

Abstract
The intake of added sugars, such as from table sugar (sucrose) and high-fructose corn syrup has increased dramatically in the last hundred years and correlates closely with the rise in obesity, metabolic syndrome, and diabetes. Fructose is a major component of added sugars and is distinct from other sugars in its ability to cause intracellular ATP depletion, nucleotide turnover, and the generation of uric acid. In this article, we revisit the hypothesis that it is this unique aspect of fructose metabolism that accounts for why fructose intake increases the risk for metabolic syndrome. Recent studies show that fructose-induced uric acid generation causes mitochondrial oxidative stress that stimulates fat accumulation independent of excessive caloric intake. These studies challenge the long-standing dogma that “a calorie is just a calorie” and suggest that the metabolic effects of food may matter as much as its energy content. The discovery that fructose-mediated generation of uric acid may have a causal role in diabetes and obesity provides new insights into pathogenesis and therapies for this important disease.
Fructose-induced weight gain and metabolic syndrome
Experimental studies from the 1950s showed the peculiar ability of fructose to induce insulin resistance in laboratory rats. Today, fructose intake has been shown to induce all features of metabolic syndrome in rats, as well as oxidative stress, endothelial dysfunction, fatty liver, microalbuminuria and kidney disease (rev. in 1). Similar findings can be shown when animals are fed sucrose or high-fructose corn syrup (HFCS), both which contain fructose (2,3). In contrast, administration of glucose or starch results in fewer features of metabolic syndrome when provided equivalent intake (4,5).
Fructose may increase the risk for obesity by altering satiety, resulting in increased food intake. The intake of fructose is not effective in stimulating insulin and leptin secretion in humans, and hence may not induce a satiety response (6). Other mechanisms may also be operative. For example, a high intake of fructose induces leptin resistance in rats (7). Fructose also encourages food intake due to stimulation of dopamine in the mesolimbic system and effects on the hypothalamus (8,9). Food intake is also stimulated by hepatic ATP depletion (10), which occurs in animals and humans administered fructose (11). Fructose may also affect metabolic rate. A recent study in humans documented a reduction in resting energy expenditure in overweight and obese subjects fed fructose but not glucose (12).
Fructose-induced metabolic syndrome does not require increased energy intake
The ability for fructose (and sucrose, which contains fructose) to stimulate food intake and to lower metabolism provides a mechanism for how a high fructose intake may encourage weight gain and visceral fat accumulation. However, fructose or sucrose also alters fat stores and metabolism independent of excessive energy intake. Although weight gain is largely controlled by overall energy intake, other features of metabolic syndrome can occur independent of weight gain. For example, rats fed fructose develop fatty liver, hypertriglyceridemia, and insulin resistance when compared with rats fed isocaloric glucose or starch-enriched diets (4,5). Indeed, hypertriglyceridemia, fatty liver, and type 2 diabetes can be induced in metabolic syndrome–prone rats with caloric restriction provided the diet is high (40%) in sucrose (which contains fructose) (5). A recent epidemiological analysis in humans also found an association of diabetes prevalence with sugar availability that was independent of total energy intake (13).
A role for uric acid in fructose-induced fat accumulation
The observation that fructose-fed rats develop fatty liver and metabolic syndrome without requiring increased energy intake suggests that the metabolism of fructose may be different from that of other carbohydrates. Fructose is distinct from glucose only in its initial metabolism. The first enzyme to metabolize fructose is fructokinase (also known as ketohexokinase [KHK]). The metabolism of fructose to fructose-1-phosphate by KHK occurs primarily in the liver, is rapid and without any negative feedback, and results in a fall in intracellular phosphate and ATP levels (14–16). This has been shown to occur in the liver in humans with relatively small doses of oral fructose (60 g fructose alone or 39 g fructose with 39 g glucose) (11). The decrease in intracellular phosphate stimulates AMP deaminase (AMPD), which catalyzes the degradation of AMP to inosine monophosphate and eventually uric acid (15) (Fig. 1). The increase in intracellular uric acid is followed by an acute rise in uric acid in the circulation likely due to its release from the liver (14). Fructose also stimulates uric acid synthesis from amino acid precursors, such as glycine (17).
FIG. 1.

Fructose-induced nucleotide turnover. Fructose is rapidly phosphorylated in the hepatocyte by KHK to fructose-1-phosphate (F-1-P), which uses ATP as a phosphate donor. Intracellular phosphate (PO4) levels decrease, stimulating the activity of AMP deaminase 2 (AMPD2). AMPD2 converts AMP to inosine monophosphate (IMP). IMP is metabolized to inosine by 5′ nucleotidase (5′NT), which is further degraded to xanthine and hypoxanthine by xanthine oxidase (XO), ultimately generating uric acid.
Recent studies suggest that this “side event” in fructose metabolism may be critical for how fructose induces metabolic syndrome. First, there are actually two KHK isoforms, and they differ in their ability to activate this pathway. KHK-C phosphorylates fructose rapidly, consuming ATP with the generation of uric acid. In contrast, KHK-A phosphorylates fructose slowly and consumes minimal ATP (18). When both KHK-C and KHK-A are deleted, mice are fully protected from fructose-induced metabolic syndrome and fatty liver (18); however, when KHK-A is selectively deleted, there is increased fructose available for metabolism by KHK-C, and the metabolic syndrome and fatty liver are worsened compared with wild-type mice despite the same intake of total calories and fructose (18). These studies suggest that differences in nucleotide turnover might influence the metabolic response.
To examine the purine nucleotide pathway in the metabolic response, we silenced aldolase B in a hepatocyte line (HepG2 cells) (19). Aldolase B is the second enzyme in fructose metabolism, and the genetic loss of aldolase B is the cause of hereditary fructose intolerance. When aldolase B is inhibited, fructose is phosphorylated by ATP but cannot be further metabolized, nevertheless fructose can be metabolized by other routes such as hexokinase. In this regard, subjects with hereditary fructose intolerance, are known to have hyperactive KHK and show enhanced ATP depletion and uric acid generation in response to fructose. As such, this is an interesting condition in which marked nucleotide turnover and ATP depletion occur but without the ability to be further metabolized by this primary enzymatic pathway to glucose, glycogen, or triglycerides (20). Nevertheless, the feeding of fructose to HepG2 cells lacking aldolase B resulted in a rapid accumulation of triglycerides, consistent with our findings that uric acid itself can induce triglyceride accumulation (19). These experiments explain why fatty liver and hyperuricemia are common complications of this disease (21) and also why fatty liver and diabetes are complications in subjects with glycogen storage disease I, in which hepatic intracellular ATP depletion and hyperuricemia also occur (22–25). Finally, it provides an explanation for why fructose is lipogenic (based on acetate labeling studies) despite little of the fructose molecule being incorporated into the triglyceride molecule itself (19).
We next addressed how the degradation of nucleotides might lead to fat accumulation. Specifically, our group and others have shown that AMPD counters the effects of AMP-activated protein kinase (AMPK) (26,27). Whereas activation of AMPK in hepatocytes induces oxidation of fatty acids and ATP generation, AMPD has opposite effects. Overexpression of AMPD in HepG2 cells blocks fatty acid oxidation and increases fat accumulation, whereas silencing AMPD blocks fructose-induced fat accumulation. The mechanism is mediated in part by the generation of uric acid, which inhibits AMPK (27).
In addition to inhibiting AMPK, uric acid may stimulate hepatic lipogenesis (28). The mechanism appears to be mediated by uric acid–dependent intracellular and mitochondrial oxidative stress (28). Although uric acid is a potent antioxidant in the extracellular environment, when uric acid enters cells via specific organic anion transporters, it induces an oxidative burst that has been shown in vascular smooth muscle cells, endothelial cells, adipocytes, islet cells, renal tubular cells, and hepatocytes (29–31). Uric acid–induced oxidative stress appears to be mediated by the stimulation of NADPH oxidase, which translocates to mitochondria (28,29,32). Uric acid can also generate triuretcarbonyl and aminocarbonyl radicals as well as alkylating species upon reaction with peroxynitrite and can also directly inactivate nitric oxide (NO) to 6-aminouracil (33,34).
The induction of oxidative stress in the mitochondria causes a reduction in aconitase-2 activity in the Krebs cycle, resulting in citrate accumulation that is transported into the cytoplasm where it activates ATP citrate lyase, acetyl CoA carboxylase, and fatty acid synthase, leading to fat synthesis (19). Uric acid also causes a reduction in enoyl CoA hydratase-1, a rate-limiting enzyme in β-fatty acid oxidation (35). The consequence is fat accumulation in the hepatocyte (19,35).
Recently, we identified another mechanism by which uric acid may increase the risk for hepatic fat accumulation and metabolic syndrome. Fructose (or sucrose) ingestion is known to increase hepatic KHK levels (5). The increased expression of KHK is driven in part by the production of uric acid from fructose (35) A rise in intracellular uric acid activates the nuclear transcription factor, carbohydrate responsive element–binding protein (35). When KHK expression is increased in HepG2 cells by uric acid exposure, the triglyceride accumulation in response to fructose is enhanced (35).
This is relevant to subjects with nonalcoholic fatty liver disease (NAFLD). Subjects with NAFLD ingest more fructose-containing soft drinks than age, sex, and BMI-matched control subjects and have increased KHK expression in their liver (36). Subjects with NAFLD who have the highest fructose intake also show the greatest ATP depletion in response to a fructose load, and those subjects with the highest uric acid levels show a greater nadir in the ATP depletion (37). These data are consistent with an induction of KHK in the liver with subsequent increased sensitivity to the effects of fructose via a uric acid–dependent mechanism.
Rodents have lower serum uric acid than humans due to the presence of uricase in their liver, and hence show a lesser rise in serum uric acid in response to fructose (38). Nevertheless, lowering uric acid has also been found to block the development of hepatic steatosis in fructose-fed rats (35). Lowering uric acid also reduces hepatic steatosis in the desert gerbil (which spontaneously develops fatty liver on a normal diet) (39), in alcoholic fatty liver (in which increased intrahepatic uric acid occurs) (40), and in the Pound mouse (a mouse model of metabolic syndrome manifesting fatty liver, obesity, insulin resistance, and hypertension caused by a leptin receptor mutation) (28). These studies supported the tight association of hyperuricemia with fatty liver; prospective studies have also reported that an elevated uric acid independently predicts the development of NAFLD (41). The ability of hyperuricemia to predict fatty liver is independent of obesity. Hyperuricemia is even associated with NAFLD in hemodialysis subjects who have a BMI below 20 (19). A summary of how fructose and uric acid induce fatty liver is shown in Fig. 2.
FIG. 2.

Classic and alternative lipogenic pathways of fructose. In the classical pathway, triglycerides (TG) are a direct product of fructose metabolism by the action of multiple enzymes including aldolase B (Aldo B) and fatty acid synthase (FAS). An alternative mechanism was recently shown (30). Uric acid produced from the nucleotide turnover that occurs during the phosphorylation of fructose to fructose-1-phosphate (F-1-P) results in the generation of mitochondrial oxidative stress (mtROS), which causes a decrease in the activity of aconitase (ACO2) in the Krebs cycle. As a consequence, the ACO2 substrate, citrate, accumulates and is released to the cytosol where it acts as substrate for TG synthesis through the activation of ATP citrate lyase (ACL) and fatty acid synthase. AMPD2, AMP deaminase 2; IMP, inosine monophosphate; PO4, phosphate.
Fructose-induced hyperuricemia, insulin resistance, and diabetes
The observation that inhibition of uric acid synthesis prevented metabolic syndrome and hepatic steatosis leads to the question of how uric acid might contribute to insulin resistance and diabetes.
Hepatic effects.
The observation that uric acid can induce mitochondrial oxidative stress and fatty liver may explain how fructose induces insulin resistance. Mitochondrial oxidative stress has a role in driving insulin resistance (42). In turn, the development of fatty liver is also linked with insulin resistance (43).
Effects in the white adipose tissue.
Uric acid may also induce insulin resistance via effects on adipocytes. Uric acid is taken up in adipocytes by an organic anion transporter where it induces oxidative stress via activation of NADPH oxidase, generating oxidized lipids and inflammatory mediators such as monocyte chemoattractant protein-1 (MCP-1) (29,44). Adiponectin synthesis is also inhibited (44). In the hyperuricemic Pound mouse, the inhibition of uric acid synthesis by allopurinol attenuates the local inflammatory response in the visceral fat, reduces the expression of inflammatory cytokines, and enhances circulating levels of adiponectin in association with an improvement in insulin resistance (44). Likewise, the reduction of uric acid by either allopurinol or benzbromarone in the fructose-fed rat results in less insulin resistance and decreases the leptin overexpression that occurs in the visceral fat (4,45).
Vascular effects.
Fructose may also induce insulin resistance via effects on the vasculature. One of the major effects of insulin is to stimulate the release of NO from endothelial cells, where it causes vasodilation that aids delivery of glucose to the skeletal muscle. Mice that cannot generate endothelial NO develop features of metabolic syndrome and insulin resistance (46). In this regard, uric acid inhibits endothelial NO generation, including in response to insulin (32). Uric acid reduces endothelial NO via several mechanisms, including blocking the uptake of the substrate, l-arginine (47), stimulating the degradation of l-arginine by arginase (48), and scavenging NO by uric acid or by uric acid–generated oxidants (32,34,49). Hyperuricemic rats have impaired endothelial function and hypertension that can be reversed by lowering uric acid or treating with l-arginine or antioxidants (50–52). Hyperuricemia is also associated with endothelial dysfunction in humans, and lowering uric acid with allopurinol improves endothelial dysfunction in asymptomatic hyperuricemia, congestive heart failure, diabetes, chronic kidney disease, obstructive sleep apnea, and with smoking (rev. in 53).
Islet cell effects.
Chronic administration of fructose or sucrose to animals not only causes insulin resistance but may also result in type 2 diabetes (5,54). Histologically, the islets show hyalinosis and macrophage infiltration, similar to what is observed in humans with type 2 diabetes. The mechanism by which fructose induces these changes is not known because the islet does not express GLUT5, which is the primary fructose transporter. However, we reported an upregulation of the urate transporter URAT-1 in islet cells of sucrose-fed rats in association with increased expression of MCP-1 (5). Incubation of cultured insulin-secreting islet cells with uric acid also causes oxidative stress and synthesis of MCP-1 (5). Oxidative stress in islets is considered to have a major role in causing the islet dysfunction of type 2 diabetes.
Evidence that fructose mediates fatty liver and insulin resistance in humans
The major source of fructose in the Western diet is from soft drinks and fruit drinks, and this accounts for approximately 7% of caloric intake in the adult, and upward to 15% of total caloric intake in adolescents. Intake of sugar and soft drinks are higher in populations at increased risk for insulin resistance and diabetes, including the African Americans, Hispanics, Native Americans, and subjects with lower income. A meta-analysis concluded that the intake of sugary soft drinks is an independent predictor for the development of metabolic syndrome and/or diabetes (55). Genetic factors enhance the risk for developing diabetes from soft drinks (56).
Clinical studies have documented the metabolic effects of fructose. Studies from the 1960s through the 1980s showed that sucrose, or fructose, can worsen hypertriglyceridemia and insulin resistance, especially if subjects were hyperinsulinemic (57,58). More recently Stanhope et al. (59) fed 25% of diet as fructose or glucose to overweight individuals for 10 weeks. Although some features of metabolic syndrome were induced with glucose, the fructose-fed subjects showed worse postprandial hypertriglyceridemia, increased hepatic de novo synthesis of fatty acids, a decrease in insulin sensitivity (noted by elevations in fasting glucose and insulin levels), increased total and visceral fat (among men), higher 24-h uric acid levels, increased systemic inflammatory mediators (MCP-1), and lower resting energy expenditure (12,59,60). In another study, Maersk et al. (61) randomized overweight adults to drink 1 L of a sugary soft drink daily for 6 months, with control subjects receiving equivalent amount of diet soft drink, milk, or water. At the end of 6 months, the subjects receiving the sugary soft drinks displayed more visceral, skeletal muscle, and liver fat and higher serum triglycerides and cholesterol compared with the group drinking milk, with a trend toward significance in the other two groups. Tappy and colleagues (62) have also shown the ability of fructose to induce insulin resistance, hepatic lipid accumulation, and hypertriglyceridemia. Similarly, our group administered 200 g fructose to overweight men for 2 weeks and documented higher blood pressure, higher triglycerides, and lower HDL cholesterol compared with baseline, with 25% of the subjects developing de novo metabolic syndrome at 2 weeks (63). Another study showed that the administration of one 8-oz soft drink per day to adolescents results in increased body weight at 18 months compared with subjects given diet soft drinks (64).
Intervention studies have also been performed to evaluate the effect of reducing sugar intake on metabolic syndrome. For example, the Atkins diet and other low carbohydrate diets tend to improve features of the metabolic syndrome more than typical low fat diets (65). A randomized study in school children reported that reducing soft drink intake, resulting in a difference of 175 mL/day between treatment and control subjects, led to a reduction in overweight or obesity by 0.2% in the treated group compared with a 7.5% increase in the control subjects at 12 months (66). A study in California showed that the banning of soft drinks in schools resulted in a reduction in overall soft drink intake with a decrease in obesity in children 6 to 11 years of age (67). Less effect was observed in older children, possibly because the overall reduction in soft drink intake in this latter group was less effective (67). Soft drink intake in the U.S. has decreased since peaking in 1999, and this is also associated with a leveling of the rates of obesity.
Role of uric acid in insulin resistance and fatty liver in humans
As mentioned, fructose increases intracellular and circulating uric acid levels due to increased nucleotide turnover and nucleotide synthesis. Initially the rise in serum uric acid is best shown shortly (30–60 min) after fructose ingestion (or ingestion of HFCS or sucrose), but total 24-h levels are also elevated (60,68). Over time, fasting serum uric acid levels increase (58). Intake of soft drinks is also associated with increasing risk for hyperuricemia (69).
An elevated serum uric acid is also one of the best independent predictors of diabetes and commonly precedes the development of both insulin resistance and diabetes (Table 1). An elevated uric acid also independently predicts the development of fatty liver (41), obesity (70), hypertension (rev. in 71), and elevations in C-reactive protein (72). Furthermore, metabolic syndrome is associated with a high frequency of hyperuricemia, and similarly, hyperuricemia is associated with metabolic syndrome (73,74). Though hyperinsulinemia may contribute to hyperuricemia by blocking uric acid excretion, it cannot be the primary reason for the association because hyperuricemia has been reported to precede the development of hyperinsulinemia and/or diabetes (Table 1).
TABLE 1.
Serum uric acid predicts the development of diabetes
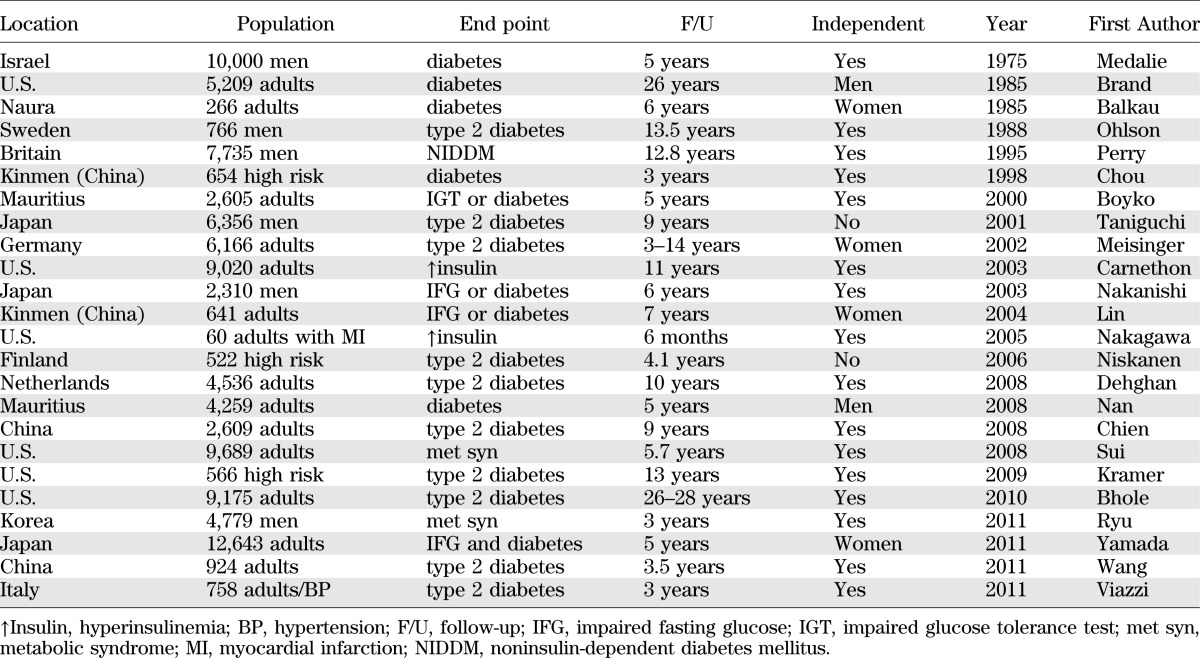
A number of conditions associated with hyperuricemia are also associated with increased risk for insulin resistance or diabetes, including chronic lead intoxication and gestational diabetes mellitus. Many drugs associated with insulin resistance are also associated with hyperuricemia, such as calcineurin inhibitors and thiazide diuretics. Indeed, lowering uric acid improves the insulin resistance induced by thiazides in rats (75).
Evidence that lowering uric acid can improve insulin resistance in humans is limited. One small study showed that lowering uric acid with benzbromarone improves insulin resistance in subjects with congestive heart failure (76). Another study reported that lowering uric acid improves HbA1c levels in normotensive diabetic subjects (77). In contrast, we were not able to show an improvement of insulin resistance with allopurinol in subjects administered fructose (63), but the doses of fructose were exceptionally high (200 g/day) raising the possibility that the doses of allopurinol we used might not have been able to block intracellular uric acid. Clearly, more studies are indicated before any definitive conclusions can be made with regards to the benefit of lowering uric acid for the treatment of insulin resistance.
Problems with the fructose and uric acid hypothesis
Concerns with animal studies.
The fructose-induced hyperuricemia hypothesis has been challenged. First, animal studies using fructose typically use pure fructose as opposed to sucrose or HFCS, which is the primary source of fructose in humans, and the dose of fructose administered to rodents is usually higher (50–60% of the diet) compared with humans (where it is typically 10–15% of the diet). Purified fructose is used, however, so one can separate the effects of fructose from glucose. Indeed, animals are more sensitive to the combination of fructose and glucose because both sugars accelerate the absorption of the other (78). Combinations of free fructose and glucose, or sucrose, induce features of metabolic syndrome with levels of fructose of 20–30% dietary intake (5,79).
Furthermore, rodents are relatively resistant to fructose in part because they generate less uric acid in response to fructose due to the presence of the uricase gene in their liver (38). Uricase degrades uric acid to allantoin, and as a consequence, rats degrade uric acid rapidly after it is formed in their liver. When uricase is inhibited, rats show a greater metabolic response to fructose with worse fatty liver and higher blood pressure (79). Indeed, there is evidence that the loss of uricase may have provided a survival advantage to ancestral apes living in Europe in the mid-Miocene and therefore may have acted like a thrifty gene (80). The subsequent rise in sugar intake over the last centuries may have acted in concert with the loss of uricase to predispose us to obesity and diabetes (80).
Clinical studies: the importance of the control group.
Recently, a number of investigators have presented meta-analyses that suggest fructose does not have a causal relationship with obesity or metabolic syndrome (81–83). Before we analyze these studies, it is important to understand the complexity related to their interpretation. First, many clinical studies use fructose alone—and often at relatively high doses—in order to evaluate the effects of fructose per se. This allows one to directly address the effects of fructose, and the use of high doses is a common experimental approach to allow one to identify metabolic effects that could otherwise take much longer periods to show. Indeed, the fact that metabolic syndrome could be induced de novo in 25% of healthy men with high doses of fructose in just 2 weeks is a statement of how strong this approach can be (63). Although studies involving HFCS or sucrose might be clinically more relevant, these types of studies will have trouble distinguishing whether the metabolic effects observed are from the fructose or the high glycemic content of these added sugars.
Nevertheless, the administration of fructose alone can be very difficult to interpret because the absorption of fructose when given alone is quite variable. As many as two-thirds of children and one-third of adults malabsorb fructose (84,85). This is likely because of variable expression of the fructose transporter GLUT5 in the gut. Expression of GLUT5 and the enzyme KHK, however, are enhanced with repeated exposure to fructose. It is interesting that studies in children have found an inverse relationship between fructose malabsorption and obesity (86). Consistent with this data, the metabolic response to fructose in children with NAFLD is greater compared with lean control subjects (87). The importance of fructose absorption has recently been highlighted in African Americans because they commonly malabsorb fructose and also have a lower frequency of NAFLD (88). The observation that NAFLD subjects may absorb fructose more efficiently is further supported by our observation of higher KHK expression in liver biopsies of NAFLD compared with other liver disease (36) and could be the reason why ATP depletion in response to fructose is greater in NAFLD subjects with a higher prior exposure to dietary fructose (37). Our observation that hyperuricemia may regulate KHK (35) also provides an explanation for why studies in which fructose is given to young athletic lean individuals are often negative and why they may not carry over to older and heavier individuals.
Another key issue is whether studies evaluating fructose should include fructose from natural fruits. One can argue that fructose is fructose regardless of source, but natural fruits also contain numerous substances that block fructose effects, including potassium, vitamin C, and antioxidants such as resveratrol, quercetin, and other flavonols. We found, for example, that whereas fructose from added sugars is associated with hypertension, fructose from natural fruits is not (89). We further showed that caloric restriction involving a reduction in fructose intake from added sugars could markedly improve metabolic syndrome in obese Mexican adults, and that this occurred even if natural fruits were administered (90).
Another important issue is whether glucose itself is the right control for fructose. Outwardly it would seem true, but we recently discovered that glucose may act to induce obesity and insulin resistance by being converted to fructose in the liver (91). Specifically, high concentrations of glucose, such as occurs in soft drinks, can induce the activation of the polyol pathway in the liver, resulting in the generation of fructose. In turn, the fructose is then metabolized by KHK, resulting in fructose-dependent effects. Indeed, glucose-induced weight gain, fat accumulation, fatty liver, and insulin resistance are all dependent on KHK. While some visceral fat and weight gain occur in glucose-fed mice lacking KHK, the development of fatty liver and hyperinsulinemia are almost entirely dependent on glucose-induced fructose metabolism (91). Hence, although fructose itself will have more metabolic effects than glucose, the glucose itself may also be inducing metabolic changes via fructose.
Meta-analyses that argue fructose is not a risk factor for metabolic syndrome
Weight gain.
A meta-analysis recently reported that fructose intake does not cause weight gain compared with other sugars in short-term studies if both groups are given the same number of total calories (isocaloric diets) (81). However, no food will cause weight gain under these conditions, as weight gain is driven primarily by increased energy intake as opposed to a reduction in metabolic rate, at least in the short-term. Indeed, the mechanism by which fructose increases weight is likely via its ability to stimulate hunger and block satiety responses (7,9), so if food intake is controlled this would not be observed.
Blood pressure.
It is a scientific fact that the administration of clinically relevant doses (60 g) of fructose acutely raises blood pressure in humans (92), and similar increases in blood pressure have been observed following ingestion of 24 ounces of HFCS or sucrose-containing beverages (68). It has also been reported that high doses of fructose raises 24-h ambulatory blood pressure in humans and can be blocked by lowering uric acid with allopurinol (63). However, the recent meta-analysis by Ha et al. (82) addressed whether short-term isocaloric fructose diets can increase blood pressure after an overnight fast. Since the acute effects of fructose to raise blood pressure occur during the ingestion of fructose (and are likely mediated by uric acid), it is not surprising that the authors did not show an effect on blood pressure; indeed, a similarly designed study would conclude that glucose-rich diets do not increase insulin levels.
An important question is whether chronic fructose ingestion may be responsible for persistent elevations in blood pressure. It is known that the greatest risk for persistent hypertension is borderline hypertension in which intermittent blood pressure elevations occur. There is also evidence that fructose causes microvascular disease in the kidney, which is known to predispose to persistent salt-sensitive hypertension. Indeed, persistent hypertension can be induced with fructose and high-salt diet in rats. Furthermore, chronic fructose ingestion over time is associated with elevations in fasting uric acid levels (58,93), in part because fructose also stimulates uric acid synthesis. Epidemiological studies have also linked fructose intake with hypertension and elevated serum uric acid levels (94). Reduction in sugar intake is also strongly associated with a reduction in blood pressure (95). Indeed, the DASH (Dietary Approaches to Stop Hypertension) diet is in essence a diet low in fructose from added sugars (while containing natural fruits, see above).
Uric acid.
Wang et al. (83) also reported that short-term isocaloric trials do not show an effect of fructose on fasting uric acid levels. Again, the design of the study would not be expected to show a rise in uric acid because the initial rise in uric acid is transient and occurs within minutes of the ingestion of fructose. However, as mentioned, there is some evidence that over time continued ingestion of fructose will result in chronic elevations of uric acid. A more detailed discussion is provided elsewhere.
Another issue with all three metanalyses is that they included control groups that ingested sucrose, which can be questioned because sucrose is a disaccharide that contains fructose (81–83).
Other issues related to uric acid.
Other aspects of the uric acid studies have also been questioned. One paradox is that the acute elevation of serum uric acid by infusion often results in an improvement in endothelial function (96). However, while uric acid is an antioxidant in the extracellular environment, it has prooxidative effects inside the cell (28,29). Several investigators have also suggested that it is not uric acid that is driving metabolic syndrome, but rather xanthine oxidase, since this enzyme generates oxidants in addition to uric acid, and it may be the former that is responsible for the metabolic syndrome. For example, high-dose allopurinol improves endothelial dysfunction in subjects with heart failure whereas the lowering of uric acid with probenecid was ineffective (97). However, this could simply relate to the relative superiority of allopurinol to lower intracellular uric acid as it blocks synthesis. Although xanthine oxidase–induced oxidants could be important, the observation that raising intracellular uric acid, even in the presence of allopurinol, can increase hepatic fat suggests that it is the uric acid that is responsible (35).
Genetic studies.
A final argument relates to the genetics of uric acid and fructose metabolism. While some genetic polymorphisms in various enzymes involved in fructose metabolism and urate transport have been linked with metabolic syndrome and hypertension (98–100), several genome-wide association studies (GWAS) could not show such associations (101,102). However, the primary polymorphism driving serum uric acid in the GWAS studies is SLC2A9; this polymorphism mediates the transport of uric acid out of tubular cells, and so it may not predict the development of diabetes because it is likely to dissociate the serum from intracellular uric acid levels, the latter of which may be more important in driving insulin resistance.
Conclusions
Searching for the cause of type 2 diabetes has been a prime area of research since Etienne Lancereaux described fat diabetes (diabetes gras) in 1880. Though more studies are needed, the evidence that fructose-induced hyperuricemia may have a contributory role is gaining ground. While it still remains a hypothesis (103), increasing evidence suggests uric acid may have a fundamental role in the manifestations of metabolic syndrome (Fig. 3). Given that hyperuricemia is a remediable risk factor, we recommend both basic and clinical studies to address this important possibility.
FIG. 3.

Uric acid: potential mechanisms for insulin resistance and diabetes. Uric acid may contribute to insulin resistance in the liver by inducing mitochondrial oxidative stress and steatosis (28). Uric acid also blocks the ability of insulin to stimulate vasodilation of blood vessels, which is important for the delivery of glucose to the skeletal muscle (4,32). Uric acid also induces local inflammation in the adipose tissue with a reduction in the production of adiponectin (44). Finally, uric acid may also have direct effects on the islet cells leading to local oxidative stress and islet dysfunction (5). Mt, mitochondria; PO4, phosphate.
ACKNOWLEDGMENTS
Support for this study was provided by ADA Basic Science Award 7-12-BS-16 to Y.Y.S.
R.J.J. has patent applications related to lowering uric acid as a means to prevent or treat insulin resistance and features of metabolic syndrome; holds stock in XORT Pharma Corp.; and is on the Scientific Board of Amway. T.N. has patent applications related to lowering uric acid as a means to prevent or treat insulin resistance and features of metabolic syndrome and holds stock in XORT Pharma Corp. No other potential conflicts of interest relevant to this article were reported.
R.J.J. wrote the first draft of the manuscript. T.N., L.G.S.-L., M.S., S.S., M.L., T.I., Y.Y.S., and M.A.L. improved the manuscript with their comments and suggestions. In addition, all of the authors have made scientific contributions that provided the groundwork for this Perspective.
REFERENCES
- 1.Johnson RJ, Perez-Pozo SE, Sautin YY, et al. Hypothesis: could excessive fructose intake and uric acid cause type 2 diabetes? Endocr Rev 2009;30:96–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sánchez-Lozada LG, Mu W, Roncal C, et al. Comparison of free fructose and glucose to sucrose in the ability to cause fatty liver. Eur J Nutr 2010;49:1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bocarsly ME, Powell ES, Avena NM, Hoebel BG. High-fructose corn syrup causes characteristics of obesity in rats: increased body weight, body fat and triglyceride levels. Pharmacol Biochem Behav 2010;97:101–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nakagawa T, Hu H, Zharikov S, et al. A causal role for uric acid in fructose-induced metabolic syndrome. Am J Physiol Renal Physiol 2006;290:F625–F631 [DOI] [PubMed] [Google Scholar]
- 5.Roncal-Jimenez CA, Lanaspa MA, Rivard CJ, et al. Sucrose induces fatty liver and pancreatic inflammation in male breeder rats independent of excess energy intake. Metabolism 2011;60:1259–1270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teff KL, Elliott SS, Tschöp M, et al. Dietary fructose reduces circulating insulin and leptin, attenuates postprandial suppression of ghrelin, and increases triglycerides in women. J Clin Endocrinol Metab 2004;89:2963–2972 [DOI] [PubMed] [Google Scholar]
- 7.Shapiro A, Mu W, Roncal C, Cheng KY, Johnson RJ, Scarpace PJ. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am J Physiol Regul Integr Comp Physiol 2008;295:R1370–R1375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bernal SY, Dostova I, Kest A, et al. Role of dopamine D1 and D2 receptors in the nucleus accumbens shell on the acquisition and expression of fructose-conditioned flavor-flavor preferences in rats. Behav Brain Res 2008;190:59–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lane MD, Cha SH. Effect of glucose and fructose on food intake via malonyl-CoA signaling in the brain. Biochem Biophys Res Commun 2009;382:1–5 [DOI] [PubMed] [Google Scholar]
- 10.Friedman MI, Harris RB, Ji H, Ramirez I, Tordoff MG. Fatty acid oxidation affects food intake by altering hepatic energy status. Am J Physiol 1999;276:R1046–R1053 [DOI] [PubMed] [Google Scholar]
- 11.Bawden S.J, Stephenson MC, Marciani L, Aithal GP, Macdonald IA, Gowland P, Morris PA. Investigating alterations in hepatic atp levels following fructose and fructose+glucose ingestion: a simple non-invasive technique to assess liver function using 31P MRS. Proc Intl Soc Magn Reson Med Sci Meet Exhib 2012;20:1369 [Google Scholar]
- 12.Cox CL, Stanhope KL, Schwarz JM, et al. Consumption of fructose-sweetened beverages for 10 weeks reduces net fat oxidation and energy expenditure in overweight/obese men and women. Eur J Clin Nutr 2012;66:201–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Basu S, Yoffe P, Hills N, Lustig RH. The relationship of sugar to population-level diabetes prevalence: an econometric analysis of repeated cross-sectional data. PLoS ONE 2013;8:e57873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mäenpää PH, Raivio KO, Kekomäki MP. Liver adenine nucleotides: fructose-induced depletion and its effect on protein synthesis. Science 1968;161:1253–1254 [DOI] [PubMed] [Google Scholar]
- 15.van den Berghe G, Bronfman M, Vanneste R, Hers HG. The mechanism of adenosine triphosphate depletion in the liver after a load of fructose. A kinetic study of liver adenylate deaminase. Biochem J 1977;162:601–609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bode JC, Zelder O, Rumpelt HJ, Wittkamp U. Depletion of liver adenosine phosphates and metabolic effects of intravenous infusion of fructose or sorbitol in man and in the rat. Eur J Clin Invest 1973;3:436–441 [DOI] [PubMed] [Google Scholar]
- 17.Emmerson BT. Effect of oral fructose on urate production. Ann Rheum Dis 1974;33:276–280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ishimoto T, Lanaspa MA, Le MT, et al. Opposing effects of fructokinase C and A isoforms on fructose-induced metabolic syndrome in mice. Proc Natl Acad Sci USA 2012;109:4320–4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lanaspa MA, Sanchez-Lozada LG, Choi YJ, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem 2012;287:40732–40744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steinmann B, Gitzelmann R, Van den Berghe G. Disorders of Fructose Metabolism. In The Metabolic and Molecular Basis of Inherited Disease. Scriver C, Beaudet A, Sly W, Valle D, Eds. New York, McGraw-Hill, 2001, p. 1489–1520 [Google Scholar]
- 21.Odièvre M, Gentil C, Gautier M, Alagille D. Hereditary fructose intolerance in childhood. Diagnosis, management, and course in 55 patients. Am J Dis Child 1978;132:605–608 [DOI] [PubMed] [Google Scholar]
- 22.Bandsma RH, Smit GP, Kuipers F. Disturbed lipid metabolism in glycogen storage disease type 1. Eur J Pediatr 2002;161(Suppl. 1):S65–S69 [DOI] [PubMed] [Google Scholar]
- 23.Greene HL, Wilson FA, Hefferan P, et al. ATP depletion, a possible role in the pathogenesis of hyperuricemia in glycogen storage disease type I. J Clin Invest 1978;62:321–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mundy HR, Lee PJ. Glycogenosis type I and diabetes mellitus: a common mechanism for renal dysfunction? Med Hypotheses 2002;59:110–114 [DOI] [PubMed] [Google Scholar]
- 25.Spiegel R, Rakover-Tenenbaum Y, Mandel H, Lumelski D, Admoni O, Horovitz Y. Secondary diabetes mellitus: late complication of glycogen storage disease type 1b. J Pediatr Endocrinol Metab 2005;18:617–619 [DOI] [PubMed] [Google Scholar]
- 26.Ouyang J, Parakhia RA, Ochs RS. Metformin activates AMP kinase through inhibition of AMP deaminase. J Biol Chem 2011;286:1–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lanaspa MA, Cicerchi C, Garcia G, et al. Counteracting roles of AMP deaminase and AMP kinase in the development of fatty liver. PLoS ONE 2012;7:e48801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lanaspa MA, Sanchez-Lozada LG, Choi YJ, et al. Uric acid induces hepatic steatosis by generation of mitochondrial oxidative stress: potential role in fructose-dependent and -independent fatty liver. J Biol Chem 2012;287:40732–40744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sautin YY, Nakagawa T, Zharikov S, Johnson RJ. Adverse effects of the classic antioxidant uric acid in adipocytes: NADPH oxidase-mediated oxidative/nitrosative stress. Am J Physiol Cell Physiol 2007;293:C584–C596 [DOI] [PubMed] [Google Scholar]
- 30.Yu MA, Sánchez-Lozada LG, Johnson RJ, Kang DH. Oxidative stress with an activation of the renin-angiotensin system in human vascular endothelial cells as a novel mechanism of uric acid-induced endothelial dysfunction. J Hypertens 2010;28:1234–1242 [PubMed] [Google Scholar]
- 31.Corry DB, Eslami P, Yamamoto K, Nyby MD, Makino H, Tuck ML. Uric acid stimulates vascular smooth muscle cell proliferation and oxidative stress via the vascular renin-angiotensin system. J Hypertens 2008;26:269–275 [DOI] [PubMed] [Google Scholar]
- 32.Sanchez-Lozada LG, Lanaspa-Garcia MA, Cristobal M, et al. Uric acid-induced endothelial dysfunction is associated with mitochondrial alterations and decreased intracellular ATP concentrations. Nephron Exp Nephrol 2012;121:e71–e78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imaram W, Gersch C, Kim KM, Johnson RJ, Henderson GN, Angerhofer A. Radicals in the reaction between peroxynitrite and uric acid identified by electron spin resonance spectroscopy and liquid chromatography mass spectrometry. Free Radic Biol Med 2010;49:275–281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gersch C, Palii SP, Kim KM, Angerhofer A, Johnson RJ, Henderson GN. Inactivation of nitric oxide by uric acid. Nucleosides Nucleotides Nucleic Acids 2008;27:967–978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lanaspa MA, Sanchez-Lozada LG, Cicerchi C, et al. Uric acid stimulates fructokinase and accelerates fructose metabolism in the development of fatty liver. PLoS ONE 2012;7:e47948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ouyang X, Cirillo P, Sautin Y, et al. Fructose consumption as a risk factor for non-alcoholic fatty liver disease. J Hepatol 2008;48:993–999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abdelmalek MF, Lazo M, Horska A, et al. Fatty Liver Subgroup of Look AHEAD Research Group Higher dietary fructose is associated with impaired hepatic adenosine triphosphate homeostasis in obese individuals with type 2 diabetes. Hepatology 2012;56:952–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stavric B, Johnson WJ, Clayman S, Gadd RE, Chartrand A. Effect of fructose administration on serum urate levels in the uricase inhibited rat. Experientia 1976;32:373–374 [DOI] [PubMed] [Google Scholar]
- 39.Xu CF, Yu CH, Xu L, Sa XY, Li YM. Hypouricemic therapy: a novel potential therapeutic option for nonalcoholic fatty liver disease. Hepatology 2010;52:1865–1866 [DOI] [PubMed] [Google Scholar]
- 40.Kono H, Rusyn I, Bradford BU, Connor HD, Mason RP, Thurman RG. Allopurinol prevents early alcohol-induced liver injury in rats. J Pharmacol Exp Ther 2000;293:296–303 [PubMed] [Google Scholar]
- 41.Ryu S, Chang Y, Kim SG, Cho J, Guallar E. Serum uric acid levels predict incident nonalcoholic fatty liver disease in healthy Korean men. Metabolism 2011;60:860–866 [DOI] [PubMed] [Google Scholar]
- 42.Hoehn KL, Salmon AB, Hohnen-Behrens C, et al. Insulin resistance is a cellular antioxidant defense mechanism. Proc Natl Acad Sci USA 2009;106:17787–17792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Samuel VT, Petersen KF, Shulman GI. Lipid-induced insulin resistance: unravelling the mechanism. Lancet 2010;375:2267–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baldwin W, McRae S, Marek G, et al. Hyperuricemia as a mediator of the proinflammatory endocrine imbalance in the adipose tissue in a murine model of the metabolic syndrome. Diabetes 2011;60:1258–1269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lanaspa M, Sautin Y, Ejaz A, et al. Uric acid and metabolic syndrome: what is the relationship? Curr Rheum Rev 2011;7:162–169 [Google Scholar]
- 46.Duplain H, Burcelin R, Sartori C, et al. Insulin resistance, hyperlipidemia, and hypertension in mice lacking endothelial nitric oxide synthase. Circulation 2001;104:342–345 [DOI] [PubMed] [Google Scholar]
- 47.Schwartz IF, Grupper A, Chernichovski T, et al. Hyperuricemia attenuates aortic nitric oxide generation, through inhibition of arginine transport, in rats. J Vasc Res 2011;48:252–260 [DOI] [PubMed] [Google Scholar]
- 48.Zharikov S, Krotova K, Hu H, et al. Uric acid decreases NO production and increases arginase activity in cultured pulmonary artery endothelial cells. Am J Physiol Cell Physiol 2008;295:C1183–C1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang DH, Park SK, Lee IK, Johnson RJ. Uric acid-induced C-reactive protein expression: implication on cell proliferation and nitric oxide production of human vascular cells. J Am Soc Nephrol 2005;16:3553–3562 [DOI] [PubMed] [Google Scholar]
- 50.Khosla UM, Zharikov S, Finch JL, et al. Hyperuricemia induces endothelial dysfunction. Kidney Int 2005;67:1739–1742 [DOI] [PubMed] [Google Scholar]
- 51.Sánchez-Lozada LG, Soto V, Tapia E, et al. Role of oxidative stress in the renal abnormalities induced by experimental hyperuricemia. Am J Physiol Renal Physiol 2008;295:F1134–F1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sánchez-Lozada LG, Tapia E, López-Molina R, et al. Effects of acute and chronic L-arginine treatment in experimental hyperuricemia. Am J Physiol Renal Physiol 2007;292:F1238–F1244 [DOI] [PubMed] [Google Scholar]
- 53.Kanbay M, Segal M, Afsar B, Kang DH, Rodriguez-Iturbe B, Johnson RJ. The role of uric acid in the pathogenesis of human cardiovascular disease. Heart 2013;99:759–766 [DOI] [PubMed] [Google Scholar]
- 54.Cummings BP, Stanhope KL, Graham JL, et al. Dietary fructose accelerates the development of diabetes in UCD-T2DM rats: amelioration by the antioxidant, alpha-lipoic acid. Am J Physiol Regul Integr Comp Physiol 2010;298:R1343–R1350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Malik VS, Popkin BM, Bray GA, Després JP, Willett WC, Hu FB. Sugar-sweetened beverages and risk of metabolic syndrome and type 2 diabetes: a meta-analysis. Diabetes Care 2010;33:2477–2483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Qi Q, Chu AY, Kang JH, et al. Sugar-sweetened beverages and genetic risk of obesity. N Engl J Med 2012;367:1387–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hallfrisch J, Reiser S, Prather ES. Blood lipid distribution of hyperinsulinemic men consuming three levels of fructose. Am J Clin Nutr 1983;37:740–748 [DOI] [PubMed] [Google Scholar]
- 58.Reiser S, Powell AS, Scholfield DJ, Panda P, Ellwood KC, Canary JJ. Blood lipids, lipoproteins, apoproteins, and uric acid in men fed diets containing fructose or high-amylose cornstarch. Am J Clin Nutr 1989;49:832–839 [DOI] [PubMed] [Google Scholar]
- 59.Stanhope KL, Schwarz JM, Keim NL, et al. Consuming fructose-sweetened, not glucose-sweetened, beverages increases visceral adiposity and lipids and decreases insulin sensitivity in overweight/obese humans. J Clin Invest 2009;119:1322–1334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cox CL, Stanhope KL, Schwarz JM, et al. Consumption of fructose- but not glucose-sweetened beverages for 10 weeks increases circulating concentrations of uric acid, retinol binding protein-4, and gamma-glutamyl transferase activity in overweight/obese humans. Nutr Metab (Lond) 2012;9:68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Maersk M, Belza A, Stødkilde-Jørgensen H, et al. Sucrose-sweetened beverages increase fat storage in the liver, muscle, and visceral fat depot: a 6-mo randomized intervention study. Am J Clin Nutr 2012;95:283–289 [DOI] [PubMed] [Google Scholar]
- 62.Lê KA, Ith M, Kreis R, et al. Fructose overconsumption causes dyslipidemia and ectopic lipid deposition in healthy subjects with and without a family history of type 2 diabetes. Am J Clin Nutr 2009;89:1760–1765 [DOI] [PubMed] [Google Scholar]
- 63.Perez-Pozo SE, Schold J, Nakagawa T, Sánchez-Lozada LG, Johnson RJ, Lillo JL. Excessive fructose intake induces the features of metabolic syndrome in healthy adult men: role of uric acid in the hypertensive response. Int J Obes (Lond) 2010;34:454–461 [DOI] [PubMed] [Google Scholar]
- 64.de Ruyter JC, Olthof MR, Seidell JC, Katan MB. A trial of sugar-free or sugar-sweetened beverages and body weight in children. N Engl J Med 2012;367:1397–1406 [DOI] [PubMed] [Google Scholar]
- 65.Gardner CD, Kiazand A, Alhassan S, et al. Comparison of the Atkins, Zone, Ornish, and LEARN diets for change in weight and related risk factors among overweight premenopausal women: the A TO Z Weight Loss Study: a randomized trial. JAMA 2007;297:969–977 [DOI] [PubMed] [Google Scholar]
- 66.James J, Thomas P, Cavan D, Kerr D. Preventing childhood obesity by reducing consumption of carbonated drinks: cluster randomised controlled trial. BMJ 2004;328:1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shi L, van Meijgaard J. Substantial decline in sugar-sweetened beverage consumption among California’s children and adolescents. Int J Gen Med 2010;3:221–224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Le MT, Frye RF, Rivard CJ, et al. Effects of high-fructose corn syrup and sucrose on the pharmacokinetics of fructose and acute metabolic and hemodynamic responses in healthy subjects. Metabolism 2012;61:641–651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Choi JW, Ford ES, Gao X, Choi HK. Sugar-sweetened soft drinks, diet soft drinks, and serum uric acid level: the Third National Health and Nutrition Examination Survey. Arthritis Rheum 2008;59:109–116 [DOI] [PubMed] [Google Scholar]
- 70.Masuo K, Kawaguchi H, Mikami H, Ogihara T, Tuck ML. Serum uric acid and plasma norepinephrine concentrations predict subsequent weight gain and blood pressure elevation. Hypertension 2003;42:474–480 [DOI] [PubMed] [Google Scholar]
- 71.Feig DI, Madero M, Jalal DI, Sanchez-Lozada LG, Johnson RJ. Uric acid and the origins of hypertension. J Pediatr 2013;162:896–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ruggiero C, Cherubini A, Miller E, 3rd, et al. Usefulness of uric acid to predict changes in C-reactive protein and interleukin-6 in 3-year period in Italians aged 21 to 98 years. Am J Cardiol 2007;100:115–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Choi HK, Ford ES. Prevalence of the metabolic syndrome in individuals with hyperuricemia. Am J Med 2007;120:442–447 [DOI] [PubMed] [Google Scholar]
- 74.Facchini F, Chen YD, Hollenbeck CB, Reaven GM. Relationship between resistance to insulin-mediated glucose uptake, urinary uric acid clearance, and plasma uric acid concentration. JAMA 1991;266:3008–3011 [PubMed] [Google Scholar]
- 75.Reungjui S, Roncal CA, Mu W, et al. Thiazide diuretics exacerbate fructose-induced metabolic syndrome. J Am Soc Nephrol 2007;18:2724–2731 [DOI] [PubMed] [Google Scholar]
- 76.Ogino K, Kato M, Furuse Y, et al. Uric acid-lowering treatment with benzbromarone in patients with heart failure: a double-blind placebo-controlled crossover preliminary study. Circ Heart Fail 2010;3:73–81 [DOI] [PubMed] [Google Scholar]
- 77.Dogan A, Yarlioglues M, Kaya MG, et al. Effect of long-term and high-dose allopurinol therapy on endothelial function in normotensive diabetic patients. Blood Press 2011;20:182–187 [DOI] [PubMed] [Google Scholar]
- 78.Truswell AS, Seach JM, Thorburn AW. Incomplete absorption of pure fructose in healthy subjects and the facilitating effect of glucose. Am J Clin Nutr 1988;48:1424–1430 [DOI] [PubMed] [Google Scholar]
- 79.Tapia E, Cristóbal M, García-Arroyo FE, et al. Synergistic effect of uricase blockade plus physiological amounts of fructose-glucose on glomerular hypertension and oxidative stress in rats. Am J Physiol Renal Physiol 2013;304:F727–F736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Johnson RJ, Andrews P. Fructose, uricase, and the back-to-Africa Hypothesis. Evol Anthropol 2010;19:250–257 [Google Scholar]
- 81.Sievenpiper JL, de Souza RJ, Mirrahimi A, et al. Effect of fructose on body weight in controlled feeding trials: a systematic review and meta-analysis. Ann Intern Med 2012;156:291–304 [DOI] [PubMed] [Google Scholar]
- 82.Ha V, Sievenpiper JL, de Souza RJ, et al. Effect of fructose on blood pressure: a systematic review and meta-analysis of controlled feeding trials. Hypertension 2012;59:787–795 [DOI] [PubMed] [Google Scholar]
- 83.Wang DD, Sievenpiper JL, de Souza RJ, et al. The effects of fructose intake on serum uric acid vary among controlled dietary trials. J Nutr 2012;142:916–923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ravich WJ, Bayless TM, Thomas M. Fructose: incomplete intestinal absorption in humans. Gastroenterology 1983;84:26–29 [PubMed] [Google Scholar]
- 85.Kneepkens CM, Vonk RJ, Fernandes J. Incomplete intestinal absorption of fructose. Arch Dis Child 1984;59:735–738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Disse S, Buelow A, Boedeker RH, et al. Reduced prevalence of obesity in children with primary fructose malabsorption: a multicenter, retrospective cohort study. Pediatr Obes; 2013;8:255–258 [DOI] [PubMed] [Google Scholar]
- 87.Jin R, Le NA, Liu S, et al. Children with NAFLD are more sensitive to the adverse metabolic effects of fructose beverages than children without NAFLD. J Clin Endocrinol Metab 2012;97:E1088–E1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Walker RW, Lê KA, Davis J, et al. High rates of fructose malabsorption are associated with reduced liver fat in obese African Americans. J Am Coll Nutr 2012;31:369–374 [DOI] [PubMed] [Google Scholar]
- 89.Madero M, Arriaga JC, Jalal D, et al. The effect of two energy-restricted diets, a low-fructose diet versus a moderate natural fructose diet, on weight loss and metabolic syndrome parameters: a randomized controlled trial. Metabolism 2011;60:1551–1559 [DOI] [PubMed] [Google Scholar]
- 90.Jalal DI, Smits G, Johnson RJ, Chonchol M. Increased fructose associates with elevated blood pressure. J Am Soc Nephrol 2010;21:1543–1549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lanaspa MA, Ishimoto T, Li N, et al. Glucose-induced obesity, fatty liver and insulin resistance is mediated by endogenous fructose. Nature Commun. In press [Google Scholar]
- 92.Brown CM, Dulloo AG, Yepuri G, Montani JP. Fructose ingestion acutely elevates blood pressure in healthy young humans. Am J Physiol Regul Integr Comp Physiol 2008;294:R730–R737 [DOI] [PubMed] [Google Scholar]
- 93.Hallfrisch J, Ellwood K, Michaelis OE, 4th, Reiser S, Prather ES. Plasma fructose, uric acid, and inorganic phosphorus responses of hyperinsulinemic men fed fructose. J Am Coll Nutr 1986;5:61–68 [DOI] [PubMed] [Google Scholar]
- 94.Nguyen S, Choi HK, Lustig RH, Hsu CY. Sugar-sweetened beverages, serum uric acid, and blood pressure in adolescents. J Pediatr 2009;154:807–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chen L, Caballero B, Mitchell DC, et al. Reducing consumption of sugar-sweetened beverages is associated with reduced blood pressure: a prospective study among United States adults. Circulation 2010;121:2398–2406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Waring WS, McKnight JA, Webb DJ, Maxwell SR. Uric acid restores endothelial function in patients with type 1 diabetes and regular smokers. Diabetes 2006;55:3127–3132 [DOI] [PubMed] [Google Scholar]
- 97.George J, Carr E, Davies J, Belch JJ, Struthers A. High-dose allopurinol improves endothelial function by profoundly reducing vascular oxidative stress and not by lowering uric acid. Circulation 2006;114:2508–2516 [DOI] [PubMed] [Google Scholar]
- 98.Shafiu M, Johnson RJ, Turner ST, et al. Urate transporter gene SLC22A12 polymorphisms associated with obesity and metabolic syndrome in Caucasians with hypertension. Kidney Blood Press Res 2012;35:477–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Parsa A, Brown E, Weir MR, et al. Genotype-based changes in serum uric acid affect blood pressure. Kidney Int 2012;81:502–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Le MT, Lobmeyer MT, Campbell M, et al. Impact of genetic polymorphisms of SLC2A2, SLC2A5, and KHK on metabolic phenotypes in hypertensive individuals. PLoS ONE 2013;8:e52062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pfister R, Barnes D, Luben R, et al. No evidence for a causal link between uric acid and type 2 diabetes: a Mendelian randomisation approach. Diabetologia 2011;54:2561–2569 [DOI] [PubMed] [Google Scholar]
- 102.Yang Q, Köttgen A, Dehghan A, et al. Multiple genetic loci influence serum urate levels and their relationship with gout and cardiovascular disease risk factors. Circ Cardiovasc Genet 2010;3:523–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Nakagawa T, Tuttle KR, Short RA, Johnson RJ. Hypothesis: fructose-induced hyperuricemia as a causal mechanism for the epidemic of the metabolic syndrome. Nat Clin Pract Nephrol 2005;1:80–86 [DOI] [PubMed] [Google Scholar]