

Abstract
Eating a “Westernized” diet high in fat and sugar leads to weight gain and numerous health problems, including the development of type 2 diabetes mellitus (T2DM). Rodent studies have shown that resveratrol supplementation reduces blood glucose levels, preserves β-cells in islets of Langerhans, and improves insulin action. Although rodent models are helpful for understanding β-cell biology and certain aspects of T2DM pathology, they fail to reproduce the complexity of the human disease as well as that of nonhuman primates. Rhesus monkeys were fed a standard diet (SD), or a high-fat/high-sugar diet in combination with either placebo (HFS) or resveratrol (HFS+Resv) for 24 months, and pancreata were examined before overt dysglycemia occurred. Increased glucose-stimulated insulin secretion and insulin resistance occurred in both HFS and HFS+Resv diets compared with SD. Although islet size was unaffected, there was a significant decrease in β-cells and an increase in α-cells containing glucagon and glucagon-like peptide 1 with HFS diets. Islets from HFS+Resv monkeys were morphologically similar to SD. HFS diets also resulted in decreased expression of essential β-cell transcription factors forkhead box O1 (FOXO1), NKX6–1, NKX2–2, and PDX1, which did not occur with resveratrol supplementation. Similar changes were observed in human islets where the effects of resveratrol were mediated through Sirtuin 1. These findings have implications for the management of humans with insulin resistance, prediabetes, and diabetes.
In many developing countries, a rapid nutritional transition has occurred from a healthy diet, high in fiber and low in fat and calories, to calorie-dense meals, containing refined carbohydrates, red meats, sugary desserts and drinks, and high-fat foods (1). When “Westernized” diets are introduced, the population responds with weight gain leading to numerous health problems, including hypertension, coronary artery disease and strokes, respiratory effects, cancers, reproductive abnormalities, and type 2 diabetes mellitus (T2DM) (2). Obesity, a result of sedentary lifestyles and Westernized diets, has reached epidemic proportions and is the major risk factor for developing T2DM.
Currently 25.8 million Americans have adult-onset or T2DM and an additional 79 million exhibit a metabolic profile that is considered a precursor to T2DM (3). Several years ago, the Diabetes Prevention Program Trial (DPPT), targeting persons with prediabetes, found that the incidence of T2DM was reduced by 31% with metformin therapy and by 58% with lifestyle interventions, including exercise and weight reduction, over an average follow-up period of 2.8 years (4). However, in absolute terms, metformin had only a minor impact on the transition to diabetes, with diabetes developing in 13 of every 14 individuals receiving metformin. Although lifestyle intervention in a highly controlled setting faired twice as well, long-term modification of diet and activity patterns are very difficult for most adults to achieve. Further, despite the findings from the DPPT and recommendations for people with prediabetes to use metformin, lose weight, and start an exercise regimen, rates of diabetes continue to escalate. Therefore, it is extremely important to investigate the metabolic and physiologic alterations that occur in prediabetic states and elucidate the underlying etiopathology in order to develop effective strategies to prevent the transition to T2DM.
In the last several years, rodent studies and experiments in vitro have provided evidence that resveratrol, a naturally occurring phytoalexin found in numerous plant species, exerts beneficial effects in organisms and may be helpful in preventing some metabolic diseases, including diabetes (5). In animals given a high-fat diet, resveratrol has been shown to increase their survival and motor function (6), reduce visceral fat and liver mass indexes (7), and induce beneficial changes in lipid parameters (8). In addition, several different rodent models of diabetes have shown that resveratrol can reduce blood glucose levels (9), preserve β-cells (10), and improve insulin action (6). However, limited human data on metabolic effects of resveratrol are more controversial. A few studies have reported that resveratrol improves insulin sensitivity in adults who are obese (11), have T2DM (12), or have impaired glucose tolerance (13), whereas other studies have supported the idea that resveratrol supplementation does not improve metabolic function in nonobese women (14) or in obese men (15).
Although rodent models are helpful for understanding β-cell biology and some aspects of the pathogenesis of T2DM, they cannot reproduce the complexity of the human disease as well as that of nonhuman primates. T2DM occurs spontaneously in ad libitum–fed nonhuman primates (16). Also, monkey and human endocrine pancreata are similar with β- and α-cells interspersed, unlike rodent islets, and with similar islet size and cellular distribution of islet cell types (17). The metabolic and hormonal changes observed in humans with insulin resistance, prediabetes, and diabetes also occur in monkeys (18,19). Therefore, monkeys serve as ideal models for studying islets as they likely display evolving changes in response to increased insulin requirement. Additionally, these changes occur within a reasonable time frame of experimentation (18). We undertook a long-term study of monkeys given a high-fat/high-sugar diet to evaluate changes in islet function and morphology resulting from unremitting increased insulin requirement in the absence of dysglycemia and whether these changes were mitigated by resveratrol.
RESEARCH DESIGN AND METHODS
Animals.
Twenty-four adult male rhesus monkeys (Macaca mulatta) were housed continuously at the National Institutes of Health (NIH) Animal Center (Poolesville, MD). The animal center is fully accredited by the American Association for Accreditation of Laboratory Animal Care, and all procedures were approved by the Animal Care and Use Committee of the National Institute on Aging Intramural Program.
Diet.
During baseline assessments, all monkeys were maintained on standard NIH monkey chow (Purina Mills, St. Louis, MO). After baseline assessment, they were randomized into one of three groups: a healthy standard diet (SD; 4 monkeys) or a high-fat/high-sugar diet in combination with either placebo (HFS; 10 monkeys) or resveratrol (HFS+Resv; 10 monkeys). The SD was a purified diet consisting of 13% kcal in fat and <5% sucrose by weight. The HFS diet was a specially formulated purified ingredient diet with 42% kcal in fat and ∼27% sucrose by weight (Teklad; Harlan, Indianapolis, IN). The monkeys were gradually switched to the HFS diet over a 3-week period. Monkeys received two meals per day at estimated ad libitum levels throughout the study, and water was always available. The average food consumption for weekly periods was the same for all three groups.
Resveratrol dosing.
Resveratrol was supplied by DSM Nutritional Products North America (Parsippany, NJ). The dose for monkeys was derived from the protective dose reported in mice (22 mg/kg) (6) and was adjusted by allometric scaling to an average monkey body weight of 12.1 kg. The monkey equivalent dose was determined to be 40.7 mg. The resveratrol was used to formulate a flavored primate treat (Bio-Serv, Frenchtown, NJ) that was given to the monkeys prior to each meal. Thus, the monkeys received a total dose of 80 mg/day for the first year, and the resveratrol dose was increased to 240 mg twice a day during the second year. Monkeys in non-resveratrol groups received a cherry-flavored placebo treat (sugar pill) that was identical in looks and taste to the resveratrol treats.
Serum resveratrol levels.
The extraction of resveratrol and its metabolites was performed with modifications of a previously published method (20). Briefly, 90 µL of methanol and 10 µL of hexestrol (internal standard) were added to 100 µL of serum, which was vortexed and centrifuged at 20,800 relative centrifugal force for 10 min at 4°C. The supernatant was transferred to an autosampler vial for further analysis. The chromatographic separation of resveratrol and its metabolites was carried out on a Shimadzu Prominence high-performance liquid chromatography system (Columbia, MD). The samples were introduced to the analytical column in 20-µL injections using a Shimadzu SIL 20A autosampler, which was maintained at 4°C. The separation of resveratrol, resveratrol-3-O-sulfate, resveratrol-3-O-glucuronide, and resveratrol-4′-O-glucuronide was accomplished using an Eclipse XDB-C18 guard column (4.6 × 12.5 mm) and a Discovery C18 column (150 × 4.6 mm; internal diameter 5 µm). The mobile phase consisted of water containing 0.1% acetic acid and 0.07% triethylamine as component A and acetonitrile as component B. A linear gradient was run as follows: 0–3 min, 20% B; 3–25 min, 20–60% B; 25–30 min, 60–20% B at a flow rate of 1.0 mL/min. Tandem mass spectrometry analysis was performed using the API4000 system with turbo ion spray (TIS) from Applied Biosystems (Foster City, CA). The data were acquired and analyzed using Analyst version 1.4.2. Negative electrospray ionization data were acquired using the following multiple reaction monitoring transitions: resveratrol (227–185); resveratrol–sulfate (307–227); resveratrol–glucuronide (403–227); and hexestrol (269–134). The TIS instrumental source settings for temperature (500°C), curtain gas (10 ψ), IS1 (60 ψ), IS2 (70 ψ), entrance potential (−10 V), and ion spray voltage (−4,500 V). The TIS compound parameter settings for declustering potential, collision energy, and collision cell exit potential were −70, −25, and −7 V for resveratrol; −50, −28, and −9 V for resveratrol metabolites; and −82, −20, and −8 V for hexesterol.
Hormone assays.
Intravenous glucose (IVG) challenge was performed with 300 mg/kg of 50% dextrose solution (Hospire, Inc., Lake Forest, IL). Blood samples were obtained by venipuncture of the femoral vein using a vacutainer and vacuum tubes. Glucose was measured in whole blood using the Ascensia Elite glucose meter (Bayer, Mishawaka, IN). Serum glucagon was measured by radioimmunoassay (Millipore, Billerica, MA), and serum insulin and media from human islets were measured by enzyme-linked immunosorbent assay (ELISA) (Mercodia, Uppsala, Sweden) according to the manufacturer’s instructions. The insulin area under the curve (AUC) was determined using Prism (GraphPad, La Jolla, CA). The insulin sensitivity index (ISI) was calculated as previously described (21).
Immunofluorescence and densitometry.
Dr. Frederic B. Askin from the Department of Pathology at The Johns Hopkins University School of Medicine (Baltimore, MD) provided anonymous human pancreas in paraffin blocks. Pancreata from two diabetic monkeys in a separate cohort were procured when animals were killed for medical reasons. Our monkeys were fasted overnight, and, after blood draw and euthanasia, the pancreata were excised. Human islets were provided by the National Institute of Diabetes and Digestive and Kidney Diseases–funded Integrated Islet Distribution Program at City of Hope and were embedded in Histogel (Thermo Scientific, Waltham, MA) after treatment. Both human islets and monkey pancreata were fixed in 4% paraformaldehyde, embedded in paraffin blocks, and sectioned (5 µm). After deparaffinization, sections were treated with antigen retrieval solution (BioGenex, Fremont, CA), washed, permeabilized, and blocked, and primary antibodies were added overnight at 4°C as follows: insulin (1:200; Millipore or Sigma, St. Louis, MO); glucagon (1:500; Millipore or Sigma); prohormone convertase (PC) 1/3 (1:200; provided by Dr. Donald Steiner, The University of Chicago, Chicago, IL); glucagon-like peptide 1 (GLP-1) (1:100; Sigma or US Biological, Swampscott, MA); Ki-67 (1:50; Dako, Carpinteria, CA); forkhead box O1 (FOXO1) (1:100; Cell Signaling Technology, Danvers, MA); pancreatic polypeptide (1:200; Millipore); somatostatin, phosphorylated insulin receptor (P-IR), and phosphorylated Bad (P-Bad; serine 136) (1:100; Santa Cruz Biotechnology, Santa Cruz, CA); and NKX2–2 and NKX6–1 (1:100; Novus Biologicals, Littleton, CO). After washing, sections were incubated with fluorescently labeled secondary antibodies (1:500; Rhodamine Red-X, FITC, Cy5; Jackson ImmunoResearch, West Grove, PA) with or without TO-PRO-3 (1:5,000; Molecular Probes, Grand Island, NY) for nuclear staining. Imaging was performed at 40× using a Zeiss (Jena, Germany) LSM-710 confocal microscope. Densitometry of staining was performed using ImageJ (NIH). Images were separated by color and inverted to black and white, and the integrated density was calculated for each stain using the tracing function.
Pancreatic islet size quantification.
Multiple images (200 µm apart) from each monkey were assessed for signal intensity. Islet sizing was performed using the MATLAB (MathWorks, Natick, MA) Image Processing Toolbox. Each islet was carefully isolated using the roipoly tracing function. The various immunostains were separated by color into variables for processing. The resulting total islet, α-cell, and β-cell sizes were calculated based on total traced size, percentage of glucagon immunostain, and percentage of insulin immunostain, respectively. Somatostatin- and pancreatic polypeptide–positive cells were sized similarly. The percentage of cells per islet was determined by counting the number of insulin-, glucagon-, and TO-PRO-3–positive cells in each diet intervention.
Quantitative PCR.
RNA was extracted using TRIzol (Invitrogen, Grand Island, NY) and an RNeasy Mini kit (Qiagen, Valencia, CA), and cDNA was synthesized using qScript cDNA Supermix (Quanta Biosciences, Gaithersburg, MD). Gene expression in monkey islets was quantified using SYBR green (Quanta Biosciences), and values were compared with standard curves and normalized to 18S (Ambion, Austin, TX). The monkey primers were: FOXO1 (forward: 5′-GGATGTGCATTCTATGGTGTACC-3′; and reverse: 5′-TTTCGGGATTGCTTATCTCAGAC-3′), NKX2–2 (forward: 5′-CCGGGCCGAGAAAGGTATG-3′; and reverse: 5′-GTTTGCCGTCCCTGACCAA-3′), NKX6–1 (forward: 5′-ATCTTCTGGCCCGGAGTGA-3′; and reverse: 5′-CGCCAAGTATTTTGTTTGTTCG-3′), and PDX1 (forward: 5′-CGGAACTTTCTATTTAGGATGTGG-3′; and reverse: 5′-AAGATGTGAAGGTCATACTGGCTC-3′) (Integrated DNA Technologies, Coralville, IA). Gene expression in human islets was quantified using TaqMan Fast Advanced Master Mix (Invitrogen) and normalized to actin labeled with VIC. Human primers were labeled with FAM and purchased from Invitrogen (Taqman Gene Expression Assays). Quantitative PCR was performed on an ABI Prism 7300 (Applied Biosystems) detection system.
GLP-1 secretion assay.
Human islets were incubated in Dulbecco’s modified Eagle’s medium (Gibco, Grand Island, NY) containing 5 mmol/L glucose (Sigma), 50 μmol/L dipeptidyl pepsidease-IV inhibitor (Millipore), and 3% bovine serum albumin at 37°C with 5% CO2 overnight. Islets were then washed with medium, separated into several dishes containing 20 islets each, and placed in fresh medium. The islets were then incubated for 1 h at 37°C, after which medium was collected and secreted GLP-1 levels were measured by ELISA (Alpco Diagnostics, Salem, NH). Levels were normalized to total protein content as determined by a BCA Protein Assay (Pierce, Rockford, IL).
cAMP assay.
Human islets were prepared as described above. Total protein was extracted using lysis buffer (0.01 mol/L TrisHcl, 1% Triton X-100, 0.15 mol/L NaCl, 0.5 mmol/L EDTA, and protease and phosphatase inhibitor cocktails). GLP-1 levels were measured by ELISA to determine the amount in total protein, and the islet extract was diluted to yield the different concentrations of GLP-1 (22). Chinese hamster ovary (CHO)/K1 cells were stably transfected with rat GLP-1 receptor (GLP-1R) as previously described (23). Both CHO/K1 and CHO/GLP-1R cells were treated with increasing concentrations (0–20 pmol/L) of full-length GLP-1 (Bachem, Torrance, CA) as well as GLP-1 islet extract for 30 min. After treatment, the cell supernatant was assayed using a Direct cAMP ELISA kit (Enzo Life Sciences, Farmingdale, NY) following the manufacturer’s protocol.
Treatment of human islets.
Human islets were prepared as described above 1 day prior to experimentation. Islets were then separated into several dishes containing 100 islets in fresh medium supplemented with 3% bovine serum albumin and 5 mmol/L glucose for the control condition or 13 mmol/L glucose and 500 μmol/L sodium palmitate (Sigma) for the HFS condition. Islets incubated under the HFS condition were also treated with 1 μmol/L EX-527 and/or 1 nmol/L resveratrol (TOCRIS Bioscience, Minneapolis, MN) for 24 h. Media were then collected for measuring insulin secretion, and islets were processed for quantitative PCR and immunofluorescence as described above.
Statistical analysis.
Quantitative data are represented as the mean ± SEM. Differences between mean values were compared statistically by one-way ANOVA followed by Bonferroni post hoc comparison. A P value of <0.05 was considered statistically significant.
RESULTS
Monkey assessment and hormone levels.
Adult male rhesus monkeys (average age at baseline 10.5 ± 0.4 years) were fed an SD, or an HFS or HFS+Resv diet for 24 months. Prior to euthanasia, the average serum concentrations of resveratrol and resveratrol-3-O-sulfate in HFS+Resv were 27.7 ± 8.6 and 239.1 ± 82.2 ng/mL, respectively, whereas the levels of resveratrol-4′-O-glucuronide and 3-O-glucuronide were below our quantitation limits; all were below detection levels in SD and HFS. SD monkeys gained very little weight, whereas HFS and HFS+Resv monkeys gained a significant amount over the 24-month period (Table 1). Based on fasting glucose and insulin levels at euthanasia, no metabolic dysregulation had occurred over 24 months within the feeding regimens (Table 1). All three groups had similar glucose levels after IVG challenge both at baseline and after 24 months (Fig. 1A). However, in both HFS and HFS+Resv monkeys, the serum insulin levels and the insulin AUC were significantly greater after IVG challenge compared with baseline levels, consistent with weight gain and insulin resistance (Fig. 1B and Table 1). The ISI of the SD animals was stable for the duration of the study, whereas both HFS and HFS+Resv animals exhibited decreased insulin sensitivity after 24 months on an HFS diet (Table 1). The fasting serum glucagon level was not significantly altered over 24 months in any group (Table 1). Within all of these parameters, there was a lot of heterogeneity in the monkeys in response to the diet intervention, both at 0 and 24 months (Supplementary Fig. 1).
TABLE 1.
Monkey characteristics and hormone levels at baseline (0 months) and after 24 months for indicated diets
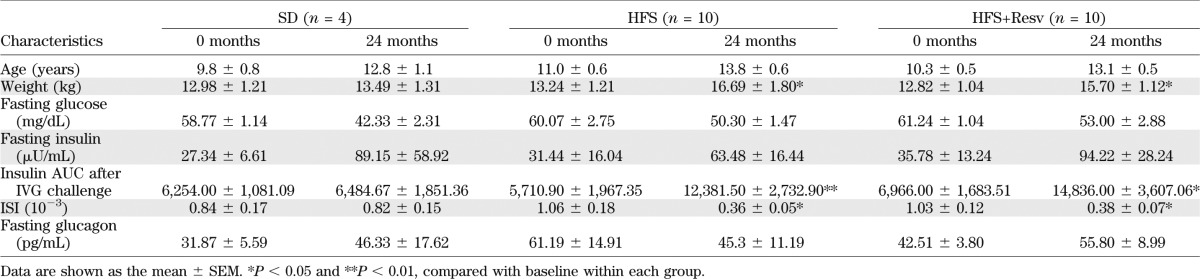
FIG. 1.

HFS diet–induced morphological changes in pancreatic islets. A and B: Glucose (A) and insulin (B) levels at fasting and after IVG challenge (300 mg/kg) before (0, open bars) and after (24, closed bars) 24 months on SD (blue circle, n = 4), HFS diet (red square, n = 10), or HFS+Resv diet (green triangle, n = 10) interventions in adult rhesus monkeys. C: Representative images of immunostaining for insulin (red), glucagon (green), and TO-PRO-3 (blue) in islets of monkeys after 24 months on the indicated diet intervention, and in two nonstudy monkeys in which T2DM spontaneously developed. Scale bar = 20 µm. D–F: Quantification of total islet size (D), β-cell mass (insulin) (E), and α-cell mass (glucagon) (F). G: Ratio of α-cell mass to β-cell mass. H and I: The percentage of cells that were insulin-positive (H) and glucagon-positive (I) in each islet. Data are shown as the mean ± SEM. *P < 0.05.
An HFS diet resulted in morphological changes in islets that were prevented by resveratrol.
On evaluating islet morphology, we found that although total islet size remained unchanged across the groups (Fig. 1D), α-cell mass was significantly increased with HFS at the expense of the β-cell mass compared with SD, with heterogeneity among the HFS islets in their numbers of α- and β-cells (Fig. 1E and F). This caused a significant alteration in the ratio of α-cell/β-cell mass (Fig. 1G). The total number of α- and β-cells per islet was also counted and corroborated the α- and β-cell mass (Fig. 1H and I). The increase in α-cell mass did not occur with resveratrol treatment. HFS+Resv monkeys exhibited a significant decrease in α-cell mass and a lower ratio of α-cell/β-cell mass compared with SD monkeys. The islet changes in HFS monkeys were similar to those of two nonstudy rhesus monkeys from our colony in which T2DM spontaneously developed, though the T2DM monkeys appeared to have even fewer β-cells (Fig. 1C, T2DM).
Lack of evidence for β-cell apoptosis or α-cell mitosis in islets from monkeys given an HFS diet.
Because the change in the α-cell/β-cell ratio without dysglycemia was unexpected, we looked for β-cell apoptosis and α-cell mitosis. No detectable evidence of β-cell apoptosis was observed because we were unable to detect TUNEL-stained nuclei or cleaved caspase 1 or 3 in ∼2,000 islets. Additionally, the expression of a prosurvival protein, P-Bad, was similar in all three groups (Fig. 2A). Despite the obvious increase in α-cell numbers with an HFS diet, no mitotic Ki-67–positive α-cells were found. Abundant Ki-67–positive cells were present in exocrine pancreas and small intestine (Fig. 2B). There were no differences in the numbers of somatostatin or pancreatic polypeptide islet cell types among diet interventions (Fig. 2C).
FIG. 2.

Lack of apoptosis in β-cells, mitosis in α-cells, or changes in the number of other intraislet hormone cell types. A: Immunostaining for insulin (green), glucagon (blue), and P-Bad (red) in islets of monkeys after 24 months on SD (top panel), HFS diet (middle panel), or HFS+Resv diet (bottom panel). Scale bar = 20 µm. B: Ki-67 (red), glucagon (green), and TO-PRO-3 (blue) staining in islets of monkeys after 24 months on the indicated diets. Red arrows indicate Ki-67–positive cells that were detected in nonendocrine tissue. Ki-67 staining in the duodenum was used as a positive control. Scale bar = 20 µm. C: Quantification of somatostatin (white bars) and pancreatic polypeptide (black bars) cell mass in the different diet interventions. Data are shown as the mean ± SEM.
Resveratrol protected depletion of β-cell–specific transcription factors by an HFS diet.
Next, we examined the effect of an HFS diet on insulin/IGF-I signaling and β-cell–specific transcription factors. HFS islets exhibited decreased numbers of tyrosine phosphorylated IR (activated insulin receptor), including within the remaining β-cells (Fig. 3A and B). P-IR in SD and HFS+Resv monkeys were similar, indicating that resveratrol was protective of P-IR in islets. Loss of β-cell P-IR in rodents causes profound defects in β-cell growth (24,25). Forkhead box protein O1 (FOXO1) is a downstream effector in β-cells of IR signaling, and total FOXO1 protein (Fig. 3C and D) and mRNA (Fig. 3E) levels were depleted in HFS islets. In β-cells of SD and HFS+Resv monkeys, total FOXO1 was similar. We next studied factors that are known to be affected by loss of FOXO1 and that are essential in maintaining β-cell phenotype (26). NKX6–1 is a transcriptional repressor that is tightly restricted to β-cell nuclei in adult islets and is known to suppress glucagon expression (27), and NKX2–2 is a transcriptional factor necessary for optimal insulin gene expression (28). Both NKX6–1 (Fig. 4A and B) and NKX2–2 (Fig. 4D and E) proteins and mRNA (Fig. 4C and F, respectively) were severely affected in HFS monkeys. Islets with the fewest number of β-cells had greatly diminished nuclear NKX6–1 expression, with a gradient of decreased expression of NKX6–1 in β-cells of different HFS monkeys (Fig. 4A, bottom panel). Similarly, the mRNA levels of PDX1, a transcription factor necessary for pancreatic development and β-cell maturation (29), were also significantly downregulated in HFS monkeys (Fig. 4G). Resveratrol appeared to protect β-cells from loss of P-IR, FOXO1, NKX6–1, NKX2–2, and PDX1, thereby preserving β-cell mass in HFS+Resv monkeys.
FIG. 3.

Decreased P-IR and total FOXO1 levels in an HFS diet. A: Immunostaining for insulin (green), glucagon (blue), and tyrosine P-IR (red) in islets of monkeys after 24 months on SD (top panel), HFS diet (middle panel), or HFS+Resv diet (bottom panel). Scale bar = 20 µm. B: Quantitation of signal intensity for P-IR. Data are shown as the mean ± SEM. *P < 0.05. C: Immunostaining for insulin (green), TO-PRO-3 (blue), and total FOXO1 (red) in islets of monkeys after 24 months on SD (top panel), HFS (middle panel), or HFS+Resv (bottom panel) diets. Scale bar = 20 µm. D and E: Quantitation of signal intensity (D) and mRNA levels (E) for FOXO1. Data are shown as the mean ± SEM. *P < 0.05.
FIG. 4.

Decreased nuclear expression of NKX6–1, NKX2–2, and PDX1 after an HFS diet. A: Immunostaining for insulin (green), glucagon (blue), and NKX6–1 (red) in monkeys after 24 months on the indicated diet. The islets from different HFS monkeys are shown in order of declining β-cell numbers (bottom panel); the red arrows indicate depletion of nuclear NKX6–1 in insulin-expressing cells. Scale bar = 20 µm. B and C: Quantitation of signal intensity (B) and mRNA levels (C) for NKX6–1. Data are shown as the mean ± SEM. *P < 0.05. D: Immunostaining for insulin (green), glucagon (blue), and NKX2–2 (red) in islets of monkeys after 24 months on SD (top panel), HFS diet (middle panel), or HFS+Resv diet (bottom panel). Scale bar = 20 µm. E and F: Quantitation of signal intensity (E) and mRNA levels (F) for NKX2–2. Data are shown as the mean ± SEM. *P < 0.05. G: Quantitation of mRNA levels for PDX1. Data are shown as the mean ± SEM. *P < 0.05.
Resveratrol protected human islets from HFS-induced changes in morphology and loss of β-cell–specific transcription factors.
Similar to observations in monkey islets after an HFS diet, when human islets were treated with high glucose and palmitate to mimic HFS conditions, the number of insulin-positive β-cells was decreased and the number of glucagon-positive α-cells was increased (Fig. 5A). No TUNEL- or Ki-67–positive nuclei were detected in the islets. Human islets that were incubated with HFS+Resv exhibited islet morphology similar to control islets. Insulin secretion was increased 2.6-fold with an HFS diet and 6.2-fold with an HFS+Resv diet, and treatment with EX-527, a selective Sirtuin 1 (SIRT1) inhibitor, significantly decreased insulin secretion (Fig. 5B). This indicates that the increase in insulin secretion observed with resveratrol treatment is mediated through SIRT1. Additionally, resveratrol treatment resulted in a significant increase in the mRNA expression of PDX1 (Fig. 5C), NKX6–1 (Fig. 5D), FOXO1 (Fig. 5E), and SIRT1 (Fig. 5F). When human islets were treated with EX-527, all of these transcription factors were expressed at levels similar to that of the control group, confirming that the effect of resveratrol in increasing β-cell–specific transcription factors in human islets is mediated, at least in part, through SIRT1.
FIG. 5.

Resveratrol protects islet morphology, increases insulin secretion, and increases mRNA expression of PDX1, NKX6–1, FOXO1, and SIRT1 in human islets. A: Immunostaining for insulin (green), glucagon (red), and TO-PRO-3 (blue) in human islets. Scale bar = 20 µm. B: Insulin secretion per human islet after 24 h in low-glucose (Control), HFS, HFS+Resv, or HFS+Resv and a SIRT1 inhibitor (HFS+Resv + EX-527). C–F: Quantitation of mRNA levels for PDX1 (C), NKX6–1 (D), FOXO1 (E), and SIRT1 (F) in human islets after the indicated treatments. Data are shown as the mean ± SEM. *P < 0.05, **P < 0.01.
Identification of PC1/3 and GLP-1 in α-cells.
Because we observed a dramatic increase in α-cell numbers in HFS compared with SD monkeys, we closely examined the α-cell compartment. Glucagon, which functions to maintain fasting glucose levels by stimulating hepatic glucose production, is produced in α-cells resulting from the enzymatic cleavage of proglucagon by PC2 (30). The glucagon-positive cells in monkey islets costained with PC2, as expected (data not shown), and additionally expressed PC1/3 (Fig. 6A). Proglucagon is also produced in L-cells of the intestine and taste cells in taste buds, and the expression of PC1/3 in those cells leads to GLP-1 production (22,31). The α-cells in our monkeys expressed GLP-1 (Fig. 6B). There was a significant increase in the amount of GLP-1 staining with HFS because of the increased number of α-cells (Fig. 6C). Although PC1/3, as expected, was also present in some insulin-containing cells, GLP-1, similar to glucagon, never colocalized with insulin. The presence of PC1/3 and GLP-1 has been reported in human pancreatic islet cells, including those from T2DM patients (32). We also detected PC1/3 (Fig. 7A) and GLP-1 (Fig. 7B) immunostaining in α-cells of the human pancreas.
FIG. 6.

Identification of PC1/3 and GLP-1 in monkey islets. A: Immunostaining for insulin (green), glucagon (blue), and PC1/3 (red) in SD monkey islets. B: Immunostaining for insulin (green), glucagon (blue), and GLP-1 (red) in islets of monkeys after 24 months on SD (top panel), HFS diet (middle panel), or HFS+Resv diet (bottom panel) interventions. Scale bar = 20 µm. C: Quantitation of signal intensity for GLP-1. Data are shown as the mean ± SEM. *P < 0.05.
FIG. 7.

Secretion of biologically active GLP-1 from human islets. A and B: Immunostaining for insulin (green) and glucagon (blue) along with PC1/3 (red) (A) or GLP-1 (red) (B) in human islets. Scale bar = 20 µm. C: Levels of secreted GLP-1 from medium alone or from 20 human islets into medium over 1 h as measured by ELISA. D: cAMP concentrations after treatment of CHO/K1 (black bars) and CHO-GLP-1R (white bars) cells with either increasing concentration of recombinant GLP-1 as a positive control or GLP-1 extracted from islets (Islet extract). Data are shown as the mean ± SEM from at least three independent experiments. *P < 0.05.
GLP-1 secreted from islets is biologically active.
Because we did not have access to fresh monkey islets to ascertain whether the GLP-1 present in primate islets was biologically active, freshly isolated human islets were used. GLP-1 was detectably secreted into culture medium after 1 h from as little as 20 human islets (Fig. 7C). In order to determine whether the GLP-1 in islets was biologically active, we extracted total islet protein, measured GLP-1 by ELISA, and normalized it to protein content. The extract was then diluted to give the indicated concentrations of GLP-1 and added to CHO cells, and CHO cells were stably transfected with GLP-1R. Nontransfected CHO cells did not respond to GLP-1 treatment as measured by a direct cAMP assay (Fig. 7D). However, CHO cells stably transfected with GLP-1R responded to recombinant GLP-1 and islet extract GLP-1 in a dose-dependent manner. Islet-extracted GLP-1 actually resulted in a significant increase in cAMP levels (Fig. 7D), demonstrating that the GLP-1 present in human islets, and presumably monkey islets, is biologically active.
DISCUSSION
After 24 months on an HFS diet, monkeys had significant weight gain, increased serum insulin levels and insulin AUC after an IVG challenge, and decreased insulin sensitivity. Moreover, significant changes in islet morphology as a result of an HFS diet, independent of dysglycemia, were also present. Hyperglycemia or the diabetic state itself was not a secondary cause of these intraislet cellular changes. Most notably, the α-cell/β-cell ratio was significantly altered because of decreased numbers of insulin-containing cells and increased numbers of glucagon- and GLP-1–containing cells, whereas total islet size was unaltered. These morphological changes are similar to those observed in monkeys in which T2DM spontaneously developed (Fig. 1C), in vervet monkeys administered an 18-month atherogenic diet (33), and in the pathologies of diabetic human pancreata (34). Additionally, an HFS diet resulted in the depletion of β-cell–specific transcription factors FOXO1, NKX6–1, NKX2–2, and PDX1. Remarkably, monkeys given resveratrol supplementation in addition to an HFS diet had islets that were similar in morphology to SD and maintained the critical transcription factors. The effect of resveratrol was independent of any effect it may have had on insulin-mediated glucose uptake (i.e., peripheral insulin sensitivity), because it did not prevent the decline in insulin sensitivity due to the HFS diet.
There are two possibilities for the increase in α-cell/β-cell ratio observed with HFS monkeys. The first is that β-cells underwent apoptosis and α-cells proliferated, but we did not capture either phenomenon because of the brevity of both. One possible signal for α-cell mitosis is that the neighboring β-cells act as brakes on α-cell turnover and this brake is lifted once a neighboring β-cell undergoes apoptosis. Additionally, apoptotic β-cells may shed microparticles/products that cause proliferation of neighboring α-cells. Indeed, apoptosis of β-cells in rodents stimulates proliferation of neighboring cells (35), and insulin itself is trophic in α-cell lines (36).
A decrease in β-cell mass has also been reported in humans with T2DM, and this was associated with an increase in islet amyloid deposition and β-cell apoptosis (37,38). Baboons, which serve as another model for T2DM (39), also exhibit severe islet amyloidosis, which is associated with increased β-cell apoptosis and decreased relative β-cell volume, as well as α-cell replication, hypertrophy, and increased relative volume (40). The increased α-cell proliferation correlated with hyperglucagonemia and hyperglycemia, neither of which was observed in our monkeys after 24 months on an HFS diet.
A second possibility for the islet findings in HFS monkeys is that β-cells dedifferentiated and became α-cells, thus explaining how islet size was unaltered. Our findings resemble the phenotype seen in FOXO1-null mice in that those mice, placed under metabolic stress, had reduced β-cell numbers due to dedifferentiation, not death, and some dedifferentiated cells became α-cells (26). We suggest that the loss of NKX6–1 and PDX1 due to depletion of FOXO1 lifted the brake on the repression of glucagon transcription, and, as a consequence, β-cells, once dedifferentiated, became α-cells. Single human β-cells normally express the glucagon gene, albeit at low levels compared with the insulin gene (41). In no instance did we find coexpression of insulin and glucagon or glucagon and NKX6–1, illustrating that regression of β-cells to a non–β-cell phenotype is necessary prior to conversion to α-cells. We conclude that unremitting hyperstimulation due to HFS food consumption, and not dysglycemia, is the so-called “metabolic stress” that causes depletion of FOXO1 and loss of β-cell phenotype. β-Cell loss of FOXO1, NKX6–2, and PDX1 did not occur with HFS consumption when resveratrol was added to the diet. Similar effects have been described in vitro where resveratrol treatment of insulinoma INS-1E cells and human islets resulted in the upregulation of key genes for β-cell function, including PDX1, ultimately potentiating glucose-stimulated insulin secretion (42). We also found that resveratrol treatment resulted in a significant increase in PDX1, NKX6–1, and FOXO1 mRNA expression in human islets incubated under HFS conditions. The increase in β-cell transcription factors was also associated with an increase in insulin secretion, as HFS+Resv-treated islets secreted insulin at levels 6.2-fold greater than control and 2.4-fold greater than HFS alone.
GLP-1 is a hormone with pleiotropic effects that helps to maintain blood glucose homeostasis. It is a powerful stimulant to insulin secretion in a glucose-dependent manner (43), increases proinsulin synthesis (44), modulates insulin sensitivity (45), and, in rodents, increases β-cell turnover (46). Additionally, glucagon secretion is inhibited by insulin (47) and by GLP-1 independently of insulin (48). We suggest that increased α-cell–derived GLP-1 promoted enhanced glucose-mediated insulin secretion and contributed to the suppression of glucagon secretion in HFS monkeys. This allowed the remaining β-cells to secrete sufficient insulin to maintain euglycemia and suppress glucagon secretion. This mechanism would be successful as long as adequate numbers of β-cells remained. Eventually β-cell numbers would have declined to a limiting amount and circulating glucagon levels would rise because of lack of sufficient insulin for inhibition of secretion. At this time, dysglycemia would become evident.
Although nonhuman primate studies are crucial to more accurately elucidate the pathogenesis of T2DM in humans, several limitations are evident because of the nature of this model. In the current study, none of the monkeys in the HFS cohort developed overt diabetes, so islets from monkeys under metabolic stress were compared with those from monkeys in which diabetes spontaneously developed to show increasing progression of β-cell depletion. Additionally, we do not know whether the α-cell/β-cell ratio seen with HFS would revert to that of SD if the animals were returned to a healthy SD, so we cannot attest to the adaptability of α-cells once metabolic stress is alleviated. Finally, although we show that daily resveratrol supplementation prevented islet changes in the presence of an HFS diet, we were unable to show the biochemical pathways by which resveratrol was protective. Its effects may be through the activation of AMP-activated protein kinase or Sirtuins, and thus FOXO1 (49,50), through the inhibition of phosphodiesterases in β-cells (51), or through a combination of many pathways. In human islets, we were able to show that blocking SIRT1 prevented the resveratrol-induced increase in insulin secretion and upregulation of PDX1, NKX6–1, FOXO1, and SIRT1 mRNA expression. This suggests that the positive effects of resveratrol on β-cell function in human islets are mediated through SIRT1.
This study was carried out in adult primates and has direct relevance to the human population that is currently facing a diabetic epidemic caused by obesity. Screening patients for early T2DM and prediabetes is a worthy goal in order to aggressively treat and preserve β-cell function (52,53). We submit, however, that islet changes occur before overt evidence of β-cell compromise and any dysglycemia are uncovered with usual testing, such as fasting glucose levels and hemoglobin A1c measurements. Although lifestyle interventions and pharmacotherapy to maintain normal glucose homeostasis and prevent conversion of prediabetes to diabetes (54) are laudatory, they are expensive, require intense input from healthcare providers, and need to be persistent. They may also be too late because morphological changes causing predisposition to diabetes could have already occurred in islets. The addition of resveratrol and resveratrol-containing foods to one’s daily diet may be a simple, inexpensive way to protect β-cells.
ACKNOWLEDGMENTS
This work was supported by the Intramural Research Program of the National Institute on Aging and by a grant from the Office of Dietary Supplements awarded to K.J.P. W.K. was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF) and funded by the Ministry of Science, ICT, and Future Planning (2012R1A1A1041352). M.E.D. was supported by the JDRF and The Sanford Project, Sioux Falls, SD.
No potential conflicts of interest relevant to this article were reported.
J.L.F. designed and performed experiments, analyzed data, and wrote the manuscript. Y.-K.S., W.K., S.M.K.-W., I.G.-M., and O.D.C. designed and performed experiments and analyzed data. M.S. and R.M. developed and performed the assays to determine serum resveratrol levels. K.F. and S.K.G. analyzed data. M.E.D. was responsible for procuring human islets from the Integrated Islet Distribution Program and for the treatment of human islets. K.J.P., J.A.M., and R.d.C. contributed to the design, execution, and analysis of experiments. J.M.E. contributed to the design of experiments, interpretation of data, and writing of the manuscript. All authors edited and reviewed the manuscript. J.M.E. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented at the 73rd Scientific Sessions of the American Diabetes Association, Chicago, Illinois, 21–25 June 2013.
The authors thank the animal care staff at the National Institutes of Health Animal Center: Edward Tilmont, Joanne Allard, Theresa Ward, Kaitlyn Lewis, Caitlin Younts, and veterinarian Dr. Rick Herbert. The authors also thank Hyekyung Yang for her assistance with processing human islets for staining. The authors are grateful to Fred E. Indig at the National Institute on Aging for his invaluable contributions with confocal imaging; and to DSM Nutritional Products for providing ResVida (resveratrol).
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db13-0266/-/DC1.
REFERENCES
- 1.Cordain L, Eaton SB, Sebastian A, et al. Origins and evolution of the Western diet: health implications for the 21st century. Am J Clin Nutr 2005;81:341–354 [DOI] [PubMed] [Google Scholar]
- 2.Haslam DW, James WPT. Obesity. Lancet 2005;366:1197–1209 [DOI] [PubMed] [Google Scholar]
- 3.Centers for Disease Control and Prevention. 2011 National Diabetes Fact Sheet [article online], 2011. Available from http://www.cdc.gov/diabetes/pubs/pdf/ndfs_2011.pdf Accessed 15 February 2013
- 4.Knowler WC, Barrett-Connor E, Fowler SE, et al. Diabetes Prevention Program Research Group Reduction in the incidence of type 2 diabetes with lifestyle intervention or metformin. N Engl J Med 2002;346:393–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Szkudelska K, Szkudelski T. Resveratrol, obesity and diabetes. Eur J Pharmacol 2010;635:1–8 [DOI] [PubMed] [Google Scholar]
- 6.Baur JA, Pearson KJ, Price NL, et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature 2006;444:337–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shang J, Chen LL, Xiao FX, Sun H, Ding HC, Xiao H. Resveratrol improves non-alcoholic fatty liver disease by activating AMP-activated protein kinase. Acta Pharmacol Sin 2008;29:698–706 [DOI] [PubMed] [Google Scholar]
- 8.Rivera L, Morón R, Zarzuelo A, Galisteo M. Long-term resveratrol administration reduces metabolic disturbances and lowers blood pressure in obese Zucker rats. Biochem Pharmacol 2009;77:1053–1063 [DOI] [PubMed] [Google Scholar]
- 9.Su H-C, Hung L-M, Chen J-K. Resveratrol, a red wine antioxidant, possesses an insulin-like effect in streptozotocin-induced diabetic rats. Am J Physiol Endocrinol Metab 2006;290:E1339–E1346 [DOI] [PubMed] [Google Scholar]
- 10.Lee J-H, Song M-Y, Song E-K, et al. Overexpression of SIRT1 protects pancreatic beta-cells against cytokine toxicity by suppressing the nuclear factor-kappaB signaling pathway. Diabetes 2009;58:344–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Timmers S, Konings E, Bilet L, et al. Calorie restriction-like effects of 30 days of resveratrol supplementation on energy metabolism and metabolic profile in obese humans. Cell Metab 2011;14:612–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Brasnyó P, Molnár GA, Mohás M, et al. Resveratrol improves insulin sensitivity, reduces oxidative stress and activates the Akt pathway in type 2 diabetic patients. Br J Nutr 2011;106:383–389 [DOI] [PubMed] [Google Scholar]
- 13.Crandall JP, Oram V, Trandafirescu G, et al. Pilot study of resveratrol in older adults with impaired glucose tolerance. J Gerontol A Biol Sci Med Sci 2012;67:1307–1312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yoshino J, Conte C, Fontana L, et al. Resveratrol supplementation does not improve metabolic function in nonobese women with normal glucose tolerance. Cell Metab 2012;16:658–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poulsen MM, Vestergaard PF, Clasen BF, et al. High-dose resveratrol supplementation in obese men: an investigator-initiated, randomized, placebo-controlled clinical trial of substrate metabolism, insulin sensitivity, and body composition. Diabetes 2013;62:1186–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hansen BC. Investigation and treatment of type 2 diabetes in nonhuman primates. Methods Mol Biol 2012;933:177–185 [DOI] [PubMed] [Google Scholar]
- 17.Brissova M, Fowler MJ, Nicholson WE, et al. Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J Histochem Cytochem 2005;53:1087–1097 [DOI] [PubMed] [Google Scholar]
- 18.Bremer AA, Stanhope KL, Graham JL, et al. Fructose-fed rhesus monkeys: a nonhuman primate model of insulin resistance, metabolic syndrome, and type 2 diabetes. Clin Transl Sci 2011;4:243–252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ortmeyer HK, Sajan MP, Miura A, et al. Insulin signaling and insulin sensitizing in muscle and liver of obese monkeys: peroxisome proliferator-activated receptor gamma agonist improves defective activation of atypical protein kinase C. Antioxid Redox Signal 2011;14:207–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Boocock DJ, Faust GES, Patel KR, et al. Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol Biomarkers Prev 2007;16:1246–1252 [DOI] [PubMed] [Google Scholar]
- 21.Swarbrick MM, Havel PJ, Levin AA, et al. Inhibition of protein tyrosine phosphatase-1B with antisense oligonucleotides improves insulin sensitivity and increases adiponectin concentrations in monkeys. Endocrinology 2009;150:1670–1679 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Shin Y-K, Martin B, Golden E, et al. Modulation of taste sensitivity by GLP-1 signaling. J Neurochem 2008;106:455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Montrose-Rafizadeh C, Wang Y, Janczewski AM, Henderson TE, Egan JM. Overexpression of glucagon-like peptide-1 receptor in an insulin-secreting cell line enhances glucose responsiveness. Mol Cell Endocrinol 1997;130:109–117 [DOI] [PubMed] [Google Scholar]
- 24.Kulkarni RN, Brüning JC, Winnay JN, Postic C, Magnuson MA, Kahn CR. Tissue-specific knockout of the insulin receptor in pancreatic beta cells creates an insulin secretory defect similar to that in type 2 diabetes. Cell 1999;96:329–339 [DOI] [PubMed] [Google Scholar]
- 25.Ueki K, Okada T, Hu J, et al. Total insulin and IGF-I resistance in pancreatic beta cells causes overt diabetes. Nat Genet 2006;38:583–588 [DOI] [PubMed] [Google Scholar]
- 26.Talchai C, Xuan S, Lin HV, Sussel L, Accili D. Pancreatic β cell dedifferentiation as a mechanism of diabetic β cell failure. Cell 2012;150:1223–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watada H, Mirmira RG, Leung J, German MS. Transcriptional and translational regulation of beta-cell differentiation factor Nkx6.1. J Biol Chem 2000;275:34224–34230 [DOI] [PubMed] [Google Scholar]
- 28.Anderson KR, Torres CA, Solomon K, et al. Cooperative transcriptional regulation of the essential pancreatic islet gene NeuroD1 (beta2) by Nkx2.2 and neurogenin 3. J Biol Chem 2009;284:31236–31248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.McKinnon CM, Docherty K. Pancreatic duodenal homeobox-1, PDX-1, a major regulator of beta cell identity and function. Diabetologia 2001;44:1203–1214 [DOI] [PubMed] [Google Scholar]
- 30.Rouillé Y, Bianchi M, Irminger JC, Halban PA. Role of the prohormone convertase PC2 in the processing of proglucagon to glucagon. FEBS Lett 1997;413:119–123 [DOI] [PubMed] [Google Scholar]
- 31.Rouillé Y, Kantengwa S, Irminger JC, Halban PA. Role of the prohormone convertase PC3 in the processing of proglucagon to glucagon-like peptide 1. J Biol Chem 1997;272:32810–32816 [DOI] [PubMed] [Google Scholar]
- 32.Marchetti P, Lupi R, Bugliani M, et al. A local glucagon-like peptide 1 (GLP-1) system in human pancreatic islets. Diabetologia 2012;55:3262–3272 [DOI] [PubMed] [Google Scholar]
- 33.Louw J, Woodroof C, Seier J, Wolfe-Coote SA. The effect of diet on the Vervet monkey endocrine pancreas. J Med Primatol 1997;26:307–311 [DOI] [PubMed] [Google Scholar]
- 34.Folli F, Okada T, Perego C, et al. Altered insulin receptor signalling and β-cell cycle dynamics in type 2 diabetes mellitus. PLoS One 2011;6:e28050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bonner C, Bacon S, Concannon CG, et al. INS-1 cells undergoing caspase-dependent apoptosis enhance the regenerative capacity of neighboring cells. Diabetes 2010;59:2799–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Z, Kim W, Chen Z, et al. Insulin and glucagon regulate pancreatic α-cell proliferation. PLoS One 2011;6:e16096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jurgens CA, Toukatly MN, Fligner CL, et al. β-cell loss and β-cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. Am J Pathol 2011;178:2632–2640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Westermark P, Andersson A, Westermark GT. Islet amyloid polypeptide, islet amyloid, and diabetes mellitus. Physiol Rev 2011;91:795–826 [DOI] [PubMed] [Google Scholar]
- 39.Chavez AO, Lopez-Alvarenga JC, Tejero ME, et al. Physiological and molecular determinants of insulin action in the baboon. Diabetes 2008;57:899–908 [DOI] [PubMed] [Google Scholar]
- 40.Guardado-Mendoza R, Davalli AM, Chavez AO, et al. Pancreatic islet amyloidosis, beta-cell apoptosis, and alpha-cell proliferation are determinants of islet remodeling in type-2 diabetic baboons. Proc Natl Acad Sci USA 2009;106:13992–13997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kirkpatrick CL, Marchetti P, Purrello F, et al. Type 2 diabetes susceptibility gene expression in normal or diabetic sorted human alpha and beta cells: correlations with age or BMI of islet donors. PLoS One 2010;5:e11053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Vetterli L, Brun T, Giovannoni L, Bosco D, Maechler P. Resveratrol potentiates glucose-stimulated insulin secretion in INS-1E beta-cells and human islets through a SIRT1-dependent mechanism. J Biol Chem 2011;286:6049–6060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nauck MA, Bartels E, Orskov C, Ebert RCW, Creutzfeldt W. Additive insulinotropic effects of exogenous synthetic human gastric inhibitory polypeptide and glucagon-like peptide-1-(7-36) amide infused at near-physiological insulinotropic hormone and glucose concentrations. J Clin Endocrinol Metab 1993;76:912–917 [DOI] [PubMed] [Google Scholar]
- 44.Alarcon C, Wicksteed B, Rhodes CJ. Exendin 4 controls insulin production in rat islet beta cells predominantly by potentiation of glucose-stimulated proinsulin biosynthesis at the translational level. Diabetologia 2006;49:2920–2929 [DOI] [PubMed] [Google Scholar]
- 45.Zander M, Madsbad S, Madsen JL, Holst JJ. Effect of 6-week course of glucagon-like peptide 1 on glycaemic control, insulin sensitivity, and beta-cell function in type 2 diabetes: a parallel-group study. Lancet 2002;359:824–830 [DOI] [PubMed] [Google Scholar]
- 46.Buteau J, Foisy S, Rhodes CJ, Carpenter L, Biden TJ, Prentki M. Protein kinase Czeta activation mediates glucagon-like peptide-1-induced pancreatic beta-cell proliferation. Diabetes 2001;50:2237–2243 [DOI] [PubMed] [Google Scholar]
- 47.Kaneko K, Shirotani T, Araki E, et al. Insulin inhibits glucagon secretion by the activation of PI3-kinase in In-R1-G9 cells. Diabetes Res Clin Pract 1999;44:83–92 [DOI] [PubMed] [Google Scholar]
- 48.Kawamori D, Akiyama M, Hu J, Hambro B, Kulkarni RN. Growth factor signalling in the regulation of α-cell fate. Diabetes Obes Metab 2011;13(Suppl. 1):21–30 [DOI] [PubMed] [Google Scholar]
- 49.Brunet A, Sweeney LB, Sturgill JF, et al. Stress-dependent regulation of FOXO transcription factors by the SIRT1 deacetylase. Science 2004;303:2011–2015 [DOI] [PubMed] [Google Scholar]
- 50.Wang A, Liu M, Liu X, et al. Up-regulation of adiponectin by resveratrol: the essential roles of the Akt/FOXO1 and AMP-activated protein kinase signaling pathways and DsbA-L. J Biol Chem 2011;286:60–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Park S-J, Ahmad F, Philp A, et al. Resveratrol ameliorates aging-related metabolic phenotypes by inhibiting cAMP phosphodiesterases. Cell 2012;148:421–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phillips LS, Olson DE. Diabetes: normal glucose levels should be the goal. Nat Rev Endocrinol 2012;8:510–512 [DOI] [PubMed] [Google Scholar]
- 53.Fradkin JE, Roberts BT, Rodgers GP. What’s preventing us from preventing type 2 diabetes? N Engl J Med 2012;367:1177–1179 [DOI] [PubMed] [Google Scholar]
- 54.American Diabetes Association Standards of medical care in diabetes—2012. Diabetes Care 2012;35(Suppl. 1):S11–S63 [DOI] [PMC free article] [PubMed] [Google Scholar]