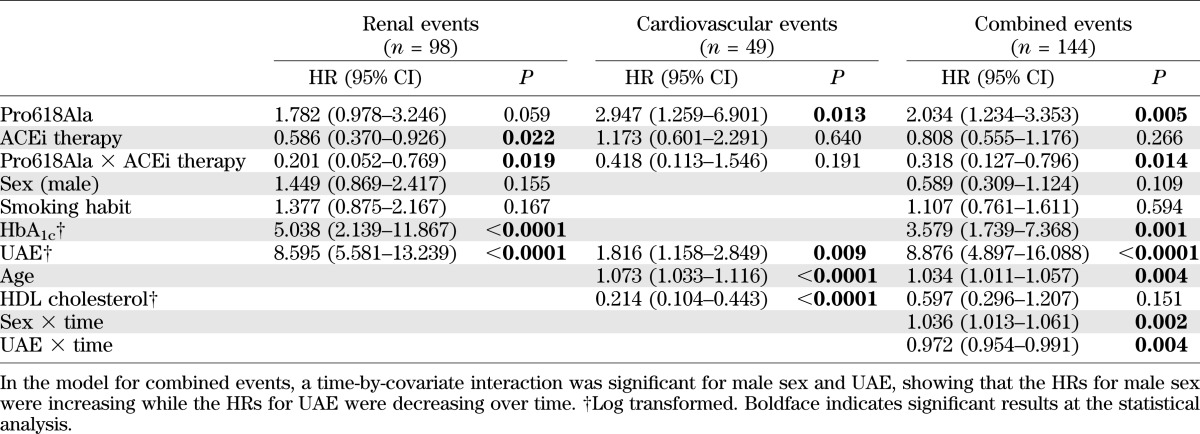
At multivariable Cox analyses, the Pro618Ala genotype was found to significantly interact with ACEi therapy in the prediction of renal events (P = 0.019) or of the combined end point (P = 0.014), whereas the interaction failed to achieve the statistical significance (P = 0.191) when cardiovascular events were considered as a single end point (Table 2 and Supplementary Table 5).
TABLE 2.
Multivariable Cox analysis with genotype–ACEi treatment interaction for renal, cardiovascular, and combined end points
Patient outcome according to Pro618Ala polymorphism and ACEi therapy
Renal events.
Progression to microalbuminuria was observed in 3 of 105 (2.9%) Ala carriers on ACEi vs. 14 of 96 (14.6%) Ala carriers on non-ACEi (unadjusted HR 0.161 [95% CI 0.046–0.560], P = 0.004) and in 30 of 471 (6.4%) Pro/Pro homozygotes on ACEi vs. 51 of 491 (10.4%) Pro/Pro homozygotes on non-ACEi (0.563 [0.359–0.884], P = 0.013] (Figs. 1 and 2). Results were confirmed after adjustment for baseline covariates (Ala carriers, ACEi yes vs. ACEi no, HR 0.12 [95% CI 0.333–0.414], P = 0.001; Pro/Pro homozygotes, ACEi yes vs. ACEi no, 0.59 [0.370–0.926], P = 0.022) (Table 3). The larger treatment effect in Ala carriers was confirmed by finding that the ratio between the two HRs for the comparison ACEi yes vs. ACEi no observed in Ala carriers and Pro/Pro homozygotes (0.201 [0.125–0.321]) was significantly lower than 1. The proportion of patients with the end point progressively increased from Ala carriers (2.9%) to Pro/Pro homozygotes (6.4%) on ACEi to Pro/Pro homozygotes (10.4%) and to Ala carriers (14.6%) on non-ACEi therapy. This was consistent with evidence that the HR for progression to the event progressively increased from 2.80 (95% CI 0.849–9.216) to 4.77 (1.484–15.357) and to 8.50 (2.416–29.962) in Pro/Pro homozygotes on ACEi and Pro/Pro homozygotes and Ala carriers on non-ACEi therapy, respectively, compared with Ala carriers on ACEi taken as the reference group (Fig. 3). All of the above analyses were adjusted for all baseline covariates that at univariable analyses were significantly associated with the event.
FIG. 2.

Impact of Pro618Ala polymorphism and ACEi therapy on considered events. Kaplan-Meyer curves show the percentages of Ala carriers or Pro/Pro homozygotes with or without ACEi therapy progressing to renal (A) or combined renal and/or cardiovascular (B) events throughout the study period. P values of unadjusted Cox analyses are shown. Boldface P values indicate statistical significance.
TABLE 3.
HRs of the comparisons between ACEi-treated and non–ACEi-treated patients within the two genotype groups and HRs of the comparisons between Ala carriers and Pro/Pro homozygotes in ACEi or non-ACEi arms
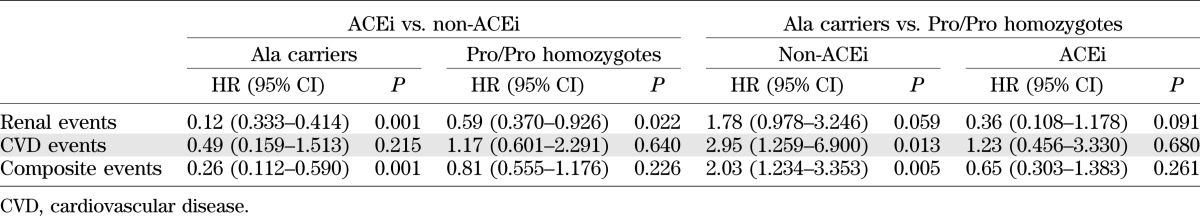
FIG. 3.
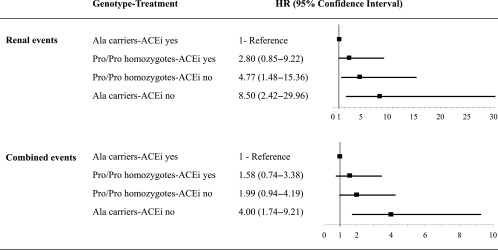
HR for considered events according to Pro618Ala genotype and ACEi treatment. HRs (95% CI) for renal and combined renal and/or cardiovascular events according to Pro618Ala polymorphism and ACEi therapy compared with Ala carriers on ACEi taken as the reference group are shown.
Thus, Ala carriers were the patients with the highest risk of renal events while on non-ACEi therapy but were also those with the largest benefit from ACEi therapy—benefit that fully offset the excess risk associated with this genotype. Pro/Pro homozygotes had a lower risk but also gained less benefit from treatment. This may explain why, independently of treatment allocation, the proportion of patients with the end point was similar among Ala carriers (8.5%) and Pro/Pro homozygotes (8.4%) considered as a whole (Fig. 1, Supplementary Fig. 4A, and Supplementary Tables 3 and 4).
Similar findings were obtained when albuminuria measured at the end of the study or at progression to the end point was considered as a continuous variable. Thus, the lowest UAE (geometric mean 5.33 μg/min) was observed in Ala carriers on ACEi, and progressively increasing excretions were observed in Pro/Pro homozygotes on ACEi (5.49 μg/min) and in Pro/Pro homozygotes and Ala carriers on non-ACEi (6.36 μg/min and 7.46 μg/min, respectively; P = 0.0029 for trend [Fig. 4A]). The trend was significant (P = 0.0002) even when data were adjusted for albuminuria at baseline. Thus, Ala carriers were the patients with the highest final albuminuria while on non-ACEi but were also those with the lowest final albuminuria while on ACEi. Again, this explained why final albuminuria in Ala carriers and Pro/Pro homozygotes (6.25 vs. 5.91 μg/min, respectively; P = 0.421) was similar when data were compared independently of treatment allocation.
FIG. 4.

UAE rate, BP, and HbA1c according to Pro618Ala polymorphism and ACEi therapy. A: UAE rate (geometric mean and 95% CI) at basal and final visit. B: Mean SBP and DBP. C: HbA1c throughout the whole observation period. Differences in UAE rate between treatment groups are adjusted for baseline values by ANCOVA. Differences in BP and HbA1c between different groups are not significant.
Cardiovascular events.
Major cardiovascular events were observed in 5 of 105 (4.8%) Ala carriers on ACEi vs. 8 of 96 (8.3%) Ala carriers on non-ACEi (HR 0.49 [95% CI 0.159–1.513], P = 0.215) and in 19 of 471 (4.0%) Pro/Pro homozygotes on ACEi vs. 17 of 491 (3.5%) Pro/Pro homozygotes on non-ACEi (1.17 [0.601–2.291], P = 0.640) (Fig. 1, Supplementary Fig. 5, and Table 3). Thus, ACEi tended to reduce the risk of events in Ala carriers (an effect that conceivably failed to achieve the statistical significance because of the relatively small number of events) but had no effect in Pro/Pro homozygotes.
Among non–ACEi-treated patients, Ala carriers had a significantly higher risk of events vs. Pro/Pro homozygotes (HR 2.95 [95% CI 1.259–6.900], P = 0.013) (Tables 2 and 3 and Supplementary Table 5)—an excess risk that was not appreciable among patients on ACEi (1.23 [0.456–3.330]) (Table 3). Independently of treatment, the overall risk of cardiovascular events was higher in Ala carriers than in Pro/Pro homozygotes (1.95 [1.028–3.713], P = 0.041) (Supplementary Fig. 4B and Supplementary Table 4).
Combined events.
Progression to combined events was observed in 8 of 105 (7.6%) Ala carriers on ACEi vs. 21 of 96 (21.9%) Ala carriers on non-ACEi (unadjusted HR 0.285 [95% CI 0.126–0.644], P = 0.003) and in 49 of 471 (10.4%) Pro/Pro homozygotes on ACEi vs. 66 of 491 (13.4%) Pro/Pro homozygotes on non-ACEi therapy (0.722 [0.499–1.045], P = 0.084) (Figs. 1 and 2B). Similar results were found after adjustment for baseline covariates (Ala carriers, ACEi yes vs. ACEi no, HR 0.26 [95% CI 0.112–0.590], P = 0.001; Pro/Pro homozygotes, ACEi yes vs. ACEi no, 0.81 [0.555–1.176], P = 0.226) (Table 3). Thus, ACEi significantly reduced the risk of events in Ala carriers but had no effect in Pro/Pro homozygotes. The difference in treatment effect between the two genotype groups was confirmed by finding that the ratio between the two HRs for the comparison ACEi yes versus ACEi no observed in Ala carriers and Pro/Pro homozygotes (0.32 [0.128–0.792]) was significantly lower than 1.
The proportion of patients with combined events was the lowest among Ala carriers on ACEi (7.6%) and progressively increased among Pro/Pro homozygotes on ACEi (10.4%) and Pro/Pro homozygotes (13.4%) and Ala carriers (21.9%) on non-ACEi therapy. This was consistent with finding that the HR for progression to the event progressively increased from 1.58 (95% CI 0.737–3.379) to 1.99 (0.944–4.187) and to 4.00 (1.739–9.207) in Pro/Pro homozygotes on ACEi and Pro/Pro homozygotes and Ala carriers on non-ACEi, respectively, versus Ala carriers on ACEi taken as the reference group (Fig. 3). Analyses were adjusted for baseline covariates that at univariable analyses were significantly associated with the event.
Thus, Ala carriers were the patients with the highest risk of combined events while on non-ACEi but were also those with the largest benefit from ACEi—benefit that fully offset the excess risk associated with this genotype. Pro/Pro homozygotes had a lower risk but gained no appreciable benefit from treatment. This explained why the overall proportion of events among Ala carriers (14.4%) and Pro/Pro homozygotes (12.0%) was similar (P = 0.333) (Fig. 1, Supplementary Fig. 4C, and Supplementary Tables 3 and 4).
Sensitivity analyses.
Multivariable models with genotype × ACEi interaction terms showed that among all considered baseline covariates only DBP and MAP significantly predicted new-onset microalbuminuria and SBP the combined renal and cardiovascular end point.
BP and metabolic control.
Baseline and follow-up SBP, DBP, and HbA1c (Fig. 4B and C) were similar among the four subgroups of Ala carriers or Pro/Pro homozygotes either on ACEi or on non-ACEi. Consistently, results of multivariable Cox analyses with genotype-treatment interaction considering renal, cardiovascular, or combined events did not change after adjustment for baseline (Supplementary Table 5) or follow-up (Supplementary Table 6) SBP, DBP, or HbA1c.
Substudy: impact of Pro618Ala polymorphism on ADAMTS13 activity levels and association between ADAMTS13 activity and outcomes.
Consistently with preliminary data in healthy subjects (Supplementary Fig. 3A), among diabetic patients with serum ADAMTS13 activity evaluation Ala carriers had significantly lower ADAMTS13 activity levels than Pro/Pro homozygotes (Supplementary Fig. 6A). Among Ala carriers, serum ADAMTS13 activity was significantly higher in those on ACEi therapy than in those without ACEi, whereas among Pro/Pro homozygotes no significant difference was observed between the two treatment groups (Supplementary Fig. 6B). Among the 487 diabetic patients, the 23 case subjects progressing to persistent microalbuminuria, the 20 with cardiovascular events, and the 41 with the combined events had significantly lower ADAMTS13 activity levels than matched diabetic control subjects without events and matched nondiabetic control subjects (Fig. 5A and B). Of relevance, ADAMTS13 activity levels in diabetic control subjects were similar to those of nondiabetic control subjects (Fig. 5B).
FIG. 5.
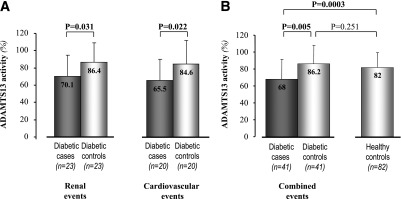
Serum ADAMTS13 activity in case and control subjects. Diabetic case subjects with renal or cardiovascular events were compared with matched diabetic control subjects without events (A). B: Diabetic case subjects with renal and/or cardiovascular combined events were compared with matched diabetic control subjects without events and nondiabetic healthy control subjects. Data are expressed as means (SD). Boldface P values indicate statistical significance.
In the case-control group considered as a whole, the point biserial correlation coefficient showed that ADAMTS13 levels negatively correlated with renal (rpb = −0.326, P = 0.027), cardiovascular (rpb = −0.351, P = 0.026), and combined (rpb = −0.288, P = 0.009) events.
DISCUSSION
In this outcome analysis of 1,163 normoalbuminuric type 2 diabetic patients prospectively followed in the setting of a randomized clinical trial (4), we found a strong interaction between the Pro618Ala ADAMTS13 polymorphism and ACEi therapy in the prediction of renal events (defined as the new onset of microalbuminuria) or the combined end point of renal and/or cardiovascular events. Thus, Ala carriers on non-ACEi therapy were the patients at the highest risk but, at the same time, were those who gained the largest benefit from ACEi therapy, whereas Pro/Pro homozygotes were at lower risk of events, but treatment effect was relatively smaller in this population. Actually, when genotype × ACEi treatment interaction was included in the multivariable model, Pro618Ala predicted renal events. Consistently, when patients were categorized according to genotype and treatment, we observed an incremental risk for renal events from Ala carriers on ACEi therapy—the subgroup with the lowest risk—to Ala carriers on non-ACEi therapy, who showed the highest risk. The renoprotective effect of ACEi therapy in Ala carriers was strong enough to fully offset the excess risk of renal events associated with this genotype. Consequently, when genotype × ACEi treatment interaction was not included in the model and thus outcome data were considered independently of ACEi therapy, the risk of renal events in Ala carriers and Pro/Pro homozygotes was comparable. A similar trend was observed when cardiovascular events were considered as outcome. Ala carriers were those at highest risk who, however, benefited the most from ACEi therapy, whereas this therapy had no appreciable effect in Pro/Pro homozygotes. In this setting, however, the predisposing effect of the Pro618Ala polymorphism was so strong that Ala carriers continued to be at higher risk of events compared with Pro/Pro homozygotes even when outcome data were considered independently of treatment. Finally, when renal and cardiovascular events were considered as a single combined end point, outcome data were largely driven by the renal events. Thus, there was an incremental risk of reaching the combined end point from Ala carriers on ACEi to Pro/Pro homozygotes on ACEi and then to Pro/Pro homozygotes and Ala carriers on non-ACEi, respectively.
Finding that metabolic and BP control during the whole observation period was similar among genotype groups and that outcome data did not appreciably change when analyses were adjusted for baseline and follow-up HbA1c, SBP, and DBP can be taken to suggest that in normoalbuminuric type 2 diabetic patients, the 618Ala variant independently affects development of renal and cardiovascular complications and, at the same time, the reno- and cardioprotective effect of ACEi therapy. Of note, the proportion of patients on antiplatelet treatment was similar between the considered groups.
Evidence from prespecified subgroup analyses that ADAMTS13 activity was lower in Ala carriers than in Pro/Pro homozygotes as well as in patients with events compared with those without suggested that the excess risk of renal and cardiovascular events observed in Ala carriers could be explained by decreased ADAMTS13 activity in this subgroup. This possibility was consistent with finding that lower ADAMTS13 activity significantly associated with an excess risk of all considered outcomes. In this regard, it is worth mentioning that VWF multimers purified from type 2 diabetic patients are partially resistant to ADAMTS13 proteolysis because of oxidative modifications by peroxynitrite in Met1606 at the ADAMTS13 cleavage site (41). Thus, impaired cleavage of oxidized VWF may eventually result in increased endothelial exposure to highly prothrombotic ULVWF multimers (41), which might sustain a further excess risk of events in particular in patients with genetically determined reduced ADAMTS13 activity associated with the 618Ala variant. Actually, the multimeric composition of VWF is a key determinant of its platelet-tethering function. In turn, platelet activation may contribute to the microvascular complications of diabetes (7), as confirmed by finding that thromboxane A2 accelerated renal disease progression and its inhibitors reduced glomerular thrombosis and glomerulosclerosis, thereby preventing proteinuria, in experimental diabetes (42). Along the same line, aspirin and/or dipyridamole significantly reduced proteinuria in type 2 diabetic patients with overt nephropathy (43). Thus, evidence that larger VWF multimers have greater potential for VWF-ligand interaction and formation of a competent platelet thrombus led to the hypothesis that excess exposure to ULVWF multimers resulting in microvascular platelet thrombosis could be relevant to the onset and progression of diabetic renal disease (25). On the other hand, the key role of defective ADAMTS13 activity in the pathogenesis of coronary and cerebrovascular events is well established (25,26) and has been convincingly confirmed by recent evidence that recombinant ADAMTS13 is cardioprotective in mouse models of acute myocardial infarction (44).
Why ACEi therapy was particularly effective in Ala carriers is purely a matter of speculation. In addition to reducing BP and proteinuria, ACEi have been consistently found to reduce the oxidative stress and endothelial dysfunction (45,46) associated with the diabetic milieu (47,48). Such an effect limits endothelial VWF production, as documented by decreased plasma VWF levels in patients with IgA nephropathy or chronic hearth failure after ACEi treatment (49,50). Finding that ADAMT13 activity levels were higher in Ala carriers with than in those without ACEi therapy suggests that ACEi might also enhance ADAMTS13 bioavailability. Endothelial cells are a major source of ADAMTS13 (12), and it is tempting to speculate that by limiting endothelial dysfunction, ACEi therapy might prevent reduction of ADAMTS13 bioavailablity of diabetes (25). Such an effect is expected to be particularly relevant in patients with genetically determined reduced ADAMTS13 activity. Altogether, the above actions might explain at least part of the renal and cardiovascular protective effects of ACEi therapy, in particular in diabetic patients with genetically determined reduced ADAMTS13 activity, such as those carrying the 618Ala allele.
The major limitation of the main study is that data were generated from post hoc exploratory analyses of a trial that was originally designed for other purposes. As for any post hoc observational analysis, data must be considered as hypothesis generating and study findings need to be confirmed in ad hoc designed prospective clinical trials.
A strength of our study was the hypothesis-driven selection of the Pro618Ala ADAMTS13 SNP that was based on previous evidence that mutations in this gene cause a rare monogenic disease of microvascular thrombosis (14) and that the 618Ala variant associates with severely impaired protein secretion and activity either in vitro (29) and in vivo (Supplementary Data). This is a qualifying aspect compared with most previous association studies between common candidate gene variants and diabetes complications. This may possibly explain the strong and clinically relevant interactions we found among Pro618Ala SNP, the end points, and treatment effect. Availability of a large cohort of patients that was representative of the average Caucasian diabetic population and was prospectively monitored according to standardized guidelines and gold-standard techniques further increased the generalizability and robustness of the study findings. However, whether our present results can be extended to other ethnicities needs further scrutiny, since ADAMTS13 SNPs other than the Pro618Ala may play a role in modulating ADAMTS13 activity in non-Caucasian populations (30).
In summary, here we show that in subjects with type 2 diabetes and normal UAE the Pro618Ala ADAMTS13 polymorphism is a major independent predictor of renal and cardiovascular events as well as of reno- and cardioprotective effects of ACEi therapy. Ala carriers appear to be those with the highest risk of events and, at the same time, with the largest benefit from ACEi therapy. Excess risk of events appears to be associated with defective ADAMTS13 activity. Based on our present data, we speculate that among the several mechanisms involved in the reno- and cardioprotective effects of ACEi therapy, increased ADAMTS13 bioavailability and, conceivably, reduced patient exposure to the prothrombotic effects of ULVWF multimers might also play a role. These findings have a wide generalizability, since patients under evaluation in our present study represent at least 90% of the average population of Caucasians with type 2 diabetes. Hypotheses generated from our present study may help planning of future trials to optimize therapy in this context.
ACKNOWLEDGMENTS
This study was partially supported by grants from the Chiesi Foundation (Milan, Italy), Fondazione ART per la Ricerca Sui Trapianti ONLUS (Milan, Italy), and Fondazione ARMR ONLUS Aiuti per la Ricerca sulle Malattie Rare (Bergamo, Italy).
This study was also partially supported by an unrestricted educational grant (577/5896) from F. Hoffmann-La Roche, Ltd. BENEDICT was supported by Abbott (Ludwigshafen, Germany). At the time the study was performed, C.H. was an employee and stockholder of F. Hoffmann-La Roche. No other potential conflicts of interest relevant to this article were reported.
E.R. designed the research, performed the experiments, undertook statistical analyses, interpreted and discussed the results, drafted the manuscript, and approved the final version of the manuscript. M.N. designed the research, interpreted and discussed the results, drafted the manuscript, and approved the final version of the manuscript. A.C. implemented statistical analyses, wrote statistical methods, interpreted statistical results, and approved the final version of the manuscript. R.D. designed the research, performed the experiments, and approved the final version of the manuscript. F.B. and M.G. performed the experiments and approved the final version of the manuscript. G.G. acquired clinical data and approved the final version of the manuscript. S.G. performed the experiments and approved the final version of the manuscript. A.P., I.I., and A.B. acquired clinical data and approved the final version of the manuscript. C.H. discussed the results and approved the final version of the manuscript. R.T. acquired clinical data and approved the final version of the manuscript. G.R. and P.R. acquired clinical data; interpreted and discussed the results; wrote, reviewed, and edited the manuscript; and approved the final version of the manuscript. M.N. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors are indebted to the staff of the Nephrology and Diabetology units and of the Clinical Research Center for Rare Diseases “Aldo & Cele Daccò” of the IRCCS—Istituto di Ricerche Farmacologiche “Mario Negri” for their assistance in the selection of and care for the subjects of this study, to Annalisa Perna for supervising the statistical analyses, to Ariela Benigni for useful discussion of the results, to Paola Boccardo for interaction with all involved ethics committees, to Elisabetta Valoti and Silvia Nosari for their contribution in genetic tests, and to Manuela Passera for help in the preparation of the manuscript (all from the IRCCS—Istituto di Ricerche Farmacologiche “Mario Negri”).
Footnotes
Clinical trial reg. no. NCT00235014, clinicaltrials.gov.
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0530/-/DC1.
A complete list of members of the BENEDICT Study Group can be found in the Supplementary Data.
E.R. and M.N. contributed equally as first author, and G.R. and P.R. contributed equally as last author of this work.
See accompanying commentary, p. 3331.
REFERENCES
- 1.Schramm TK, Gislason GH, Køber L, et al. Diabetes patients requiring glucose-lowering therapy and nondiabetics with a prior myocardial infarction carry the same cardiovascular risk: a population study of 3.3 million people. Circulation 2008;117:1945–1954 [DOI] [PubMed] [Google Scholar]
- 2.Adler AI, Stevens RJ, Manley SE, Bilous RW, Cull CA, Holman RR, UKPDS GROUP Development and progression of nephropathy in type 2 diabetes: the United Kingdom Prospective Diabetes Study (UKPDS 64). Kidney Int 2003;63:225–232 [DOI] [PubMed] [Google Scholar]
- 3.Ruggenenti P, Fassi A, Ilieva AP, et al. BENEDICT-B Study Investigators Effects of verapamil added-on trandolapril therapy in hypertensive type 2 diabetes patients with microalbuminuria: the BENEDICT-B randomized trial. J Hypertens 2011;29:207–216 [DOI] [PubMed] [Google Scholar]
- 4.Ruggenenti P, Fassi A, Ilieva AP, et al. Bergamo Nephrologic Diabetes Complications Trial (BENEDICT) Investigators Preventing microalbuminuria in type 2 diabetes. N Engl J Med 2004;351:1941–1951 [DOI] [PubMed] [Google Scholar]
- 5.Brenner BM, Cooper ME, de Zeeuw D, et al. RENAAL Study Investigators Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001;345:861–869 [DOI] [PubMed] [Google Scholar]
- 6.Investigators TH, Heart Outcomes Prevention Evaluation Study Investigators Effects of ramipril on cardiovascular and microvascular outcomes in people with diabetes mellitus: results of the HOPE study and MICRO-HOPE substudy. Lancet 2000;355:253–259 [PubMed] [Google Scholar]
- 7.Mustand JF, Packham MA. Platelets and diabetes mellitus. N Engl J Med 1984;311:665–667 [DOI] [PubMed] [Google Scholar]
- 8.Yu Y, Suo L, Yu H, Wang C, Tang H. Insulin resistance and endothelial dysfunction in type 2 diabetes patients with or without microalbuminuria. Diabetes Res Clin Pract 2004;65:95–104 [DOI] [PubMed] [Google Scholar]
- 9.Meigs JB, O’Donnell CJ, Tofler GH, et al. Hemostatic markers of endothelial dysfunction and risk of incident type 2 diabetes: the Framingham Offspring Study. Diabetes 2006;55:530–537 [DOI] [PubMed] [Google Scholar]
- 10.Kistorp C, Chong AY, Gustafsson F, et al. Biomarkers of endothelial dysfunction are elevated and related to prognosis in chronic heart failure patients with diabetes but not in those without diabetes. Eur J Heart Fail 2008;10:380–387 [DOI] [PubMed] [Google Scholar]
- 11.Sporn LA, Marder VJ, Wagner DD. von Willebrand factor released from Weibel-Palade bodies binds more avidly to extracellular matrix than that secreted constitutively. Blood 1987;69:1531–1534 [PubMed] [Google Scholar]
- 12.Turner NA, Nolasco L, Ruggeri ZM, Moake JL. Endothelial cell ADAMTS-13 and VWF: production, release, and VWF string cleavage. Blood 2009;114:5102–5111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dong JF, Moake JL, Nolasco L, et al. ADAMTS-13 rapidly cleaves newly secreted ultralarge von Willebrand factor multimers on the endothelial surface under flowing conditions. Blood 2002;100:4033–4039 [DOI] [PubMed] [Google Scholar]
- 14.Galbusera M, Noris M, Remuzzi G. Inherited thrombotic thrombocytopenic purpura. Haematologica 2009;94:166–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lemkes BA, Hermanides J, Devries JH, Holleman F, Meijers JC, Hoekstra JB. Hyperglycemia: a prothrombotic factor? J Thromb Haemost 2010;8:1663–1669 [DOI] [PubMed] [Google Scholar]
- 16.Vazzana N, Ranalli P, Cuccurullo C, Davì G. Diabetes mellitus and thrombosis. Thromb Res 2012;129:371–377 [DOI] [PubMed] [Google Scholar]
- 17.Hirano T, Ookubo K, Kashiwazaki K, Tajima H, Yoshino G, Adachi M. Vascular endothelial markers, von Willebrand factor and thrombomodulin index, are specifically elevated in type 2 diabetic patients with nephropathy: comparison of primary renal disease. Clin Chim Acta 2000;299:65–75 [DOI] [PubMed] [Google Scholar]
- 18.Fioretto P, Stehouwer CD, Mauer M, et al. Heterogeneous nature of microalbuminuria in NIDDM: studies of endothelial function and renal structure. Diabetologia 1998;41:233–236 [DOI] [PubMed] [Google Scholar]
- 19.Persson F, Rossing P, Hovind P, et al. Endothelial dysfunction and inflammation predict development of diabetic nephropathy in the Irbesartan in Patients with Type 2 Diabetes and Microalbuminuria (IRMA 2) study. Scand J Clin Lab Invest 2008;68:731–738 [DOI] [PubMed] [Google Scholar]
- 20.Frankel DS, Meigs JB, Massaro JM, et al. Von Willebrand factor, type 2 diabetes mellitus, and risk of cardiovascular disease: the Framingham Offspring study. Circulation 2008;118:2533–2539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelras S, Delmas Y, Lamireau D, et al. Severe transient ADAMTS13 deficiency in pneumococcal-associated hemolytic uremic syndrome. Pediatr Nephrol 2011;26:631–635 [DOI] [PubMed] [Google Scholar]
- 22.Zhao BQ, Chauhan AK, Canault M, et al. von Willebrand factor-cleaving protease ADAMTS13 reduces ischemic brain injury in experimental stroke. Blood 2009;114:3329–3334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gandhi C, Motto DG, Jensen M, Lentz SR, Chauhan AK. ADAMTS13 deficiency exacerbates VWF-dependent acute myocardial ischemia/reperfusion injury in mice. Blood 2012;120:5224–5230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doi M, Matsui H, Takeda H, et al. ADAMTS13 safeguards the myocardium in a mouse model of acute myocardial infarction. Thromb Haemost 2012;108:1236–1238 [DOI] [PubMed] [Google Scholar]
- 25.Taniguchi S, Hashiguchi T, Ono T, et al. Association between reduced ADAMTS13 and diabetic nephropathy. Thromb Res 2010;125:e310–e316 [DOI] [PubMed] [Google Scholar]
- 26.Kaikita K, Soejima K, Matsukawa M, Nakagaki T, Ogawa H. Reduced von Willebrand factor-cleaving protease (ADAMTS13) activity in acute myocardial infarction. J Thromb Haemost 2006;4:2490–2493 [DOI] [PubMed] [Google Scholar]
- 27.Chion CK, Doggen CJ, Crawley JT, Lane DA, Rosendaal FR. ADAMTS13 and von Willebrand factor and the risk of myocardial infarction in men. Blood 2007;109:1998–2000 [DOI] [PubMed] [Google Scholar]
- 28.Kokame K, Matsumoto M, Soejima K, et al. Mutations and common polymorphisms in ADAMTS13 gene responsible for von Willebrand factor-cleaving protease activity. Proc Natl Acad Sci USA 2002;99:11902–11907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Plaimauer B, Fuhrmann J, Mohr G, et al. Modulation of ADAMTS13 secretion and specific activity by a combination of common amino acid polymorphisms and a missense mutation. Blood 2006;107:118–125 [DOI] [PubMed] [Google Scholar]
- 30.Jang MJ, Kim NK, Chong SY, et al. Frequency of Pro475Ser polymorphism of ADAMTS13 gene and its association with ADAMTS-13 activity in the Korean population. Yonsei Med J 2008;49:405–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De Cosmo S, Motterlini N, Prudente S, et al. BENEDICT Study Group Impact of the PPAR-gamma2 Pro12Ala polymorphism and ACE inhibitor therapy on new-onset microalbuminuria in type 2 diabetes: evidence from BENEDICT. Diabetes 2009;58:2920–2929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maeda S, Kobayashi MA, Araki S, et al. A single nucleotide polymorphism within the acetyl-coenzyme A carboxylase beta gene is associated with proteinuria in patients with type 2 diabetes. PLoS Genet 2010;6:e1000842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kathiresan S, Melander O, Anevski D, et al. Polymorphisms associated with cholesterol and risk of cardiovascular events. N Engl J Med 2008;358:1240–1249 [DOI] [PubMed] [Google Scholar]
- 34.Krolewski AS, Poznik GD, Placha G, et al. A genome-wide linkage scan for genes controlling variation in urinary albumin excretion in type II diabetes. Kidney Int 2006;69:129–136 [DOI] [PubMed] [Google Scholar]
- 35.BENEDICT Group The BErgamo NEphrologic DIabetes Complications Trial (BENEDICT): design and baseline characteristics. Control Clin Trials 2003;24:442–461 [DOI] [PubMed] [Google Scholar]
- 36.Noris M, Bucchioni S, Galbusera M, et al. International Registry of Recurrent and Familial HUS/TTP Complement factor H mutation in familial thrombotic thrombocytopenic purpura with ADAMTS13 deficiency and renal involvement. J Am Soc Nephrol 2005;16:1177–1183 [DOI] [PubMed] [Google Scholar]
- 37.Galbusera M, Bresin E, Noris M, et al. Rituximab prevents recurrence of thrombotic thrombocytopenic purpura: a case report. Blood 2005;106:925–928 [DOI] [PubMed] [Google Scholar]
- 38.Concato J, Feinstein AR, Holford TR. The risk of determining risk with multivariable models. Ann Intern Med 1993;118:201–210 [DOI] [PubMed] [Google Scholar]
- 39.Altman DG, Bland JM. Interaction revisited: the difference between two estimates. BMJ 2003;326:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bellera CA, MacGrogan G, Debled M, de Lara CT, Brouste V, Mathoulin-Pélissier S. Variables with time-varying effects and the Cox model: some statistical concepts illustrated with a prognostic factor study in breast cancer. BMC Med Res Methodol 2010;10:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lancellotti S, De Filippis V, Pozzi N, et al. Formation of methionine sulfoxide by peroxynitrite at position 1606 of von Willebrand factor inhibits its cleavage by ADAMTS-13: A new prothrombotic mechanism in diseases associated with oxidative stress. Free Radic Biol Med 2010;48:446–456 [DOI] [PubMed] [Google Scholar]
- 42.Okumura M, Imanishi M, Okamura M, et al. Role for thromboxane A2 from glomerular thrombi in nephropathy with type 2 diabetic rats. Life Sci 2003;72:2695–2705 [DOI] [PubMed] [Google Scholar]
- 43.Khajehdehi P, Roozbeh J, Mostafavi H. A comparative randomized and placebo-controlled short-term trial of aspirin and dipyridamole for overt type-2 diabetic nephropathy. Scand J Urol Nephrol 2002;36:145–148 [DOI] [PubMed] [Google Scholar]
- 44.De Meyer SF, Savchenko AS, Haas MS, et al. Protective anti-inflammatory effect of ADAMTS13 on myocardial ischemia/reperfusion injury in mice. Blood 2012;120:5217–5223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hershon KS. Mechanistic and clinical aspects of renin-angiotensin-aldosterone system blockade in the prevention of diabetes mellitus and cardiovascular disease. Endocr Pract 2011;17:430–440 [DOI] [PubMed] [Google Scholar]
- 46.Napoli C, Bruzzese G, Ignarro LJ, et al. Long-term treatment with sulfhydryl angiotensin-converting enzyme inhibition reduces carotid intima-media thickening and improves the nitric oxide/oxidative stress pathways in newly diagnosed patients with mild to moderate primary hypertension. Am Heart J 2008;156:1154.e1151–1158 [DOI] [PubMed]
- 47.Bravi MC, Armiento A, Laurenti O, et al. Insulin decreases intracellular oxidative stress in patients with type 2 diabetes mellitus. Metabolism 2006;55:691–695 [DOI] [PubMed] [Google Scholar]
- 48.Giugliano D, Ceriello A, Paolisso G. Oxidative stress and diabetic vascular complications. Diabetes Care 1996;19:257–267 [DOI] [PubMed] [Google Scholar]
- 49.Hernández E, Toledo T, Alamo C, Mon C, Rodicio JL, Praga M. Elevation of von Willebrand factor levels in patients with IgA nephropathy: effect of ACE inhibition. Am J Kidney Dis 1997;30:397–403 [DOI] [PubMed] [Google Scholar]
- 50.Gibbs CR, Blann AD, Watson RD, Lip GY. Abnormalities of hemorheological, endothelial, and platelet function in patients with chronic heart failure in sinus rhythm: effects of angiotensin-converting enzyme inhibitor and beta-blocker therapy. Circulation 2001;103:1746–1751 [DOI] [PubMed] [Google Scholar]