

Abstract
OBJECTIVE
The Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial demonstrated similar long-term clinical effectiveness of insulin-sensitizing (IS) versus insulin-providing (IP) treatments for type 2 diabetes on cardiovascular outcomes in a cohort with documented coronary artery disease. We evaluated the effects of randomized glycemic control strategy (IS vs. IP) on the prevalence and incidence of diabetic peripheral neuropathy (DPN).
RESEARCH DESIGN AND METHODS
DPN (defined as Michigan Neuropathy Screening Instrument [MNSI] clinical examination score >2) was assessed at baseline and yearly for 4 years. DPN prevalence and incidence were compared by intention-to-treat modeling by logistic generalized estimating equation models for prevalence and Kaplan-Meier estimates and Cox regression models for incidence rates.
RESULTS
Results are reported for 2,159 BARI 2D participants (70% males) with valid baseline and at least one follow-up MNSI score (mean age 62 ± 9 years, mean HbA1c 7.7 ± 1.6%, diabetes duration 10 ± 9 years). There were no differences in the prevalence of DPN between the IS and the IP groups throughout the 4 years of follow-up. In 1,075 BARI 2D participants with no DPN at baseline, the 4-year cumulative incidence rate of DPN was significantly lower in the IS (66%) than in the IP (72%) strategy group (P = 0.02), which remained significant after adjusting for the in-trial HbA1c (P = 0.04). In subgroup analyses, IS strategy had a greater benefit in men (hazard ratio 0.75 [99% CI 0.58–0.99], P < 0.01).
CONCLUSIONS
Among patients with type 2 diabetes followed for up to 4 years during BARI 2D, a glycemic control therapy with IS significantly reduced the incidence of DPN compared with IP therapy and may add further benefit for men.
Diabetic peripheral neuropathies (DPNs) are consequences of diabetes-induced large and small, myelinated and unmyelinated, nerve fiber injury and are among the most common and perplexing complications of diabetes. Although the clinical manifestations, pattern of neurological deficits, symptoms, and clinical course are quite heterogeneous, DPN ultimately affects >50% of patients with diabetes (1). DPN is a major cause of disability and is associated with high mortality and poor quality of life (1). Patients with DPN have a 25% cumulative risk of a lower-extremity amputation (2). The 3-year survival rate in patients with DPN is 20% less than in age- and sex-matched diabetic patients without this complication (1,2).
Intensive glucose control has proven efficacy in delaying or preventing DPN in type 1 diabetes (T1DM) (3–5) but with less evidence for benefit in patients with type 2 diabetes (T2DM). In any case, most people with diabetes do not reach and maintain the glycemic levels needed to achieve these benefits (5–7). Despite promising preclinical data, large-scale pharmacologic interventions for established DPN have been disappointing. To date, no disease-modifying treatment other than glycemic control is available for DPN.
The Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial enrolled 2,368 participants with both T2DM and angiographically documented coronary artery disease (CAD). Participants were randomly assigned in a factorial design to either prompt revascularization or initial intensive medical therapy for CAD and to either insulin-sensitizing (IS) or insulin-providing (IP) drugs for glycemic control (8). The primary outcomes of BARI 2D have been reported (8). BARI 2D offered a unique opportunity to compare the effects of IS versus IP strategies on DPN outcomes among participants with T2DM and stable CAD.
In a previous cross-sectional analysis of the baseline DPN evaluations, we found that ∼50% of BARI 2D participants had DPN at baseline. Multivariate analysis showed that insulin use was associated with a higher prevalence of DPN, whereas IS use was associated with a trend for lower prevalence of DPN (9). The aim of the present longitudinal analysis was to determine whether after a 4-year follow-up, an IP-based strategy differed from an IS-based strategy with regard to the incidence of DPN onset and remission in the BARI 2D participants according to the absence or presence of DPN at baseline, respectively.
RESEARCH DESIGN AND METHODS
Study population
The design of BARI 2D has been previously described (10). Briefly, 2,368 participants with both T2DM and stable CAD were enrolled at 49 clinical sites in the U.S., Canada, Brazil, Mexico, the Czech Republic, and Austria between January 2001 and March 2005. Treatment continued until the 6-year visit or until the last annual visit before 1 December 2008. Eligibility criteria included patients with both T2DM and angiographically documented CAD suitable for a revascularization, percutaneous coronary intervention, or coronary artery bypass graft surgery ; with a glycated hemoglobin (HbA1c) ≤13.0%;with a creatinine level <2.0 mg/dL; and without class III or IV heart failure. The institutional review board at each participating site approved the protocol. All participants provided written informed consent. All data were analyzed at the coordinating center at the University of Pittsburgh. An independent data and safety monitoring board approved the study protocol and monitored the safety of the participants.
Treatment strategies
Participants were randomly assigned to two treatment strategies in a two-by-two factorial design, as follows: 1) prompt coronary revascularization plus intensive medical therapy strategy or intensive medical therapy with initially deferred revascularization and 2) an IS or IP strategy to achieve a target HbA1c <7.0%. Participants assigned to the IS strategy were treated with metformin, thiazolidinediones (TZDs), or both if needed. Participants assigned to the IP strategy were treated with sulfonylureas or meglitinides and insulin if needed. During follow-up, drug agents in the opposite strategy could be added by the site diabetologist if believed necessary to achieve the target HbA1c goal. Therefore, participants randomized to the IP strategy could be prescribed IS drugs and participants randomized to the IS strategy could be prescribed IP drugs to achieve an HbA1c <7.0% as detailed in the study protocol. At least one drug from each of the major antidiabetic drug classes was available during the study at no cost to the participants, and rosiglitazone was the TZD provided at no cost.
DPN assessment in BARI 2D
In BARI 2D, DPN was assessed with the Michigan Neuropathy Screening Instrument (MNSI) at baseline and at annual examinations thereafter for an average of 4.5 years. The MNSI is a validated clinical screening assessment for DPN and has been widely used in clinical trials and longitudinal cohort studies, including the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications (11,12). It includes two separate assessments: a 15-item interviewer-administered symptom score questionnaire and a lower-extremity clinical examination that includes foot inspection and assessment of vibratory sensation and ankle reflexes (13). A symptom score of ≥7 (of 15) has been shown to have high specificity but low sensitivity in identifying DPN (11,13). The MNSI clinical examination score has been validated to confirm the presence of DPN as defined by clinical symptoms, physical examination signs, and nerve conduction abnormalities consistent with distal symmetrical peripheral neuropathy (11,13). A cutoff score of >2 was shown to have the highest sensitivity and specificity of correctly classifying DPN (11,13); therefore, in the present analysis, DPN was defined as an MNSI clinical examination score of >2. Although the overall BARI 2D trial had an average 5.3-year follow-up, the present analysis focused on data from entry to 4-year follow-up because only a few sites started participant enrollment earlier.
DPN outcomes
The main outcomes of this analysis were to determine the incidence of DPN in the subset without DPN at baseline and the incidence of DPN remission in those with DPN at baseline. The incidence of DPN was defined as the first occurrence of MNSI score >2 during follow-up among participants with baseline MNSI scores ≤2. The incidence of DPN remission was defined as the first reversal of an MNSI score to ≤2 during follow-up among participants with baseline MNSI scores >2. The prevalence of DPN was defined as the proportion of participants with MNSI score >2 at baseline and at each annual visit. The prevalence of symptoms was defined as the proportion of participants with a symptom score ≥7.
By the BARI 2D protocol, the time window of acceptance for each annual visit was from 12 weeks before the randomization anniversary date through 36 weeks after the randomization anniversary date. The BARI 2D database showed that 97% of the participants accomplished their annual visits within 3 months after each anniversary date. Therefore, the incidence of DPN onset and remission were determined at the end of the first quarter of each follow-up year.
Statistical methods
Data were analyzed on an intention-to-treat basis between randomized IS strategy and randomized IP strategy. The randomization balance at baseline was assessed within two baseline groups: the group with baseline MNSI score ≤2 (no DPN) and the group with baseline MNSI score >2 (DPN). Chi-square tests were performed for categorical variables, and t tests were performed for continuous variables. In addition, per-protocol analyses of only patients who remained in the assigned treatment arm are reported because of concern about the rate of crossover between treatment arms.
The prevalence of DPN at each year was compared with χ2 tests. A logistic model with a generalized estimating equation approach was constructed to estimate the overall odds ratios between IS and IP strategies. The interaction effect between the randomized IS/IP strategy and follow-up time were tested and dropped when not significant.
The rates of DPN onset and remission were computed with Kaplan-Meier estimates. The differences were tested by a log-rank test. A Cox regression model was constructed to estimate the hazard ratios (HRs) of IS versus IP strategies after adjustment for in-trial HbA1c as a time-varying covariate. In the subgroup analyses, the HRs of DPN were evaluated in the prespecified subgroups as defined by age, sex, baseline HbA1c, baseline triglyceride level, and baseline MNSI score to demonstrate the different effects of the IS versus IP strategy.
The statistical significance level was set at 0.05 for the general prevalence and cumulative rate comparisons. In subgroup analyses, the significance level was set at 0.01 to control for multiple comparisons. The statistical analyses were performed with SAS version 9.2 (SAS Institute Inc., Cary, NC) software, and all figures were plotted with R version 2.8 (R Development Core Team, www.R-project.org). Data are presented as mean ± SD.
RESULTS
Results are reported for 2,159 BARI 2D participants with valid baseline and at least one follow-up MNSI examination score (BARI 2DN) (Table 1), who represent >91% of the BARI 2D study population. Death or withdrawal during the first year of the trial was the most common reason for exclusion. The excluded participants were slightly older (64 ± 10 vs. 62 ± 9 years, P = 0.01), had a slightly higher prevalence of hypertension (89 vs. 82%, P = 0.007), had a higher prevalence of macroalbuminuria (16 vs. 9%, P = 0.002), and consumed a higher proportion of alcohol beyond recommended levels (9 vs. 3%, P < 0.0001). The groups were similar in many other characteristics, including percentage of females, race/ethnicity, diabetes duration, HbA1c, systolic and diastolic blood pressure (BP), lipid variables, and various classes of medication at baseline (Supplementary Table 1).
Table 1.
Baseline characteristics of BARI 2D participants randomized to IS vs. IP strategy stratified by baseline DPN
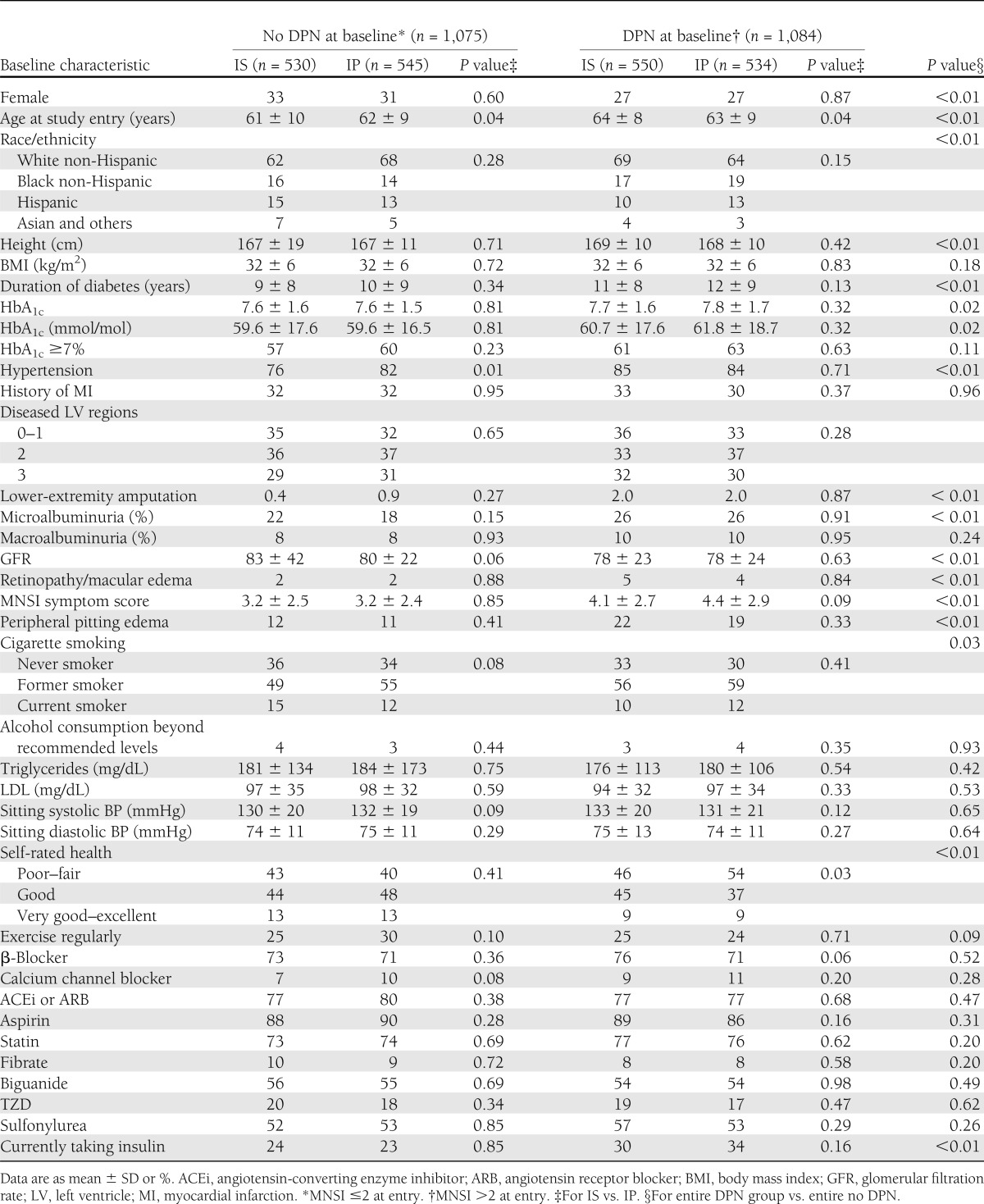
As a whole, the BARI 2DN cohort included participants 62 ± 9 years of age with a mean HbA1c of 7.7 ± 1.6% and mean diabetes duration of 10 ± 9 years at baseline. They were ∼30% women and well represented by minorities (∼37%), with ∼17% black non-Hispanics and ∼13% Hispanics. At baseline, 50% of participants had DPN (MNSI clinical score >2). Approximately 16% of the BARI 2 DN participants had a MNSI symptom score ≥7. Those with DPN compared with those without DPN at baseline were older (63 ± 9 vs. 61 ± 9 years, P < 0.01), were more likely to be male (73 vs. 68%, P < 0.01), had longer diabetes duration (11 ± 9 vs. 10 ± 8 years, P < 0.01), had slightly higher HbA1c (7.7 ± 1.6 vs. 7.6 ± 1.6%, P = 0.02), and had a higher prevalence of microalbuminuria (26 vs. 20%, P < 0.01). Participants with DPN had a significantly greater number of symptoms (P < 0.001) and were twice as likely to have an MNSI symptom score ≥7 (21 vs. 10%, P < 0.001) compared with those without DPN. There were no differences in the symptoms score by treatment strategy group assignment (IS vs. IP) (Table 1).
Table 1 presents clinical characteristics of the BARI 2DN cohort stratified by DPN status as follows: without DPN at baseline (no DPN) and with DPN at baseline (DPN). The IS and IP strategy groups were balanced at baseline in most variables. In the no DPN group, participants randomized to IP were slightly older and had a slightly greater incidence of hypertension than those randomized to IS. In the DPN group, participants randomized to IS were slightly older than those randomized to IP.
Similar to observations seen in the entire BARI 2D cohort, by 6 months, HbA1c was significantly lower in the IS strategy arm than in the IP strategy arm, and this difference was maintained through study end (7.1 ± 1.4 vs. 7.6 ± 1.4% in IS vs. IP at year 4, P < 0.01). At study end, there were no differences between the two treatments arms in the systolic and diastolic BP and LDL cholesterol and triglyceride levels. However, body mass index and waist circumference were slightly lower in IS arm than in the IP arm (32 ± 7 vs. 33 ± 6 kg/m2 [P = 0.01] and 108 ± 15 vs. 110 ± 14 cm [P = 0.04], respectively). At the end of the first year, 36% of the BARI 2D participants randomized to IS were also taking IP drugs, and 8% in IP were also taking IS drugs. At the end of year 4, 48% of participants in IS were also taking IP drugs, and 14% in IP were also taking IS drugs.
The overall prevalence of DPN throughout the 4 years of follow-up was similar between IS (51%) and IP (53%) strategies (P = 0.43). The overall odds ratio of DPN during the 4-year follow-up between IS and IP strategy was 0.99 (95% CI 0.87–1.12, P = 0.83).
Among the 1,075 participants with no DPN at entry, the 4-year cumulative incidence rate of DPN was significantly lower in the IS strategy group (66%) than in the IP strategy group (72%) (P = 0.02) (Fig. 1A). After adjusting for in-trial HbA1c, the HR of incident DPN for IS versus IP was 0.84 (95% CI 0.71–0.99, P = 0.04) (Fig. 2). Among the 1,084 participants with DPN at entry, the 4-year remission rate of DPN was not different between IS and IP strategy (P = 0.2).
Figure 1.

A: Four-year incidence rates of DPN in the IS vs. IP treatment group in BARI 2D. B: Per-protocol 4-year incidence rates of DPN in the IS vs. IP treatment group in BARI 2D.
Figure 2.
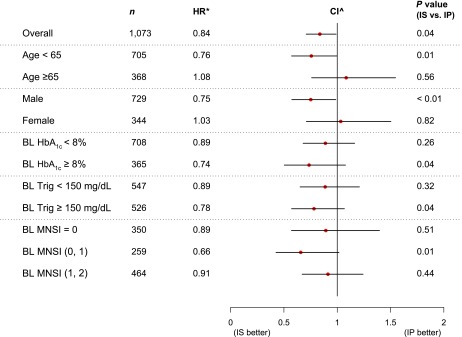
Subgroup analysis for 4-year DPN incidence. *Adjusted for in-trial HbA1c as a time-varying covariate; ^95% CI for overall, 99% CI for subgroups. BL, baseline; Trig, triglyceride level.
Considering the effect of crossover from IS to additional IP drugs, as described previously, we performed a separate per-protocol analysis restricted only to participants who remained on the designated treatment without the addition of treatment from the other arm. However, the characteristics of the participants who remained on their assigned treatment for the duration of the trial were different from those who required the addition of treatment from the other arm. In particular, IS participants who remained on per-protocol treatment had a shorter duration of diabetes and lower HbA1c and were less likely to be taking insulin at baseline than were IP participants who remained on per-protocol treatment (P < 0.001 for all) (Supplementary Table 2).
The per-protocol analysis showed no significant treatment difference in DPN prevalence at the year 1 visit (P = 0.26), but a significant separation between IS and IP arms was observed in year 2 (45 vs. 56%, P < 0.001) and year 3 (46 vs. 57%, P < 0.001), and a marginally significant separation was observed in year 4 (48 vs. 54%, P = 0.08). The overall odds ratio of any DPN during the 4-year follow-up in the per-protocol analysis between IS and IP strategy was 0.69 (95% CI 0.54–0.89, P < 0.001). In multivariate analyses adjusting also for duration of diabetes and insulin use at study entry, the odds ratio for DPN was 0.85 (0.65–1.11, P = 0.22) between IS and IP strategy.
Among participants with no DPN at study entry, the per-protocol cumulative incidence was 69% overall (61 and 74% in the IS and IP groups, respectively, at year 4, P < 0.001). The odds ratio of incident DPN among those with no DPN at study entry was 0.59 (95% CI 0.43–0.82, P < 0.001) between the IS and the IP per-protocol treatment groups (Fig. 1B). After adjusting for diabetes duration and insulin use at study entry, the odds ratio for incident DPN was 0.68 (0.49–0.95, P = 0.02) between IS and IP strategy.
Subgroup analyses demonstrated the different effects of the IS and IP strategies in specific subgroups (Fig. 2). After adjusting for the in-trial HbA1c as a time-varying covariate, the IS strategy had a greater effect in males (P < 0.01). A trend for benefit with the IS strategy was observed in participants in the following baseline subgroups: <65 years of age (P = 0.01), MNSI score ≤1 (P = 0.01), HbA1c ≥8%, and triglyceride level ≥150 mg/dL (P = 0.04).
CONCLUSIONS
Among patients with T2DM and stable CAD, these data demonstrate that a glycemic control therapy with IS significantly reduced the cumulative incidence of the new onset of DPN compared with IP therapy among those without DPN at baseline who were followed for up to 4.5 years during BARI 2D trial. This effect was also observed in a per-protocol analysis restricted only to participants who remained on the designated treatment. However, the cumulative incidence of DPN remission among those with DPN at baseline was not different between IS and IP strategy groups.
Hyperglycemia has been regarded as the major culprit for initiating the cascade of metabolic and molecular abnormalities that result in a degenerative phenomena and progressive neurological deficits. Although tight glucose control was shown to prevent neuropathy in patients with T1DM (3,4,14), trials designed to achieve similar glycemic control in patients with T2DM reported less efficacy (5–7). These studies suggest that factors other than hyperglycemia, including metabolic factors such as dyslipidemia or other components of the metabolic syndrome and chronic inflammation, are involved in the pathophysiology of DPN. The BARI 2D trial was designed to maintain similar glycemic targets in the two medical arms (HbA1c <7.0%) to allow a comparison between mechanistically distinct treatment strategies. Although the HbA1c was lower in the IS arm throughout BARI 2D, the beneficial effects of IS treatment on DPN incidence persisted after adjusting for the in-trial HbA1c. However, the adjustment for HbA1c does not completely exclude a potential glycemic-mediated effect in this difference because other factors such as glucose variability and the timing of changes in glucose level could have also played a role.
Emerging data suggest that most of the available agents used to treat hyperglycemia may promote additional effects that could directly interact with the development of complications independently of glucose lowering. These effects include lipid metabolism, body weight, oxidative stress, or chronic inflammation.
Evidence for an important role of low-grade inflammation in the pathogenesis of DPN is emerging from both experimental and clinical studies. Experimental evidence obtained in animal models of diabetes show an enhanced inflammatory response in diabetic nerves mediated by nuclear factor-κB (NF-κB) activation (15,16) and upregulation of multiple inflammatory mediators, including tumor necrosis factor-α, in the sensory neurons from T2DM models early in the course of DPN (17). Human data show that subjects with DPN have increased serum levels of inflammatory cytokines compared with those without DPN (18). Microarray experiments detected differentially expressed genes functionally enriched in pathways involving inflammatory responses and lipid metabolism in patients with progressive DPN (19). In this respect, experimental and human data demonstrate that TZDs may reduce inflammatory responses through NF-κB blockade (20,21). Metformin has also been shown to exert direct anti-inflammatory effects by inhibiting NF-κB–derived inflammatory cytokines through blockade of the phosphatidylinositol 3-kinase–Akt pathways (22). Because evidence from the present cohort of the BARI 2 D trial clearly shows that the IS treatment strategy leads to diminished intensity of the systemic inflammatory state compared with the IP strategy (23), it is possible that one of the beneficial effects on DPN observed in the IS arm may be associated with these effects on chronic inflammation.
Additional effects of IS agents on other DPN-related mechanisms are possible. For instance, TZDs may reduce oxidative stress by modulating the altered expression of reduced NADPH oxidase and by promoting improvement of mitochondrial function (24,25) independently from their glucose-lowering and IS properties. TZD may also reduce generation of advanced glycation end products and may prevent the activation of protein kinase C (26,27). They were shown to slow or prevent the development of neuropathy in animal models of T1DM by maintaining normal myelinated fiber architecture and number, reducing macrophage infiltration in the sciatic nerve, and modulating key regulatory elements in genes involved in DPN progression (28,29).
Treatment with metformin may also have a protective effect in the development of diabetes complications independently of their conventional antihyperglycemic effects. Metformin is associated with a favorable effect on weight gain that could have offset the usual weight gain associated with TZD use, explaining the lack of weight gain observed in the BARI 2D participants randomized to IS arm compared with weight gain in the IP arm. Metformin also has pleiotropic actions with direct vascular effects, such as improvement in lipid profiles (30), prevention of oxidative stress-induced endothelial cell death (31), and direct neuroprotective effects in primary neurons through inhibition of apoptotic cell death related to oxidative stress (32). Conversely, long-term metformin exposure may be an iatrogenic cause for exacerbation of peripheral neuropathy in patients with T2DM because metformin is associated with lower serum vitamin B12 levels, higher serum homocysteine and methylmalonic acid levels, and clinically more severe peripheral neuropathy than in similar patients with no metformin exposure (33).
An alternate hypothesis for these findings is that the IP agents are harmful to peripheral nerves. Although not in keeping with the burden of evidence in patients with T2DM (5) and with theoretical mechanisms of benefit to peripheral nerves (34), experimental evidence suggests that the hyperinsulinemia induced by the exogenous administration of insulin—and by extension, the hyperinsulinemia induced by insulin secretagogues—may result in neuronal insulin resistance, which in turn may cause neuronal injury through the impairment of mitochondrial fission (35). In addition, the weight gain observed with the IP treatment could have contributed to an increase in chronic inflammation and oxidative stress (36). Finally, one can speculate that the higher incidence of hypoglycemia in the IP arm throughout the trial and the possibly higher glucose variability in these participants may also have contributed to a higher rate of diabetes complications (37).
The strengths of this study include the large sample size, large proportion of women and minorities, and characterization for multiple complications and vascular risk factors. In addition, the randomized design targeting the same values of HbA1c allowed for the evaluation of antihyperglycemic strategies acting through distinct mechanisms but with comparable tight glucose control.
The limitations of this study include the post hoc nature of the analysis, the fact that the BARI 2D trial was not powered to detect an effect on DPN outcomes, and the crossover between IP and IS arms during the trial. Although the crossover could not be controlled in the design of the study, we expected the results of an impact of one treatment strategy over the other would be biased toward the null, which was confirmed in the per-protocol analysis. In addition, although we found an association of the IS strategy with lower neuropathy incidence, we were unable to identify whether the benefit was specific to biguanides or TZDs. The MNSI is a meaningful and highly predictive clinical test, but it provides information on only a large-fiber dysfunction; therefore, changes consistent with small-fiber neuropathy, an important component of diabetic neuropathies, could not be adequately assessed. Finally, the subjective nature of the MNSI may have contributed to the sizable number of participants with incident reversal of neuropathy score, although overall, the cumulative incidence of neuropathy showed a significant net increase.
In summary, this analysis suggests that in patients with T2DM and stable CAD, a therapeutic algorithm favoring an IS over an IP regimen is protective against the new onset of DPN but is not of sufficient benefit in DPN remission. Future research is warranted to confirm these findings and to better understand the mechanisms associated with a possible beneficial effect of IS agents on peripheral nerve function as well as a putative injurious effect of insulin administration and insulin secretagogues.
Acknowledgments
The trial was sponsored by the National Heart, Lung, and Blood Institute and the National Institute of Diabetes and Digestive and Kidney Diseases (U01-HL-061744, U01-HL-061746, U01-HL-061748, U01-HL-063804), with additional support from multiple industry sponsors, including the manufacturer of rosiglitazone GlaxoSmithKline (a detailed list is provided in Supplementary Funding Sources).
R.P.-B. received a research grant from Amylin Pharmaceuticals (now Bristol-Myers Squibb) unrelated to the manuscript. J.G. received a research grant from Amylin Pharmaceuticals (now Bristol-Myers Squibb) and Merck unrelated to this manuscript and is a member of the Merck speakers’ bureau. B.A.P. received speaker honoraria from Medtronic Inc., Johnson and Johnson, Roche, GlaxoSmithKline Canada, Novo Nordisk, Eli Lilly, and sanofi-aventis; has received research grant support from Medtronic and Boehringer Ingelheim; and serves on an advisory board for Neurometrix, Inc. No other potential conflicts of interest relevant to this article were reported.
Industry sponsors had no role in the collection or analysis of the data, had no access to outcome data at any time during the trial, and did not participate in the design or drafting of this manuscript or its content. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute; the National Institute of Diabetes and Digestive and Kidney Diseases; or the National Institutes of Health.
R.P.-B. researched data and wrote the manuscript. J.L. researched data and contributed to the results and discussion. M.M.B., J.E., P.P., and F.W. reviewed and edited the manuscript. S.A. and J.G. contributed to the discussion and reviewed and edited the manuscript. A.D.A. and T.L.Z.J. researched data and reviewed and edited the manuscript. B.A.P. researched data and contributed to discussion. R.P.-B. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented in abstract form at the 72nd Scientific Sessions of the American Diabetes Association, Philadelphia, Pennsylvania, 8–12 June 2012, and at the 22nd Annual Meeting of the Diabetic Neuropathy Study Group of the European Association for the Study of Diabetes, Dresden, Germany, 27–30 September 2012.
Footnotes
Clinical trial reg. no. NCT00006305, clinicaltrials.gov.
This article contains Supplementary Data online at http://care.diabetesjournals.org/lookup/suppl/doi:10.2337/dc13-0012/-/DC1.
A slide set summarizing this article is available online.
References
- 1.Tesfaye S, Boulton AJ, Dyck PJ, et al. Toronto Diabetic Neuropathy Expert Group Diabetic neuropathies: update on definitions, diagnostic criteria, estimation of severity, and treatments. Diabetes Care 2010;33:2285–2293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boulton AJ, Vileikyte L, Ragnarson-Tennvall G, Apelqvist J. The global burden of diabetic foot disease. Lancet 2005;366:1719–1724 [DOI] [PubMed] [Google Scholar]
- 3.The Diabetes Control and Complications Trial Research Group The effect of intensive treatment of diabetes on the development and progression of long-term complications in insulin-dependent diabetes mellitus. N Engl J Med 1993;329:977–986 [DOI] [PubMed] [Google Scholar]
- 4.Effect of intensive diabetes treatment on nerve conduction in the Diabetes Control and Complications Trial. Ann Neurol 1995;38:869–880 [DOI] [PubMed] [Google Scholar]
- 5.Callaghan BC, Little AA, Feldman EL, Hughes RA. Enhanced glucose control for preventing and treating diabetic neuropathy. Cochrane Database Syst Rev 2012;6:CD007543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Azad N, Emanuele NV, Abraira C, et al. The effects of intensive glycemic control on neuropathy in the VA cooperative study on type II diabetes mellitus (VA CSDM). J Diabetes Complications 1999;13:307–313 [DOI] [PubMed] [Google Scholar]
- 7.UK Prospective Diabetes Study (UKPDS) Group Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998;352:837–853 [PubMed] [Google Scholar]
- 8.Frye RL, August P, Brooks MM, et al. BARI 2D Study Group A randomized trial of therapies for type 2 diabetes and coronary artery disease. N Engl J Med 2009;360:2503–2515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pop-Busui R, Lu J, Lopes N, Jones TL, BARI 2D Investigators Prevalence of diabetic peripheral neuropathy and relation to glycemic control therapies at baseline in the BARI 2D cohort. J Peripher Nerv Syst 2009;14:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brooks MM, Frye RL, Genuth S, et al. Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) Trial Investigators Hypotheses, design, and methods for the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) Trial. Am J Cardiol 2006;97(12A):9G–19G [DOI] [PubMed] [Google Scholar]
- 11.Herman WH, Pop-Busui R, Braffett BH, et al. DCCT/EDIC Research Group Use of the Michigan Neuropathy Screening Instrument as a measure of distal symmetrical peripheral neuropathy in type 1 diabetes: results from the Diabetes Control and Complications Trial/Epidemiology of Diabetes Interventions and Complications. Diabet Med 2012;29:937–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martin CL, Albers J, Herman WH, et al. DCCT/EDIC Research Group Neuropathy among the diabetes control and complications trial cohort 8 years after trial completion. Diabetes Care 2006;29:340–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Feldman EL, Stevens MJ, Thomas PK, Brown MB, Canal N, Greene DA. A practical two-step quantitative clinical and electrophysiological assessment for the diagnosis and staging of diabetic neuropathy. Diabetes Care 1994;17:1281–1289 [DOI] [PubMed] [Google Scholar]
- 14.Albers JW, Herman WH, Pop-Busui R, et al. Diabetes Control and Complications Trial /Epidemiology of Diabetes Interventions and Complications Research Group Effect of prior intensive insulin treatment during the Diabetes Control and Complications Trial (DCCT) on peripheral neuropathy in type 1 diabetes during the Epidemiology of Diabetes Interventions and Complications (EDIC) Study. Diabetes Care 2010;33:1090–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wang Y, Schmeichel AM, Iida H, Schmelzer JD, Low PA. Enhanced inflammatory response via activation of NF-kappaB in acute experimental diabetic neuropathy subjected to ischemia-reperfusion injury. J Neurol Sci 2006;247:47–52 [DOI] [PubMed] [Google Scholar]
- 16.Cameron NE, Cotter MA. Pro-inflammatory mechanisms in diabetic neuropathy: focus on the nuclear factor kappa B pathway. Curr Drug Targets 2008;9:60–67 [DOI] [PubMed] [Google Scholar]
- 17.Cheng HT, Dauch JR, Oh SS, Hayes JM, Hong Y, Feldman EL. p38 mediates mechanical allodynia in a mouse model of type 2 diabetes. Mol Pain 2010;6:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Doupis J, Lyons TE, Wu S, Gnardellis C, Dinh T, Veves A. Microvascular reactivity and inflammatory cytokines in painful and painless peripheral diabetic neuropathy. J Clin Endocrinol Metab 2009;94:2157–2163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hur J, Sullivan KA, Pande M, et al. The identification of gene expression profiles associated with progression of human diabetic neuropathy. Brain 2011;134:3222–3235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Haffner SM, Greenberg AS, Weston WM, Chen H, Williams K, Freed MI. Effect of rosiglitazone treatment on nontraditional markers of cardiovascular disease in patients with type 2 diabetes mellitus. Circulation 2002;106:679–684 [DOI] [PubMed] [Google Scholar]
- 21.Agarwal R. Anti-inflammatory effects of short-term pioglitazone therapy in men with advanced diabetic nephropathy. Am J Physiol Renal Physiol 2006;290:F600–F605 [DOI] [PubMed] [Google Scholar]
- 22.Isoda K, Young JL, Zirlik A, et al. Metformin inhibits proinflammatory responses and nuclear factor-kappaB in human vascular wall cells. Arterioscler Thromb Vasc Biol 2006;26:611–617 [DOI] [PubMed] [Google Scholar]
- 23.Sobel BE, Hardison RM, Genuth S, et al. BARI 2D Investigators Profibrinolytic, antithrombotic, and antiinflammatory effects of an insulin-sensitizing strategy in patients in the Bypass Angioplasty Revascularization Investigation 2 Diabetes (BARI 2D) trial. Circulation 2011;124:695–703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ghosh S, Patel N, Rahn D, et al. The thiazolidinedione pioglitazone alters mitochondrial function in human neuron-like cells. Mol Pharmacol 2007;71:1695–1702 [DOI] [PubMed] [Google Scholar]
- 25.Hwang J, Kleinhenz DJ, Rupnow HL, et al. The PPARgamma ligand, rosiglitazone, reduces vascular oxidative stress and NADPH oxidase expression in diabetic mice. Vascul Pharmacol 2007;46:456–462 [DOI] [PubMed] [Google Scholar]
- 26.Haneda M, Koya D, Kikkawa R. Cellular mechanisms in the development and progression of diabetic nephropathy: activation of the DAG-PKC-ERK pathway. Am J Kidney Dis 2001;38(Suppl. 1):S178–S181 [DOI] [PubMed] [Google Scholar]
- 27.Martens FM, Visseren FL, de Koning EJ, Rabelink TJ. Short-term pioglitazone treatment improves vascular function irrespective of metabolic changes in patients with type 2 diabetes. J Cardiovasc Pharmacol 2005;46:773–778 [DOI] [PubMed] [Google Scholar]
- 28.Yamagishi S, Ogasawara S, Mizukami H, et al. Correction of protein kinase C activity and macrophage migration in peripheral nerve by pioglitazone, peroxisome proliferator activated-gamma-ligand, in insulin-deficient diabetic rats. J Neurochem 2008;104:491–499 [DOI] [PubMed] [Google Scholar]
- 29.Wiggin TD, Kretzler M, Pennathur S, Sullivan KA, Brosius FC, Feldman EL. Rosiglitazone treatment reduces diabetic neuropathy in streptozotocin-treated DBA/2J mice. Endocrinology 2008;149:4928–4937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu MS, Johnston P, Sheu WH, et al. Effect of metformin on carbohydrate and lipoprotein metabolism in NIDDM patients. Diabetes Care 1990;13:1–8 [DOI] [PubMed] [Google Scholar]
- 31.Detaille D, Guigas B, Chauvin C, et al. Metformin prevents high-glucose-induced endothelial cell death through a mitochondrial permeability transition-dependent process. Diabetes 2005;54:2179–2187 [DOI] [PubMed] [Google Scholar]
- 32.El-Mir MY, Detaille D, R-Villanueva G, et al. Neuroprotective role of antidiabetic drug metformin against apoptotic cell death in primary cortical neurons. J Mol Neurosci 2008;34:77–87 [DOI] [PubMed] [Google Scholar]
- 33.Wile DJ, Toth C. Association of metformin, elevated homocysteine, and methylmalonic acid levels and clinically worsened diabetic peripheral neuropathy. Diabetes Care 2010;33:156–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Qiang X, Satoh J, Sagara M, et al. Gliclazide inhibits diabetic neuropathy irrespective of blood glucose levels in streptozotocin-induced diabetic rats. Metabolism 1998;47:977–981 [DOI] [PubMed] [Google Scholar]
- 35.Kim B, Feldman EL. Insulin resistance in the nervous system. Trends Endocrinol Metab 2012;23:133–141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Xu H, Barnes GT, Yang Q, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 2003;112:1821–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirsch IB, Brownlee M. Beyond hemoglobin A1c—need for additional markers of risk for diabetic microvascular complications. JAMA 2010;303:2291–2292 [DOI] [PubMed] [Google Scholar]