

Abstract
Purpose
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK), one of the most prevalent and procarcinogenic compounds in tobacco, is bioactivated by respiratory cytochrome P450 (CYP) 2A13, forming DNA adducts and initiating lung cancer. CYP2A13 inhibition offers a novel strategy for chemoprevention of tobacco-associated lung cancer.
Methods
Twenty-four analogs of a 4-benzylmorpholine scaffold identified by high throughput screening were evaluated for binding and inhibition of both functional human CYP2A enzymes, CYP2A13 and the 94%-identical hepatic CYP2A6, whose inhibition is undesirable. Thus, selectivity is the major challenge in compound design.
Results
A key feature resulting in CYP2A13-selective binding and inhibition was substitution at the benzyl ortho position, with three analogs being >25-fold selective for CYP2A13 over CYP2A6.
Conclusions
Two such analogs were negative for genetic and hERG toxicities and metabolically stable in human lung microsomes, but displayed rapid metabolism in human liver and in mouse and rat lung and liver microsomes, likely due to CYP2B-mediated degradation. A specialized knockout mouse mimicking the human lung demonstrates compound persistence in lung and provides an appropriate test model. Compound delivered by inhalation may be effective in the lung but rapidly cleared otherwise, limiting systemic exposure.
Keywords: cytochrome P450 2A13, cytochrome P450 2A6, tobacco, lung cancer, chemopreventative
INTRODUCTION
The cytochrome P450 (CYP) superfamily of heme-containing monooxygenase enzymes metabolizes a variety of exogenous and endogenous substrates which can be manipulated for therapeutic benefit.(1) For example, the selective inhibition of androgen and estrogen biosynthetic CYP enzymes has afforded significant progress in the treatment of prostate and breast cancer.(2, 3) In contrast, inhibition of xenobiotic-metabolizing CYP enzymes has often been viewed as an unproductive approach to drug discovery due to selectivity issues expected for such agents. Despite this, a selective CYP3A4 inhibitor has now been deliberately developed from retonavir for explicit use as pharmacokinetic booster.(4-6) Similarly, inhibition of CYP enzymes involved in the activation of procarcinogens can offer chemopreventative opportunities; such is the case in tobacco-associated lung cancer. Lung cancer is the leading cause of all cancer-related deaths,(7) and >80% of all lung cancer incidence results from tobacco use.(8) According to the CDC, 69% of tobacco users desire to quit,(9) but due to the addictive properties of nicotine and ineffectiveness of current tobacco cessation techniques, only 5% of users successfully cease tobacco use.(10) A complementary strategy to reduce lung cancer prevalence is to decrease the in vivo formation of tobacco carcinogens. 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) is one of the most prevalent and, when activated, one of the most carcinogenic compounds in tobacco. NNK is selectively biotransformed by cytochrome P450 2A13 (CYP2A13) in the respiratory tract to form one of two diazonium ions that generate DNA adducts in genes like p53 and ras and can initiate lung cancer.(11, 12)
A CYP2A13 inhibitor, therefore, offers a novel therapeutic strategy to reducing the risk of lung cancer in tobacco users. This target is validated by CYP2A13 polymorphisms and animal studies. Tobacco users with natural-occurring polymorphisms resulting in reduced CYP2A13 activity exhibit normal physiological characteristics but have a decreased risk for lung adenocarcinoma.(13, 14) Mouse studies have substantiated the role of the respiratory tract(15) and the CYP2A13 ortholog, CYP2A5,(16) in NNK bioactivation. NNK-induced tumors were also reduced from 70% to 5% in rats with the known pan-CYP2A inhibitor, phenethyl isothiocyanate (PEITC)(17). Finally, CYP2A13 inhibition has been correlated to a decrease in NNK bioactivation and an increase in NNK clearance via nontoxic glucuronidation and excretion.(18) Thus, once CYP2A13 is inhibited, an increased amount of NNK is cleared via nontoxic, non-carcinogenic metabolites.
The most significant difficulty in developing effective P450 inhibitors arises in designing selectivity for an individual enzyme in this superfamily. In contrast to biosynthetic CYP enzymes like aromatase that are fairly specific for their substrates, most xenobiotic-metabolizing CYP enzymes have evolved to recognize and act upon a wide range of chemically diverse small molecules. This is often accomplished via large and/or flexible active sites that frustrate structure-based predictions of ligand binding. For example, the large active site of the hepatic CYP3A4 enzyme is responsible for the metabolism of 34%(19) of clinically used drugs, so that even a selective CYP3A4 inhibitor such as the pharmacokinetic booster cobicistat can frequently have substantial adverse drug-drug interactions. Extrahepatic CYP enzymes such as CYP2A13 that are much less involved in drug metabolism are also much less likely to have adverse effects, so long as the inhibitors are selective and do not inhibit other CYP enzymes.
The largest challenge in developing CYP2A13 inhibition as a chemopreventative approach is likely to be nonselective inhibition of the only other functional human CYP2A enzyme, the 94% identical hepatic CYP2A6. CYP2A6 shares a number of substrates and inhibitors with CYP2A13, but is primarily involved in vivo in the metabolism of drugs in systemic circulation, including nicotine, and does not effectively bioactivate NNK or contribute to DNA adduct formation in the respiratory tract(20, 21). The emphasis of this work was the identification and evaluation of small molecule inhibitors of CYP2A13 that are selective vs. CYP2A6. High throughput screening was initially used to identify a 4-benzylmorpholine scaffold that might have the potential to effectively and selectively inhibit CYP2A13. A library of twenty-four 4-benzylmorpholine analogs was then generated. Analysis of differential binding to and inhibition of recombinant, purified CYP2A13 and CYP2A6 enzymes in vitro led to the determination of structure-activity relationships for the 4-benzylmorpholine scaffold with the human CYP2A enzymes that may form the basis for future studies toward lung cancer chemoprevention.
MATERIALS AND METHODS
Protein Expression and Purification
Human CYP2A enzymes were truncated, His-tagged, expressed, and purified as previously described.(22, 23) Rat cytochrome P450 oxidoreductase (POR) was purified using published procedures.(24)
High Throughput Screening
The spectral shift assay (vide infra) reflecting interactions of compounds with CYP2A13 was adapted to HTS format for screening ChemBridge library of 35,200 compounds. Each 384-well screening plate contained 320 library compounds (20 uM in 0.71% DMSO) and a set of standards comprising 32 wells: 16-wells each of 0.71% DMSO (negative control), and 16-wells of ositive control (30 μM phenethyl isothiocyanate or PEITC). Purified CYP2A13 (3 μM) was added to all the wells of the assay plates and the protein/compound mixtures were allowed to equilibrate at room temperature for 5 min. The plates were read on SpectraMax (Molecular Devices, CA) at 385 nm and 420 nm. Primary screen actives were identified as compounds that resulted in difference of 0.1 (plate median + 5 standard deviation) in raw absorbance values at 385 nm and 420 nm.
Spectral Ligand Binding Assay
Ligand binding was monitored by following spectral changes upon titration with increasing concentrations of ligand and the data analyzed as previously described,(25) except that all compounds were dissolved in DMSO. However, the imidazole-containing compounds bound tightly enough to require a reduction in protein concentration to 0.1 μM, use of 5 cm path length cuvette, and analysis with the tight binging equation as described previously.(26)
Coumarin Metabolism Assay(27, 28)
The 50 pmol of purified CYP2A enzyme was reconstituted with 200 pmol of POR by coincubation on ice for 50 minutes before addition to the reaction samples containing buffer (50 mM Tris, HCl, pH 7.4, 5 mM MgCl), 0-90 μM coumarin substrate, and varying inhibitor concentrations. Samples were warmed for 3 minutes at 37 °C before addition of 1 mM NADPH to initiate the 250 μl reaction. Catalysis proceeded for 10 min before termination with 75 μl of 20% trichloroacetic acid (TCA) to precipitate protein. TCA was added to all standard samples prior to NADPH addition. Additional buffer (200 μl) and 100 μl of 1.6 M glycine were added to each tube before centrifugation at 4,400 × g for 6 minutes. CYP2A6 samples required an additional 5-fold dilution due to detector limitations. Formation of the 7-hydroxycoumarin metabolite was monitored via fluorescence detection with an excitation wavelength of 355 nm and an emission wavelength of 460 nm following chromatographic separation with a reverse phase C18 column on an HPLC. A mobile phase of 40% methanol and 0.12% acetic acid was used with a flow rate of 0.8 mL min−1. A small amount of a second product, likely the 3,4-epoxide metabolite,(29) was observed at the lowest substrate concentration (5 μM) in the absence of inhibitor and automated outlier exclusion analysis with the data analysis software Prism 5 was used to handle this data point. Prism 5 was also used for data analysis of the six substrate and four inhibitor concentrations resulting in the kcat and Km values that were used to determine the mode of inhibition and ultimately Ki value for each compound with the CYP2A enzymes with the appropriate formula shown below. Competitive inhibition: and where Km Obs is the observed Km, [I] is the inhibitor concentration, Ki is the inhibition constant, and [S] is the substrate concentration. Mixed model inhibition: , , and
Inhibitors
The benzylmorpholine analogs 4-(2-fluorobenzyl)morpholine (2), 4-(2-chlorobenzyl)morpholine (3), 4-(2-bromobenzyl)morpholine (4), 4-(2-methylbenzyl)morpholine (5), 4-(2-chloro-6-fluorobenzyl)morpholine (7), 4-(2,6-dichlorobenzyl)morpholine (8), 4-(2,3-dichlorobenzyl)morpholine (12), 4-(2,4-dichlorobenzyl)morpholine (13), 4-(2,3-dichlorobenzyl)-2,6-dimethylmorpholine (14), 4-(naphthalen-1-ylmethyl)morpholine (15), and 4-(2-chlorobenzyl)thiomorpholine (16) were purchased from ChemBridge. The stereochemistry of the methyl substituents on 14 is unknown but the supplier indicates it is believed to be racemic.
Benzylmorpholine (1),(30) 4-(2-ethylbenzyl)morpholine (6), 4-(2-chloro-6-methylbenzyl)morpholine (9), 4-(3-chlorobenzyl)morpholine (10), 4-(4-chlorobenzyl)morpholine (11),(31) 1-(2-chlorobenzyl)-4-methylpiperazine (17), 1-(2,6-dichlorobenzyl)-4-methylpiperazine (18), 4-(thiophen-2-ylmethyl)morpholine (19), 4-((3-chlorothiophen-2-yl)methyl)morpholine (20), 1-(2-chlorobenzyl)-4-methylpiperazine (21), and 1-(2,6-dichlorobenzyl)-4-methylpiperazine (22) were synthesized in a MiniBlock® apparatus using published protocols.(32) Synthesis of 4-(1-(2-chlorophenyl)ethyl)morpholine (23) followed the same procedure(32) in a reaction flask, which resulted in a racemic mixture. Production of 1-(2-chlorobenzyl)-1H-imidazole (17) and 1-(2,6-dichlorobenzyl)-1H-imidazole (18) also used previously established methods.(33) 4-(2-Chlorophenethyl)morpholine (24) was synthesized by combining 2-chlorophenethyl bromide (1.3 mmol), morpholine (1.0 mmol), potassium carbonate (2.0mmol) in 4 mL of dry acetonitrile and stirred at 80 °C overnight. Product was extracted with ethyl acetate, washed with sodium bicarbonate, dried with magnesium sulfate, and solvent was removed under vacuum.
Compound analysis by 1H and 13C NMR was performed on a Bruker 400 MHz instrument. TopSpin and MestReNova were used to analyze the NMR data, which is given in ppm. LCMS was completed with a Waters 2795 HPLC and Waters LCT Premier TOF mass spectrometer. Compound purity was determined to be >95% by HPLC reverse phase chromatography. The HPLC mobile phase consisted of a 5-95% organic gradient with solvents A (99% water (pH 9.8 ammonium hydroxide), 1% acetonitrile) and B (99% acetonitrile, 1% water) and a flow rate of 0.6 mL/min.
4-(2-Ethylbenzyl)morpholine (6)
The synthesis afforded a clear oil with a yield of 77.9 mg (35%). HRMS (m/z): [M+H] 206.15; calculated 206.15. 1H NMR (400 MHz, chloroform-d) δ 7.35 - 7.12 (m, 4H), 3.79 - 3.67 (m, 4H), 3.52 (s, 2H), 2.78 (q, J = 7.5 Hz, 2H), 2.48 (dd, J = 5.8, 3.7 Hz, 4H), 1.27 (t, J = 7.5 Hz, 3H).
4-(2-Chloro-6-methylbenzyl)morpholine (9)
The synthesis yielded 55.6 mg (27%) of an oily product. HRMS (m/z): [M+H] 226.10; calculated 226.09. 1H NMR (400 MHz, chloroform-d) δ 7.31 - 7.19 (m, 1H), 7.14 - 7.07 (m, 2H), 3.73 - 3.60 (m, 6H), 2.57 - 2.49 (m, 4H), 2.47 (s, 3H).
4-(3-Chlorobenzyl)morpholine (10)
The synthesis resulted in 25 mg (44.7%) of a yellow oil. HRMS (m/z): [M+H] 212.09; calculated 212.08. 1H NMR (400 MHz, chloroform-d) δ 7.37 (d, J = 2.0 Hz, 1H), 7.33 - 7.05 (m, 3H), 3.81 - 3.62 (m, 4H), 3.49 (s, 2H), 2.46 (dd, J = 5.7, 3.7 Hz, 4H).
1-(2-Chlorobenzyl)-1H-imidazole (17)
The synthesis produced 30.7 mg (17%) of a yellow oil. HRMS (m/z): [M+H] 193.05; calculated 193.05. 1H NMR (400 MHz, chloroform-d) δ 7.57 (s, 1H), 7.41 (dd, J = 7.7, 1.6 Hz, 1H), 7.33 - 7.18 (m, 2H), 7.10 (s, 1H), 6.99 - 6.88 (m, 2H), 5.23 (s, 2H).
1-(2,6-Dichlorobenzyl)-1H-imidazole (18)
The resulting synthetic product was a yellow oil with a yield of 171.3 mg (75%). HRMS (m/z): [M+H] 227.01; calculated 227.01. 1H NMR (400 MHz, chloroform-d) δ 7.61 (s, 1H), 7.39 - 7.24 (m, 2H), 7.17 (dd, J = 8.8, 7.4 Hz, 1H), 6.98 (d, J = 14.9 Hz, 2H), 5.33 (s, 2H).
4-(Thiophen-2-ylmethyl)morpholine (19)
The synthesis resulted in 130 mg (71%) of a yellow oil. HRMS (m/z): [M+H] 184.08; calculated 184.07. 1H NMR (400 MHz, chloroform-d) δ 7.24 (dd, J = 5.0, 1.3 Hz, 1H), 6.94 (ddd, J = 7.4, 4.4, 2.2 Hz, 2H), 3.75 - 3.68 (m, 6H), 2.53 - 2.45 (m, 4H).
4-((3-Chlorothiophen-2-yl)methyl)morpholine (20)
The synthesis afforded a yellow oil with a yield of 150 mg (70%). HRMS (m/z): [M+H] 218.04; calculated 218.03. 1H NMR (400 MHz, Chloroform-d) δ 7.23 (d, J = 5.3 Hz, 1H), 6.87 (d, J = 5.3 Hz, 1H), 3.85 - 3.65 (m, 6H), 2.67 - 2.38 (m, 4H).
1-(2-Chlorobenzyl)-4-methylpiperazine (21)
The synthesis of 21 yielded a yellow oil with a yield of 168.8 mg (75%). HRMS (m/z): [M+H] 225.12; calculated 225.11. 1H NMR (400 MHz, chloroform-d) δ 7.49 (dd, J = 7.5, 1.9 Hz, 1H), 7.36 (dd, J = 7.7, 1.5 Hz, 1H), 7.31 - 7.15 (m, 2H), 3.65 (s, 2H), 2.58 (s, 4H), 2.48 (s, 4H), 2.31 (s, 3H).
1-(2,6-Dichlorobenzyl)-4-methylpiperazine (22)
The synthesis resulted in 184.1 mg (71.0%) of an amorphous solid. HRMS (m/z): [M+H] 259.08; calculated 259.07. 1H NMR (400 MHz, chloroform-d) δ 7.26 (d, J = 8.0 Hz, 2H), 7.09 (dd, J = 8.5, 7.5 Hz, 1H), 3.73 (s, 2H), 2.60 (s, 5H), 2.43 - 2.34 (m, 3H), 2.24 (s, 3H).
4-(1-(2-Chlorophenyl)ethyl)morpholine (23)
The synthesis afforded a yield of 48.1 mg (21%) of an oil. HRMS (m/z): [M+H] 226.10; calculated 226.09. 1H NMR (400 MHz, chloroform-d) δ 7.60 (dd, J = 7.8, 1.8 Hz, 1H), 7.35 (dd, J = 7.9, 1.4 Hz, 1H), 7.31 - 7.23 (m, 1H), 7.18 (dd, J = 7.7, 1.8 Hz, 1H), 3.89 (q, J = 6.6 Hz, 1H), 3.72 (dt, J = 5.8, 3.6 Hz, 4H), 2.57 (dt, J = 9.8, 4.6 Hz, 2H), 2.41 (ddd, J = 11.3, 5.8, 3.3 Hz, 2H), 1.31 (d, J = 6.6 Hz, 3H).
4-(2-Chlorophenethyl)morpholine (24)
The synthesis resulted in 129 mg (58%) of a yellow oil. HRMS (m/z): [M+H] 226.10; calculated 226.09. 1H NMR (400 MHz, Chloroform-d) δ 7.39 - 7.03 (m, 4H), 3.88 - 3.62 (m, 4H), 3.00 - 2.83 (m, 2H), 2.65 - 2.40 (m, 6H).
Compound solubility
For compounds evaluated in binding and inhibition studies, solubility was evaluated in buffer by monitoring turbidity in the UV/Vis cuvettes. For test compounds advanced to toxicity and metabolic stability studies, aqueous solubility was explicitly determined in phosphate-buffered saline at pH 7.4 using the shake-flask incubation method (24 hours at room temperature). The mean solubilities of 4-(2-chlorobenzyl)morpholine (3) and 4-(2-chloro-6-fluorobenzyl)morpholine (7) thus determined were 180 and 184 μM, respectively. These values are near the 200 μM maximum for the assay conditions and established that solubility is generally high for these compounds.
Metabolic Stability Phenotyping with recombinantly expressed CYPs
The half-lives of select compounds were determined with a panel of recombinantly expressed cytochrome P450 enzymes including 1A2, 2C9, 2D6, 3A4, and 2C19 (Pan Vera Corp., Madison, WI) and 2C8 and 2B6 (BD Gentest, San Jose, CA) in a plate assay format. Incubations consisted of 100 pmol/ml of one of the above P450 enzymes and 1 μM of the inhibitor in 0.1 mM potassium phosphate, pH 7.4, 3.33 mM MgCl2 in a total of 200 μl. These solutions were prewarmed to 37 °C for 10 min. prior to initiating the reaction with the addition of 1 mM NADPH regenerating system (NADP+, glucose-6-phosphate, glucose-6-phosphate dehydrogenase in 0.1 mM postassium phosphate). Aliquots of 25 μl were removed at various times (0, 5, 10, 20, 30, 45 min) and immediately quenched by addition to 75 μl of methanol containing buspirone as an internal standard. Controls substituted potassium phosphate buffer for the NADPH-generating system. Samples were centrifuged at 3,000 rpm and 50 μl of the supernatant diluted 1:1 with water. Mass spectrometry was used to determine the peak areas for the inhibitors and internal standard, from which the peak area ratios were calculated. The ln of this ratio vs. time gave the slope, which was converted to T1/2. All reactions were run in duplicate.
Metabolic Stability in Rat, Mouse, and Human Lung and Liver Microsomes
The half-lives of select compounds were determined with microsomes from pools of male B6C2F1 mouse liver, male B6C2F1 mouse lung, male Sprague-Dawley rat liver, male Sprague-Dawley rat lung, human liver and human lung (Xenotech) and from transgenic B6 Cyp2a(4/5)bgs-Null mice (35) as a gift from Dr. Xinxin Ding (Wadsworth Center). In each case 1.0 or 0.1 mg protein/ml, respectively, was added to 50 mM potassium phosphate buffer, pH 7.4, with 3 mM MgCl2, and 1 mM EDTA with and without the NADPH-generating system and 1 μM test compound (in 0.1% DMSO) to a total of 200 μl volume in a 96-well plate using a Tecan Liquid Handling System at 37 °C in triplicate. Reactions were initiated by the addition of the NADPH-generating system (1 mM NADP, pH 7.4, 5 mM glucose-6-phosphate, and 1 U/ml glucose-6-phosphate dehydrogenase) and were terminated at 0, 5, 10, and 20 minutes by the addition of 200 μl acetonitrile containing 0.1 μM antipyrene (v/v). Samples were centrifuged at 920 × g for 10 min. at 10 °C and the supernatant analyzed by LC/MS/MS. The time course of disappearance of the parent compound was evaluated as a single exponential decay and used to determine the rate constant of elimination values (kel, min−1) and converted to T1/2. The rate constant of elimination was used to calculate the in vitro intrinsic clearance, Clint microsomes: Clint-microsomes (μl/min/mg protein) = kel(min−1) × incubation volume (μl)/mg protein per incubation.
RESULTS AND DISCUSSION
To initiate a discovery effort to identify potential scaffolds for selective inhibitors of human CYP2A13, a high throughput screening campaign was conducted. A cuvette-based assay based on UV/Vis absorbance changes upon ligand binding (described in Methods) was miniaturized and adapted for screening in 384-well format. Fig. 1 summarizes data from the high throughput screen. An average Z’ score of 0.89 ± 0.08 was obtained from positive (CYP2A13+PEITC) and negative control wells (CYP2A13+DMSO), which is an acceptable dynamic range for a high throughput screening assay. Primary screening of 35,200 Chembridge library compounds resulted in a high hit rate of 1.4%. This relatively high hit rate likely reflects the nature of the target. Xenobiotic-metabolizing cytochrome P450 enzymes have evolved to be promiscuous and would be expected to bind a higher number of library compounds than most proteins. Of the 508 primary screen actives that were repurchased and tested for binding with both CYP 2A13 and the 94% identical CYP2A6, 93 compounds were found to bind to CYP2A13 specifically (ΔAbs 385-419 nm > 0.2) compared with CYP2A6 (ΔAbs 385-419 nm <0.03). Several CYP2A13-specific scaffolds were tentatively identified. Analysis of few representative compounds at varying concentrations resulted in identification of 4-(2-chloro-6-fluorobenzyl) morpholine as a true selective ligand of CYP2A13 vs. CYP2A6. As shown in the Figure 1D, 4-(2-chloro-6-fluorobenzyl) morpholine bound CYP2A13 with a Kd of approximately 2 μM, but caused no spectral shift with 2A6.
Figure 1.

Identification of CYP2A13-specific ligands via high throughput screening. A. Distribution of positive (PEITC) and negative (DMSO) controls. B. Scattergram of the UV/Vis spectral shifts observed for 35,200 compounds in the screen. C. Schematic representation of compound workflow. D. Identification of 4-benzylmorpholine scaffold as a specific ligand for CYP2A13 vs. CYP2A6. While CYP2A13 (left) shows increasing absorbance changes in response to ligand titration, no absorbance changes are observed upon titration with CYP2A6 (right).
Thus, this study was designed to evaluate 4-benzylmorpholine as a potential scaffold for CYP2A13-selective inhibitors. A pilot library of twenty-four 4-benzomorpholine analogs was designed to obtain structure–activity relationships in the parent scaffold and constructed using standard chemistry. Both ligand binding and enzyme inhibition were used to evaluate the interactions of these analogs with purified, recombinant CYP2A enzymes. Ligand binding is conveniently measured by observing, in difference mode, shifts in the electronic absorbance spectrum as ligands enter the active site. Inhibition was measured as the disruption of hydroxylation of the prototypical CYP2A substrate coumarin to 7-hydroxycoumarin. The resulting Kd and Ki values are shown in Table 1, along with the selectivity, as calculated by the ratio of the CYP2A6 value to the CYP2A13 value.
Table 1.
Spectral equilibrium dissociation constants (Kd) and inhibition constants (Ki) for the benzylmorpholine library with the human CYP2A6 and CYP2A13 enzymes, along with selectivity ratios for each.a
| Compound | Structure | Kd (μM)b,c | Selectivity | Ki (μM)d | Selectivity | ||
|---|---|---|---|---|---|---|---|
| 2A6 | 2A13 |
Kd (2A6)/ Kd (2A13) |
2A6 | 2A13 |
Ki (2A6)/ Ki (2A13) |
||
| 1 |
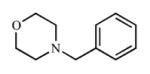
|
≥300 (I  II) II) |
116 (I) |
≥2.6 | 675 α=3.4 (M) |
477 (C) |
1.4 |
| 2 |
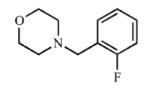
|
No** Spectral Shift |
71.7 (I) |
NA | ≥1000 | 379 (C) |
≥2.6 |
| 3 |
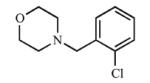
|
No** Spectral Shift |
5.9 ± 3.4 (I) |
NA | ≥1000 | 39.7 ± 4.3 (C) |
≥25.2 |
| 4 |
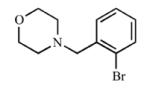
|
No** Spectral Shift |
6.3 (I) |
NA | ≥1000 | 17.6 ± 3.1 (C) |
≥56.8 |
| 5 |
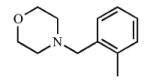
|
No** Spectral Shift |
18.5 (I) |
NA | ≥1000 | 66.9 (C) |
≥14.9 |
| 6 |
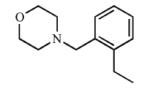
|
≥575 (I) |
4.5 (I) |
128 | 818 α=13.3 (M) |
16.9 (C) |
48.4 |
| 7 |
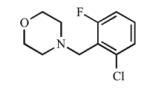
|
No** Spectral Shift |
5.2 (I) |
NA | 287 (C) |
38.1 (C) |
7.5 |
| 8 |
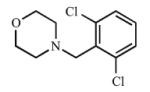
|
≥522 (I) |
1.3 (I) |
401 | 237 (C) |
18.1 (C) |
13.7 |
| 9 |
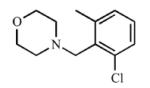
|
515.0 (I) |
1.1 (I) |
468 | 296 (C) |
19.7 ± 1.2* (C) |
15.0 |
| 10 |
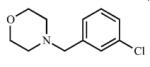
|
≥400 (I  II) II) |
69.1 ± 30.2 (I) |
≥5.8 | 352 (C) |
260 (C) |
1.4 |
| 11 |
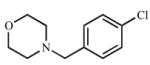
|
≥245 (I) |
166.8 ± 60.8 (I) |
≥1.5 | 448 (C) |
458 (C) |
1.0 |
| 12 |
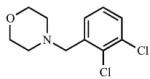
|
≥305 (I) |
30.8 ± 8.1 (I) |
9.9 | 836 (C) |
88.9 (C) |
9.4 |
| 13 |
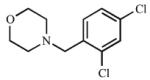
|
≥198 (I) |
34.2 (I) |
≥5.8 | 995.0 (C) |
113 ± 14 (C) |
8.9 |
| 14 |
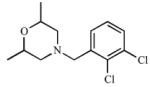
|
≥462 (I) |
37.0 (I) |
12.5 | ≥1000 | 646 α=19.6 (M) |
≥2.6 |
| 15 |
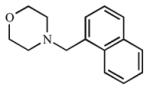
|
Weak Spectral Shift |
41.8+ (I) |
NA | 500 α=3.1 (M) |
219 ± 18.7* (C) |
2.3 |
| 16 |
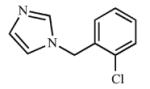
|
0.1 (II) |
<0.1 (II) |
≥1.0 | 2.9 (C) |
2.8 α=3.9 (M) |
1.0 |
| 17 |
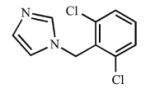
|
0.9 (II) |
<0.1 (II) |
≥9.0 | 0.9 (C) |
2.1* α=2.0 (M) |
0.4 |
| 18 |
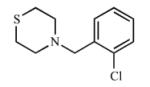
|
154 (II) |
8.3 ± 4.1 (I) |
18.6 | 483 α=3.1 (M) |
59.0 (C) |
8.2 |
| 19 |
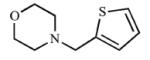
|
≥464 (I) |
30.8 (I) |
15.1 | 270 (C) |
131 (C) |
2.1 |
| 20 |
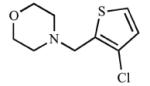
|
≥239 (I) |
10.7 (I) |
22.3 | 433 (C) |
54.7 (C) |
7.9 |
| 21 |
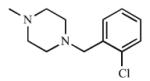
|
Weak Spectral Shift |
306 (I) |
NA | ≥1000 | ≥1000 | NA |
| 22 |
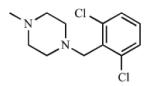
|
No Spectral Shift |
≥362 (I) |
NA | ≥1000 | ≥1000 | NA |
| 23 |
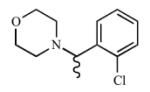
|
≥538 (I) |
7.6 ± 2.8 (I) |
≥70.8 | 98.5 (C) |
15.9 (C) |
6.2 |
| 24 |
|
≥604 (I) |
160 (I) |
≥3.8 | ≥1000 | 604 (C) |
≥1.7 |
Standard error is reported for data with ≥ 3 trials, but the majority of data is presented as the average of duplicate assays
NA: Not applicable
Spectral shift notes: (I), Type I spectral shift; (II), Type II spectral shift; (I II), Change from type I to type II with a spectral shift at inhibitor concentrations ≥300 μM; ≥ Minimal Kd value, except with 1 and 10 which had spectral shifts at ≥300 nm but no Kd value could be determined;
II), Change from type I to type II with a spectral shift at inhibitor concentrations ≥300 μM; ≥ Minimal Kd value, except with 1 and 10 which had spectral shifts at ≥300 nm but no Kd value could be determined;
Kd value from average of duplicate samples with <4% variation, but an additional assay reported a value of 121 μM;
Evaluated concentrations greater than or equal to the maximum CYP2A13 concentration tested
Inhibition notes: (C), Competitive Inhibitor; (M), Mixed Inhibitor;
Outlier exclusion
Titration of CYP2A13 and CYP2A6 with most of the 4-benzylmorpholine analogs revealed binding primarily resulted in type I spectral shifts in the UV/Vis difference spectra, as indicated by (I) in the Kd column of Table 1. This transition from the Fe hexacoordinate low spin, water bound state (λmin = ~420 nm) to a pentacoordinate high spin state (λmax = ~385 nm), reflects ligand binding in the P450 active site so as to displace the water molecule normally coordinated to the ferric heme iron in the resting state of the enzyme. In contrast, binding of compounds 1, 10, and 18 to CYP2A6 and binding of compounds 16 and 17 to both CYP2A13 and CYP2A6 resulted in difference spectra with decreases in absorbance at 393-413 nm and increases in absorbance at ~433 nm called type II spectra shifts. Such transitions upon ligand binding indicate conversion from the hexacoordinate, low-spin, water-bound iron to a hexacoordinate form in which the ligand typically coordinates to the iron via its lone pair electrons. Thus, the spectra of these compounds give an indication of the orientation of some ligands with respect to the catalytic heme iron. Some compounds yielded very limited or no spectral shifts with CYP2A6 which indicates the compound is either not binding or not binding in the active site in such a way that displaces water on the heme iron, which are desirable features of a CYP2A13-selective inhibitor.
Inhibition studies indicated that all but seven 4-benzylmorpholine analogs were purely competitive inhibitors of both CYP2A13 and CYP2A6. However, mixed inhibition resulting from inhibitor binding to both the free and substrate-bound form of the enzyme was observed for compounds 14, 16, and 17 with CYP2A13, and compounds 1, 6, 15, and 18 with CYP2A6. In all cases the alpha value was greater than 1, indicating a preference for inhibitor binding to free enzyme over the enzyme/substrate complex. Compounds 6 and 14 had the strongest preference for the free enzyme with alpha values of 13.3 and 19.6, respectively, while the remaining compounds demonstrating mixed inhibition had alpha values between 2.0-3.9.
Ortho (C2) substitution on the benzene ring
The parent, unsubstituted 4-benzylmorpholine compound (1), demonstrated a type I spectral shift with CYP2A13 (Kd = 116 μM), but a definitive Kd value could not be determined for CYP2A6. Binding of (1) to CYP2A6 resulted in spectral shifts at concentrations that converted from type I to type II over the course of the titration and were consistent with a Kd ≥300 μM. Not only did this parent compound bind poorly, it was also a poor inhibitor of both CYP2A13 (Ki = 477 μM) and CYP2A6 (Ki = 675 μM) and was not selective for either enzyme.
Addition of a fluorine atom on the benzene ring (2) would be expected to maintain the overall size of the analog in comparison to 1, but enhance the lipophilicity, electronegativity, and overall inductive effects of the compound. This modification did not have a significant impact on the binding and inhibition of CYP2A13, but interactions with CYP2A6 were greatly affected as no spectral shift, consistent with no binding, and poor inhibition (Ki = ≥1000) was observed with 2, illustrating an increase in selectivity for CYP2A13. Exchanging the fluorine atom for a chlorine atom (3) further enhanced the affinity and inhibition for CYP2A13 resulting in low micromolar Kd (5.9 ± 3.4 μM) and Ki (39.7 ± 4.3 μM) values, but without any apparent binding to CYP2A6. Thus, this compound is highly selective for CYP2A13. This same trend with selective binding and inhibition was observed for the remaining ortho substituted compounds (4-6), which contained either electron withdrawing or electron donating substituents. Compounds 4 and 6 were the most selective inhibitors of CYP2A13 and were similar in both size and lipophilicity but varied in electronic contributions from the substituent. These results illustrated that size and lipophilicity at the ortho position may be more important than electronics in conferring selective binding and inhibition of CYP2A13.
Disubstitution at the ortho position resulted in comparable Kd values or slight increases in affinity for CYP2A13 over 3, with Kd values of 5.2 μM (7), 1.3 μM (8), and 1.1 μM (9), while maintaining low micromolar inhibition with Ki values between 18.1 μM and 38.1 μM with CYP2A13. While 7 did not cause a spectral shift at all with CYP2A6, compounds 8 and 9 demonstrated some binding to CYP2A6 with Kd values of ~500 μM but were still >400-fold selective for binding to CYP2A13. Overall, the disubstituted compounds were not as selective as the singly substituted compounds with regard to enzyme inhibition. For CYP2A6, Ki values ranged from 237 μM to 296 μM, thus reducing the selectivity to 7.5- to 15-fold in comparison to ≥25-fold for (3). These results were interesting as CYP2A6 has an overall smaller active site volume than CYP2A13(22) and did not demonstrate a spectral shift with 7 and weakly bound 8 and 9, yet competitive inhibition was observed with CYP2A6 and the larger disubstituted analogs. Thus, while disubstitution increased the overall lipophilicity of the compound and maintained selective binding and inhibition of CYP2A13, it enabled weak binding and inhibition of CYP2A6, perhaps via a small conformational change in the CYP2A6 active site.
Meta and Para (C3 and C4) substitution on the benzyl ring
Single substitutions at the meta (10) and para (11) positions of the benzyl ring both decreased affinity (Kd = 69.1 ± 30.2 μM (10), 168 ± 60.8 μM (11)) and inhibition (Ki = 260 μM (10), and 458 μM (11)) for CYP2A13 in comparison to the ortho position (3). As with compound 1, definitive equilibrium dissociation constants could not be determined for 10 and 11 with CYP2A6, but weak spectral shifts are consistent with Kd values of ≥400 μM (10) and ≥ 245 μM (11). Similarly both compounds weakly inhibited CYP2A6 with Ki values of 352 μM (10) and 448 μM (11). Evaluation of the binding and inhibition selectivity ratios indicated that compounds 10 and 11 were not selective for either CYP2A enzyme. However, disubstitution with a chlorine atom at the ortho position (12, 13) rescued selectivity for CYP2A13 as illustrated by the inhibition results with an increase from ~1- to ~9-fold selectivity for CYP2A13, further demonstrating the importance of the ortho substitution. Additional di-methyl substitution on the morpholine ring as shown in 14 had minimal effects on Kd values, but this change had a greater effect in decreasing the inhibition of both CYP2A13 and CYP2A6.
Ring Substitutions
Multiple ring analogs demonstrated a wide range of binding and inhibition constants. A naphthalene substitution for benzene (15) weakly inhibited both CYP2A enzymes. Replacing the morpholine ring with imidazole (16, 17) resulted in extremely potent but non-selective analogs with submicromolar Kd and Ki values. Not surprisingly, the nitrogen of the imidazole ring coordinated to the heme as demonstrated by the type II spectral shift. Interestingly, both 16 and 17 were mixed inhibitors with low α values with CYP2A13, but purely competitive inhibitors with CYP2A6. While the type II binding and potent inhibition were anticipated for the imidazole compounds (16 and 17) due to the accessibility of the nitrogen for coordination to the heme iron, it was unexpected to observe a type II spectral shift for 18 with CYP2A6 (Kd = 154 μM) versus a type I spectral shift with CYP2A13 (Kd = 8.3 ± 4.1 μM). It is at present unknown as to which active site features resulted in the distinct binding modes. Additionally, unlike the imidazole containing ligands, compound 18 was still selective for CYP2A13 with both binding (18-fold) and inhibition (8-fold).
The sulfur atoms in the thiophene rings of compounds 19 and 20 differed from 18 in that only type I spectral shifts were observed for these compounds. The thiophene ring mimicked a benzene ring, and it was expected that 19 and 20 would correlate with results from compounds 1 and 3, respectively. Bioisosteric replacement of the benzene ring with a thiophene ring, 19, increased the affinity and inhibition of CYP2A13 (Kd = 30.8 μM, Ki = 131 μM) over the parent compound, 1, but had similar results for CYP2A6 (Kd = ≥464 μM, Ki = 270 μM). The ortho position of the sulfur may rationalize why 19 had a >3-fold selectivity for CYP2A13 versus the parent compound (1) even though 19 had the same spatial arrangement. As was observed for the parent compound, addition of a chlorine substituent at the ortho position on the thiophene analog, 20, increased the selectivity for CYP2A13 in both binding (22.3-fold) and inhibition (7.9-fold). This was achieved by decreasing the equilibrium binding constant to 10.7 μM for CYP2A13 but did not significantly impact the binding to CYP2A6 (Kd = ≥239 μM). Additionally, the chlorine atom increased selectivity for CYP2A13 by reducing the Ki value to 54.7 μM (20) while simultaneously decreasing inhibition of CYP2A6 (Ki = 433 μM).
The remaining ring analogs had a methylpiperazine ring in place of the morpholine ring (21, 22), which resulted in poor affinity and inhibition of both CYP2A enzymes as the Kd values were ≥300 μM for CYP2A13 with weak or no spectral shifts with CYP2A6, and the Ki values were ≥1000 μM. The weak interactions between 21 and 22 with the CYP2A enzymes were due to either the disruption of optimal interactions within the active site and/or charge addition. It was hypothesized that the methyl group could disrupt a hydrogen bond between the nitrogen atom and N297, one of two polar residues within the active site. In addition, incorporation of the methylpiperazine ring would have introduced a second positive charge on the ligand at physiological pH. This charge may not have been well tolerated, resulting in the poor binding and inhibition that was observed.
Linker Substitutions
Attachment of a methyl group to the one carbon linker (23) yielded a compound that maintained comparable affinity and inhibition of CYP2A13 (Kd = 7.6 ± 2.8 μM, Ki = 15.9 μM) as 3, but decreased selectivity since 23 also bound and inhibited CYP2A6 (Kd = ≥538 μM, Ki = 98.5 μM). These trends were not observed with 24 when the linker was extended to an ethyl group between the morpholine and benzene ring. Instead, a decrease in both affinity and inhibition with the CYP2A enzymes was observed. Decreased affinity for CYP2A13 could be due to the poor fit of the longer molecule within the active site, while the methyl group on the linker in 23 did not significantly clash within the active site but rather may have had additional van der Waals interactions with hydrophobic residues.
Compilation of the binding and inhibition studies has enabled the determination of preliminary structure-activity relationships (SAR) for 4-benzylmorpholine-based agents against CYP2A13 (Figure 2). We conclude that substitution at the ortho position of the benzyl ring was the most vital feature of the benzylmorpholine scaffold, promoting selective binding and inhibition of CYP2A13. Disubstitution at both ortho positions on the benzyl ring slightly enhanced affinity for both CYP2A enzymes resulting in a reduction in the selectivity for CYP2A13 versus the singly substituted analogs. Meta and para substitutions on the benzyl ring, linkage extension between the rings, and exchange of the morpholine ring for a methylpiperazine ring proved to be detrimental alterations in achieving selective binding and inhibition for CYP2A13.
Figure 2.
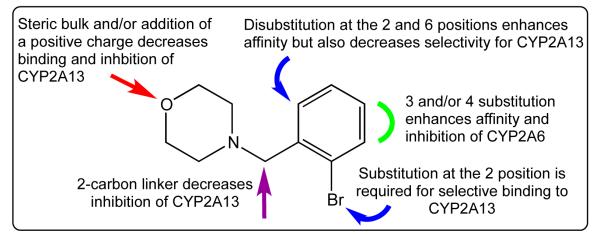
Structure-activity relationships for the 4-benzylmorpholine scaffold with human CYP2A enzymes.
Toxicity and Metabolic Stability Evaluation
The in vitro results led to the conclusion that 4-benzylmorpholine analogs could selectively inhibit CYP2A13. The next stage in the development of these analogs as potential chemopreventative agents was the determination of toxicity and metabolic stability. First, compounds 3 and 7, which represented both the singly and disubstituted compounds, were evaluated for genetic and cardiac toxicity (Cerep, Redmond, WA). The Ames test was employed to determine genetic toxicity, and few or no revertants were detected at compound concentrations ranging from 5-100 μM. The potential of 3 and 7 to cause cardiac toxicity via inhibition of the hERG channel was evaluated because this can be a major reason for drug withdrawal or limitation. In a patch clamp assay, compounds 3 and 7 inhibited the tail current by 13.9% and 24.8%, respectively, at the highest concentration tested, 10 μM. These results suggest the hERG IC50 is >10 μM, placing the potency in the low category and suggesting that toxicity associated with long QT intervals is unlikely to be arise from these compounds.
The metabolic stability of 3 and 7 were evaluated with human, mouse, and rat lung and liver microsomes over a 20-minute incubation (Xenotech, Overland Park, KS). Since the CYP2A13 enzyme targeted by these inhibitors is located in the respiratory tract, it was reassuring to observe that the benzylmorpholine analogs were very stable in human lung microsomes with minimal or no loss of substrate observed (Table 2). However, under the same conditions in human liver microsomes, at the end of 20 min only 53% or 14% of the substrates (3 and 7, respectively) remained and in vitro T1/2 values of 22.9 and 7.6 minutes were calculated. Corresponding in vitro intrinsic clearance values were 30.3 and 91.4 μl/min/mg protein, respectively. Thus the 4-benzylmorpholine compounds are metabolized at substantially different rates between tissues. Parallel evaluations from mouse and rat microsomes indicated ≤3% compound remaining in either the lung or liver microsomes after 20 min. Thus 4-benzylmorpholine compounds are rapidly metabolized in mouse and rat lung and liver and human liver microsomes, but persistent in human lung, the target tissue. This data brought to light substantial differences in metabolic stability of the samples between species and tissues, suggesting that dosing in human lung through inhalation would be optimal and that systematic exposure might be low due to rapid hepatic clearance. However, these results also brought to light metabolic concerns over preclinical compound testing in rat or mice model systems.
Table 2.
Metabolic stability of compounds 3 and 7 with human, mouse, and rat lung and liver microsomes. Data for (3) is in normal font, while data for (7) is in bold.
| Species | Test System |
Percent Loss of Substrate (%) |
Estimated Half life (min) |
Estimated in vitro intrinsic clearance (μl/min/mg protein) |
|---|---|---|---|---|
| Human | Liver | 47.0/85.5 | 22.9/7.6 | 30.3/91.4 |
| Lung | No Loss/No Loss | NA/NA | NA/NA | |
| Mouse | Liver | 100/100 | 1.0*/0.8* | 687/916 |
| Lung | 99.9/100 | 1.6*/1.3* | 433/551 | |
| Rat | Liver | 96.7/97.3 | 4.7*/4.0* | 149/173 |
| Lung | 99.8/100 | 2.4*/1.5* | 283/478 |
Values were less than the shortest incubation time and are therefore rough estimates
NA, Not applicable due to the lack of substrate depletion
In an effort to determine the enzyme(s) likely to be involved in metabolism of these compounds, compounds 3 and 7 (1 μM) were screened against a panel of the major human xenobiotic metabolizing CYP enzymes (CYP1A2, CYP2C8, CYP2C9, CYP2D6, CYP3A4, CYP2C19, and CYP2B6) and human liver microsomes (HLM) for in vitro half-life determination (Table 3). The percent compound remaining was detected at 0, 5, 10, 20, 30, and 45 minutes. The average in vitro half-life for the compounds ranged from <5 minutes with CYP2B6 (3 and 7) to essentially no loss with CYP1A2 (3). CYP2B6 most quickly reduced concentrations of benzylmorpholine compounds with 0% of both compounds remaining after only 5 minutes. Substantial metabolism also occurred with CYP2C19 and to a lesser extent CYP3A4 and CYP2C8.
Table 3.
Results for the average half-life of compounds 3 and 7 with a panel of human recombinant hepatic cytochromes P450.
| Compound | Average T1/2 (min) | ||||||
|---|---|---|---|---|---|---|---|
| CYP1A2 | CYP2C8 | CYP2C9 | CYP2D6 | CYP3A4 | CYP2C19 | CYP2B6 | |
| 3 | 2317 | 62 | 476 | 61 | 33 | 13 | <5 |
| 7 | 1214 | 51 | 230 | 78 | 25 | 18 | <5 |
The identification of CYP2B6 as a potentially significant component in rapid 4-benzylmorpholine metabolism also suggested a possible rationale for the differences between compound metabolic stability in mouse lung and human lung. While human lung tissues have lower expression levels of the functional CYP2B enzyme, CYP2B6,(34) mice have five CYP2B enzymes which might be active in the degradation of 4-benzylmorpholine compounds. We tested this hypothesis by evaluating the metabolic stability of 7 in lung and liver microsomes from knockout mice vs. wild type mice (Figure 3). These knockout mice had a deletion of a DNA segment containing the genes coding for the CYP2A4, CYP2A5, CYP2G1, CYP2S1, and all five CYP2B enzymes (Cyp2a(4/5)bgs mice) (35). It should be noted that these experiments were performed with 1/10 the amount of protein (0.1 mg/ml) as the studies above due to limited sample, so the clearance and t1/2 values can only be compared with the side-by-side controls for wild type mice. In these transgenic mouse lung microsomes metabolic stability of 7 was increased 5-fold compared to wild type lung microsomes (in vitro t1/2 = 669 min vs. 124 min for wild type mice; in vitro intrinsic clearance 10.4 vs. 55.7 for wild type mice). In liver microsomes from transgenic mice the metabolic stability of 7 increased by 1.7-fold (in vitro t1/2 = 127 min vs. 73 minutes for wild type mice; in vitro intrinsic clearance 54.8 vs. 95.3 for wild type mice). The increased metabolic stability in the knockout mouse lung microsomes was more similar to the metabolic stability in the human lung, suggesting the knockout mouse as an appropriate model for preclinical studies of inhibitor effects on NNK bioactivation.
Figure 3.

Metabolic stability of 7 in A) lung and B) liver microsomes generated from wild type and transgenic CYP2A/CYP2B knockout mice.
CONCLUSION
By altering various parts of the 4-benzylmorpholine small molecule scaffold, we were able to identify the key structural aspects that contributed to the selective binding and inhibition between the two closely related human CYP2A enzymes. This determination of structure-activity relationships demonstrated that ortho-substituted 4-benzylmorpholine analogs have the most promise as selective inhibitors of CYP2A13. Additionally testing for genetic and cardio toxicities revealed no obstacles to further development and testing. Metabolic stability testing demonstrated these compounds are stable in human lung microsomes but would be rapidly cleared upon circulation, which would be expected to reduce systemic exposure and any side effects. Administration through inhalation would reduce the first pass effects while delivering the CYP2A13 inhibitors directly to the target tissue where they are designed to block NNK bioactivation to carcinogens in tobacco users. While such a chemopreventative agent would not be a replacement for tobacco cessation efforts, it could be an effective complementary therapy to reduce the risk of lung cancer in tobacco users who are unable, unwilling, or in the process of ceasing tobacco use. Identification of CYP2A13-selective 4-benzylmorpholine compounds and an appropriate model system reflecting human lung lay the groundwork for pre-clinical in vivo testing of these compounds.
ACKNOWLEDGMENTS
Thanks are due to Kelin Li, who provided synthetic training to LB. We are grateful for purity analysis by Patrick Porubsky and Ben Neuenswander at The University of Kansas Center for Chemical Methodologies and Library Design. Toxicity studies were undertaken at Cerep. Metabolic stability in microsomes was undertaken at Xenotech, LLC. Mike Wester facilitated metabolic screening with the major human xenobiotic metabolizing CYP enzymes. Transgenic microsomes were a gift from Xinxin Ding at the Wadsworth Center, New York State Department of Health and State University of New York at Albany. This work was supported by the Kansas Masonic Cancer Research Institute, the University of Kansas General Research Fund, NIH grant NIGMS GM076343, and the Institute for Advancing Medical Innovation.
Abbreviations
- CYP
cytochrome P450
- hERG
human ether-a-go-go-related gene
- SAR
structure activity relationship
- NNK
4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone
- DMSO
dimethyl sulfoxide
- PEITC
phenethyl isothiocyanate
- POR
NADPH-cytochrome P450 oxidoreductase
- Ki
inhibition constant
- Kd
equilibrium dissociation constant
Footnotes
AUTHOR CONTRIBUTIONS AR: HTS screening; JA and FS: library design and synthetic routes; L.B: synthesis, NMR characterization, binding and inhibition assays, data analysis; D.N.: metabolic stability with recombinant enzymes; E.S. conceived and designed the study, participated in data analysis; L.B. and E.S. wrote the manuscript, which all authors edited and approved.
REFERENCES
- 1.Guengerich FP. Human Cytochrome P450 Enzymes. In: Ortiz de Montellano PR, editor. Cytochrome P450: Structure, Mechanism, and Biochemistry. Kluwer Academic/Plenum; New York: 2005. pp. 377–530. [Google Scholar]
- 2.Tomao F, Spinelli G, Vici P, Pisanelli GC, Cascialli G, Frati L, Panici PB, Tomao S. Current role and safety profile of aromatase inhibitors in early breast cancer. Expert Rev Anticancer Ther. 2011;11(8):1253–1263. doi: 10.1586/era.11.96. [DOI] [PubMed] [Google Scholar]
- 3.Vasaitis TS, Bruno RD, Njar VC. CYP17 inhibitors for prostate cancer therapy. J Steroid Biochem Mol Biol. 2011;125(1-2):23–31. doi: 10.1016/j.jsbmb.2010.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cvetkovic RS, Goa KL. Lopinavir/ritonavir: a review of its use in the management of HIV infection. Drugs. 2003;63(8):769–802. doi: 10.2165/00003495-200363080-00004. [DOI] [PubMed] [Google Scholar]
- 5.Elion R, Cohen C, Gathe J, Shalit P, Hawkins T, Liu HC, Mathias AA, Chuck SL, Kearney BP, Warren DR. Phase 2 study of cobicistat versus ritonavir each with once-daily atazanavir and fixed-dose emtricitabine/tenofovir df in the initial treatment of HIV infection. AIDS. 2011;25(15):1881–1886. doi: 10.1097/QAD.0b013e32834b4d48. [DOI] [PubMed] [Google Scholar]
- 6.Mathias AA, German P, Murray BP, Wei L, Jain A, West S, Warren D, Hui J, Kearney BP. Pharmacokinetics and pharmacodynamics of GS-9350: a novel pharmacokinetic enhancer without anti-HIV activity. Clin Pharmacol Ther. 2010;87(3):322–329. doi: 10.1038/clpt.2009.228. [DOI] [PubMed] [Google Scholar]
- 7.World Health Organization [accessed 11/14/08];Cancer. http://www.who.int/mediacentre/factsheets/fs297/en/
- 8.American Cancer Society . Cancer Facts and Figures 2012. Atlanta: 2012. [Google Scholar]
- 9.Center for Disease Control Smoking and Tobacco Use. http://www.cdc.gov/tobacco/data_statistics/fact_sheets/index.htm.
- 10.Hecht SS, Kassie F, Hatsukami DK. Chemoprevention of lung carcinogenesis in addicted smokers and ex-smokers. Nat Rev Cancer. 2009;9(7):476–488. doi: 10.1038/nrc2674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hecht SS. Biochemistry, biology, and carcinogenicity of tobacco-specific N-nitrosamines. Chem Res Toxicol. 1998;11(6):559–603. doi: 10.1021/tx980005y. [DOI] [PubMed] [Google Scholar]
- 12.Hecht SS. DNA adduct formation from tobacco-specific N-nitrosamines. Mutat Res. 1999;424(1-2):127–142. doi: 10.1016/s0027-5107(99)00014-7. [DOI] [PubMed] [Google Scholar]
- 13.Wang H, Tan W, Hao B, Miao X, Zhou G, He F, Lin D. Substantial reduction in risk of lung adenocarcinoma associated with genetic polymorphism in CYP2A13, the most active cytochrome P450 for the metabolic activation of tobacco-specific carcinogen NNK. Cancer Res. 2003;63(22):8057–8061. [PubMed] [Google Scholar]
- 14.Zhang X, Su T, Zhang QY, Gu J, Caggana M, Li H, Ding X. Genetic polymorphisms of the human CYP2A13 gene: identification of single-nucleotide polymorphisms and functional characterization of an Arg257Cys variant. J Pharmacol Exp Ther. 2002;302(2):416–423. doi: 10.1124/jpet.302.2.416. [DOI] [PubMed] [Google Scholar]
- 15.Weng Y, Fang C, Turesky RJ, Behr M, Kaminsky LS, Ding X. Determination of the role of target tissue metabolism in lung carcinogenesis using conditional cytochrome P450 reductase-null mice. Cancer Res. 2007;67(16):7825–7832. doi: 10.1158/0008-5472.CAN-07-1006. [DOI] [PubMed] [Google Scholar]
- 16.Zhou X, D’Agostino J, Xie F, Ding X. Role of CYP2A5 in the Bioactivation of the Lung Carcinogen 4-(Methylnitrosamino)-1-(3-Pyridyl)-1-Butanone in Mice. J Pharmacol Exp Ther. 2012;341(1):233–241. doi: 10.1124/jpet.111.190173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hecht SS, Trushin N, Rigotty J, Carmella SG, Borukhova A, Akerkar S, Rivenson A. Complete inhibition of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone-induced rat lung tumorigenesis and favorable modification of biomarkers by phenethyl isothiocyanate. Cancer Epidemiol Biomarkers Prev. 1996;5(8):645–652. [PubMed] [Google Scholar]
- 18.Morse MA, Eklind KI, Toussaint M, Amin SG, Chung FL. Characterization of a glucuronide metabolite of 4-(methyl-nitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and its dose-dependent excretion in the urine of mice and rats. Carcinogenesis. 1990;11(10):1819–1823. doi: 10.1093/carcin/11.10.1819. [DOI] [PubMed] [Google Scholar]
- 19.Rendic S. Summary of information on human CYP enzymes: human P450 metabolism data. Drug Metab Rev. 2002;34(1-2):83–448. doi: 10.1081/dmr-120001392. [DOI] [PubMed] [Google Scholar]
- 20.He XY, Shen J, Ding X, Lu AY, Hong JY. Identification of critical amino acid residues of human CYP2A13 for the metabolic activation of 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone, a tobacco-specific carcinogen. Drug Metab Dispos. 2004;32(12):1516–1521. doi: 10.1124/dmd.104.001370. [DOI] [PubMed] [Google Scholar]
- 21.Su T, Bao Z, Zhang Q-Y, Smith TJ, Hong J-Y, Ding X. Human cytochrome P450 CYP2A13: predominant expression in the respiratory tract and its high efficiency metabolic activation of a tobacco-specific carcinogen, 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone. Cancer Res. 2000;60(18):5074–5079. [PubMed] [Google Scholar]
- 22.Smith BD, Sanders JL, Porubsky PR, Lushington GH, Stout CD, Scott EE. Structure of the human lung cytochrome P450 2A13. J Biol Chem. 2007;282(23):17306–17313. doi: 10.1074/jbc.M702361200. [DOI] [PubMed] [Google Scholar]
- 23.DeVore NM, Smith BD, Urban MJ, Scott EE. Key residues controlling phenacetin metabolism by human cytochrome P450 2A enzymes. Drug Metab Dispos. 2008;36(12):2582–2590. doi: 10.1124/dmd.108.023770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shen AL, Porter TD, Wilson TE, Kasper CB. Structural analysis of the FMN binding domain of NADPH-cytochrome P-450 oxidoreductase by site-directed mutagenesis. J Biol Chem. 1989;264(13):7584–7589. [PubMed] [Google Scholar]
- 25.DeVore NM, Smith BD, Wang JL, Lushington GH, Scott EE. Key residues controlling binding of diverse ligands to human cytochrome P 450 2A enzymes. Drug Metab Dispos. 2009;37(6):1319–1327. doi: 10.1124/dmd.109.026765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DeVore NM, Scott EE. Structures of cytochrome P450 17A1 with prostate cancer drugs abiraterone and TOK-001. Nature. 2012;482(7383):116–119. doi: 10.1038/nature10743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Devore NM, Meneely KM, Bart AG, Stephens ES, Battaile KP, Scott EE. Structural comparison of cytochromes P450 2A6, 2A13, and 2E1 with pilocarpine. FEBS J. 2011 doi: 10.1111/j.1742-4658.2011.08412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rahnasto M, Raunio H, Poso A, Juvonen RO. More potent inhibition of human CYP2A6 than mouse CYP2A5 enzyme activities by derivatives of phenylethylamine and benzaldehyde. Xenobiotica. 2003;33(5):529–539. doi: 10.1080/0049825031000085979. [DOI] [PubMed] [Google Scholar]
- 29.von Weymarn LB, Murphy SE. CYP2A13-catalysed coumarin metabolism: comparison with CYP2A5 and CYP2A6. Xenobiotica. 2003;33(1):73–81. doi: 10.1080/0049825021000022302. [DOI] [PubMed] [Google Scholar]
- 30.Ohta H, Yuyama Y, Uozumi Y, Yamada YMA. In-water dehydrative alkylation of ammonia and amines with alcohols by a polymeric bimetallic catalyst. OrgLett. 2011;13(15):3892–3895. doi: 10.1021/ol201422s. [DOI] [PubMed] [Google Scholar]
- 31.Long TR, Maity PK, Samarakoon TB, Hanson PR. ROMP-Derived Oligomeric Phosphates for Application in Facile Benzylation. Org Lett. 2010;12(13):2904–2907. doi: 10.1021/ol1006604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. Reductive amination of aldehydes and ketones with sodium triacetoxyborohydride. studies on direct and indirect reductive amination procedures. J Org Chem. 1996;61(11):3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 33.Marcia de Figueiredo R, Thoret S, Huet C, Dubois J. Palladium-Catalyzed Intramolecular Arylation of N-Benzyl-2-iodoimidazoles: A Facile and Rapid Access to 5H-Imidazo[2,1-a]isoindoles. Synthesis. 2007;2007(4):529–540. [Google Scholar]
- 34.Ding X, Kaminsky LS. Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol. 2003;43(149-173) doi: 10.1146/annurev.pharmtox.43.100901.140251. [DOI] [PubMed] [Google Scholar]
- 35.Wei Y, Li L, Zhou X, Zhang Q-Y, Dunbar A, Liu F, Kleutzman K, Yang W, Ding X. Generation and characterization of a novel Cyp2A(4/5)bgs-Null Mouse Model. Drug Metab. Dispos. 2013;41(132-140) doi: 10.1124/dmd.112.048736. [DOI] [PMC free article] [PubMed] [Google Scholar]