

Abstract
Background
Macrophage migration inhibitory factor (MIF) exerts a protective effect on ischemic myocardium by activating AMP-activated protein kinase (AMPK). Small molecules that increase the affinity of MIF for its receptor have been recently designed, and we hypothesized that such agonists may enhance AMPK activation and limit ischemic tissue injury.
Methods and Results
Treatment of cardiomyocytes with the candidate MIF agonist, MIF20, augmented AMPK phosphorylation, increased by 50% the surface localization of glucose transporter, and enhanced by 25% cellular glucose uptake when compared to MIF alone. In mouse hearts perfused with MIF20 prior to no-flow ischemia and reperfusion, post-ischemic left ventricular function improved commensurately with an increase in cardiac MIF-AMPK activation and an augmentation in myocardial glucose uptake. By contrast, small molecule MIF agonism was not effective in cells or tissues genetically deficient in MIF or the MIF receptor, verifying the specificity of MIF20 for MIF-dependent AMPK signaling. The protective effect of MIF20 also was evident in an in vivo regional ischemia model. Mice treated with MIF20 followed by left coronary artery occlusion and reperfusion showed a significant reduction in infarcted myocardium.
Conclusions
These data support the pharmacologic utility of small molecule MIF agonists in enhancing AMPK activation and reducing cardiac ischemic injury.
Keywords: MIF agonist, glucose transporter, ischemia
Introduction
Recent studies have identified a unique role for the innate cytokine, MIF, in cellular metabolism. MIF is released from diverse cell types in response to activating stimuli and it regulates glycolysis and glucose utilization in target tissues 1-4. These activities initially were defined in the context of systemic inflammation, however there is also evidence that MIF exerts a more fundamental role in the metabolic response to environmental stress 5, 6. Within the heart, MIF is released by ischemic cardiomyocytes and acts by autocrine/paracrine signaling to increase the phosphorylation of AMP-activated protein kinase (AMPK), leading to enhanced glucose uptake and protection from ischemic injury and cellular apoptosis. Circulating MIF levels increase during myocardial infarction, suggesting that this pathway is active during cardiac ischemia 7, 8. Notably, functional polymorphisms in MIF occur commonly in the population and human cells that express a low MIF expression allele show reduced activation of AMPK during hypoxia that is corrected by exogenous MIF administration 5. The MIF-AMPK activation pathway also is impaired during normal aging because of a reduction in the hypoxia-inducible factor-1α mediated transcriptional activation of MIF 6.
MIF activates AMPK phosphorylation by signaling through a membrane receptor complex that is comprised of the MIF binding protein, CD74, and the signal transduction component, CD44 5, 9, 10. MIF interacts with CD74 via contacts in the N-terminal region of the cytokine; this is also the site of MIF’s intrinsic tautomerase activity, which is considered to be vestigial and nonphysiologic 11. The availability of a high resolution crystal structure of MIF has enabled the discovery of tautomerase inhibitors and a subset of such inhibitors have been found to additionally interfere with MIF’s high affinity interaction with CD74 12-15. A structure-based molecular design study that focused on the MIF tautomerase site has led to the discovery of not only small molecule MIF antagonists but also the first agonists that enhance MIF interaction with CD74. Both antagonists and agonists bind to the MIF tautomerization site and appear to differentially influence structural or dynamic features of the MIF:MIF receptor interaction 13, 16. We performed functional characterization of candidate MIF agonists and hypothesized that small molecules capable of increasing MIF interaction with its receptor could prove useful in augmenting MIF-dependent AMPK signaling and limiting ischemic cardiac injury. In the present report, we assessed the therapeutic impact of this novel pharmacologic approach for protection against cardiac stress responses and ischemic injury.
Materials and Methods
Reagents and MIF Binding Studies
Mouse recombinant macrophage migration inhibitory factor (MIF) was prepared as described previously 17 and MIF20, MIF21, and MIF33 were synthesized as described by Jorgensen et al. 16. The MIF antagonist, ISO-1, was purchased from Calbiochem (San Diego, CA). The binding of MIF to its receptor, CD74, first was measured in a cell-free assay employing biotinylated MIF (83 nM of trimer) and immobilized recombinant CD74 ectodomain (sCD74 = CD7473-232) as previously described 13. Biotinylated-MIF pre-incubated with MIF20, MIF21, or MIF33 was introduced at concentrations of 1 – 1000 nM (calculated as MIF trimer) together to plate-bound sCD74 and incubated overnight at 4°C. After washing 4 times, bound biotinylated-MIF was detected with a streptavidin-alkaline phosphatase conjugate. Separate experiments established that the addition of small molecule MIF agonists or antagonists to immobilized sCD74 followed by washing did not to influence subsequent MIF binding.
The real-time binding interaction of human MIF with sCD74 was measured by surface plasmon resonance using a BIAcore T100 optical biosensor, the Series S sensor chip CM5, and the BIA Evaluation software (Biacore GE Healthcare) 9. Recombinant sCD74 (1 μM) was immobilized by amine coupling (Biacore Amine Coupling Kit) and the remaining active carboxylic residues neutralized with ethanolamine. The derived sensor chips then were washed and equilibrated in HBS buffer (1 mM DTT, 2.5 mM MgCl2, 20 mM HEPES, 1 mM EDTA, 150 mM NaCl, 0.005% P20) at a flow rate 20 μl/min, and MIF was introduced at five serial dilutions (62.5 nM, 125 nM, 250 nM, 500 nM, 1000 nM) in HBS buffer with or without 5 μM of MIF20 in 60 μl injection volumes at a flow rate of 20 μl/min. Binding was measured at 25°C for 3 min, followed by 20 min of dissociation. One min of sensor chip regeneration was performed with 1M NaCl/50 mM NaOH. The whole process was repeated 2 times for each dilution sample. Sensorgram response data were analyzed and the kinetic constants calculated with the BIA evaluation kinetics package.
Isolation of Mouse Cardiomyocytes
MIF-KO and CD74-KO mice, both in the C57BL/6 background, were from the Yale Animal Resources Center 5. Wild type (WT) C57BL/6 mice were obtained from Charles River Laboratories (Wilmington, MA). Cardiomyocytes were isolated enzymatically 18. In brief, the hearts of male C57BL/6 WT, MIF-KO, CD74-KO mice (4-6 months) were removed and perfused with oxygenated (5% CO2/95% O2) Krebs-Henseleit bicarbonate (KHB) buffer containing 118 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 1.2 mM MgSO4, 1.2 mM KH2PO4, 25 mM NaHCO3, 10 mM HEPES and 10 mM glucose. Hearts then were perfused with a Ca2+-free KHB containing Liberase Blendzyme 4 (Hoffmann-La Roche Inc., Indianapolis, IN) for 15 min. After perfusion, the left ventricles were removed and minced to disperse cardiomyocytes in Ca2+-free KHB buffer. Extracellular Ca2+ was added incrementally back to 1.25 mM. Culture preparations with predominantly rod-shaped myocytes with clear edges were selected for pharmacological treatment.
Treatment of Mouse Cardiomyocytes with MIF, MIF Agonists, or MIF Antagonists
Isolated cardiomyocytes were treated with recombinant MIF alone or together with candidate agonists in different molar ratios with respect to MIF (1:1, 1:5 and 1:10) 19. Unless otherwise specified, MIF concentrations of 8 nM (calculated as trimer) were employed, which is similar to the 10.8 nM concentration of MIF found to activate AMPK in murine cardiac muscle 5. For MIF receptor antagonism, 1 μM of the MIF antagonist, ISO-1 15, was used to inhibit MIF binding to CD74 20. Cardiomyocytes were incubated at 37°C for 20 min and after treatment, the cells were washed with cold PBS and subjected to cold lysis buffer containing protease cocktail (Roche) to perform immunoblotting analysis. AMPK activity was determined by measuring [32P]ATP incorporation into the synthetic SAMS peptide after immunoprecipitation 5, 6.
Immunoblotting
Western blotting was performed as described previously 5, 18. Cardiomyocyte lysate proteins were resolved by SDS-PAGE and transferred onto polyvinylidene difluoride membranes. For re-probing, the membranes were stripped with 50 mM Tris-HCl, 2% SDS, and 0.1 mM β-mercaptoethanol (pH 6.8). Rabbit polyclonal antibodies directed against phospho-AMPK (pAMPK), total AMPK, and GLUT4 were purchased from Cell Signaling (Danvers, MA) 6, 18. Rabbit polyclonal antibodies against phospho-acetyl-CoA carboxylase (p-ACC) and total ACC were purchased from Millipore (Billerica, MA).
Cell Surface GLUT4 Labeling by Bio-LC-ATB-BGPA
Cell membrane GLUT4 labeling was performed as previously described 5. After treatment, cardiomyocytes were incubated with KHB buffer containing 100 μM of bio-LCBGPA and the reaction was exposed to UV irradiation to induce crosslinking between bio-LCBGPA and cell surface GLUT4. The photo-labeled GLUT4 was purified by streptavidin-agarose and quantified by immunoblotting using a GLUT4 specific antibody.
Glucose Uptake in Cardiomyocytes
2-Deoxy-D-[1-3H] glucose accumulation in cardiomyocytes was performed as described previously 21. Isolated cardiomyocytes were washed three times with Krebs-Ringer-HEPES (KRH) buffer containing 128 mM NaCl, 4.7 mM KCl, 1.25 mM CaCl2, 1.25 mM MgSO4, 10 mM Na2HPO, and 20 mM HEPES. The cells then were treated with 1 μM oligomyocin or 8 nM MIF together with 8 nM MIF20 or 1 μM ISO-1 for 20 min. Glucose uptake was initiated by the addition of 0.1 ml KRH buffer and 2-deoxy-D-[1-3H] glucose (0.2 μCi/ml, GE Healthcare, Piscataway, NJ) and 5 mM glucose as final concentrations. After 30 min, glucose uptake was terminated by washing the cells three times with cold PBS. The cells were lysed overnight with 1 ml of 0.5 M NaOH and 0.1% SDS. The radioactivity retained in the cell lysates was determined by scintillation counting (Beckmann LC 6000IC) and normalized to protein concentration measured by Bradford Protein Assay Kit (Biorad, Hercules, CA).
Perfusion of Isolated Heart Ex Vivo
Mice were anesthetized with pentobarbital (60 mg/kg i.p.), heparinized (100U i.p.), and the hearts excised and retrogradely perfused (4 ml/min, Radnoti Glass Technology, Inc.) with 95% O2/5% CO2 equilibrated KHB containing 7 mM glucose, 1% BSA and 0.4 mM oleate. For the ex vivo ischemic model, the buffer flow was cut off for 25 min and the hearts were reperfused with the same rate of buffer flow during reperfusion. For MIF agonism studies, mouse hearts were stabilized for 15 min and then treated with 8 nM MIF20 for 15 min before ischemia induction. The LabChart6 software (AD Instruments, Colorado Springs, CO) was used to monitor the heart rate and left ventricle pressure. After 30 min of reperfusion, the heart tissues were freeze-clamped for further immunoblotting analysis. For glucose uptake measurement, the rate of cardiac glucose transport was measured by the amount of tritiated water generated from [2-3H] glucose and released into the coronary effluent 22.
In Vivo Regional Ischemia and Myocardial Infarct Size Measurement
Mice were anesthetized, intubated and ventilated with a respirator 5. The body temperature was maintained at 37°C with a heating pad. After left lateral thoracotomy, the left anterior descending coronary artery was occluded for 20 min with an 8-0 nylon suture and polyethylene tubing to prevent arterial injury, and then released for reperfusion. MIF20 (0.15 μg/kg) or vehicle was administered via intra-peritoneal injection 15 min before ischemia. Electrocardiographic monitoring confirmed the development of ischemic ST-segment elevation during coronary occlusion (AD Instruments). At the end of reperfusion, the hearts were excised and the ischemic region of the left ventricle was separated before freeze clamping in liquid N2. Freeze clamped heart tissues were stored in −80°C for further immunoblotting analysis. For infarct size measurement, the hearts were reperfused for 24 hr and then excised for dual staining. Non-necrotic tissue in the ischemic region was stained red by TTC (1%, w/v) and the non-ischemic region was stained with Evan’s blue (1%, w/v). Hearts then were fixed and sectioned into 1 mm slices, photographed using a Leica MZ95 microscope and analyzed by NIH Image J software. The myocardial infarct size was calculated as the area of myocardial necrosis as a percentage of ischemic area at risk (AAR) 5.
Glucose Oxidation Analysis
Cardiac substrate metabolism was determined in the working heart model as previously described 5. The heart preload was set at 15 cm H2O and afterload at 80 cm H2O. The flow rate was maintained at 15 ml/min. Mouse hearts were cannulated through the aorta to initiate retrograde Langendorff perfusion followed by cannulation of the pulmonary vein to initiate anterograde perfusion in the working heart mode. Cardiac function was monitored by a pressure transducer connected to the aortic outflow. Glucose oxidation was determined by the production of 14CO2 from [U-14C]glucose in the perfusate. The 14CO2 in the coronary effluent was captured with hyamine hydroxide and enumerated by scintillation counting.
Statistical Analysis
Shapiro-Wilk tests were used to assess data normality, and F-tests were used for testing variances among groups. Data followed a normal distribution with constant variance and were expressed as means ± SEM. Significance was tested by two tail Student’s t tests or two-way ANOVA with post hoc analysis. A p<0.05 was considered as significant.
Results
MIF agonists enhance the MIF-AMPK signaling cascade
Recent efforts at structure-based design of small molecule inhibitors of MIF have led to the serendipitous discovery of the first MIF agonists that augment MIF interaction with its cell surface receptor, CD74 13,16. The structures of three such triazole-based agonist compounds (MIF20, MIF21, MIF33), are shown in Fig 1A; these were identified on the basis of their binding to the MIF tautomerase site and their ability to enhance MIF interaction with the MIF receptor ectodomain (sCD74=CD7473-232). Using an in vitro, ELISA-based sCD74 capture assay, each of these compounds induced a concentration-dependent increase in the amount of MIF bound to its receptor, with a rank order of MIF20>MIF21>>MIF33 (Fig. 1B). MIF20 increased MIF binding to the receptor ectodomain by ~2.5 fold at the concentration of 500 nM.
Figure 1.

Small molecule MIF agonists enhance MIF binding to its receptor, CD74, and augment MIF-dependent AMPK phosphorylation. (A) Chemical structures of MIF agonists. MIF20, MIF21, and MIF33 correspond to compounds 5a, 5c, and 3d in ref. 16. (B) Concentration-dependent binding studies of the interaction of biotinylated-MIF (83 nM) with immobilized sCD74 (CD7473-232) in the presence of increasing concentration of MIF agonist. Values shown are the mean of triplicate measurements and are representative of at least two experiments. (C) Measurement of the equilibrium dissociation constants between MIF and sCD74 in the presence of MIF20 by surface plasmon resonance (BIAcore analysis). The MIF receptor ectodomain (sCD74) was immobilized onto CM5 chips and increasing concentrations of MIF introduced into the flow phase together with vehicle (upper panel) or different concentrations of MIF20 (lower panel). (D) MIF-dependent activation of the intracellular phosphorylation of AMPK in mouse cardiomyocytes is enhanced by MIF20 and MIF21. Cultured mouse cardiomyocytes were stimulated with MIF (8 nM) together with test agonists (80 nM) for 20 mins. The cells then were washed, lysed and the intracellular content of pAMPK at Thr172 and AMPKα quantified by western blotting. Densitometric analyses show the mean ± SEM of at least 3 independent experiments. (E) Activation of AMPK complexes containing α1 and α2 isoforms of the catalytic subunit in cardiomyocytes in response to MIF and small molecule MIF agonists assessed by immunoprecipitation and phosphorylation of the target SAMS peptide in vitro. Data are mean + SEM of 3 independent experiments. (*p<0.05 vs. corresponding control, respectively; †p<0.05 vs. MIF alone).
The ability of MIF20, the most potent compound, to increase the binding of MIF to its receptor was verified by surface plasmon resonance (BIAcore analysis), which measures equilibrium binding interactions in real-time by changes in the solution refractive index (Fig. 1C). Measurement of the equilibrium dissociation constants between MIF and sCD74 in the presence of MIF20 revealed a 2.7 fold increase in the high-affinity binding interaction between MIF and its receptor versus MIF alone (MIF: KD = 6.06 × 10−9 M vs. MIF/MIF20: KD = 2.25 × 10−9 M). Of note, the main effect of MIF20 was to increase the association rate between MIF and sCD74 by 5.3 fold (MIF: ka = 1.85 × 105 M vs. MIF/MIF20: ka = 9.89 × 105 M). These data suggest that MIF20 functions to alter the conformation of MIF in a manner that enhances its initial association with the CD74 ectodomain.
MIF activates CD74-dependent AMPK phosphorylation in isolated heart muscle and in cultured cardiomyocytes 5, 6. To investigate whether candidate MIF agonists functionally modulate cellular AMPK activity, we stimulated murine cardiomyocytes with MIF together with MIF20, MIF21, or MIF33. The addition of agonist compounds alone did not influence the phosphorylation of AMPK or the total cellular content of AMPK, as assessed by western blotting for pAMPK(Thr172) and AMPKα (Fig. 1D). However, the addition of agonists to MIF in a tenfold molar excess (80 nM agonist : 8 nM MIF) augmented significantly the phosphorylation of AMPK. The mean increase in AMPK phosphorylation was ~2-fold higher for MIF20 and MIF21 when compared to MIF alone. Of note, the relative change in the phosphorylation of AMPK observed with MIF20, MIF21, and MIF33 mirrored the relative potency of the three compounds (MIF20>MIF21>>MIF33) that was observed in the MIF receptor binding assay (Fig. 1B). Cellular AMPK activity was measured by 32P-PO42− incorporation into the SAMS peptide substrate after specific immunoprecipitation of the two AMPKα isoforms, revealing that the main effects of MIF20 and MIF21 in murine cardiomyocytes on the AMPKα1 and AMPKa2 isoforms (Fig. 1E).
MIF20 augments cardiac AMPK activation
We selected the most potent agonist, MIF20, for further study. The addition of increasing concentrations of MIF20 to MIF in molar ratios of 1:1, 5:1, or 10:1 increased in a dose-dependent fashion the phosphorylation of both AMPK and its downstream effector, acetyl-CoA carboxylase (ACC) (Figs. 2A,B). Even at a molar ratio of 1:1 (8 nM of agonist and 8 nM MIF), there was an almost 2.5 fold increase in the phosphorylation ACC, suggesting that agonist treatment of cardiomyocytes is associated with a physiologically relevant activation of AMPK-dependent responses. The addition of MIF20 to a low stimulatory concentration of MIF (both at 4 nM) also augmented the phosphorylation of AMPK (Fig. 2C). By contrast, the addition of ISO-1, a small molecule that binds to the MIF tautomerase site in an antagonistic fashion to inhibit MIF interaction with its receptor 15, 20, prevented AMPK phosphorylation (Fig. 2D). Similar results were obtained by assessing cell surface GLUT4 levels and cellular glucose uptake, which are two additional downstream and tissue protective actions of AMPK signaling. Under the conditions of low stimulatory concentrations of MIF (8 nM), MIF20 increased, and the MIF antagonist ISO-1 decreased, GLUT4 cell-surface levels and glucose uptake (Figs. 2E,H). We also tested the effect of concomitant MIF20 and ISO-1 on cardiomyocytes with respect to AMPK signaling. ISO-1 treatment reduced the agonistic effect of MIF20 on MIF-dependent induction of AMPK and ACC phosphorylation, as well as membrane GLUT4 expression (Figs. 2E-H). This action was in accord with the antagonistic effect of ISO-1 on MIF20’s enhancement of MIF binding to sCD74 (Fig. 2I). We further compared the action of MIF and MIF20 in WT or MIFKO cardiomyocytes to control for any unexpected effect of genetic Mif deficiency on the AMPK pathway and to confirm the requirement for exogenously added MIF. MIF20 did not differentially influence AMPK or ACC phosphorylation in WT or MIF-KO cells and its action was dependent on the addition of exogenous, recombinant MIF (Fig. 2J).
Figure 2.


MIF20 augments MIF-dependent cardiomyocyte AMPK phosphorylation and downstream effector responses. Isolated mouse cardiomyocytes were incubated with MIF (8 nM) together with MIF20 at the indicated concentrations. The intracellular content of (A) pAMPK (Thr172), and (B) phosphorylated acetyl-CoA carboxylase (pACC at Ser79), were determined by western blotting and densitometric scanning with reference to total intracellular AMPK and ACC content. Mean ± SEM for 3 independent experiments. *p<0.05 vs. corresponding control; †p<0.05 vs. MIF alone. (C) AMPK phosphorylation in mouse cardiomyocytes stimulated with low concentrations of MIF is augmented by equimolar concentrations of MIF20 (4 nM or 8 nM). Values are the mean ± SEM for 3 independent experiments. *p<0.05 vs. corresponding 0 group; †p<0.05 vs. corresponding MIF alone. (D) The MIF antagonist ISO-1 (1 μM) blocks MIF-dependent stimulation of AMPK phosphorylation. Mean ± SEM for 3 independent experiments. *p<0.05 vs. corresponding 0 group; †p<0.05 vs. corresponding MIF alone. (E) AMPK phosphorylation and (F) ACC phosphorylation stimulated by the treatment of MIF plus MIF20 was decreased by ISO-1. *p<0.05 vs. corresponding control; †p<0.05 vs. MIF alone; #p<0.05 vs. MIF plus MIF20. (G) Cardiomyocyte cell surface GLUT4 levels increase in response to stimulation with MIF plus MIF20 (each at 8 nM), while the MIF antagonist ISO-1 (1 μM) inhibits MIF-dependent increases in cell surface GLUT4 levels. Densitometric analysis of the ratio of cell surface versus total GLUT4 (cm-GLUT4/t-GLUT4) shows the mean ± SEM for 3 independent experiments. *p<0.05 vs. corresponding control; †p<0.05 vs. MIF alone; #p<0.05 vs. MIF plus MIF20. (H) MIF-dependent glucose uptake in cardiomyocytes is augmented by MIF plus MIF20 (each at 8 nM) and inhibited by the MIF antagonist, ISO-1 (1 μM). Values are means ± SEM for 3 independent experiments. *p<0.05 vs. corresponding control; †p<0.05 vs. MIF alone. (I) Binding of biotinylated-MIF (83 nM) to immobilized sCD74 under conditions where sCD74 was pre-incubated with vehicle, MIF20 (8 nM), ISO-1 (1 M), MIF (8 nM ) plus MIF20 (8 nM), MIF (8 nM) plus ISO-1 (1 M), or MIF (8 nM) plus MIF20 (8 nM) plus ISO-1 (1 M), followed by washing and addition of biotinylated MIF. ***p<0.005 vs. MIF or MF20 alone, **p<0.02 vs. MIF, MIF20, or MIF+MIF20, *p<0.05 vs. MIF+MIF20 or MIF+ISO-1. (J) Influence of MIF (8 nM) and MIF20 (8 nM) on AMPK and ACC phosphorylation in WT or MIFKO cardiomyocytes.
MIF20 triggers AMPK mediated by MIF receptor CD74
The small molecule MIF agonists described herein bind to MIF’s intrinsic tautomerase site and are considered to influence MIF’s conformation and productive interaction with its receptor 11, 13, 16. Accordingly, both MIF and its receptor, CD74, are hypothesized to be essential for the function of these compounds. To confirm the specificity of MIF20 in the MIF/CD74 dependent activation of the AMPK pathway, we compared the action of MIF20 in cardiomyocytes obtained from mice genetically deficient in MIF or the MIF receptor, CD74 (CD74-KO). Because MIF is expressed by cardiomyocytes, any stimulatory action of MIF20 on AMPK in MIF-KO cells would be dependent on exogenously added MIF and could not reflect, for instance, an enhanced release of endogenous MIF or an unexpected intracellular action of MIF 23. MIF20 both augmented the MIF-dependent phosphorylation of AMPK and ACC in MIFKO cardiomyocytes and it enhanced the downstream GLUT4 translocation to cell surface (Fig. 3A, B). The expected inhibitory action of the MIF antagonist, ISO-1, also was observed under the experimental conditions where exogenous MIF was added. By contrast, neither MIF, MIF20, nor the combination of MIF plus MIF20 showed any effect on AMPK or ACC phosphorylation or GLUT4 translocation in cardiomyocytes genetically deficient in CD74 (Fig. 3C, D). These observations support the requirement for an interaction between MIF and its cell surface receptor in the enhancement of AMPK activation by the small molecule agonist MIF20.
Figure 3.

MIF20 action in cardiomyocytes genetically deficient in MIF (MIF-KO) or the MIF cell surface receptor, CD74 (CD74-KO). Isolated mouse cardiomyocytes from the indicated mouse strains were incubated with MIF (8 nM) together with MIF20 (8 nM) or the MIF antagonist ISO-1 (1 μM) as shown. (A) The intracellular content of pAMPK (Thr172) and pACC (Ser79), and (B) cell surface GLUT4 levels in MIF-KO cardiomyocytes. (C) The intracellular content of pAMPK (Thr172) and pACC (Ser79), and (D) cell surface GLUT4 levels in MIF receptor (CD74-KO) cardiomyocytes. All values are means ± SEM for 3 independent experiments. *p<0.05 vs. corresponding control; †p<0.05 vs. MIF alone.
MIF20 modulates cardiac glucose uptake
There is increasing evidence that activation of the AMPK pathway is beneficial in limiting cardiac damage during ischemia and reperfusion 5, 6. We examined the effect of MIF20 in a well-established ex vivo model of post-ischemic cardiac dysfunction. Isolated mouse hearts were subjected to 30 minutes of baseline perfusion, followed by 25 minutes of global, no-flow ischemia and 30 minutes of reperfusion. The addition of 8 nM of MIF20 to the cardiac perfusion buffer for 15 minutes prior to the induction of ischemia (25 min) improved recovery of function during the post-ischemic reperfusion period, as evidenced by a significant elevation in heart rate-LV pressure product (Fig. 4A). This improvement in cardiac function recovery was associated with increased phosphorylation of AMPK and ACC, as well as with an enhancement in glucose transport during the reperfusion period (Figs. 4B, C). In separate experiments to study substrate oxidation in the working mouse heart, MIF20 also enhanced glucose oxidation in heart tissue during reperfusion after 15 min of ischemia ex vivo (Fig. 4D).
Figure 4.

MIF20 stimulates cardiac AMPK activation and glucose uptake, and ameliorates post-ischemic cardiac dysfunction. Isolated hearts from C57BL/6 WT mice underwent 30 minutes of baseline perfusion, 25 minutes of global no-flow ischemia, and 30 minutes of reperfusion. The MIF agonist, MIF20, was added to the perfusion buffer during baseline perfusion 15 min prior to ischemia at a final concentration of 8 nM. (A) Heart rate-left-ventricular developed pressure product during baseline and post-ischemic reperfusion (n=4 hearts for each group. Mean ± SEM, *p<0.05 vs. vehicle controls). (B) Representative western blots for phosphorylated AMPK and ACC in cardiac tissue from two hearts during reperfusion, and after treatment with vehicle or MIF20. Tissue lysates were prepared by freeze-clamping cardiac muscle 30 mins after initiation of reperfusion. The densitometric analysis is for 4 hearts per group (Mean ± SEM, *p<0.05 vs. vehicle). (C) Measurement of glucose uptake in cardiac tissue during baseline and post-ischemic reperfusion periods after treatment with vehicle or MIF20. The perfusate was collected at 1, 5, and 10 mins after beginning reperfusion and the values averaged for each heart. (n=4 hearts for each group. Mean + SEM, *p<0.05 for reperfusion vs. corresponding baseline; †p<0.05 for MIF20 vs. vehicle). (D) Enhancement of MIF-dependent glucose oxidation rate by MIF20 (8 nM) in a separate group of anterograde perfused working mouse hearts subjected to 15 min ischemia and 30 mins reperfusion (n=4 hearts per experimental group; *p<0.05 vs. corresponding baseline; †p<0.05 for MIF20 vs. vehicle).
The MIF receptor CD74 mediates MIF20’s cardioprotection against ischemic injury
Mice genetically deficient in MIF show increased post-ischemic cardiac dysfunction and injury because of impaired activation of AMPK 5. MIF20 had no discernable cardioprotective active in the absence of endogenous MIF or the MIF receptor, as assessed by recovery of contractile function during post-ischemic reperfusion of MIF-KO and CD74-KO hearts respectively (Figs. 5A, B). Western blotting analysis also showed a reduced level of AMPK phosphorylation after MIF20 treatment of MIF-KO or CD74-KO versus WT hearts (Fig. 5C), which is in agreement with the isolated cardiomyocyte findings showing a strict dependence for MIF and the MIF receptor in the action of MIF20 (Fig. 3).
Figure 5.

The cardioprotective effect of MIF20 requires endogenous MIF and the cell surface MIF receptor, CD74. Heart rate-left ventricular developed pressure product during baseline perfusion, ischemia and post-ischemic reperfusion of MIF-KO hearts (A) and CD74-KO hearts (B) treated with or without MIF20 (8 nM) prior to ischemia (n=4 hearts for each group, P=NS). (C) Representative western blots of cardiac AMPK phosphorylation 30 mins after initiation of reperfusion in WT, MIF-KO, and CD74-KO hearts treated with vehicle or MIF20 (8 nM). Two representative tissue lysates are shown by western blotting and densitometry shows the mean + SEM for 4 hearts for each experimental group. *p<0.05 vs. WT vehicle.
To additionally test for the role of AMPK in MIF agonist mediated cardioprotection, we examined if MIF20 augmented recovery after post-ischemic reperfusion injury of hearts encoding an enzymatically inactive, AMPK-kinase “dead” transgene (AMPK-KD) 24. Such mice have been shown previously to have impaired recovery of left ventricular contractile function during post-ischemic re-perfusion associated with a reduction in downstream AMPK-mediated processes 22. Isolated hearts from AMPK-KD mice were perfused for 15 min and then treated with MIF20 or vehicle for 15 min before 25 min no-flow ischemic followed by 30 min reperfusion. The heart rate-left ventricular developed pressure product in AMPK-KD hearts, which is already reduced when compared to WT hearts treated similarly (22 and Fig. 5), was unaffected by the administration of MIF20 (Fig. 6A). As anticipated, western blotting of AMPKKD heart tissue showed that MIF20 administration failed to activate pACC as an indicator of downstream AMPK activity (Fig. 6B).
Figure 6.
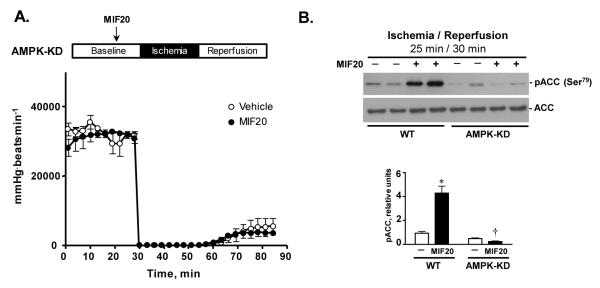
AMPK kinase-dead (KD) hearts fail to demonstrate a cardioprotective action of MIF20. (A) Heart-rate-left-ventricular-developed pressure product of AMPK-KD hearts during baseline (30 min), ischemia (25 min) and post-ischemic reperfusion (30 min) after treatment with MIF20 (8 nM) or vehicle control. (n=4 hearts for each group). (B) Representative western blots comparing pACC in two representative WT and AMPK-KD hearts sampled 30 mins after post-ischemia reperfusion. Densitometry measurements (mean + SEM) are for 4 hearts in each group. *p<0.05 vs. WT vehicle. †p<0.05 vs.WT MIF20.
Finally, we examined myocardial infarct size after regional ischemia and reperfusion in vivo in mice treated systemically with MIF20. Anesthetized mice were subjected to 20 minutes of ligation of the left anterior descending coronary artery, followed by 24 hours reperfusion. Pre-treatment of mice with MIF20 reduced infarct size by 40%, and this effect was associated with an augmentation in the content of phosphorylated AMPK and phosphorylated ACC in the ischemic region of the LV (Fig. 7A.). Labeling of heart tissue after ischemia/reperfusion with the membrane impermeable compound, bio-LC-ATB-BGPA also revealed MIF20 treatment to enhance the translocation of GLUT4 to cell surfaces (Fig. 7B). As expected, MIF20 showed no demonstrable effect on myocardial infarct size induced by in vivo regional ischemia and reperfusion when administered to mice lacking MIF (Fig. 7C) or the MIF receptor CD74 (Fig. 7D).
Figure 7.

MIF20 reduces myocardial necrosis and activates AMPK in mouse heart in vivo in a MIF and MIF receptor dependent fashion. Anesthetized mice were subjected to 20 min regional ischemia by ligation of the left coronary artery followed by 24 hours of reperfusion. MIF20 or vehicle was injected intra-peritoneally 15 min prior to ischemia. (A) Representative pictures show myocardial infarct size in heart sections defined by dual staining with 2,3,5-triphenyltetrazolium (TTC) and Evans Blue. Bar graphs quantify the ratio of the area at risk (AAR) to myocardium, and the ratio of infarct size (INF) to AAR. AMPK and ACC phosphorylation in different tme periods during ischemia and reperfusion was assessed by western blotting. (B) MIF20 treatment is associated with enhanced GLUT4 translocation to the membrane of myocardial tissue. (C) Lack of MIF20 effect on myocardial necrosis, AMPK and ACC phosphorylation in MIF-KO mice. (D) Lack of MIF20 effect on myocardial necrosis, AMPK and ACC phosphorylation in MIF receptor (CD74-KO) mice. n=4-6 hearts for each studied group. *p<0.05 vs. vehicle.
Discussion
A recently completed structure-based discovery program for small molecule antagonists of MIF capitalized on the apposition of MIF’s tautomerase site with its receptor binding domain and led to the identification of potent, drug-like inhibitors of MIF-dependent signal transduction11, 13. Remarkably, molecules were identified that while similarly designed to dock within the MIF tautomerase site were found to increase rather than decrease MIF interaction with its receptor 16. These triazole-based compounds enhanced MIF dependent ERK1/2 activation in fibroblasts and are considered to induce a conformational change in MIF that increases its receptor interaction and signaling efficiency. While these molecules may be termed MIF receptor agonists, it is important to note that their activity requires the presence of MIF ligand. Elucidation of the precise structural features induced by MIF20 that lead to enhanced MIF interaction with CD74 may be revealed by x-ray crystallography or solution NMR studies of a MIF20-MIF co-complex 25, 26, which are currently underway.
We determined the influence of the most potent compound of this series, MIF20, on the equilibrium rate constant for MIF binding to CD74 by surface plasmon resonance, which measures solution binding interactions in real-time by changes in refractive index. MIF20 enhanced MIF affinity for CD74 by 2.7 fold, with the major effect observed to be to increase ligand association rather than to decrease ligand dissociation. MIF20, as well as its less active isoquinolinyl congener, MIF21, also increased the MIF-dependent phosphorylation and enzymatic activation of the regulatory kinase AMPK in cell based assays.
AMPK is a stress signaling kinase and key regulator of energy generating and consuming pathways that protects cells against hypoxic injury and death 27, 28. The release of endogenous MIF from ischemic myocardium has been shown to stimulate AMPK activation in a CD74 dependent, autcrine/paracrine manner, leading to enhanced glucose uptake and a beneficial tissue protective response 5. Given the potential physiologic importance of this pathway, especially in aging where MIF release and subsequent AMPK activation are impaired 6, we studied the efficacy of small molecule MIF agonism as a strategy for activating MIF-dependent signal transduction in the cardiomyocyte and myocardial tissue response to ischemia. MIF20, when added to cultured cardiomyocytes at concentrations of 8 - 80 nM, increased MIF-dependent AMPK activation and its downstream effect on ACC phosphorylation, GLUT4 translocation, and glucose uptake. In these studies, recombinant MIF was added at concentrations that mimicked those observed to AMPK activation in cardiac muscle5. These effects also were strictly dependent on the presence of the MIF receptor, CD74. The physiologic benefit of this augmentation in MIF signal transduction and AMPK activation was verified by the study of post-ischemic cardiac contractile dysfunction ex vivo. The addition of MIF20 to the perfusion buffer not only increased the phosphorylation of AMPK and ACC, and enhanced glucose transport, but it improved the functional recovery of isolated hearts during the post-ischemic reperfusion period. As in the case of MIF20 action on cardiomyocytes, no changes were noted in either MIF or CD74 deficient hearts.
Finally, we tested the effect of MIF20 in a mouse model of left coronary artery ligation, which mimics clinical myocardial infarction, to assess if augmentation of the MIF-AMPK signaling pathway by pharmacologic MIF agonism translates into protection from ischemic damage. Both endogenous MIF release or exogenous MIF infusion have been shown to be beneficial in this model 5, 6, and MIF20 administration similarly reduced myocardial necrosis.
Augmentation of AMPK activity during ischemic episodes is considered to be of clinical utility to protect the heart or other tissues against ischemia-reperfusion injury 29. Prior studies have identified a significant, age-induced impairment in AMPK activation in the heart that appears due to a reduction in hypoxia-inducible factor 1α mediated MIF expression 6. Human cells with a low-expression MIF promoter polymorphism, which occurs commonly in the population, have diminished MIF release and AMPK activation in response to hypoxia 5. As systemic circulating MIF concentrations are within the range of the KD to be a significant augmentation of MIF binding to trigger AMPK activation, although the magnitude of this effect would depend on the concentration of MIF released locally by ischemic myocardium as well as the precise shape and activation threshold of the dose-response curve. We suggest that augmentation of endogenous MIF signal transduction via pharmacologic activation of the MIF ligand and the ensuing AMPK response offers an attractive approach for compensating for intrinsic age or genetic factors associated with low MIF expression and susceptibility to ischemic tissue damage.
The ischemic myocardium releases macrophage migration inhibitory factory (MIF), which acts locally to increase the cardioprotective action of AMP-activated protein kinase (AMPK). AMPK is activated by hypoxia and this response is impaired in aging. There are also common, functional polymorphisms in MIF that influence the strength of the cellular AMPK response to hypoxia. We identified pharmacologic agonists of MIF that increase its binding to its cognate cell surface receptor CD74, and in the present study, we show that a first-in-class MIF agonist, MIF20, augments the activation of the MIF receptor in cardiomyocytes and increases the downstream tissue protective action of AMPK on glucose uptake and metabolism. In mouse hearts perfused with MIF20, post-ischemic left ventricular function improved commensurately with an increase in cardiac MIF-AMPK activation. Mice treated with MIF20 followed by left coronary artery occlusion and reperfusion also showed a reduction in infarcted myocardium. Augmentation of endogenous myocardial MIF signaling by small molecular MIF agonists may offer a novel approach in selected clinical settings to compensate for intrinsic age or genetic deficiencies in protective AMPK responses.
Acknowledgments
Funding Sources: This work was supported by grants from the American Heart Association (0835169N and 12GRNT11620029), National Natural Science Foundation of China (No. 31171121), NIH GM032136, AI042310, HL063811, the American Diabetes Association (1-11-BS-92), and the Brookdale Foundation.
Footnotes
Conflict of Interest Disclosures: Yale University has applied for patents describing small molecule MIF modulators.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Benigni F, Atsumi T, Calandra T, Metz C, Echtenacher B, Peng T, Bucala R. The proinflammatory mediator macrophage migration inhibitory factor induces glucose catabolism in muscle. J Clin Invest. 2000;106:1291–1300. doi: 10.1172/JCI9900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Atsumi T, Cho YR, Leng L, McDonald C, Yu T, Danton C, Hong EG, Mitchell RA, Metz C, Niwa H, Takeuchi J, Onodera S, Umino T, Yoshioka N, Koike T, Kim JK, Bucala R. The proinflammatory cytokine macrophage migration inhibitory factor regulates glucose metabolism during systemic inflammation. J Immunol. 2007;179:5399–5406. doi: 10.4049/jimmunol.179.8.5399. [DOI] [PubMed] [Google Scholar]
- 3.Verschuren L, Kooistra T, Bernhagen J, Voshol PJ, Ouwens DM, van Erk M, de Vries-van der Weij J, Leng L, van Bockel JH, van Dijk KW, Fingerle-Rowson G, Bucala R, Kleemann R. Mif deficiency reduces chronic inflammation in white adipose tissue and impairs the development of insulin resistance, glucose intolerance, and associated atherosclerotic disease. Circ Res. 2009;105:99–107. doi: 10.1161/CIRCRESAHA.109.199166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Serre-Beinier V, Toso C, Morel P, Gonelle-Gispert C, Veyrat-Durebex C, Rohner-Jeanrenaud F, Calandra T, Roger T, James RW, Montet X, Buhler L, Bosco D, Berney T. Macrophage migration inhibitory factor deficiency leads to age-dependent impairment of glucose homeostasis in mice. J Endocrinol. 2010;206:297–306. doi: 10.1677/JOE-09-0342. [DOI] [PubMed] [Google Scholar]
- 5.Miller EJ, Li J, Leng L, McDonald C, Atsumi T, Bucala R, Young LH. Macrophage migration inhibitory factor stimulates amp-activated protein kinase in the ischaemic heart. Nature. 2008;451:578–582. doi: 10.1038/nature06504. [DOI] [PubMed] [Google Scholar]
- 6.Ma H, Wang J, Thomas DP, Tong C, Leng L, Wang W, Merk M, Zierow S, Bernhagen J, Ren J, Bucala R, Li J. Impaired macrophage migration inhibitory factor-amp-activated protein kinase activation and ischemic recovery in the senescent heart. Circulation. 2010;122:282–292. doi: 10.1161/CIRCULATIONAHA.110.953208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yu CM, Lau CP, Lai KW, Huang XR, Chen WH, Lan HY. Elevation of plasma level of macrophage migration inhibitory factor in patients with acute myocardial infarction. Am J Cardiol. 2001;88:774–777. doi: 10.1016/s0002-9149(01)01850-1. [DOI] [PubMed] [Google Scholar]
- 8.Takahashi M, Nishihira J, Katsuki T, Kobayashi E, Ikeda U, Shimada K. Elevation of plasma levels of macrophage migration inhibitory factor in patients with acute myocardial infarction. Am J Cardiol. 2002;89:248–249. doi: 10.1016/s0002-9149(01)02251-2. [DOI] [PubMed] [Google Scholar]
- 9.Leng L, Metz CN, Fang Y, Xu J, Donnelly S, Baugh J, Delohery T, Chen Y, Mitchell RA, Bucala R. Mif signal transduction initiated by binding to cd74. J Exp Med. 2003;197:1467–1476. doi: 10.1084/jem.20030286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shi X, Leng L, Wang T, Wang W, Du X, Li J, McDonald C, Chen Z, Murphy JW, Lolis E, Noble P, Knudson W, Bucala R. Cd44 is the signaling component of the macrophage migration inhibitory factor-cd74 receptor complex. Immunity. 2006;25:595–606. doi: 10.1016/j.immuni.2006.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fingerle-Rowson G, Kaleswarapu DR, Schlander C, Kabgani N, Brocks T, Reinart N, Busch R, Schutz A, Lue H, Du X, Liu A, Xiong H, Chen Y, Nemajerova A, Hallek M, Bernhagen J, Leng L, Bucala R. A tautomerase-null macrophage migration-inhibitory factor (mif) gene knock-in mouse model reveals that protein interactions and not enzymatic activity mediate mif-dependent growth regulation. Mol Cell Biol. 2009;29:1922–1932. doi: 10.1128/MCB.01907-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Orita M, Yamamoto S, Katayama N, Fujita S. Macrophage migration inhibitory factor and the discovery of tautomerase inhibitors. Curr Pharm Des. 2002;8:1297–1317. doi: 10.2174/1381612023394674. [DOI] [PubMed] [Google Scholar]
- 13.Cournia Z, Leng L, Gandavadi S, Du X, Bucala R, Jorgensen WL. Discovery of human macrophage migration inhibitory factor (mif)-cd74 antagonists via virtual screening. J Med Chem. 2009;52:416–424. doi: 10.1021/jm801100v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ouertatani-Sakouhi H, El-Turk F, Fauvet B, Roger T, Le Roy D, Karpinar DP, Leng L, Bucala R, Zweckstetter M, Calandra T, Lashuel HA. A new class of isothiocyanate-based irreversible inhibitors of macrophage migration inhibitory factor. Biochemistry. 2009;48:9858–9870. doi: 10.1021/bi900957e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lubetsky JB, Dios A, Han J, Aljabari B, Ruzsicska B, Mitchell R, Lolis E, Al-Abed Y. The tautomerase active site of macrophage migration inhibitory factor is a potential target for discovery of novel anti-inflammatory agents. J Biol Chem. 2002;277:24976–24982. doi: 10.1074/jbc.M203220200. [DOI] [PubMed] [Google Scholar]
- 16.Jorgensen WL, Gandavadi S, Du X, Hare AA, Trofimov A, Leng L, Bucala R. Receptor agonists of macrophage migration inhibitory factor. Bioorg Med Chem Lett. 2010;20:7033–7036. doi: 10.1016/j.bmcl.2010.09.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bernhagen J, Mitchell RA, Calandra T, Voelter W, Cerami A, Bucala R. Purification, bioactivity, and secondary structure analysis of mouse and human macrophage migration inhibitory factor (mif) Biochemistry. 1994;33:14144–14155. doi: 10.1021/bi00251a025. [DOI] [PubMed] [Google Scholar]
- 18.Wang J, Yang L, Rezaie AR, Li J. Activated protein c protects against myocardial ischemic/reperfusion injury through amp-activated protein kinase signaling. J Thromb Haemost. 2011;9:1308–1317. doi: 10.1111/j.1538-7836.2011.04331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hare AA, Leng L, Gandavadi S, Du X, Cournia Z, Bucala R, Jorgensen WL. Optimization of n-benzyl-benzoxazol-2-ones as receptor antagonists of macrophage migration inhibitory factor (mif) Bioorg Med Chem Lett. 2010;20:5811–5814. doi: 10.1016/j.bmcl.2010.07.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leng L, Chen L, Fan J, Greven D, Arjona A, Du X, Austin D, Kashgarian M, Yin Z, Huang XR, Lan HY, Lolis E, Nikolic-Paterson D, Bucala R. A small-molecule macrophage migration inhibitory factor antagonist protects against glomerulonephritis in lupus-prone nzb/nzw f1 and mrl/lpr mice. J Immunol. 2011;186:527–538. doi: 10.4049/jimmunol.1001767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhao P, Wang J, He L, Ma H, Zhang X, Zhu X, Dolence EK, Ren J, Li J. Deficiency in tlr4 signal transduction ameliorates cardiac injury and cardiomyocyte contractile dysfunction during ischemia. J Cell Mol Med. 2009;13:1513–1525. doi: 10.1111/j.1582-4934.2009.00798.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Russell RR, 3rd, Li J, Coven DL, Pypaert M, Zechner C, Palmeri M, Giordano FJ, Mu J, Birnbaum MJ, Young LH. Amp-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J Clin Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleemann R, Hausser A, Geiger G, Mischke R, Burger-Kentischer A, Flieger O, Johannes FJ, Roger T, Calandra T, Kapurniotu A, Grell M, Finkelmeier D, Brunner H, Bernhagen J. Intracellular action of the cytokine mif to modulate ap-1 activity and the cell cycle through jab1. Nature. 2000;408:211–216. doi: 10.1038/35041591. [DOI] [PubMed] [Google Scholar]
- 24.Mu J, Brozinick JT, Jr., Valladares O, Bucan M, Birnbaum MJ. A role for amp-activated protein kinase in contraction- and hypoxia-regulated glucose transport in skeletal muscle. Mol Cell. 2001;7:1085–1094. doi: 10.1016/s1097-2765(01)00251-9. [DOI] [PubMed] [Google Scholar]
- 25.Muhlhahn P, Bernhagen J, Czisch M, Georgescu J, Renner C, Ross A, Bucala R, Holak TA. Nmr characterization of structure, backbone dynamics, and glutathione binding of the human macrophage migration inhibitory factor (mif) Protein Sci. 1996;5:2095–2103. doi: 10.1002/pro.5560051016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho Y, Crichlow GV, Vermeire JJ, Leng L, Du X, Hodsdon ME, Bucala R, Cappello M, Gross M, Gaeta F, Johnson K, Lolis EJ. Allosteric inhibition of macrophage migration inhibitory factor revealed by ibudilast. Proc Natl Acad Sci U S A. 2010;107:11313–11318. doi: 10.1073/pnas.1002716107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nagendran J, Waller TJ, Dyck JR. Ampk signalling and the control of substrate use in the heart. Mol Cell Endocrinol. 2013;366:180–193. doi: 10.1016/j.mce.2012.06.015. [DOI] [PubMed] [Google Scholar]
- 28.Hardie DG, Ross FA, Hawley SA. Ampk: A nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim AS, Miller EJ, Wright TM, Li J, Qi D, Atsina K, Zaha V, Sakamoto K, Young LH. A small molecule ampk activator protects the heart against ischemia-reperfusion injury. J Mol Cell Cardiol. 2011;51:24–32. doi: 10.1016/j.yjmcc.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]