

Abstract
OBJECTIVE
The development of new insulin sensitizers is an unmet need for the treatment of type 2 diabetes. We investigated the effect of GFT505, a dual peroxisome proliferator–activated receptor (PPAR)-α/δ agonist, on peripheral and hepatic insulin sensitivity.
RESEARCH DESIGN AND METHODS
Twenty-two abdominally obese insulin-resistant males (homeostasis model assessment of insulin resistance >3) were randomly assigned in a randomized crossover study to subsequent 8-week treatment periods with GFT505 (80 mg/day) or placebo, followed by a two-step hyperinsulinemic-euglycemic insulin clamp with a glucose tracer to calculate endogenous glucose production (EGP). The primary end point was the improvement in glucose infusion rate (GIR). Gene expression analysis was performed on skeletal muscle biopsy specimens.
RESULTS
GFT505 improved peripheral insulin sensitivity, with a 21% (P = 0.048) increase of the GIR at the second insulin infusion period. GFT505 also enhanced hepatic insulin sensitivity, with a 44% (P = 0.006) increase of insulin suppression of EGP at the first insulin infusion period. Insulin-suppressed plasma free fatty acid concentrations were significantly reduced on GFT505 treatment (0.21 ± 0.07 vs. 0.27 ± 0.11 mmol/L; P = 0.006). Neither PPARα nor PPARδ target genes were induced in skeletal muscle, suggesting a liver-targeted action of GFT505. GFT505 significantly reduced fasting plasma triglycerides (−21%; P = 0.003) and LDL cholesterol (−13%; P = 0.0006), as well as liver enzyme concentrations (γ-glutamyltranspeptidase: −30.4%, P = 0.003; alanine aminotransferase: −20.5%, P = 0.004). There was no safety concern or any indication of PPARγ activation with GFT505.
CONCLUSIONS
The dual PPARα/δ agonist GFT505 is a liver-targeted insulin-sensitizer that is a promising drug candidate for the treatment of type 2 diabetes and nonalcoholic fatty liver disease.
Type 2 diabetes mellitus is a complex disorder under the combined control of environmental and genetic factors. Hyperglycemia in type 2 diabetes results from a combination of insulin resistance in several insulin target tissues (including liver, skeletal muscle, and adipose tissue) and β-cell dysfunction (1,2). The relative contribution of these two defects to the pathogenesis of diabetes continues to be debated. Longitudinal studies in high-risk individuals seem to indicate that insulin resistance is an early phenomenon, occurring years before any evidence of glucose intolerance, whereas β-cell failure develops later in the pathogenesis of disease (3). However, most of the genetic loci associated with the risk of type 2 diabetes mellitus, identified in genome-wide association studies, encode proteins involved in the insulin secretion pathway (4).
The development of new insulin sensitizers appears critical for an optimal therapeutic management of type 2 diabetes and insulin resistance–associated diseases, such as nonalcoholic fatty liver disease (NAFLD). Metformin and thiazolidinediones (TZDs) are the two classes of insulin sensitizers available on the market (5,6). The mechanism of action of metformin, currently used as the first-line drug in type 2 diabetes, remains poorly understood. Whereas metformin effectively reduces gluconeogenesis and hepatic glucose production, its effect on peripheral insulin resistance remains controversial (7). TZDs are ligands for the transcription factor peroxisome proliferator–activated receptor (PPAR) γ, which improve both hepatic and peripheral insulin sensitivity (8,9). However, TZDs are highly debated because of the occurrence of several adverse events (AEs) such as body weight gain, fluid retention, congestive heart failure, bone fractures, and possibly bladder cancer (10).
The PPAR nuclear receptor subfamily is composed of three members, PPARα, PPARγ, and PPARδ (also known as PPARβ). PPARα, the target of the hypolipidemic fibrates, is highly expressed in liver parenchymal cells, where it controls genes involved in lipid and lipoprotein metabolism (11). However, PPARα agonists, such as fenofibrate, do not influence glucose homeostasis in humans (12–14). PPARδ is widely expressed and plays a critical role in mitochondrial function, fatty acid oxidation, and insulin sensitivity in mice (15,16). In humans, 2-week clinical studies in healthy volunteers (17) and moderately overweight subjects (18) demonstrated that the synthetic PPARδ agonist GW501516 improves dyslipidemia (reducing plasma triglycerides [TGs] and increasing HDL cholesterol) and glucose metabolism (decreasing plasma insulin), whereas liver fat content was reduced.
GFT505 and its main active circulating metabolite, GFT1007, are PPAR modulators with preferential activity on human PPARα in vitro (half-maximal effective concentration [EC50]: 45 nmol/L for GFT505 and 15 nmol/L for GFT1007 compared with ≈30 μmol/L for fenofibrate), with additional activity on human PPARδ (EC50: 175 nmol/L for GFT505 and 75 nmol/L for GFT1007 compared with 1 nmol/L for GW501516). Studies in rodents indicated that both GFT505 and GFT1007 undergo extensive enterohepatic cycling and are liver-targeted (19). Recently, we demonstrated that GFT505 treatment improves several metabolic parameters, including fasting plasma glucose (FPG) and homeostasis model assessment of insulin resistance (HOMA-IR), in abdominally obese patients (20). To further assess the effect of GFT505 on insulin sensitivity, we performed a placebo-controlled, randomized, crossover study in abdominally obese male subjects using the gold standard hyperinsulinemic-euglycemic clamp method. Moreover, the effect of GFT505 was assessed on several other metabolic parameters, including plasma lipids and liver enzymes.
RESEARCH DESIGN AND METHODS
Clinical study design
A multicenter, randomized, single-blind (subject), placebo-controlled, crossover study was performed between 18 January 2011 (first patient, first visit) and 28 November 2011 (last patient, last visit) with GFT505 (2-[2,6 dimethyl-4-[3-[4-(methylthio)phenyl]-3-oxo-1(E)-propenyl]phenoxyl]-2-methylpropanoic acid), 80 mg (4 capsules of 20 mg each once daily before breakfast) in male patients aged 18–75 years (see Supplementary Fig. 1 for the study design). The duration of each treatment period was 8 weeks, separated by a 6-week wash-out period. The randomization was stratified by site (Center of Clinical Investigation Nantes and Centre de Recherche en Nutrition Humaine Rhône Alpes). The primary objective was to evaluate in each patient the differences in glucose infusion rate (GIR) measured at the second step of a hyperinsulinemic-euglycemic clamp performed at the end of each 8-week treatment period with GFT505 or placebo. The secondary objectives included the evaluation of differences in basal and insulin-suppressed endogenous glucose production (EGP), FPG, HbA1c, plasma lipids including free fatty acids (FFA), liver enzymes, and inflammatory markers at the end of each treatment period. Gene expression (mRNA) was analyzed in muscle tissue biopsy specimens collected once at the end of the first treatment period in the two groups.
Inclusion criteria were as follows: waist circumference ≥94 cm; BMI ≤45 kg/m2; and HOMA-IR (FPG [mmol/L] × fasting plasma insulin [µUI/mL]/22.5) >3. Patients had to be consuming stable diets and performing physical exercise within 3 months before screening. Main exclusion criteria were uncontrolled hypertension (blood pressure >160–95 mmHg), known heart failure (New York Heart Association stage I–IV), acute cardiovascular episode 6 months before the start of the study, diabetes mellitus, history of bariatric surgery, and excessive alcohol consumption (>3 alcoholic beverage/day). Biochemical exclusion criteria included fasting plasma TG >400 mg/dL or LDL cholesterol concentration >220 mg/dL, thyroid-stimulating hormone more than twice the upper limit of normal, chronic renal failure (creatinine clearance <60 mL/min according to Modification of Diet in Renal Disease formula or serum creatinine >180 µmol/L), active liver disease or hepatic dysfunction as defined by elevations in liver enzymes (alanine aminotransferase [ALT], aspartate aminotransferase, and γ-glutamyltransferase [γGT] more than three-times the upper limit of normal), and unexplained serum creatine phosphokinase more than twice the upper limit of normal. Hypolipidemic drugs were not permitted, except statins (except fluvastatin) or ezetimibe, if the dose had been constant and stable at least for 3 months before screening and remained constant during the study.
Hyperinsulinemic-euglycemic clamps
Insulin sensitivity was assessed after each treatment period by using the two-step hyperinsulinemic-euglycemic clamp procedure according to DeFronzo et al. (21). After an overnight fast, the subjects were admitted to each center (0800 h) and fitted with intravenous catheters placed into the veins in one forearm for insulin and glucose infusion and in the other forearm for blood sampling. The GIR was adjusted according to arterialized blood glucose samples measured every 5–10 min to maintain euglycemia (5.0 ± 0.5 mmol/L). After a basal period of 2 h, two different flow rates of insulin were used successively: 0.2 mU⋅kg−1⋅min−1 for the first 2 h and 1.0 mU⋅kg−1⋅min−1 for the last 2 h. Blood samplings were collected each 10 min during the last 30 min of basal period and each insulin infusion step of the clamp to determine glycemia, insulinemia, plasma FFA, TGs, and glucose isotopic enrichment in 2H. The use of nonradioactive glucose tracer (deuterated glucose [6,6 2H2]-glucose; Eurisotop, St. Aubin, France) during the clamp allowed to measure the rate of glucose appearance (Ra) in basal condition and during the steady state of each insulin infusion period. During the basal period, the amount of tracer infused was based on weight and glycemia of the subject. It included an initial bolus, adjusted by the basal glycemia (FPG [g/L] × 80 × base flow), followed by a constant base flow corresponding to 1% of the theoretical rate of glucose disappearance (3 mg⋅kg−1⋅min−1). During the hyperinsulinemic clamp, the infusion rate of tracer [6.6-2H2]-glucose was adapted to glucose infusion to maintain a constant ratio between the labeled glucose and the unlabeled glucose (1%), corresponding to 0.03 (first step) and 0.08 (second step) mg⋅kg−1⋅min−1. The exact quantity of tracer infused was determined by weighing the syringe at the beginning and the end of the test.
Calculations
The average rate of glucose infused at steady state of the first level (90–120 min) and the second level (210–240 min) of the clamp is called GIR and is expressed in mg⋅kg−1⋅min−1. Flow measurement of glucose turnover was measured in the final 20 min of the basal period (calculations for times −20, −10, and 0). With glycemia stable, the Ra corresponds to the basal peripheral glucose utilization (Rd) and also to the EGP. EGP (expressed in mg/kg⋅min−1) was calculated by the following equation:
With F indicating flow rate of glucose infused (mg/kg⋅min−1) and MR indicating molar ratio tracer/traced (MR% deuterated glucose/natural glucose).
Skeletal muscle biopsies
The biopsy of the vastus lateralis muscle was performed with a Bergstrom trocar after local anesthesia of the biopsy area with lidocaine 1%. The biopsy location was identified in the anterolateral side of the thigh, at the junction of the lower-third middle-third through the segment joining the spaced femorotibial and anterior superior iliac spine superior.
mRNA extraction and quantitative PCR
Total RNA was extracted using the mirVana miRNA isolation kit (Ambion) following the manufacturer’s instructions. After synthesis of complementary DNA using Moloney murine leukemia virus reverse transcriptase protein, the real-time PCR measurement of individual cDNAs was performed using iQ SYBR Green Supermix kit to measure duplex DNA formation with the iQ or MyiQ Real-Time PCR Detection Systems (Bio-Rad). The primers used are listed in the Supplementary Table 1. Expression levels were normalized using 18S gene as reference.
Statistical analysis
Calculation of sample size.
Based on our previous experience (22), we assumed the level of GIR at the second step of the clamp in moderately insulin-resistant obese patients to be 4.9 mg⋅kg−1⋅min−1 with SD of 1.0 mg⋅kg−1⋅min−1, and we assumed the expected GIR difference between treatments to be 20% (Δ = 0.98 mg⋅kg−1⋅min−1). With a sample size in each sequence group of 10 (a total sample size of 20), a 2 × 2 cross-over design would have 80% power to detect a difference in means of 0.980 (the difference between a treatment 1 mean, µ1, of 4.900, and a treatment 2 mean, µ2, of 3.920) assuming that the crossover ANOVA √mean squares error was 1.000 (the SD of differences, σd, would be 1.414) using a two-group t test (crossover ANOVA) with a 0.050 two-sided significance level.
The efficacy analysis was performed on an intent-to-treat basis. The main efficacy variable was the difference in GIR level after an 8-week period of treatment. A mixed model was built with treatment, period, clinical center, sequence, and interaction of treatment center as fixed factors and patient within sequence as random factor (carry-over effect). Least squares means were derived from the mixed model. The treatment effect was computed as differences between least squares means. The 95% two-sided CI was computed. Tests were two-sided and type I error risk was set at 0.05. No formal adjustment of type 1 error risk has been made. Given the number of secondary end points, P values between 0.01 and 0.05 should be interpreted cautiously. Because muscle biopsies were performed only once per patient, gene expression comparisons were performed using unpaired tests. All coauthors had access to the study data and have reviewed and approved the final manuscript.
RESULTS
GFT505 improves both hepatic and whole-body insulin sensitivity in humans
Forty-nine abdominally obese patients were selected to take part in the study. There were 27 screening failures. Twenty-six patients did not meet the inclusion criteria and one patient withdrew his consent. All 22 randomized patients received both study treatments and completed the entire study without major protocol deviation.
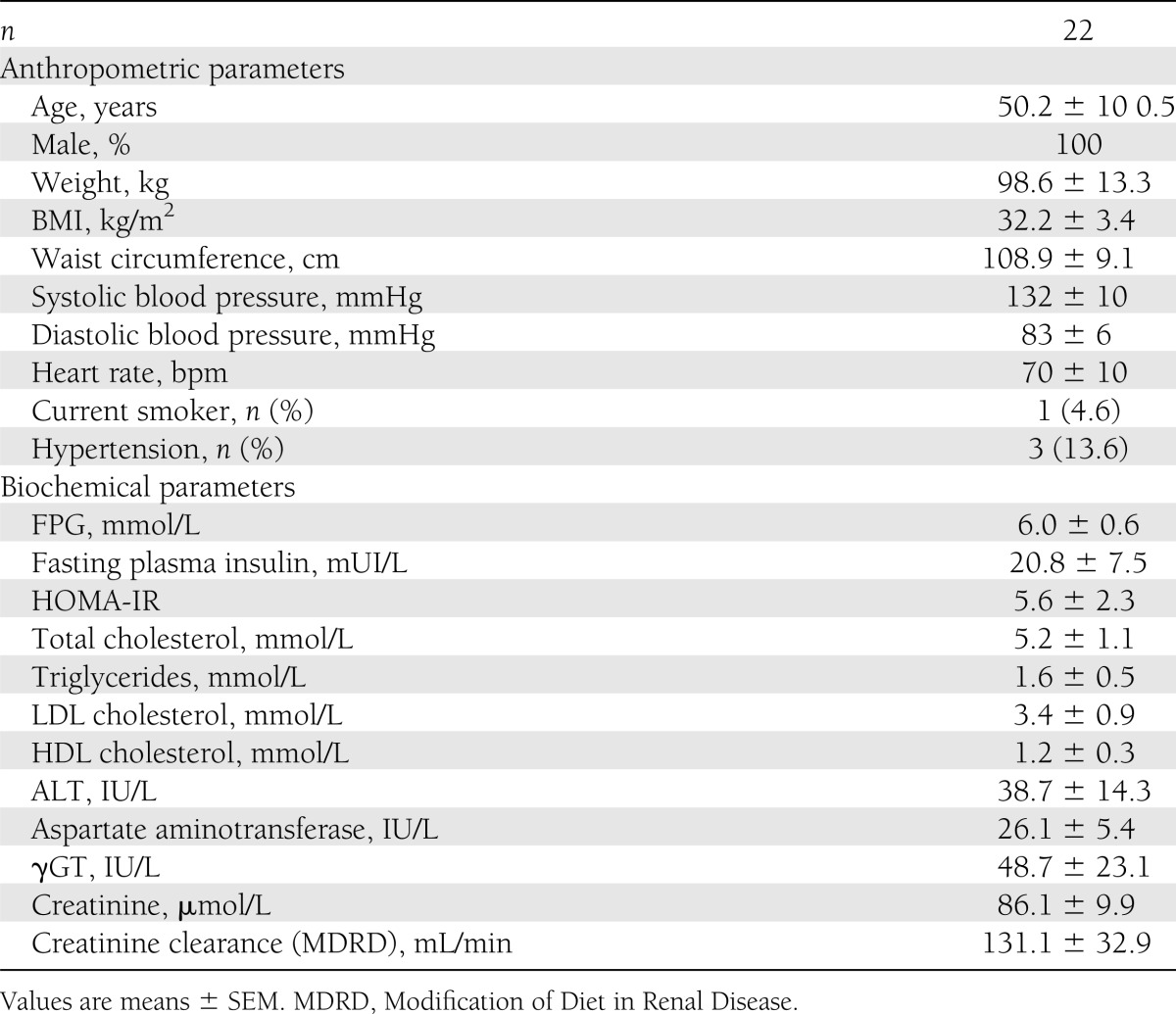
All patients were Caucasian males aged 50.2 ± 10.5 years (see Table 1 for baseline characteristics). Patients were severely insulin-resistant (mean HOMA-IR, 5.6 ± 2.3) with abdominal obesity (mean BMI, 32.2 ± 3.4 kg/m2; mean waist circumference, 108.9 ± 9.1 cm). Patients did not have type 2 diabetes but the mean FPG was moderately elevated at 6.0 ± 0.6 mmol/L.
Table 1.
Baseline characteristics at screening
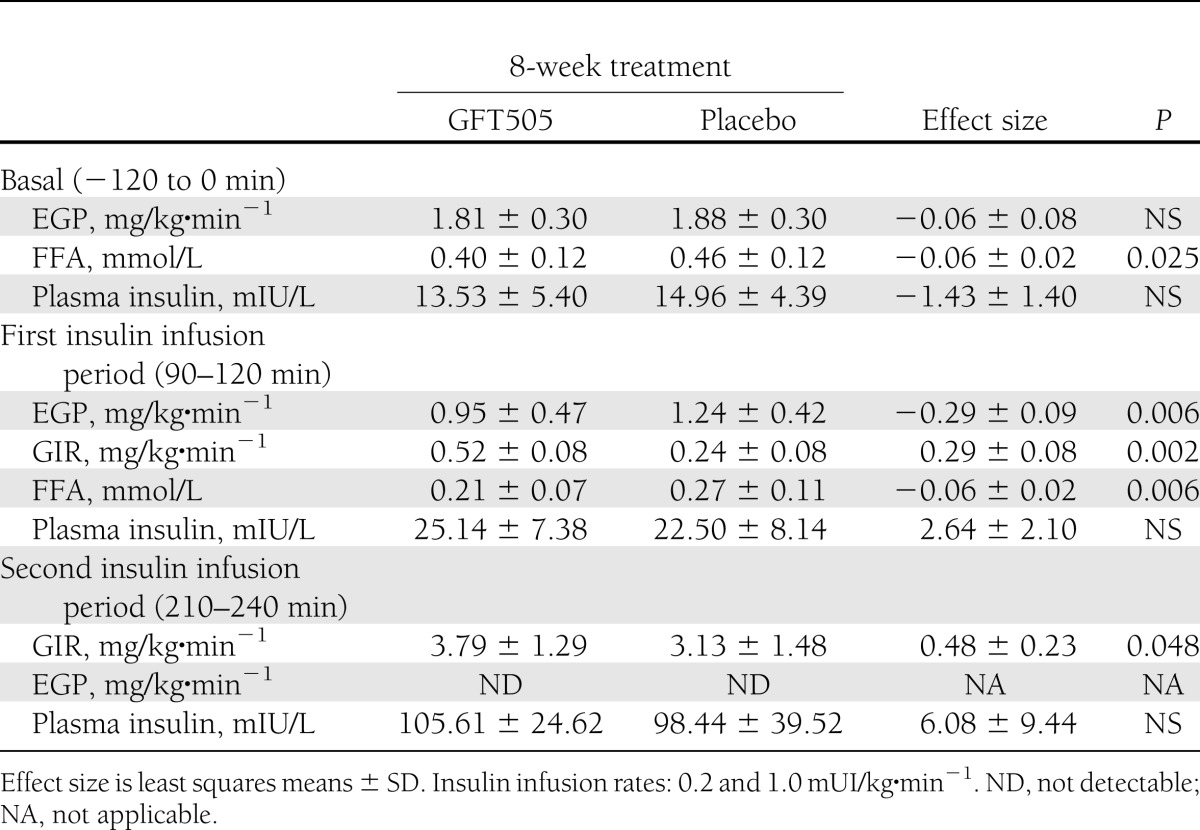
In this population of nondiabetic subjects, GFT505 did not lower FPG concentrations (Table 2). However, GFT505 significantly increased the GIR both at the first and second steps (primary end point) of the hyperinsulinemic-euglycemic clamp, respectively, by 116% (P = 0.002) and 21% (P = 0.048) when compared with placebo, demonstrating an improvement of whole-body insulin sensitivity (Table 3). After an overnight fast, the majority (95%) of EGP is derived from the liver (23), therefore reflecting the hepatic glucose production. The basal rate of EGP did not differ significantly between the two groups. However, during the first insulin infusion period, the suppression of EGP compared with baseline levels was 43.5% greater after GFT505 than after placebo (P = 0.006), further indicating an improvement of hepatic insulin action (Table 3). During the second insulin infusion period, EGP was fully suppressed in both groups, with values below the limit of detection. The baseline fasting plasma FFA concentrations were reduced on GFT505 versus placebo treatment (0.40 ± 0.12 vs. 0.46 ± 0.12 mmol/L; P = 0.025). Moreover, insulin suppression of plasma FFA levels during the first insulin infusion period was significantly higher on GFT505 treatment (0.21 ± 0.07 vs. 0.27 ± 0.11 mmol/L; P = 0.006) (Table 3). GFT505 reduced plasma adiponectin concentrations (4,247 ± 2,011 vs. 4,617 ± 2,314 mg/mL; P = 0.009) (Table 2).
Table 2.
Metabolic and anthropometric parameters after GFT505 or placebo treatment periods
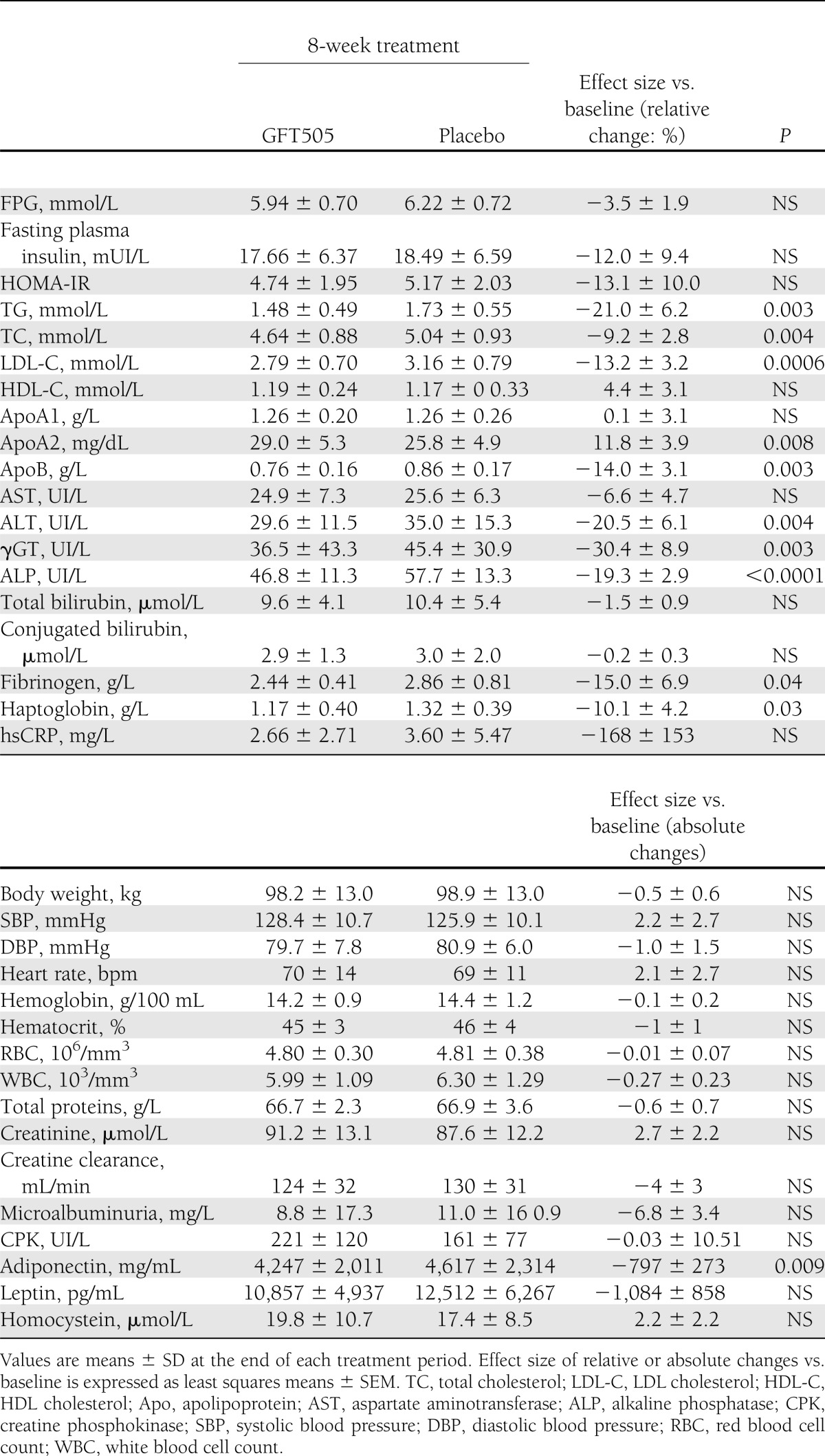
Table 3.
Hepatic and whole-body glucose metabolism during the two-step hyperinsulinemic-euglycemic clamp after 8-week treatment period with GFT505 or placebo
Effect of GFT505 on skeletal muscle gene expression
To further decipher the molecular action of GFT505, skeletal muscle biopsies were performed at the end of the first period of treatment to measure mRNA gene expression in placebo-treated (n = 9) and GFT505-treated (n = 9) patients (Fig. 1). GFT505 failed to increase significantly the expression of PPARα and PPARδ target genes, such as CPT1a, CPT1b, PDK4, and CD36. In addition, GFT505 did not alter the expression of the insulin-regulated transcription factor SREBP1-C or the insulin-responsive glucose transporter 4.
Figure 1.

Effect of GFT505 on human skeletal muscle gene expression. Gene (mRNA) expression in skeletal muscle biopsy specimens at the end of the first 8-week treatment period with GFT505 (80 mg/day) or placebo (n = 9/group). mRNA levels were measured by quantitative PCR. Values (means ± SD) are normalized to the expression of 18S and are expressed relative to placebo group. PCB, placebo group.
Effect of GFT505 on plasma lipoproteins
GFT505 also improved several lipid parameters significantly from baseline (Table 2). GFT505 reduced fasting plasma TG (relative effect size vs. placebo of −21%; P = 0.003), LDL cholesterol, and apolipoprotein B (relative effect size vs. placebo of −13.2% [P = 0.0006] and −14% [P = 0.003], respectively). HDL cholesterol and apolipoprotein A-I concentrations were not altered by GFT505 treatment. However, there was a significant increase of apolipoprotein A-II after GFT505 treatment (relative effect size vs. placebo of +11.8%; P = 0.008). All lipid parameters returned to baseline values within 2 weeks after treatment cessation (data not shown).
GFT505 improves liver enzymes
Plasma levels of γGT, a marker of liver dysfunction, were significantly reduced from baseline by GFT505 compared with placebo treatment (effect size: −30.4%; P = 0.003). In addition, GFT505 significantly decreased ALT levels (effect size: −20.5%; P = 0.004), with no effect on aspartate aminotransferase concentrations (effect size: −6.6%; P = NS), suggesting that GFT505 can improve NAFLD in these insulin-resistant subjects (Table 2). A highly significant reduction of alkaline phosphatase levels also was observed after GFT505 treatment (effect size: −19.3%; P < 0.0001) (Table 2).
GFT505 also led to a modest reduction of haptoglobin (effect size: −10.1%; P = 0.03) and fibrinogen (effect size: −15.0%; P = 0.04) levels, with no significant effect on high-sensitivity C-reactive protein concentrations (Table 2).
Safety of GFT505
GFT505 showed a good tolerance profile. A total of 37 emergent AEs were reported during the two treatment periods: 16 AEs were reported by 10 patients (45%) in the GFT505 group and 21 AEs were reported by 15 patients (61%) in the placebo group (Supplementary Table 2). Among these AEs, 12 were considered as possibly related to the study treatment by the investigators. Of them, four were reported with GFT505 (one case of upper gastrointestinal pain, one case of fatigue, one case of eczema, and one case of abnormal sweating) and eight were reported with placebo (Supplementary Table 3). No AE led to the discontinuation of GFT505 and no serious AE related to GFT505 was reported. Six additional AEs were reported during the 6-week wash-out period and eight were reported during the 2-week follow-up period; none of them was judged related to GFT505. Only one serious AE (femoral neck fracture of the left hip after an accidental fall) was reported during the course of the study concerning one patient during treatment with placebo.
No abnormal evolution of laboratory values, vital signs, or electrocardiograms was observed during the treatment periods. Notably, there was no body weight increase with GFT505 (Table 2). All safety parameters including hematological parameters, plasma creatinine, creatine phosphokinase, and homocysteine concentrations remained unaltered during the GFT505 treatment period (Table 2).
CONCLUSIONS
The present randomized study, using the gold standard hyperinsulinemic-euglycemic clamp method, clearly demonstrates that GFT505 is a new insulin-sensitizer that improves both peripheral and hepatic insulin sensitivity in insulin-resistant abdominally obese subjects. In accordance with a hepatic insulin-sensitizing action, GFT505 improved the levels of liver enzymes (γGT and ALT). GFT505 also improved plasma lipid parameters, significantly decreasing both TG and LDL cholesterol levels. Finally, the safety profile of GFT505 was good, with no reported serious AE.
In clinical practice, TZDs remain the most efficacious insulin-sensitizing drugs that improve both hepatic and peripheral insulin sensitivity (8,9,24). In parallel, TZDs reduce liver fat in patients with type 2 diabetes with or without nonalcoholic steatohepatitis (8,25–27). Metformin is a biguanide that acts by inhibiting mitochondrial complex I, leading to reduced hepatic mitochondrial ATP production and gluconeogenesis rates (7). In contrast to TZDs, several studies indicate that metformin fails to improve peripheral insulin sensitivity and to reduce liver fat content (7,24,28).
Compared with placebo, GFT505 improved simultaneously the GIR by 21% during the second insulin infusion period and the suppression of EGP by 43% during the first insulin infusion period of the hyperinsulinemic-euglycemic clamp. The extent of the GIR increase on GFT505 treatment seems to be in a range similar to that observed in two placebo-controlled studies with pioglitazone, one in prediabetic subjects with coronary heart disease (+12.5%) (29) and the second in nondiabetic subjects with combined hyperlipidemia (+33%) (30). Although our data demonstrate that GFT505 is a new potent insulin sensitizer, only a head-to-head trial will allow a reliable comparison between these two drugs. GFT505 also enhanced insulin suppression of FFA levels, indicating a global insulin-sensitizing effect on muscle, liver, and adipose tissue.
In agreement with previous clinical (20) and animal studies, there is no evidence of PPARγ activity of GFT505 in this study either. In diabetic db/db mice, 58-day treatment with GFT505 produced a similar decrease in FPG and HbA1c as rosiglitazone, but without increasing plasma adiponectin concentrations and without inducing the expression of PPARγ target genes in adipose tissue (data not shown). Here, we confirm that GFT505 does not increase plasma adiponectin concentrations in humans either. In contrast, a significant reduction of adiponectin levels is even observed on GFT505 treatment, suggesting that GFT505 either improves adiponectin signaling or exerts its effects in an adiponectin-independent manner. Thus, together with the low affinity and partial agonist activity of GFT505 on PPARγ, as well as the large enterohepatic cycling and liver-targeted pharmacodynamics of GFT505 (as illustrated by the lack of gene regulation effects in skeletal muscle), our data seem to exclude insulin-sensitizing effects resulting from PPARγ activation in adipose tissue. The insulin-sensitizing effect of GFT505 could be linked to its PPARδ or combined PPARδ/PPARα activity. In three randomized hyperinsulinemic-euglycemic clamp studies, the pure PPARα agonist fenofibrate failed to improve either peripheral or hepatic insulin sensitivity in patients with the metabolic syndrome (12), NAFLD (13), or type 2 diabetes (14). In contrast, it has been demonstrated that the pure PPARδ agonist GW501516 significantly reduces HOMA-IR in abdominally obese patients (16). However, to the best of our knowledge, the current study is the first to directly demonstrate that combined PPARδ/PPARα activation leads to an improvement of insulin sensitivity in humans.
To explore the molecular mechanisms whereby GFT505 improves insulin sensitivity, skeletal muscle biopsies were performed at the end of the first treatment period. Because muscle biopsies were performed only once per patient during the study, gene expression comparisons were unpaired and may lack power. Skeletal muscle is one of the main PPARδ target tissues (15). Pharmacological activation of PPARδ with GW501516 increases fatty acid oxidation in skeletal muscle and improves metabolism in mice (15,16). In addition, PPARδ activation with specific synthetic agonists also enhances fatty acid oxidation and oxidative gene expression (31), as well as basal and insulin-stimulated glucose uptake in primary human myotubes (32). In human skeletal muscle biopsy specimens, GW501516 has been shown to increase CPT1b mRNA expression (18). In contrast, GFT505 failed to induce the expression of several PPARδ target genes (CPT1b, PDK4, CD36) in muscle biopsy specimens. Therefore, it seems unlikely that a direct activation of PPARδ in skeletal muscle contributes to the insulin-sensitizing action of GFT505 in vivo. Moreover, GFT505 does not induce PPARα/δ target genes (CPT1 and PDK4) in skeletal muscles of diabetic db/db mice either (data not shown). These results are in accordance with the pharmacokinetics of GFT505, which is a liver-targeted drug that undergoes extensive enterohepatic cycling (19). Thus, additional studies are needed to unravel the precise molecular mechanism behind the peripheral insulin-sensitizing effect of GFT505.
Interestingly, GFT505 improved insulin suppression of plasma FFA levels during the first step of the hyperinsulinemic-euglycemic clamp, an effect potentially contributing to the improvement in peripheral insulin sensitivity, possibly by reducing intramyocellular lipotoxicity (33). A similar reduction of circulating FFA has been previously observed in subjects with abdominal obesity treated with PPARδ agonists (18,34,35). In contrast, PPARα agonists, such as fenofibrate, reduced fasting plasma FFA levels in some (12,13), but not all, studies (14). However, overall fenofibrate fails to improve glucose homeostasis in type 2 diabetes (14,36). In a direct comparison with the PPARδ agonist GW501516, the pure PPARα agonist GW590735 also failed to decrease fasting plasma FFA concentrations (18). Finally, insulin suppression of circulating FFA was not modified by fenofibrate in several clamp studies (12,14). The enhanced insulin suppression of plasma FFA levels with GFT505 suggests improved insulin sensitivity in adipocytes. Unfortunately, we did not sample adipose tissue biopsy specimens during the study; therefore, we are unable to determine whether GFT505 can directly activate PPARδ target genes in adipose tissue. Alternatively, the decrease in circulating FFA after GFT505 treatment could be attributable to an increase in FFA uptake and β-oxidation in the liver. Therefore, additional studies are needed to unravel the mechanism of action of GFT505 on FFA metabolism.
An intriguing finding of our study is that GFT505 reduces both γGT (−30%) and ALT (−20%) levels, suggesting an improvement in liver function in these severely insulin-resistant subjects. These data are in line with previous studies with GFT505 (20) and selective PPARδ agonists in pilot studies (18,35). Eight weeks of treatment with the PPARδ agonist MBX-8025 (100 mg/d) reduced γGT by 28% in overweight men with mixed dyslipidemia (35). In addition, 2 weeks of treatment with the PPARδ agonist GW501516 decreased γGT by 23% in moderately obese men, without improving ALT. Interestingly, GW501516 also reduced liver fat content by 20% (18), and this effect correlated with the change in γGT levels (18). The decrease of hepatic steatosis after PPARδ activation by GW501516 could be attributable to the reduced flux of FFA from adipose tissue or an increase in FFA uptake and β-oxidation in the liver. In contrast, there is no evidence for a beneficial role of PPARα agonists in human NAFLD because fenofibrate does not improve transaminase levels or hepatic fat content in several randomized studies (12–14).
The safety profile of GFT505 is reassuring, with no specific AEs. In contrast to what is observed with PPARγ agonists, there was neither body weight gain nor fluid retention. In addition, there were no significant increases in plasma creatinine or homocysteine levels, which are some classical PPARα-related side effects (37).
The study has several limitations. Our analysis was restricted to Caucasian men and studies in women are warranted. The duration of treatment remains too short to assess the long-term safety of the drug. In addition, statistical significance of secondary end points should be interpreted cautiously because of multiple comparisons. A measure of glucose/lipid oxidation by indirect calorimetry may allow determining the effect of GFT505 on FFA oxidation. A direct measure of liver fat content is lacking to reinforce the hypothesis that GFT505 improves NAFLD by reducing liver steatosis.
In conclusion, this study demonstrates that the dual PPARα/δ agonist GFT505 is a new insulin sensitizer that exerts a beneficial impact on multiple components of the metabolic syndrome, including hepatic and peripheral insulin resistance, dyslipidemia, inflammation, and possibly NAFLD. Many of these effects seem to be mediated by a combined activation of PPARδ and PPARα into the liver, but the precise mode of action of GFT505 remains to be further determined. Based on the results of this study and previous clinical trials, a randomized, double-blind, placebo-controlled, 1-year phase IIb study (ClinicalTrials.gov identifier NCT01694849) is currently ongoing and will assess the efficacy and safety of GFT505 in patients with histologically proven nonalcoholic steatohepatitis.
Acknowledgments
This study was funded by Genfit, Loos, France. B.C. received consultant fees from Genfit. R.H. and B.N. are employees of Genfit. B.S. received consultant fees from Genfit. No other potential conflicts of interest relevant to this article were reported.
B.S. is a member of the Institut Universitaire de France.
B.C. participated in the study as coordinating investigator, designed the study, wrote the first draft, and reviewed and edited the manuscript. R.H. designed the study and reviewed and edited the manuscript. S.L.-P. and Y.Z. participated in the study as coinvestigators. V.S. performed the stable isotopes analysis. B.N. performed the rodent experiments. L.F. prepared and provided the nonradioactive glucose tracer. H.V. performed the skeletal muscle biopsy analyses and reviewed and edited the manuscript. B.S. designed the study, contributed to the discussion, and reviewed and edited the manuscript. M.L. participated in the study as investigator, designed the study, contributed to the discussion, and reviewed and edited the manuscript. B.C. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were previously presented in poster form at the 74th Scientific Sessions of the American Diabetes Association, Philadelphia, Pennsylvania, 8–12 June 2012.
The authors acknowledge Pierre Clerson (Orgamétrie) for his advice on statistical analysis. The authors also thank Eliane Hivernaud (Center of Clinical Investigation Nantes), Jocelyne Peyrat (Centre de Recherche en Nutrition Humaine Rhône-Alpes), and Christine Maitrepierre (Centre de Recherche en Nutrition Humaine Rhône-Alpes) for their technical support during the clamp, and Laure Gabert and Corinne Louche-Pélissier (Centre de Recherche en Nutrition Humaine Rhône-Alpes) for mass spectrometry.
Footnotes
This article contains Supplementary Data online at http://care.diabetesjournals.org/lookup/suppl/doi:10.2337/dc12-2012/-/DC1.
Clinical trial reg. no. NCT01271777, www.clinicaltrials.gov.
References
- 1.Kahn CR. Insulin action, diabetogenes, and the cause of type II diabetes (Banting Lecture). Diabetes 1994;43:1066–1084 [DOI] [PubMed] [Google Scholar]
- 2.Saltiel AR, Kahn CR. Insulin signalling and the regulation of glucose and lipid metabolism. Nature 2001;414:799–806 [DOI] [PubMed] [Google Scholar]
- 3.Martin BC, Warram JH, Krolewski AS, Bergman RN, Soeldner JS, Kahn CR. Role of glucose and insulin resistance in development of type 2 diabetes mellitus: results of a 25-year follow-up study. Lancet 1992;340:925–929 [DOI] [PubMed] [Google Scholar]
- 4.Stolerman ES, Florez JC. Genomics of type 2 diabetes mellitus: implications for the clinician. Nat Rev Endocrinol 2009;5:429–436 [DOI] [PubMed] [Google Scholar]
- 5.Charbonnel B, Cariou B. Pharmacological management of type 2 diabetes: the potential of incretin-based therapies. Diabetes Obes Metab 2011;13:99–117 [DOI] [PubMed] [Google Scholar]
- 6.Inzucchi SE, Bergenstal RM, Buse JB, et al. American Diabetes Association (ADA) European Association for the Study of Diabetes (EASD) Management of hyperglycemia in type 2 diabetes: a patient-centered approach: position statement of the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetes Care 2012;35:1364–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Phielix E, Szendroedi J, Roden M. The role of metformin and thiazolidinediones in the regulation of hepatic glucose metabolism and its clinical impact. Trends Pharmacol Sci 2011;32:607–616 [DOI] [PubMed] [Google Scholar]
- 8.Juurinen L, Kotronen A, Granér M, Yki-Järvinen H. Rosiglitazone reduces liver fat and insulin requirements and improves hepatic insulin sensitivity and glycemic control in patients with type 2 diabetes requiring high insulin doses. J Clin Endocrinol Metab 2008;93:118–124 [DOI] [PubMed] [Google Scholar]
- 9.Miyazaki Y, Mahankali A, Matsuda M, et al. Effect of pioglitazone on abdominal fat distribution and insulin sensitivity in type 2 diabetic patients. J Clin Endocrinol Metab 2002;87:2784–2791 [DOI] [PubMed] [Google Scholar]
- 10.Cariou B, Charbonnel B, Staels B. Thiazolidinediones and PPARγ agonists: time for a reassessment. Trends Endocrinol Metab 2012;23:205–215 [DOI] [PubMed] [Google Scholar]
- 11.Lefebvre P, Chinetti G, Fruchart JC, Staels B. Sorting out the roles of PPAR alpha in energy metabolism and vascular homeostasis. J Clin Invest 2006;116:571–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belfort R, Berria R, Cornell J, Cusi K. Fenofibrate reduces systemic inflammation markers independent of its effects on lipid and glucose metabolism in patients with the metabolic syndrome. J Clin Endocrinol Metab 2010;95:829–836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fabbrini E, Mohammed BS, Korenblat KM, et al. Effect of fenofibrate and niacin on intrahepatic triglyceride content, very low-density lipoprotein kinetics, and insulin action in obese subjects with nonalcoholic fatty liver disease. J Clin Endocrinol Metab 2010;95:2727–2735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bajaj M, Suraamornkul S, Hardies LJ, Glass L, Musi N, DeFronzo RA. Effects of peroxisome proliferator-activated receptor (PPAR)-α and PPAR-γ agonists on glucose and lipid metabolism in patients with type 2 diabetes mellitus. Diabetologia 2007;50:1723–1731 [DOI] [PubMed] [Google Scholar]
- 15.Tanaka T, Yamamoto J, Iwasaki S, et al. Activation of peroxisome proliferator-activated receptor delta induces fatty acid beta-oxidation in skeletal muscle and attenuates metabolic syndrome. Proc Natl Acad Sci USA 2003;100:15924–15929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee CH, Olson P, Hevener A, et al. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci USA 2006;103:3444–3449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sprecher DL, Massien C, Pearce G, et al. Triglyceride:high-density lipoprotein cholesterol effects in healthy subjects administered a peroxisome proliferator activated receptor δ agonist. Arterioscler Thromb Vasc Biol 2007;27:359–365 [DOI] [PubMed] [Google Scholar]
- 18.Risérus U, Sprecher D, Johnson T, et al. Activation of peroxisome proliferator-activated receptor (PPAR)δ promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes 2008;57:332–339 [DOI] [PubMed] [Google Scholar]
- 19.Hanf R, Rubenstrunk A, Baron M, et al. GFT505, a dual PPARα/δ agonist has beneficial effects in animal models of NAFLD/NASH through PPARα-dependent and independent mechanisms (Abstract). Diabetologia 2011;54(Suppl. 1):S510 [Google Scholar]
- 20.Cariou B, Zaïr Y, Staels B, Bruckert E. Effects of the new dual PPAR α/δ agonist GFT505 on lipid and glucose homeostasis in abdominally obese patients with combined dyslipidemia or impaired glucose metabolism. Diabetes Care 2011;34:2008–2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979;237:E214–E223 [DOI] [PubMed] [Google Scholar]
- 22.Cariou B, Chetiveaux M, Zaïr Y, et al. Fasting plasma chenodeoxycholic acid and cholic acid concentrations are inversely correlated with insulin sensitivity in adults. Nutr Metab (Lond) 2011;8:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ekberg K, Landau BR, Wajngot A, et al. Contributions by kidney and liver to glucose production in the postabsorptive state and after 60 h of fasting. Diabetes 1999;48:292–298 [DOI] [PubMed] [Google Scholar]
- 24.Natali A, Ferrannini E. Effects of metformin and thiazolidinediones on suppression of hepatic glucose production and stimulation of glucose uptake in type 2 diabetes: a systematic review. Diabetologia 2006;49:434–441 [DOI] [PubMed] [Google Scholar]
- 25.Tiikkainen M, Häkkinen AM, Korsheninnikova E, Nyman T, Mäkimattila S, Yki-Järvinen H. Effects of rosiglitazone and metformin on liver fat content, hepatic insulin resistance, insulin clearance, and gene expression in adipose tissue in patients with type 2 diabetes. Diabetes 2004;53:2169–2176 [DOI] [PubMed] [Google Scholar]
- 26.Belfort R, Harrison SA, Brown K, et al. A placebo-controlled trial of pioglitazone in subjects with nonalcoholic steatohepatitis. N Engl J Med 2006;355:2297–2307 [DOI] [PubMed] [Google Scholar]
- 27.Ratziu V, Giral P, Jacqueminet S, et al. LIDO Study Group Rosiglitazone for nonalcoholic steatohepatitis: one-year results of the randomized placebo-controlled Fatty Liver Improvement with Rosiglitazone Therapy (FLIRT) Trial. Gastroenterology 2008;135:100–110 [DOI] [PubMed] [Google Scholar]
- 28.Hällsten K, Virtanen KA, Lönnqvist F, et al. Rosiglitazone but not metformin enhances insulin- and exercise-stimulated skeletal muscle glucose uptake in patients with newly diagnosed type 2 diabetes. Diabetes 2002;51:3479–3485 [DOI] [PubMed] [Google Scholar]
- 29.Rizza S, Cardellini M, Porzio O, et al. Pioglitazone improves endothelial and adipose tissue dysfunction in pre-diabetic CAD subjects. Atherosclerosis 2011;215:180–183 [DOI] [PubMed] [Google Scholar]
- 30.Naoumova RP, Kindler H, Leccisotti L, et al. Pioglitazone improves myocardial blood flow and glucose utilization in nondiabetic patients with combined hyperlipidemia: a randomized, double-blind, placebo-controlled study. J Am Coll Cardiol 2007;50:2051–2058 [DOI] [PubMed] [Google Scholar]
- 31.Debard C, Cozzone D, Ricard N, et al. Short-term activation of peroxysome proliferator-activated receptor beta/delta increases fatty acid oxidation but does not restore insulin action in muscle cells from type 2 diabetic patients. J Mol Med (Berl) 2006;84:747–752 [DOI] [PubMed] [Google Scholar]
- 32.Krämer DK, Al-Khalili L, Perrini S, et al. Direct activation of glucose transport in primary human myotubes after activation of peroxisome proliferator-activated receptor delta. Diabetes 2005;54:1157–1163 [DOI] [PubMed] [Google Scholar]
- 33.Shulman GI. Cellular mechanisms of insulin resistance. J Clin Invest 2000;106:171–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ooi EM, Watts GF, Sprecher DL, Chan DC, Barrett PH. Mechanism of action of a peroxisome proliferator-activated receptor (PPAR)-δ agonist on lipoprotein metabolism in dyslipidemic subjects with central obesity. J Clin Endocrinol Metab 2011;96:E1568–E1576 [DOI] [PubMed] [Google Scholar]
- 35.Bays HE, Schwartz S, Littlejohn T, 3rd, et al. MBX-8025, a novel peroxisome proliferator receptor-δ agonist: lipid and other metabolic effects in dyslipidemic overweight patients treated with and without atorvastatin. J Clin Endocrinol Metab 2011;96:2889–2897 [DOI] [PubMed] [Google Scholar]
- 36.Ginsberg HN, Elam MB, Lovato LC, et al. ACCORD Study Group Effects of combination lipid therapy in type 2 diabetes mellitus. N Engl J Med 2010;362:1563–1574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Taskinen MR, Sullivan DR, Ehnholm C, et al. FIELD study investigators Relationships of HDL cholesterol, ApoA-I, and ApoA-II with homocysteine and creatinine in patients with type 2 diabetes treated with fenofibrate. Arterioscler Thromb Vasc Biol 2009;29:950–955 [DOI] [PubMed] [Google Scholar]