

Abstract
OBJECTIVE
The aim of this study was to elucidate whether subclinical nerve dysfunction as reflected by neurophysiological testing predicts the development of clinical neuropathy in patients with type 1 diabetes.
RESEARCH DESIGN AND METHODS
Fifty-nine patients were studied twice with neurophysiological measurements at baseline and at follow-up. At baseline, patients were 15.5 ± 3.22 years (range 7–22 years) of age, and duration of diabetes was 6.8 ± 3.3 years. At follow-up, patients were 20–35 years of age, and disease duration was 20 ± 5.3 years (range 10–31 years).
RESULTS
At baseline, patients showed modestly reduced nerve conduction velocities and amplitudes compared with healthy subjects, but all were free of clinical neuropathy. At follow-up, clinical neuropathy was present in nine (15%) patients. These patients had a more pronounced reduction in peroneal motor nerve conduction velocity (MCV), median MCV, and sural sensory nerve action potential at baseline (P < 0.010–0.003). In simple logistic regression analyses, the predictor with the strongest association with clinical neuropathy was baseline HbA1c (R2 = 48%, odds ratio 7.9, P < 0.002) followed by peroneal MCV at baseline (R2 = 38%, odds ratio 0.6, P < 0.006). With the use of a stepwise forward analysis that included all predictors, first baseline HbA1c and then only peroneal MCV at baseline entered significantly (R2 = 61%). Neuropathy impairment assessment showed a stronger correlation with baseline HbA1c (ρ = 0.40, P < 0.002) than with follow-up HbA1c (ρ = 0.034, P < 0.007).
CONCLUSIONS
Early defects in nerve conduction velocity predict the development of diabetic neuropathy. However, the strongest predictor was HbA1c during the first years of the disease.
Peripheral neuropathy is a common complication of type 1 diabetes, which increases in frequency with the duration of disease (1). The progression of neuropathy is predicted by poor metabolic control and may be prevented or retarded during the first 5 years by near normoglycemia (2). An abnormality of nerve conduction tests is the first objective quantitative indication of the condition (3,4). Nerve conduction results deteriorate with increasing age in healthy subjects and to an even greater extent in diabetic subjects (5). Therefore, it is assumed that clinically evident neuropathy would develop earlier in subjects with subclinical neuropathy than in subjects without (6); however, it is not known whether electrophysiological abnormalities seen early in the disease predict clinical neuropathy later on. Therefore, the primary aim of the current study was to elucidate whether signs and symptoms of neuropathy develop in diabetic patients with subclinical neuropathy detectable only with electrophysiological tests later in the disease. A second aim was to study whether poor metabolic control early in the disease predicts the development of neuropathy later on, as suggested by the memory effect demonstrated in the Epidemiology of Diabetes Interventions and Complications (EDIC) study (7–9) and the legacy effect demonstrated in the UK Prospective Diabetes Study (UKPDS) (10).
RESEARCH DESIGN AND METHODS
Subjects
Unselected patients with type 1 diabetes (N = 102) examined on at least one previous occasion with nerve conduction tests were included in the current study (11,12). Table 1 shows the background data at baseline and follow-up of the 59 patients who agreed to participate. All patients had been receiving intensive therapy from disease onset, which involved the administration of insulin four to seven times daily by either injection or an external subcutaneous infusion pump. All subjects gave their informed consent before participation. The study protocol was approved by the Regional Research Ethics Committee, Linköping.
Table 1.
Background data and electrophysiological results for 59 patients with type 1 diabetes examined at baseline and follow-up
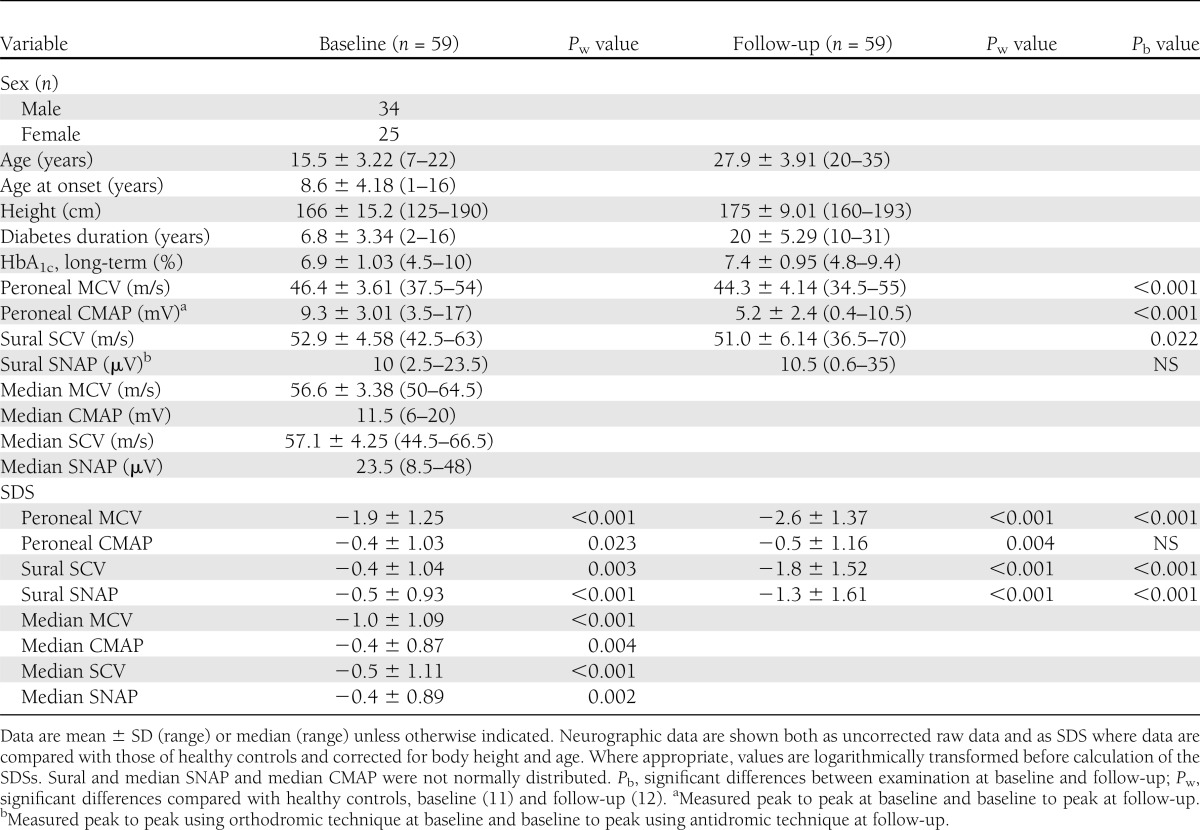
Baseline examination
At baseline, no patient had a history of neurological or metabolic disease besides diabetes, of alcohol abuse, or of taking medicine known to influence peripheral nerve function. A direct inquiry, modified from Dyck et al. (13), was made about typical symptoms of neuropathy. The tendon reflexes were examined bilaterally in the quadriceps and gastrocnemius, and vibratory sense was tested in the first metatarsal bilaterally with a 128-Hz tuning fork. Patients were asymptomatic, and tendon reflexes and vibration sense were present in all patients.
The baseline neurophysiological examination included bilateral measurements of the peroneal and median motor nerve conduction velocity (MCV) and compound muscle action potential (CMAP) amplitude and the sural and median sensory nerve conduction velocity (SCV) and sensory nerve action potential (SNAP). All amplitudes (i.e., CMAPs, SNAPs) were measured from peak to peak, and sensory nerves were studied with orthodromic recording of SNAPs. The results of the baseline examination have been published (11,12) as well as a description of the controls (n = 128) (14).
Follow-up examination
At follow-up, assessment of neurological symptoms and examination followed a previously described fixed protocol (15). Cases of numbness, allodynia, paraesthesia, and pain in the lower and upper extremities were summed to give a neuropathy symptom assessment (NSA) score. A neuropathy impairment assessment (NIA) included sensory screening for touch, pinprick, vibration, and temperature assessed on the first metatarsal, dorsum of the feet, and tibial regions. Clinical examination also included gastrocnemius and quadriceps reflexes and joint proprioception for first metatarsals. Responses were graded as normal, decreased, or absent (0, 1, and 2, respectively).
At follow-up, electroneurography was performed with surface electrodes, and digital equipment was used for stimulation and recording (Keypoint; Dantec Medical, Skovlunde, Denmark). Bilateral measurements of peroneal MCV and CMAP and sural SCV and SNAP were carried out on all patients according to standard techniques (15). Peroneal CMAPs were measured from baseline to peak, where baseline was the beginning of the response. The sural nerve was studied by antidromic technique, and the amplitude of SNAP was the peak value minus the baseline value defined by the interpolation between the level at the beginning and the level at the end of the SNAP. Sural SCV was calculated from the initial potential representing the fastest conducting axons in the nerve. Examinations were performed under standardized conditions wherein the legs were warmed with heating pads for at least 10 min to obtain skin temperatures of 32–35°C. Quantitative sensory tests (QSTs) were performed bilaterally according to standardized procedures (15). The first metatarsal and the tibia (∼10 cm below the knee) were subjected to increasing vibrations by an attached probe (Vibrameter; Somedic, Stockholm, Sweden), thus determining the vibration perception thresholds (VPTs). Warmth perception thresholds (WPTs) and cold perception thresholds (CPTs) were determined by the Marstock technique, with a probe starting at 32°C changing temperature at 1°C/s until the patient reported a feeling of heat or cold (16). The probe was applied over the dorsum of the foot and over the anterior tibial muscle. Measurements were repeated three times, and the mean was calculated and recorded.
The control group at follow-up comprised healthy volunteers (43 males and 57 females) 38 ± 9.8 years (range 21–55 years) of age and with a body height of 182 ± 7.2 cm (range 164–196 cm) for males and 168 ± 6.8 cm (range 150–181 cm) for females (17). Control subjects underwent a clinical examination and responded to a questionnaire. Exclusion criteria were 1) heredity of neurological disease, 2) presence of neurologic or metabolic disease, 3) treatment with medicine known to influence nerve function, or 4) signs of peripheral neuropathy, such as lack of tendon reflexes or decreased vibration sense.
There was no significant difference between patients examined at follow-up (n = 59) and patients who were not (n = 43). Variables compared were age at onset of diabetes, duration of diabetes, long-term HbAlc at baseline, peroneal MCV, peroneal CMAP, sural SCV, and sural SNAP (Mann-Whitney U test, data not shown).
Clinical neuropathy
The presence of diabetic neuropathy was determined by a staged approach according to established criteria (4,18) (i.e., stage 0 = no nerve conduction abnormality; 1a = nerve conduction abnormality only; 1b = nerve conduction abnormality + signs; 2a = nerve conduction + signs + symptoms; and 2b = nerve conduction abnormality + symptoms + more severe signs [i.e., >50% weakness of ankle dorsiflexion]). Nerve conduction abnormality was defined as more than one abnormal attribute in two separate nerves. An abnormal attribute with regard to nerve conduction velocity (NCV) was defined as <−2.33 SDS (first percentile or less) for the peroneal nerve (MCV and/or CMAP) and for the sural nerve (SCV and/or SNAP).
Metabolic control
Medical records were retrieved, and all HbA1c values were extracted and converted to Mono S calibration (upper reference value <5.3%). Long-term metabolic control at baseline and follow-up was estimated by calculating a weighted mean HbA1c for each patient by dividing the total HbA1c area under the curve by time elapsed. Long-term metabolic control will be mentioned and abbreviated as HbA1c herein.
Statistical analysis
SPSS version 20 (IBM Corp., Chicago, IL) statistical software was used to carry out the analysis. First, an average value from the right and left sides was calculated for each variable of nerve conduction and QST in individual patients and controls. An SDS was then calculated for each variable of nerve conduction as follows: (observed value − predicted value) / residual SD, where the predicted value and the residual SD were retrieved from linear regression analyses of healthy controls. Electrophysiological data generally are presented as SDS corrected for age and body height. When stated, raw data are also presented. Note that the calculations of SDS were based on two different populations of controls: from the baseline examination (14) and from the examination at follow-up (17) (Table 2). Several variables at both baseline and follow-up were not normally distributed; therefore, the values were logarithmically transformed before the analyses. Wilcoxon signed ranks tests were used to compare results within groups, and Mann-Whitney U test was used for between-group comparisons. In the correlation analyses, Spearman ρ was applied.
Table 2.
Equations obtained from healthy controls
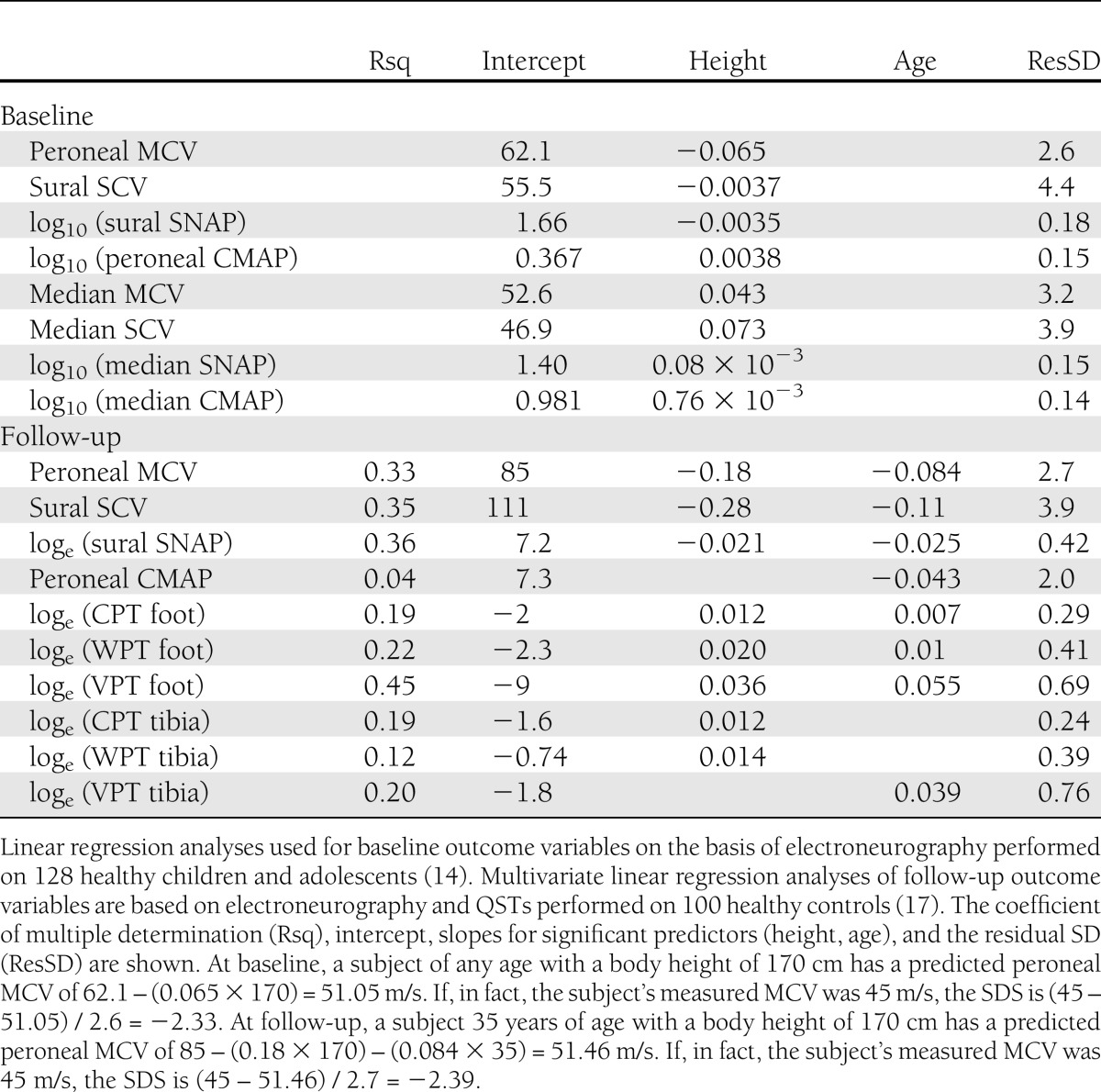
Binary logistic regression analyses were performed with the clinical neuropathy at follow-up as a dichotomous dependent variable (0 = no, 1 = yes). Predictors used were HbA1c (baseline and follow-up), sex, diabetes duration, and baseline nerve conduction variables. First, all predictors were used one at a time to create crude estimates of association with clinical neuropathy. Second, all predictors with a significant association with clinical neuropathy were tested together with HbA1c at baseline, the predictor with the strongest association to clinical neuropathy. The Nagelkerke coefficient of determination (R2) was applied. A multiple stepwise forward linear regression analysis was performed, with the total NIA score as the dependent variable and the same predictors as in the logistic regression. P < 0.05 was considered statistically significant.
RESULTS
Clinical evaluation, NSA, and NIA at follow-up
At follow-up, no patient was taking any medicine known to influence peripheral nerve function. Symptoms of neuropathy, defined as an NSA ≥1, were reported by 12 of the 59 patients (20%, 4 female, 8 male). Seven patients had an NSA of 1 and five ≥2. Paraesthesia was most common, being reported by seven patients. Four patients had symptoms of pain. On average, NIA at follow-up was 12 points.
Nerve conduction tests at baseline and follow-up
Nerve conduction variables were low already at baseline and even more so at follow-up, indicating a progression in nerve dysfunction (Table 1). Measured as SDS, there was a significant reduction over time in peroneal MCV and sural SCV and SNAP. Table 1 also shows the mean values of raw data for nerve conduction parameters. Sural SNAP, which did not show a normal distribution, was almost identical at baseline and follow-up (median 10 µV). There were significant correlations between electrophysiological findings at baseline and those at follow-up. Baseline peroneal MCV correlated with all electrophysiological variables at follow-up (peroneal MCV, ρ = 0.56, P < 0.001; sural SCV, ρ = 0.54, P < 0.001; sural SNAP, ρ = 0.61, P < 0.001; peroneal CMAP, ρ = 0.36, P < 0.006). Baseline sural SNAP correlated with sural SNAP at follow-up (ρ = 0.43, P < 0.001).
QSTs at follow-up
There were significant deviations from normal for all QST variables both on the first metatarsal and on the tibia (P < 0.001). The greatest deviations from normal were observed for CPT foot and CPT tibia, with SD scores (SDSs) of 1.8 and 1.6, respectively, followed by VPT foot with an SDS of 0.9.
HbA1c at baseline and follow-up
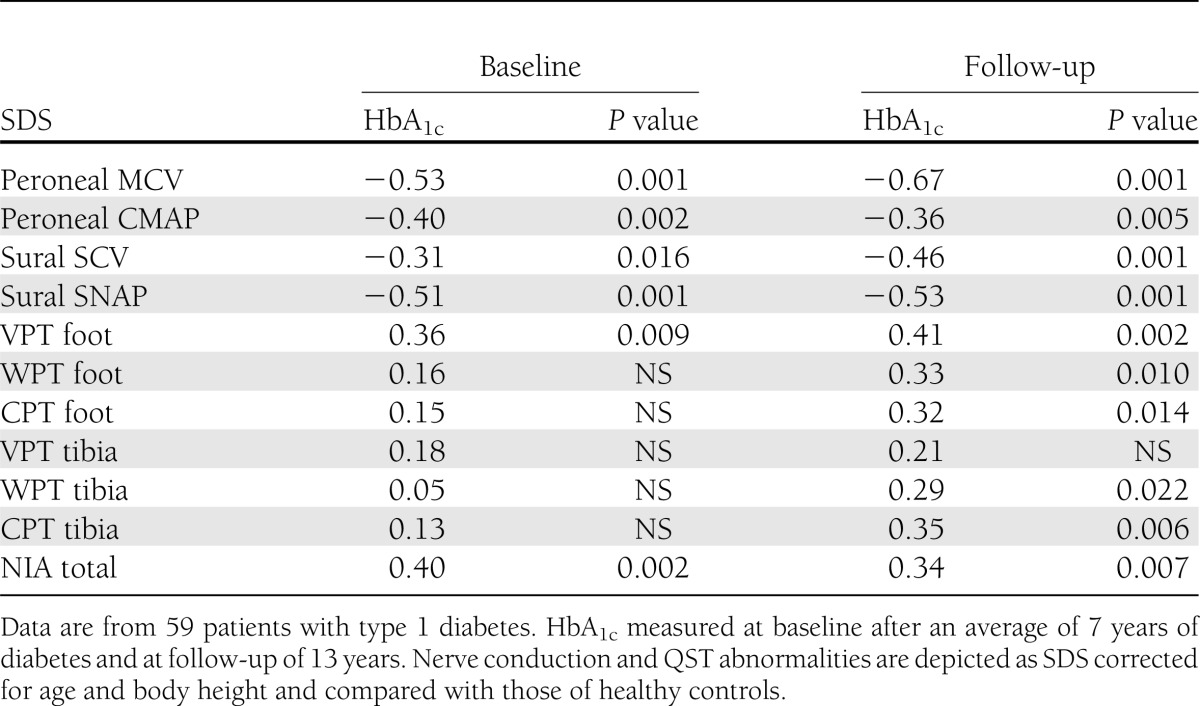
HbA1c at baseline was 6.9 ± 1.03% and at follow-up, 7.4 ± 0.94% (P < 0.001). HbA1c at baseline and follow-up correlated with several nerve conduction and QST variables at follow-up (Table 3). The strongest correlation was between follow-up HbA1c and peroneal MCV followed by sural SNAP (ρ = −0.67 and −0.53, respectively, P < 0.001). NIA and peroneal CMAP showed a stronger correlation with baseline HbA1c than with HbA1c at follow-up (Table 3).
Table 3.
Correlation (Spearman ρ) between long-term metabolic control (HbA1c) and parameters of nerve conduction, QST, and NIA at follow-up
Baseline and follow-up variables in patients with and without clinical neuropathy
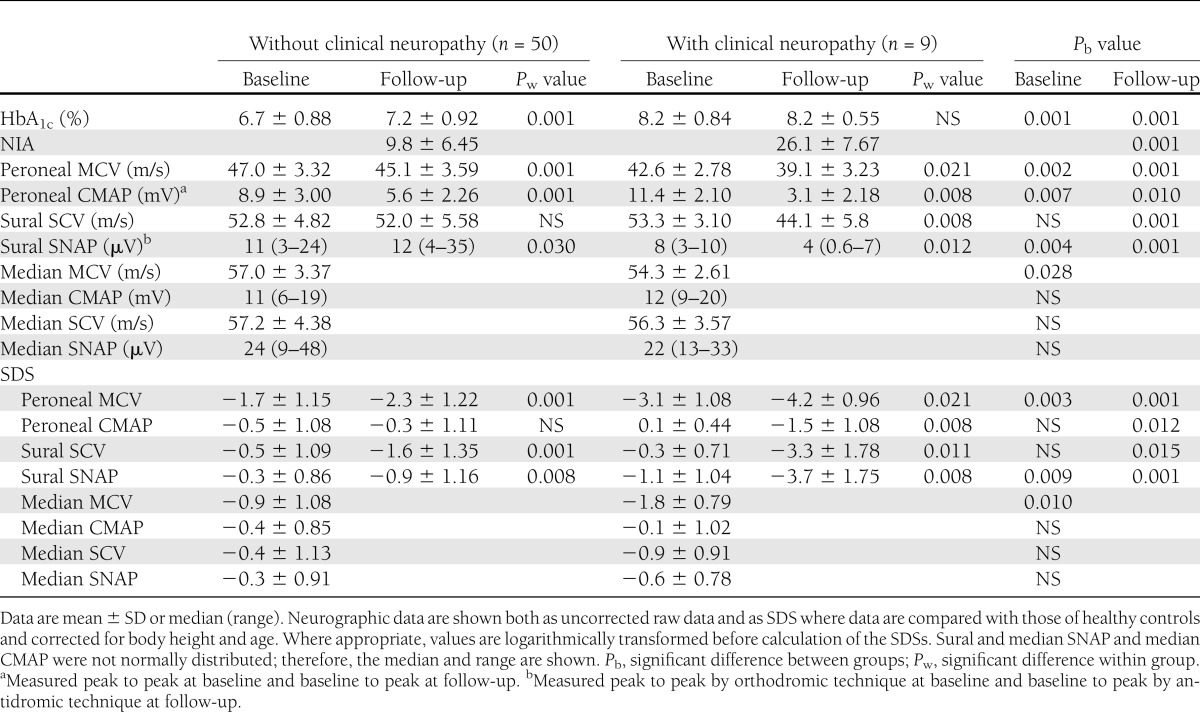
Table 4 shows that 9 of the 59 patients (15%) satisfied the criteria for clinical neuropathy at follow-up; details are presented in Table 5. Patients with clinical neuropathy had poor metabolic control measured as a significant rise in HbA1c already at baseline. On the other hand, patients without neuropathy had a less pronounced increase in HbA1c at baseline and deteriorated over time, although the mean HbA1c at follow-up was still significantly lower than for those with clinical neuropathy (Table 5). Furthermore, patients with clinical neuropathy had a more pronounced decrease in NCV and a larger increase in sensory thresholds at follow-up than patients without neuropathy. For all electrophysiological and QST variables except WPT foot and tibia and VPT tibia, this difference was significant. Table 5 also shows that patients with clinical neuropathy at follow-up had a significantly more pronounced reduction in peroneal MCV, median MCV, and sural SNAP at baseline an average of 13 years earlier (P < 0.028–0.002 for raw data and P < 0.010–0.003 for SDS). Sural SNAP showed a significant decrease between baseline and follow-up in patients with clinical neuropathy, but in those without neuropathy, sural SNAP was actually higher at follow-up compared with raw data (12 vs. 11 µV). In contrast, between baseline and follow-up, sural SNAP SDS also showed a decrease in patients without clinical neuropathy.
Table 4.
Presence and severity of neuropathy in 59 patients with type 1 diabetes examined at baseline and follow-up
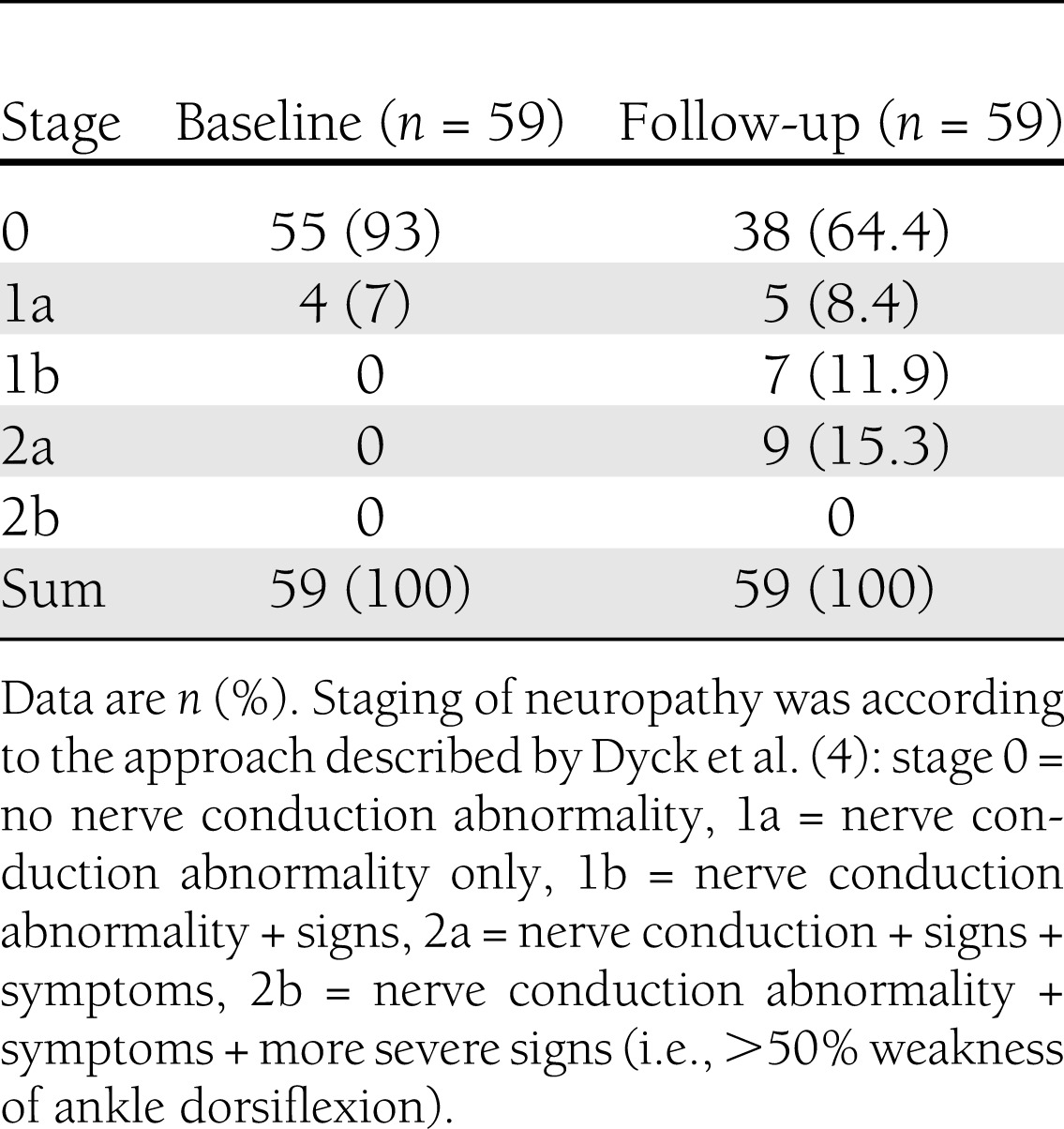
Table 5.
Baseline and follow-up nerve conduction results, HbA1c, NIA score, and QST results in 59 diabetic patients with or without clinical neuropathy at follow-up
Early predictors of clinical neuropathy
In bivariate logistic regression analysis, the predictor with the strongest association with clinical neuropathy at follow-up was baseline HbA1c (R2 = 48%, odd ratio [OR] 7.9, P < 0.002) followed by peroneal MCV (raw data, R2 = 38%, OR 0.6, P < 0.006; SDS, R2 = 25%, OR 0.3, P < 0.004), follow-up HbA1c (R2 = 27%, OR 4.3, P < 0.007), sural SNAP at baseline (raw data, R2 = 23%, OR 0.7, P < 0.016; SDS, R2 = 15%, OR 0.4, P < 0.031), and median MCV at baseline (raw data, R2 = 14%, OR 0.8, P < 0.038; SDS, R2 = 15%, OR 0.4, P < 0.021). Figure 1 shows the predictive value of baseline peroneal MCV in relation to clinical neuropathy at follow-up. The threshold at baseline was defined at 50% probability of having clinical neuropathy corresponding to a peroneal MCV of 41.5 m/s. Thus, increased HbA1c and decreased nerve conduction velocities at baseline indicated later clinical neuropathy. Sex, age at diabetes onset, and follow-up diabetes duration were not significantly correlated with clinical neuropathy.
Figure 1.

Predicted probability (from logistic regression) of having clinical neuropathy after an average of 20 years with type 1 diabetes as a function of peroneal MCV at baseline, where duration of disease was an average of 7 years. Data are from 59 patients with type 1 diabetes. DPN, diabetic peripheral neuropathy.
In a stepwise forward analysis that included all predictors, first baseline HbA1c and then only peroneal MCV at baseline entered significantly (R2 = 61%). Although significant in a logistic crude analysis, sural SNAP and median MCV at baseline did not contribute significantly to explaining clinical neuropathy later on when adjusted for baseline HbA1c. NIA correlated with HbA1c at both baseline (ρ = 0.40, P < 0.002) and follow-up (ρ = 0.34, P < 0.007). NIA also correlated (Pearson) with peroneal MCV (r = −0.26, P = 0.047) and sural SNAP SDS (r = −0.30, P = 0.022) at baseline.
CONCLUSIONS
The most important findings are 1) that subclinical nerve dysfunction, as reflected by nerve conduction data, predicts clinical neuropathy many years later and 2) that the strongest predictor for the presence of clinical neuropathy after an average of 20 years with type 1 diabetes was poor metabolic control during the first years of the disease. The concept of an asymptomatic or a subclinical form of neuropathy is well established. It is assumed that the progression of neuropathy is a continuum from normal nerve function to subclinical neuropathy detectable with electrophysiological tests to clinically evident neuropathy detectable on neurological examination (6). Indirect evidence supporting this view is that 1) patients with symptoms of neuropathy have more pathological findings in the neurological examination and more pronounced QST and nerve conduction defects (5,19,20) and 2) nerve conduction results deteriorate over time in normal subjects and even more so in diabetic patients (5). However, the transition from subclinical to clinical neuropathy in patients with type 1 diabetes has not been described previously. The current study shows that nerve dysfunction early on in the disease predicts clinical neuropathy several years later. A decrease in baseline peroneal MCV, median MCV, and sural SNAP was associated with a significantly higher risk of having a clinical neuropathy an average of 13 years after the first electrophysiological examination. Forward logistic regression analysis showed a positive predictive value of an early decrease in peroneal MCV also when long-term HbA1c was accounted for. This novel result emphasizes the role of early nerve conduction measurements in young diabetic patients.
The strongest predictor for the development of a clinical neuropathy was poor metabolic control early on in the disease (i.e., up to the baseline examination). HbA1c at follow-up showed a less pronounced correlation with the presence of clinical neuropathy. This is consistent with another report of a durable effect of prior intensive treatment (with better metabolic control) on the development of neuropathy (7). These findings suggest that poor metabolic control early in the disease is a major risk factor for the development of neuropathy, regardless of whether patients are treated with conventional or intensive therapy. Thus, the current study supports the so-called memory effect shown by the EDIC follow-up of the Diabetes Control and Complications Trial (DCCT) (7–9) and the legacy effect in the long-term follow-up of UKPDS (10). The strong correlation seen between baseline HbA1c and clinical neuropathy is consistent with the finding that good metabolic control reduces the risk for neuropathy (21).
In clinical practice, the presence of symptoms and signs defines diabetic neuropathy, but a more accurate diagnosis for research purposes requires the presence of electrophysiological abnormalities (19). Previously published consensus statements advocate nerve conduction studies as the method of choice because these are sensitive, specific, and validated measures of the presence of nerve function impairment, whereas other neurophysiological tests (e.g., QST, autonomic tests) are useful in characterizing neuropathic expression (3,4,18). Therefore, the current study defines clinical neuropathy by 1) symptoms of neuropathy, 2) signs of neuropathy, and 3) nerve conduction defects (i.e., one or fewer abnormal parameters in two separate nerves, sural and peroneal). The definition of abnormal nerve conduction is based on published consensus statements, and results of QSTs are not included in the definition of clinical neuropathy (4,18). It is emphasized that the number of patients with a clinical neuropathy depends on the definition (22). The assessment of neurological symptoms and examination in the follow-up part of the current study were identical to those published by Ekberg et al. (15). These items have not been validated and, therefore, may differ from published composite scores (23). At baseline, patients were examined with the use of a less rigorous protocol, but the baseline cohort of 59 subjects represents with certainty 59 patients without the reference standard definition of neuropathy. The dropout rate was a little >40%, and these subjects’ clinical data at baseline did not differ from those of reexamined patients. A limitation of the current study is the relatively small cohort size and small number of outcomes. Conclusions are based on findings in 59 patients, 9 of whom fulfilled the criteria for clinical neuropathy. However, because in Sweden all children and adolescents with type 1 diabetes within a geographic region attend the same hospital clinic, all patients within a geographic area were considered for participation in the original study. Patients were included in the current study because they had been examined at least on one earlier occasion.
Nerve conduction measurements followed a fixed protocol. All examinations at baseline and follow-up were performed by either one of two experienced technicians. Skin temperature was controlled for by warming all patients for at least 10 min before the examination. The results were compared with those of healthy individuals, and SDSs were calculated on the basis of height and age, which is appropriate as patients grow from adolescence into adulthood. Data on both control populations have been published, and the electrophysiological examinations at baseline and follow-up differed slightly. However, the comparison of patients and controls at baseline was based on recordings from an identical technique, and the same was true for comparisons at follow-up. Because of local protocols of different laboratories, orthodromic recordings of SNAPs were made at baseline and antidromic recordings at follow-up. In the current study, median sural amplitude was similar in size at baseline and follow-up (10 µV), which is explained by the finding that SNAPs in both healthy subjects and patients with neuropathy are larger in antidromic than in orthodromic recordings (24). A major advantage in using SDS and not raw data in the calculations is that it enabled us to follow the progression of nerve dysfunction more thoroughly, regardless of the neurophysiological technique.
Long-term metabolic control in the current study was measured as a weighted mean of all HbA1c measurements during the studied disease period (i.e., an average over many years). Several methods for measuring HbA1c are in use and have been used during the study period, and all values have been converted to Mono S calibration (upper reference value <5.3%). The number of HbA1c measurements per patient differed, as did the number of measurements in the same patient over time, but as a rule, two to four measurements of HbA1c were performed every year. Furthermore, it should be noted that multiple insulin injection therapy was introduced in the late 1970s in Sweden; all patients in the current study have undergone this treatment during their entire disease period.
The current study shows that all nerve attributes declined over time, as indicated at follow-up by a negative correlation between peroneal MCV, sural SCV, sural SNAP, and peroneal CMAP and age. At follow-up, patients were younger than controls (28 ± 3.9 vs. 38 ± 9.8 years). It is known that NCV, SNAP, and CMAP declines with age (25); therefore, an increasing patient age cannot explain the finding that neuropathy developed in a number of patients.
Earlier studies showed that clinical and subclinical neuropathies are more common in patients with poor metabolic control (1,26). With the advent of multiple insulin injection therapy, which led to improved metabolic control, the prevalence of confirmed clinical neuropathy has been significantly reduced (5). Lowering levels of HbA1c retards the deterioration in both NCV and symptoms and signs of neuropathy (7,27). The magnitude of the reduction is such that it has been proposed that clinically overt diabetic neuropathy may even be prevented (5). The current study, which included only patients under intensive treatment from disease onset, indicates that this is not the case. Clinically, overt neuropathy is still present in an unselected group of young patients with reasonably good metabolic control. The prevalence rate amounted to 15%, and patients with clinical neuropathy were characterized by symptoms of neuropathy, more pathological findings in the clinical examination, more pronounced defects in nerve conduction, and increased sensory thresholds. The prevalence of symptomatic neuropathy in the current study was higher than a reported rate of 11% in patients treated with conventional injections of insulin (28). A slightly longer duration of diabetes (20 vs. 15 years) may be one explanation of the difference, but it is likely that there are several possible explanations, such as a patient selection bias. Furthermore, the definition of what is considered a significant symptom and what is considered to be diabetic neuropathy differs among studies.
In conclusion, the present longitudinal study shows that early defects in NCV precede and predict clinical neuropathy many years later and thereby confirms what is suggested by previous epidemiology studies (5,23,29). Despite intensive therapy from disease onset with reasonably good metabolic control, clinical neuropathy is still seen in 15% of patients with type 1 diabetes after an average of 20 years. The strongest predictor for the development of a clinical neuropathy is HbA1c during the first years of the disease, which stresses the importance of good metabolic control during the early years of diabetes.
Acknowledgments
This study was supported by grants from the Swedish Child Diabetes Foundation (Barndiabetesfonden).
No potential conflicts of interest relevant to this article were reported.
L.H. contributed to the study conception and design and data acquisition, analysis, and interpretation and wrote the manuscript. N.A. contributed to the data acquisition, analysis, and interpretation and drafting and final approval of the manuscript. B.J. performed the statistical analyses and contributed to the data interpretation and critical revision and final approval of the manuscript. J.L. contributed to the study conception and design, data interpretation, and critical revision and final approval of the manuscript. G.C. contributed to the data acquisition and critical revision and final approval of the manuscript. J.W.-T. contributed to the study conception and design, data acquisition, and critical revision and final approval of the manuscript. L.H. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank Britten Winström-Nodemar, Department of Clinical Neurophysiology, Karolinska Hospital, and Gunnel Lundblad, Department of Clinical Neurophysiology, Linköping University Hospital, for their skillful assistance in the nerve function tests. The authors also thank Dr. John Wahren, Department of Molecular Medicine and Surgery, Karolinska Institutet, for valuable comments on the manuscript.
References
- 1.Pirart J. Diabetes mellitus and its degenerative complications. A prospective study of 4,400 patients observed between 1947 and 1973. Diabetes Care 1978;1:168–188, 252–263 [Google Scholar]
- 2.Ziegler D, Mayer P, Muhlen H, Gries A. The natural history of somatosensory and autonomic nerve dysfunction in relation to glycaemic control during the first 5 years after diagnosis of type 1 (insulin-dependent) diabetes mellitus. Diabetologia 1991;34:822–829 [DOI] [PubMed]
- 3.Tesfaye S, Boulton AJ, Dyck PJ, et al. Toronto Diabetic Neuropathy Expert Group Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 2010;33:2285–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dyck PJ, Albers JW, Andersen H, et al. Diabetic polyneuropathies: update on research definition, diagnostic criteria and estimation of severity. Diabetes Metab Res Rev 2011;27:620–628 [DOI] [PubMed] [Google Scholar]
- 5.The Diabetes Control and Complications Trial Research Group The effect of intensive diabetes therapy on the development and progression of neuropathy. Ann Intern Med 1995;122:561–568 [DOI] [PubMed] [Google Scholar]
- 6.Albers JW, Herman WH, Pop-Busui R, Martin CL, Cleary P, Waberski B; Diabetes Control and Complications Trial (DCCT)/Epidemiology of Diabetes Intervention and Complications (EDIC) Research Group. Subclinical neuropathy among Diabetes Control and Complications Trial participants without diagnosable neuropathy at trial completion: possible factors of incipient neuropathy? Diabetes Care 2007;30:2613–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin CL, Albers J, Herman WH, et al. DCCT/EDIC Research Group Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes Care 2006;29:340–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.White NH, Sun W, Cleary PA, et al. DCCT-EDIC Research Group Effect of prior intensive therapy in type 1 diabetes on 10-year progression of retinopathy in the DCCT/EDIC: comparison of adults and adolescents. Diabetes 2010;59:1244–1253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.de Boer IH, Sun W, Cleary PA, et al. DCCT/EDIC Research Group Intensive diabetes therapy and glomerular filtration rate in type 1 diabetes. N Engl J Med 2011;365:2366–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Holman RR, Paul SK, Bethel MA, Matthews DR, Neil HA. 10-year follow-up of intensive glucose control in type 2 diabetes. N Engl J Med 2008;359:1577–1589 [DOI] [PubMed] [Google Scholar]
- 11.Hyllienmark L, Brismar T, Ludvigsson J. Subclinical nerve dysfunction in children and adolescents with IDDM. Diabetologia 1995;38:685–692 [DOI] [PubMed] [Google Scholar]
- 12.Hyllienmark L, Maltez J, Dandenell A, Ludvigsson J, Brismar T. EEG abnormalities with and without relation to severe hypoglycaemia in adolescents with type 1 diabetes. Diabetologia 2005;48:412–419 [DOI] [PubMed] [Google Scholar]
- 13.Dyck PJ, Karnes J, O’Brien PC, Swanson CJ. Neuropathy Symptom Profile in health, motor neuron disease, diabetic neuropathy, and amyloidosis. Neurology 1986;36:1300–1308 [DOI] [PubMed] [Google Scholar]
- 14.Hyllienmark L, Ludvigsson J, Brismar T. Normal values of nerve conduction in children and adolescents. Electroencephalogr Clin Neurophysiol 1995;97:208–214 [DOI] [PubMed] [Google Scholar]
- 15.Ekberg K, Brismar T, Johansson BL, et al. C-Peptide replacement therapy and sensory nerve function in type 1 diabetic neuropathy. Diabetes Care 2007;30:71–76 [DOI] [PubMed] [Google Scholar]
- 16.Hansson P, Lindblom U, Lindström P. Graded assessment and classification of impaired temperature sensibility in patients with diabetic polyneuropathy. J Neurol Neurosurg Psychiatry 1991;54:527–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hyllienmark L, Jonsson B, Ekberg K, Lindström P. Abnormal cold perception in the lower limbs: a sensitive indicator for detection of polyneuropathy in patients with type 1 diabetes mellitus. Diabetes Res Clin Pract 2009;85:298–303 [DOI] [PubMed] [Google Scholar]
- 18.England JD, Gronseth GS, Franklin G, et al. American Academy of Neurology. American Association of Electrodiagnostic Medicine. American Academy of Physical Medicine and Rehabilitation Distal symmetric polyneuropathy: a definition for clinical research: report of the American Academy of Neurology, the American Association of Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology 2005;64:199–207 [DOI] [PubMed] [Google Scholar]
- 19.Boulton AJ, Vinik AI, Arezzo JC, et al. ; American Diabetes Association. Diabetic neuropathies: a statement bythe American Diabetes Association Diabetes Care 2005;28:956–962 [DOI] [PubMed] [Google Scholar]
- 20.Dyck PJ. Detection, characterization, and staging of polyneuropathy: assessed in diabetics. Muscle Nerve 1988;11:21–32 [DOI] [PubMed] [Google Scholar]
- 21.Tesfaye S, Chaturvedi N, Eaton SE, et al. EURODIAB Prospective Complications Study Group Vascular risk factors and diabetic neuropathy. N Engl J Med 2005;352:341–350 [DOI] [PubMed] [Google Scholar]
- 22.Dyck PJ, Carter RE, Litchy WJ. Modeling nerve conduction criteria for diagnosis of diabetic polyneuropathy. Muscle Nerve 2011;44:340–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dyck PJ, Davies JL, Litchy WJ, O’Brien PC. Longitudinal assessment of diabetic polyneuropathy using a composite score in the Rochester Diabetic Neuropathy Study cohort. Neurology 1997;49:229–239 [DOI] [PubMed] [Google Scholar]
- 24.Burke D, Skuse NF, Lethlean AK. Sensory conduction of the sural nerve in polyneuropathy. J Neurol Neurosurg Psychiatry 1974;37:647–652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Taylor PK. Non-linear effects of age on nerve conduction in adults. J Neurol Sci 1984;66:223–234 [DOI] [PubMed] [Google Scholar]
- 26.Gregersen G. Variations in motor conduction velocity produced by acute changes of the metabolic state in diabetic patients. Diabetologia 1968;4:273–277 [DOI] [PubMed] [Google Scholar]
- 27.Amthor KF, Dahl-Jørgensen K, Berg TJ, et al. The effect of 8 years of strict glycaemic control on peripheral nerve function in IDDM patients: the Oslo Study. Diabetologia 1994;37:579–584 [DOI] [PubMed] [Google Scholar]
- 28.Boulton AJ, Knight G, Drury J, Ward JD. The prevalence of symptomatic, diabetic neuropathy in an insulin-treated population. Diabetes Care 1985;8:125–128 [DOI] [PubMed] [Google Scholar]
- 29.Dyck PJ, O’Brien PC, Litchy WJ, Harper CM, Klein CJ, Dyck PJB. Monotonicity of nerve tests in diabetes: subclinical nerve dysfunction precedes diagnosis of polyneuropathy. Diabetes Care 2005;28:2192–2200 [DOI] [PubMed] [Google Scholar]