

Abstract
Spinal muscular atrophy (SMA) is caused by mutations of the survival motor neuron 1 (SMN1) gene, retention of the survival motor neuron 2 (SMN2) gene and insufficient expression of full-length survival motor neuron (SMN) protein. Quinazolines increase SMN2 promoter activity and inhibit the ribonucleic acid scavenger enzyme DcpS. The quinazoline derivative RG3039 has advanced to early phase clinical trials. In preparation for efficacy studies in SMA patients, we investigated the effects of RG3039 in severe SMA mice. Here, we show that RG3039 distributed to central nervous system tissues where it robustly inhibited DcpS enzyme activity, but minimally activated SMN expression or the assembly of small nuclear ribonucleoproteins. Nonetheless, treated SMA mice showed a dose-dependent increase in survival, weight and motor function. This was associated with improved motor neuron somal and neuromuscular junction synaptic innervation and function and increased muscle size. RG3039 also enhanced survival of conditional SMA mice in which SMN had been genetically restored to motor neurons. As this systemically delivered drug may have therapeutic benefits that extend beyond motor neurons, it could act additively with SMN-restoring therapies delivered directly to the central nervous system such as antisense oligonucleotides or gene therapy.
INTRODUCTION
The inherited motor neuron disease spinal muscular atrophy (SMA) causes motor neuron degeneration and profound muscle weakness. Although disease severity varies, many patients die in infancy or early childhood. SMA is caused by homozygous mutations of the survival motor neuron 1 (SMN1) gene (1), but all patients retain the SMN2 gene in variable copy number. SMN2 contains a single nucleotide change such that most SMN2-derived mRNAs lack exon 7 thus producing a truncated, highly unstable SMN protein (2,3). A minority of SMN2 transcripts are full-length encoding functional SMN protein. Importantly, SMA disease severity correlates inversely with SMN2 copy number in patients (4) and, in rare individuals, five copies of SMN2 in the absence of SMN1 are sufficient to prevent disease manifestations (5). Currently, there is no disease-modifying treatment for SMA; however, increasing SMN expression in SMA patients has been a principal goal of therapeutics development (6).
SMA mouse models have been invaluable for investigating mechanisms of motor neuron dysfunction in SMA and for the preclinical testing of potential therapeutics (7,8). The most frequently studied severe SMA delta 7 (SMAΔ7) mice are null for the endogenous mouse Smn gene, but express the human SMN2 gene (and a delta 7 transgene) and thus reduced SMN protein levels (7,8). These mice exhibit severe motor behavioral abnormalities and death by 2 weeks of life. Several studies have documented abnormalities of both neuromuscular junction function and structure (9–15) as well as central synaptic abnormalities with resulting hypotrophy of muscle (12,16). Importantly, global restoration of SMN expression to these mice within the first postnatal week results in a marked extension of survival (17,18), providing proof of principle that restoration of SMN expression even after SMA disease onset is beneficial. Interestingly, recent studies using conditional SMA mice in which SMN expression is restored selectively to motor neurons suggest that targeting to motor neurons alone may be insufficient (19–21) and that other neurons and/or non-neuronal cell types play important roles in disease pathogenesis (19,22,23). Although both gene therapy and antisense oligonucleotides (ASOs) have also shown efficacy in severe mouse models (23–28), these biologics will likely need to be delivered directly to the CNS in patients with SMA. Thus, small molecules with widespread tissue bioavailability are also needed in this disease.
The quinazoline compound series was first identified during a high throughput screen to identify transcriptional activators of the SMN2 gene in a motor neuron-like cell line (29). Medicinal chemistry efforts generated several 2,4-diaminoquinazoline derivatives, some of which showed increased SMN mRNA and protein levels and gem number in treated SMA patient-derived fibroblast cell lines (29,30), and modestly increased survival in severe SMA mice (31). Using an unbiased protein microarray strategy, the molecular target of these compounds was identified as DcpS (32), an RNA decapping enzyme that hydrolyzes the m7GpppN cap structure following 3′ to 5′ mRNA degradation (33). RG3039 was selected for further development as the lead clinical candidate due to reduced off-target liabilities compared with other derivatives. Recently, a phase I clinical trial was completed with preliminary data indicating good safety results in healthy volunteers. In preparation for efficacy clinical trials in SMA patients, we investigated the effects of RG3039 in severe SMA mice.
Results
RG3039 is well-tolerated at doses that robustly inhibit DcpS in the CNS
RG3039 has a similar chemical structure to previously studied 2,4-diaminoquinazoline derivatives including D156844 (29,30). Quinazolines have been shown to inhibit the RNA decapping enzyme DcpS with the potency of SMN2 promoter activation strongly correlating with DcpS enzyme inhibition (32). To verify the ability of RG3039 to inhibit DcpS, we examined DcpS enzyme activity in protein extracts isolated from a P10 SMA mouse brain treated in vitro with various doses of drug. As expected, RG3039 robustly inhibited DcpS enzyme activity in a dose-dependent manner with an IC50 = 3.4 nm (Fig. 1A).
Figure 1.
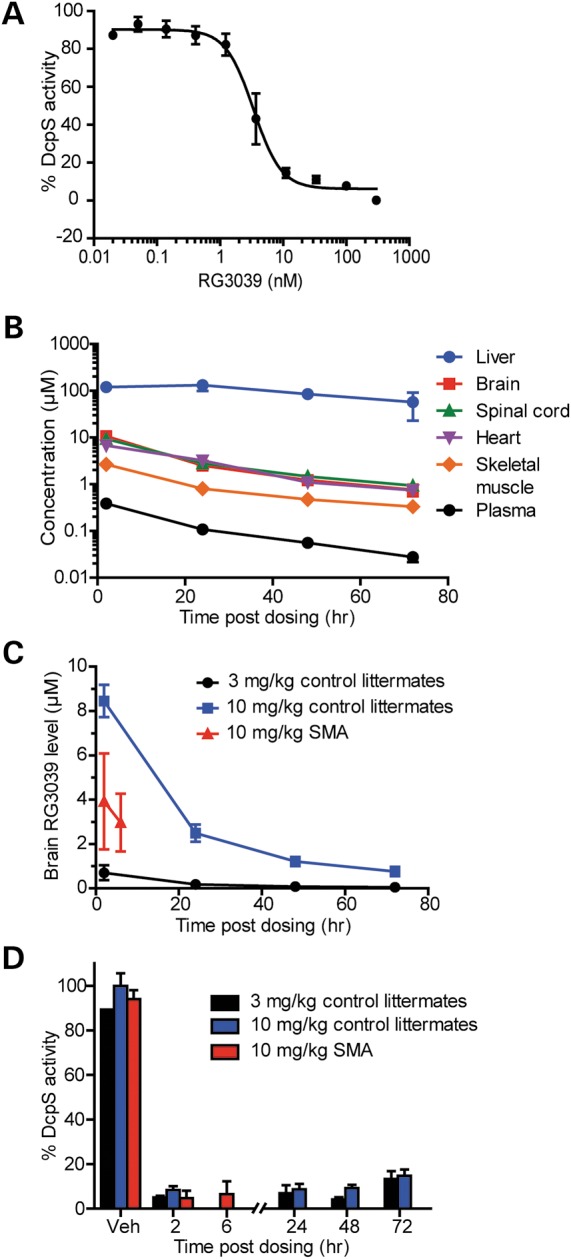
RG3039 crosses the blood brain barrier and inhibits the RNA decapping enzyme DcpS in the CNS. (A) DcpS enzyme activity in brain protein extracts treated in vitro with increasing doses of RG3039 (n = 3 per data point). (B) RG3039 drug levels in various tissues harvested from normal littermate mice treated from P1 to P10 with 10 mg/kg of RG3039 and harvested at various times after the last dose (liver, brain, plasma: n = 4 or 5 mice per time point and heart, spinal cord and muscle: n = 4–5 mice pooled). (C) RG3039 drug levels in brain and plasma of control littermate and SMA mice dosed with multiple doses of either 3 or 10 mg/kg of RG3039 (CNTL 3 mg/kg: n = 4 per time point, CNTL 10 mg/kg: n = 4–5 per time point, SMA 10 mg/kg: n = 13–14 per time point). (D) DcpS enzyme activity in the brains of control littermate and SMA mice dosed with 3 or 10 mg/kg of RG3039 (n = at least 4–5 per data point, except vehicle controls n = 1–2).
In order to explore the tolerability of RG3039 in vivo, we treated cohorts of SMAΔ7 mice daily with drug doses ranging from 3 to 40 mg/kg beginning on the day of birth (P1). Mice treated with RG3039 at 10 mg/kg/day showed increased weights and survival, which were not further increased by the 20 mg/kg dose (Supplementary Material, Fig. S1A and B). A dose of 40 mg/kg/day caused impaired weight gain and shortened survival suggesting some drug toxicity (Supplementary Material, Fig. S1A and B). To determine the biodistribution of RG3039, we examined drug levels of RG3039 in CNS tissues (brain and spinal cord) as well as in skeletal muscle, heart, liver and plasma in normal littermate mice dosed i.p. with 10 mg/kg daily from P1 until P10. RG3039 distributed to the brain, spinal cord, heart and skeletal muscle at approximately equivalent concentrations (Fig. 1B). The highest drug levels were evident in liver and lowest levels in plasma (Fig. 1C). We also examined drug levels in the brains of control littermates receiving 3 versus 10 mg/kg of RG3039 and of SMA mice receiving 10 mg/kg of RG3039 (Fig. 1C). As expected, the 10 mg/kg dose was associated with significantly higher brain drug concentrations than the lower dose. SMA mice dosed with 10 mg/kg showed increased drug levels relative to the 3 mg/kg-dosed control littermates, but reduced levels compared with littermates receiving the higher dose (Fig. 1C). In order to confirm the ability of RG3039 to inhibit DcpS enzyme activity in vivo, we examined DcpS enzyme activity in normal littermate and SMA mice dosed with either 3 or 10 mg/kg of RG3039 from P1 to P10 and harvested brain tissues at various time points after the last dose. DcpS activity was inhibited by ∼90% within 2 h and remained ∼80% inhibited 72 h after the last dose in both control littermates and SMA mice (Fig. 1D), despite the lower drug levels in SMA mice. Together these data indicate that RG3039 has excellent CNS bioavailability in vivo and robustly inhibits DcpS enzyme activity both in vitro and in vivo.
RG3039 increases survival and motor function of severe SMA mice despite modest effects on SMN expression or function
We next evaluated the effects of RG3039 on disease outcomes in SMA mice. SMA mice were treated with RG3039 at 10 mg/kg daily from P1 until death (Fig. 2A). Drug-treated SMA mice demonstrated a 26% increase in median survival compared with vehicle-treated mice (log rank P = 0.0001) (Fig. 2B) and a 16% increase in maximal weight achieved (P < 0.0001) (Fig. 2C). Improved righting time latencies evident after P11 (P < 0.0001) and improved ambulatory index scores evident between P13 and P17 (P < 0.0001) indicated significant improvements in motor function (Fig. 2D and E). These data are consistent with a previous study that showed a 21% increase in median survival of SMA mice treated postnatally with the quinazoline derivative D156844 (31).
Figure 2.

RG3039 improves survival and motor function of SMA mice. (A) Photo of vehicle-treated and 10 mg/kg RG3039-treated SMA mice and an untreated normal littermate at P13. (B) Kaplan–Meier survival curves, (C) weights, (D) righting time latencies, and (E) ambulatory index composite scores of vehicle-treated and 10 mg/kg RG3039-treated SMA mice (vehicle: n = 12, RG3039 10 mg/kg: n = 13, except ambulatory index n = 5–8 per group).
Given that RG3039 has been shown to activate the SMN2 promoter in vitro (29), we examined whether drug treatment increased SMN2 gene expression in SMA mice. SMA mice were treated with RG3039 at 10 mg/kg from P1 to P10 and full-length and truncated SMN transcript levels were determined by RT-qPCR in spinal cord, brain, skeletal muscle, liver and heart tissue 6 h after the last dose. Both full-length and truncated SMN transcript levels showed modest increases with full-length levels increasing by ∼30–40% in neural tissues (Fig. 3A and B). Brain tissues harvested at other time points (1, 2, 4, 12, 24 and 48 h) showed no greater change in transcript levels and similar patterns were seen with an alternative endogenous control (18 s) (data not shown). Interestingly, when we compared SMN transcript levels in vehicle-treated littermates to SMA mice, we observed that full-length levels (arising solely from SMN2) were reduced in SMA mice (Supplementary Material, Fig. S2) consistent with impaired exon 7 inclusion in the setting of SMN protein deficiency as previously reported by others (34). The modest increase in full-length SMN transcript levels observed after RG3039 treatment was sufficient to restore them to control littermate levels (Supplementary Material, Fig. S2).
Figure 3.

RG3039 minimally effects SMN levels in vivo. (A) Full-length (FL) and (B) SMNΔ7 SMN transcript levels in tissues from vehicle and RG3039-treated SMA mice at P10 6 h after the last dose, *P < 0.05, **P < 0.01 (brain: n = 13–15 per group, spinal cord: n = 5 per group, liver, heart, muscle: n = 10 per group). (C and D) Western blot analysis of SMN protein levels in (C) spinal cord and (D) brain extracts from vehicle and RG3039-treated SMA mice (n = 5 per group). (E and F) In vitro snRNP assembly analysis with radioactive U1 snRNA in (E) spinal cord or (F) brain extracts. U1 snRNA was immunoprecipitated with anti-SmB antibodies from each snRNP assembly reaction and analyzed by denaturing gel electrophoresis and autoradiography. (G and H) Quantification of U1 snRNA levels immunoprecipitated from snRNP assembly reactions with (G) spinal cord and (H) brain extracts from the experiments in (E) and (F) (n = 5 per group).
We next analyzed the effect of RG3039 treatment on SMN protein expression and found that the modest changes in full-length SMN transcript levels did not correlate with detectable increases in SMN protein levels in the CNS (Fig. 3C and D). Lastly, we measured whether RG3039 increased the activity of SMN in the assembly of spliceosomal snRNPs (35)—which is disrupted in SMA mice (36)—and found no significant changes in either spinal cord or brain (Fig. 3E–H). Thus, the phenotypic benefit of RG3039 in severe SMA mice appears to occur in the presence of limited effects on SMN protein expression or snRNP assembly.
RG3039 improves motor unit function in SMA mice
To explore the underlying cellular basis of the improved phenotype in SMA mice, we examined the structure and function of motor units. Motor neuron loss and associated muscle atrophy are the classical pathologies of SMA and we have previously shown particular vulnerability at the lumbar spinal segment 1 (L1) with ∼60% motor neuron loss occurring between P1 and P4 (16,20). To assess whether RG3039 can mitigate this loss, we quantified the number of axons in L1 ventral roots at P13 (Fig. 4A and B). Motor axon number was significantly reduced in vehicle-treated SMA mice compared with WT mice (SMA: 234.0 ± 22.3 versus WT: 372.0 ± 12.1); however, RG3039 treatment did not result in any improvement (SMA + RG3039: 238.0 ± 11.7) (Fig. 4B).
Figure 4.
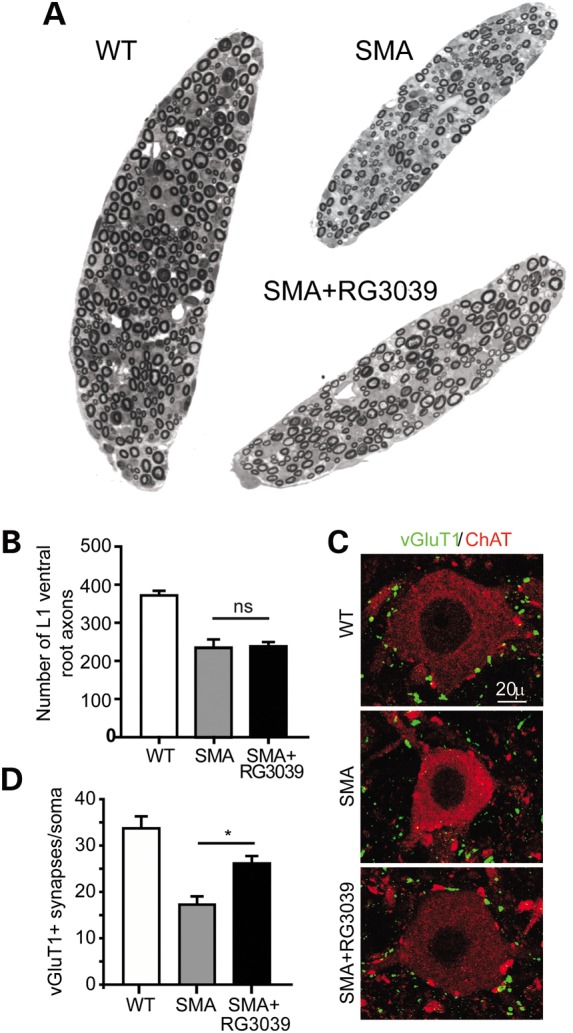
RG3039 does not change ventral root axon number, but does improve motor neuron somal synaptic input. (A) Image of L1 ventral roots from control littermate, vehicle-treated SMA and RG3039-treated SMA mice. (B) Quantification of ventral root number. (C) Representative images of motor neurons (red = ChAT) and somal VGLUT1 synapses (green = VGLUT1) from control littermate, vehicle-treated SMA and RG3039-treated SMA mice. (D) Quantification of VGLUT1 motor neuron somal synapses (*P < 0.05, n = 3 per group).
Although specific motor neuron populations degenerate in SMA mice, others are retained at disease end-stage, albeit they may have impaired synaptic connectivity. Lower lumbar motor neurons show a decrease in the number of proprioceptive primary afferents abutting onto the soma and proximal dendrites at a time when there is little motor neuron loss in SMA mice (12,16). To assess whether RG3039 treatment can increase the number of central synapses, we sectioned L3–L5 spinal cord segments from P13 mice and immunolabeled motor neurons and proprioceptive primary afferents with choline acetyltransferase (ChAT) and VGLUT1, respectively. RG3039 treatment resulted in a ∼50% increase in the number of VGLUT1 synapses on L3–L5 motor neurons (SMA: 17.2 ± 1.8; SMA + RG3039: 26.1 ± 1.6; WT: 33.7 ± 2.6, P = 0.0006) (Fig. 4C and D). Together these results indicate that although RG3039 may not prevent motor neuron loss, the synaptic input to remaining motor neurons is significantly improved.
In order to examine the functional output of motor neurons after RG3039 treatment, we examined the morphology and physiology of NMJ synapses. Denervation of NMJs is evident in several vulnerable muscles in SMA mice, but NMJs remain well innervated, despite a reduction in quantal content, in resistant muscles such as the EDL (37). To assess whether RG3039 can prevent denervation, we evaluated NMJs in the longissimus capitis, splenius capitis and serratus posterior inferior (SPI) muscles (Fig. 5A). The percentage of fully innervated NMJs was increased in both the SPI (SMA: 37.8 ± 2.9%; SMA + RG3039: 57.4 ± 3.0%) and longissimus (SMA: 20.4 ± 2.9%; SMA + RG3039: 40.6 ± 6.7%) muscles with RG3039 treatment (Fig. 5B), while denervation in the splenius capitis muscle was not significantly ameliorated (SMA: 52.7 ± 5.7%; SMA + RG3039: 63.6 ± 3.1%; Fig. 5B). No differences in neurofilament accumulation in the presynaptic terminals of NMJs from the SPI or EDL muscles were found (Fig. 5C and D), suggesting that neurofilament accumulation is not a determinant of NMJ denervation as we have previously reported (37).
Figure 5.

RG3039 prevents NMJ denervation and improves NMJ synaptic transmission. (A) Image of NMJs in the SPI muscle of WT, vehicle-treated SMA and RG3039-treated SMA mice. Blue (neurofilament), green (synaptophysin) and red (α-bungarotoxin). (B) Quantification of percentage of fully innervated NMJs in the longissimus, SPI and splenius muscles, *P < 0.05 (n = 7 per group). (C) Image of NMJs demonstrating neurofilament accumulations in presynaptic terminals. Green (neurofilament) and red (α-bungarotoxin). (D) Quantification of NF accumulation in SPI and EDL muscle (n = 4 per group). (E) MEPP amplitudes, (F) MEPP frequencies, (G) EPP amplitudes, and (H) quantal content in the EDL muscle of WT, SMA vehicle-treated and RG3039-treated SMA mice, *P < 0.05 (n = 6 per group). (I) The percentage of silent NMJs in the splenius muscle in WT, SMA vehicle-treated and RG3039-treated SMA mice, *P < 0.05 (n = 3–5 per group).
In order to assess the synaptic transmission of these NMJs, intracellular recording was performed in the denervation-resistant EDL and vulnerable splenius muscles from vehicle- and RG3039-treated SMAΔ7 mice at P12–13 (Fig. 5E–I). Consistent with our prior observations (12), quantal content in the EDL muscle was reduced by 25% in SMA mice compared with WT mice (Fig. 5H). With RG3039 treatment, SMAΔ7 NMJs showed an increase in the amplitude of endplate potentials (EPPs) and a corresponding 30% increase of quantal content. In the vulnerable splenius muscle in non-treated SMA mice, only ∼50% of the NMJs displayed EPPs upon nerve stimulation indicating that 50% of NMJs were silent (Fig. 5I). This result is consistent with our morphological analysis that only ∼56% of NMJs are fully innervated in SMAΔ7 splenius muscle (Fig. 5B). With RG3039 treatment, the percentage of silent junctions decreased to ∼20% (Fig. 5I). These data suggest that although of the number of fully innervated NMJs in the splenius muscle did not increase with RG3039 treatment, the function of partially innervated NMJs was significantly improved in this muscle. Together, our findings demonstrate that RG3039 improves NMJ synaptic transmission efficacy in both resistant and vulnerable muscles.
To determine whether the improvement of NMJ innervation and function was accompanied by increased muscle size, we measured the total cross-sectional area of the longissimus capitis. At P12, the total cross-sectional area was reduced by ∼8-fold in SMAΔ7 mice compared with WT littermates (Fig. 6A and B). Consistent with improved innervation of longissimus muscle NMJs (Fig. 5B), treatment with RG3039 resulted in a ∼80% increase in muscle cross-sectional area (Fig. 6A and B). This increase in muscle size can be principally attributed to a ∼95% increase in individual myofiber size (Fig. 6C) as there was no significant increase in myofiber number in treated mice (Fig. 6D).
Figure 6.
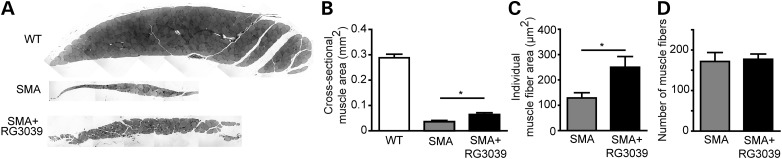
RG3039 increases myofiber size. (A) Image of cross-sections of the longissimus muscle from SMA vehicle-treated, SMA RG3039-treated and WT littermate mice. Quantification of (B) muscle cross sectional area, (C) myofiber size, and (D) myofiber number, *P < 0.05 (n = 6–7 per group).
RG3039 extends survival of ChATCre SmnRes conditional SMA mice
Several recent studies have highlighted that SMA disease pathology may extend beyond motor neurons and that increased SMN expression in motor neurons or in the CNS alone may be insufficient to provide substantial reversal of the SMA phenotype (19–21,23). Given the widespread tissue bioavailability of RG3039, we asked whether this compound could provide further therapeutic benefits to SMA mice in which SMN expression had already been restored to motor neurons. We utilized a line of SMA mice (ChATCreSmnRes SMA mice) in which increased SMN expression in motor neurons is driven by Cre under control of the ChAT promoter (20). We have previously reported that motor neuron expression of SMN in these mice results in only an 8-day increase in survival (median survival extended from 15 days in ChATCre−SmnRes SMA mice to 23 days in ChATCre+SmnRes SMA mice) (20). To explore whether ChATCre+SmnRes SMA mice would further benefit from RG3039, we treated these mice with vehicle or RG3039 at 10 mg/kg starting at P1. RG3039-treated ChATCre+SmnRes SMA mice showed a 66% increase in median survival (vehicle-treated: 25 days and RG3039-treated: 41.5 days, P < 0.0001) (Fig. 7A). In addition, drug-treated mice showed a 55% increase in maximal weight achieved (P < 0.0001) (Fig. 7B). These data indicate that RG3039 treatment combined with motor neuron restoration of SMN had better than additive effects and suggest that while RG3039 can improve motor neuron function, it may also have beneficial effects that are independent of motor neurons.
Figure 7.

RG3039 further extends survival of SMA mice expressing increased SMN levels in motor neurons. (A) Kaplan–Meier survival and (B) weight curves of ChATCre+ SmnRes SMA mice treated with vehicle or RG3039 (n = 8 per group).
DISCUSSION
RG3039 is the first small molecule developed specifically for SMA patients that has reached early stage clinical trials. Here, we demonstrate that RG3039 has widespread tissue bioavailability including in the CNS where it robustly inhibits its target enzyme, DcpS, and results in improved survival and motor behavior in SMAΔ7 mice. Importantly, this is accompanied by increased NMJ synaptic transmission as well as retained synaptic connectivity at both central and NMJ synapses, outcomes that would be predicted to have a meaningful impact on muscle power in SMA patients. In a co-submitted study, Gogliotti et al. demonstrate that RG3039 also increases motor function and survival in 2B/- SMA mice and in 5058 Hemi SMA mice confirming drug efficacy in three independent mouse models of SMA. In the milder 2B/- SMA model, a 600% increase in survival was also associated with improved NMJ structure and connectivity.
Multiple studies have demonstrated widespread morphological abnormalities of NMJs in SMA mice that include simplification of endplates, presynaptic neurofilament accumulations and denervation of specific muscle groups relevant to feeding, breathing and head movement (9–15,37). These abnormalities are associated with impaired synaptic transmission as evidenced by reduced quantal content. In this study, we show that RG3039 improves the function of fully innervated NMJs in the EDL muscle, but also likely improves the function of partially innervated NMJs in the splenius muscle with a 30% decrease in silent junctions despite a lack of increased number of fully innervated NMJs. These improvements are associated with both increased muscle size as well as reduced motor behavioral deficits in the absence of an increase in motor axon number. This suggests that improved function of remaining motor units in SMA mice can have a significant impact on muscle strength and survival. Recently, similar morphological abnormalities of NMJs have been documented in human SMA patients (38). NMJ transmission abnormalities have also been reported in human patients with 49% of type II and III SMA patients showing a decremental response with 3 Hz repetitive nerve stimulation (39). These studies indicate that benefits to NMJ connectivity and function observed in SMA mice could also have meaningful effects in SMA patients and that future clinical trials of RG3039 should include measures of NMJ function such as repetitive nerve stimulation as well as measures of muscle fatigue.
Several recent studies have suggested that SMN protein deficiency causes abnormalities of other neuronal types and other tissue types, which play important roles in SMA disease pathogenesis. Specifically, abnormalities of Ia afferent inputs to motor neurons arising from DRG neurons have been suggested to play a central role in motor neuron dysfunction and degeneration (12,16,40). It has also been suggested that abnormalities of liver, gut and heart contribute to disease in SMA mice. One of the most promising treatment strategies for SMA is ASOs, which enhance exon 7 inclusion into SMN2-derived mRNAs by specific binding to a splicing motif in SMN2 pre-mRNAs. The ISIS Pharmaceuticals SMNRx ASO is currently being tested in clinical trials in SMA patients. Because of poor distribution to CNS tissue from blood, it is being administered by intrathecal delivery despite limited demonstrated benefit in SMA mice compared with systemic delivery (23). Given the widespread biodistribution of RG3039, we evaluated whether it could have further benefit to SMA mice in which SMN had already been genetically restored to motor neurons. Strikingly, RG3039 provided even more survival benefit to ChATCre+SmnRes SMA mice than to SMAΔ7 mice, indicating that RG3039 might act additively with SMN-restoring therapies delivered only to the CNS.
The quinazoline compounds were initially identified during a screen for SMN2 promoter activators (29). Subsequent studies indicated that the target of the drug is the RNA decapping enzyme DcpS with quinazolines able to bind and hold DcpS in an open, catalytically incompetent conformation (32). Our studies indicate that DcpS enzyme activity is robustly inhibited in SMA mouse tissues and thus DcpS activity in circulating white blood cells can serve as a useful pharmacodynamic marker of drug activity in clinical trials of SMA patients. In contrast, the ability of RG3039 to activate SMN2 gene expression in vivo is modest and is not associated with detectable changes in SMN protein expression nor in snRNP assembly. These results may indicate that the beneficial effects of RG3039 are SMN independent, influencing aspects of RNA metabolism affected by low SMN levels. The best established function for DcpS is in hydrolyzing the m7GpppN cap structure of mRNAs (33). Nonetheless, DcpS also localizes to the nucleus and has been described as a nucleocytoplasmic shuttling protein (41). DcpS has also been shown to interact with components of the cap-binding complex, and reduced cellular levels of DcpS alters first intron mRNA splicing (41). It is intriguing to postulate that modulation of RNA metabolism by inhibiting the DcpS enzyme may be beneficial in SMA, especially given the observation of pre-mRNA splicing defects in severe SMA mice (42–44). Further investigation is needed to further understand the therapeutic mechanisms of action of RG3039.
MATERIALS AND METHODS
Mice
Experiments were carried out in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by Johns Hopkins University and University of Southern California Animal Care Committees. Breeder pairs for SMAΔ7 mice (7) on a FVB background (Stock number 005025) were purchased from Jackson Laboratories. Conditional SMA mice expressing increased SMN in motor neurons (ChATCreSmnRes SMA mice) were generated as described (20). Mice were genotyped by PCR of tail DNA as previously reported (20,45). SMA mice were null (mSmn−/−) and control littermates were heterozygous (mSmn+/−) or wild-type (mSmn+/+) at the mSmn allele.
Mice received intraperitoneal (i.p.) injections starting on post natal day 1 (P1) defined as the day of birth. All treated litters were culled at P4 to a total of six pups. RG3039 was dissolved in distilled water (or saline in Ko laboratory) at appropriate concentrations for 2.5 µl volume/gram injection volume. Daily weights and righting-time were determined starting on P1 as previously described (45). Ambulation index was performed between P13 and P23. A compilation score, calculated by multiplying the percentage of time spent upright by the number of 5.5×5.5 cm grid squares crossed, was determined during two 60 s trials as previously described (20).
RG3039 drug levels
Tissues were homogenized in 0.1% formic acid (1:5 w/v). Plasma samples were analyzed without further treatment. All samples were analyzed on an LC/MS/MS system, which consisted of an Agilent 1100 series quaternary pump and micro degasser (Agilent, Santa Clara, CA, USA), a CTC PAL autosampler (Leap Technologies, Carrboro, NC, USA) and an API 4000 triple quadrupole mass spectrometer equipped with an ESI TurboV source (AB Sciex, Foster City, CA, USA). The system was controlled by Analyst 1.4.2 (AB Sciex). MRM transitions were 432.3/256.2 and 382.2/206.2 for the RG3039 and internal standard, respectively. An Agilent ZORBAX Eclipse XDB-CN 2.1×50 mm 5-Micron was used for liquid chromatography using an acetonitrile gradient elution. The quantitation was performed by using Analyst 1.4.2 with a calibration curve fitted using a linear regression and 1/x2 weighting.
DcpS activity
Mouse brains were homogenized in lysis buffer (PBS, pH 7.4, 1% BSA, 0.1% IGEPAL CA-630 with protease and phosphatase inhibitors). For the DcpS enzyme decapping assay, 20 µg of tissue extract was mixed with 3 µm of biotinylated CAP substrate m7GpppA(N6) (kindly provided by Edward Darzynkiewicz) in the decapping buffer (50 mm Tris, pH 7.9, 100 mm MgCl2, 150 mm (NH4)2SO4) with a 10 µl total reaction volume. The reaction was terminated by adding 10 µl of 10 µm RG3039 to the reaction mixture. The resulting enzymatic reaction product, biotinylated ADP, was quantified using ELISA. Streptavidin coated plates (Pierce, #15501) were used to capture the ADP-biotin. The bound ADP-biotin was detected using ADP2 antibody (BellBrook Labs, # 2015) and HRP-conjugated goat anti-mouse IgG (Invitrogen, # 626520) incubated with TMB substrate (Cell Signaling, # 7004L) followed by measuring absorbance at 450 nm on a SpectraMax plate reader (Molecular Devices). A standard curve was generated with biotin-ADP.
Quantitative reverse transcription PCR (RT-qPCR)
RNA was isolated from tissues (brain, spinal cord, quadriceps muscle, liver or heart) using the RNeasy kit (Qiagen) and converted to cDNA and analyzed as previously described (20,45). Reactions were run in triplicate using the ABI Prism 7900 Sequence Detector System as previously described (45). Full-length (from SMN2) and truncated SMN (potentially arising from either the SMN2 or delta 7 SMN transgenes) transcript levels were expressed relative to the endogenous controls GusB or 18S rRNA mRNA. Data for each transcript were normalized to one vehicle-treated heterozygous mouse brain sample.
In vitro snRNP assembly and western blot
In vitro snRNP assembly assays with radioactive U1 snRNA and mouse tissue extracts followed by immunoprecipitation with anti-SmB antibodies were carried out according to established procedures (45–47). The levels of immunoprecipitated U1 snRNA were quantified using a Typhoon Phosphorimager (Molecular Dynamics). Western blot analysis with anti-SMN clone 8 (BD Transduction Laboratories) and anti-Tubulin DM 1A (Sigma) was performed as previously described (48).
Ventral root and muscle histology
Mice were intracardially perfused with Ringer's solution followed by 4% paraformaldehyde. The L1 ventral roots and longissimus muscle were dissected, post-fixed in osmium tetroxide and embedded in Epon. One µm thick sections were stained with toluidine blue and imaged by light microscopy. The number of ventral root axons, largest muscle cross-sectional area, individual myofiber area and the number of myofibers were quantified using Adobe Photoshop and ImageJ software.
Immunohistochemistry
The longissimus capitis, splenius capitis and SPI muscles were dissected and teased into layers 5–10 fibers thick. Pre-synaptic nerve terminals were labeled with anti-neurofilament (Millipore) and anti-synaptophysin (Invitrogen) antibodies, and postsynaptic acetylcholine receptors were labeled with α-bungarotoxin (Invitrogen). NMJs were visualized and quantified by epifluorescence or confocal microscopy. NMJs were considered fully innervated if the presynaptic nerve terminal completely co-localized with the postsynaptic endplate, and the innervation percentage was calculated as the number of fully innervated NMJs divided by the total number of NMJs quantified. Neurofilament accumulation was defined as neurofilament staining that occupied more than 1/4 of the motor endplate area (12).
To examine central synapses, lumbar cord spinal segments 3–5 (L3–5) were dissected and sectioned at 80 µm. Spinal cord sections were stained with anti-ChAT (Millipore) and anti-VGLUT1 (Synaptic Systems) antibodies, and imaged at 1 µm intervals using ×100 oil-immersion objectives on the Zeiss LSM confocal microscope. The number of primary proprioceptive afferents was quantified as VGLUT1+ puncta abutting on the soma and proximal dendrites of motor neurons.
Electrophysiological recordings from NMJs
Conventional intracellular recordings were performed as described previously (12). The EDL and splenius muscles with their nerves attached were dissected in normal Ringer's solution and were pre-incubated in 2–3 μm μ-conotoxin for 30 min to block muscle contraction. The recording was then performed in toxin-free Ringer's solution. EPPs were elicited by 1 Hz train through a suction electrode and recorded via a glass pipette filled with 3 m KCl. The EPP amplitudes were normalized to −50 mV and corrected for nonlinear summation. Data were acquired and analyzed using pClamp8 software and Minianalysis software. The mean quantal content was calculated by the direct method (49). The Student's t-test was used for comparison of means between groups.
Statistics
All data are expressed as mean ± standard error of the mean. Morphological and biochemical data were analyzed using Excel and Statistica software. Statistical significance was determined using either Student's t-test or a two-way analysis of variance (ANOVA).
SUPPLEMENTARY MATERIAL
Supplementary Material is available at HMG online.
Conflict of Interest statement. H.L.P., W.M., B.X., S.S., V.J. and J.R.R. are employees of Repligen Corporation, which has a financial interest in RG3039.
FUNDING
This work was supported in part by funding from Repligen Corporation. In addition, C.J.S. was supported by National Institutes of Health (R01NS062869). C.-P.K. was supported by the Families of Spinal Muscular Atrophy and the Spinal Muscular Atrophy Foundation. L.P. was supported by National Institues of Health (R01NS069601). Families of SMA supported and directed the identification and generation of the quinazoline series of compounds, including RG3039.
Supplementary Material
REFERENCES
- 1.Lefebvre S., Burglen L., Reboullet S., Clermont O., Burlet P., Viollet L., Benichou B., Cruaud C., Millasseau P., Zeviani M., et al. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- 2.Lorson C.L., Strasswimmer J., Yao J.M., Baleja J.D., Hahnen E., Wirth B., Le T., Burghes A.H., Androphy E.J. SMN oligomerization defect correlates with spinal muscular atrophy severity. Nat. Genet. 1998;19:63–66. doi: 10.1038/ng0598-63. [DOI] [PubMed] [Google Scholar]
- 3.Monani U.R., Lorson C.L., Parsons D.W., Prior T.W., Androphy E.J., Burghes A.H., McPherson J.D. A single nucleotide difference that alters splicing patterns distinguishes the SMA gene SMN1 from the copy gene SMN2. Hum. Mol. Genet. 1999;8:1177–1183. doi: 10.1093/hmg/8.7.1177. [DOI] [PubMed] [Google Scholar]
- 4.Feldkotter M., Schwarzer V., Wirth R., Wienker T.F., Wirth B. Quantitative analyses of SMN1 and SMN2 based on real-time lightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am. J. Hum. Genet. 2002;70:358–368. doi: 10.1086/338627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Prior T.W., Swoboda K.J., Scott H.D., Hejmanowski A.Q. Homozygous SMN1 deletions in unaffected family members and modification of the phenotype by SMN2. Am. J. Med. Genet. A. 2004;130:307–310. doi: 10.1002/ajmg.a.30251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Van Meerbeke J.P., Sumner C.J. Progress and promise: the current status of spinal muscular atrophy therapeutics. Discov. Med. 2011;12:291–305. [PubMed] [Google Scholar]
- 7.Le T.T., Pham L.T., Butchbach M.E., Zhang H.L., Monani U.R., Coovert D.D., Gavrilina T.O., Xing L., Bassell G.J., Burghes A.H. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum. Mol. Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- 8.Park G.H., Kariya S., Monani U.R. Spinal muscular atrophy: new and emerging insights from model mice. Curr. Neurol. Neurosci. Rep. 2010;10:108–117. doi: 10.1007/s11910-010-0095-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kariya S., Park G.H., Maeno-Hikichi Y., Leykekhman O., Lutz C., Arkovitz M.S., Landmesser L.T., Monani U.R. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008;16:2552–2269. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Murray L.M., Comley L.H., Thomson D., Parkinson N., Talbot K., Gillingwater T.H. Selective vulnerability of motor neurons and dissociation of pre- and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum. Mol. Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- 11.Kong L., Wang X., Choe D.W., Polley M., Burnett B.G., Bosch-Marce M., Griffin J.W., Rich M.M., Sumner C.J. Impaired synaptic vesicle release and immaturity of neuromuscular junctions in spinal muscular atrophy mice. J. Neurosci. 2009;29:842–851. doi: 10.1523/JNEUROSCI.4434-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ling K.K., Lin M.Y., Zingg B., Feng Z., Ko C.P. Synaptic defects in the spinal and neuromuscular circuitry in a mouse model of spinal muscular atrophy. PLoS ONE. 2010;5:e15457. doi: 10.1371/journal.pone.0015457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruiz R., Casanas J.J., Torres-Benito L., Cano R., Tabares L. Altered intracellular Ca2+ homeostasis in nerve terminals of severe spinal muscular atrophy mice. J. Neurosci. 2010;30:849–857. doi: 10.1523/JNEUROSCI.4496-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dachs E., Hereu M., Piedrafita L., Casanovas A., Caldero J., Esquerda J.E. Defective neuromuscular junction organization and postnatal myogenesis in mice with severe spinal muscular atrophy. J. Neuropathol. Exp. Neurol. 2011;70:444–461. doi: 10.1097/NEN.0b013e31821cbd8b. [DOI] [PubMed] [Google Scholar]
- 15.Lee Y.I., Mikesh M., Smith I., Rimer M., Thompson W. Muscles in a mouse model of spinal muscular atrophy show profound defects in neuromuscular development even in the absence of failure in neuromuscular transmission or loss of motor neurons. Dev. Biol. 2011;356:432–444. doi: 10.1016/j.ydbio.2011.05.667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mentis G.Z., Liu W., Blivis D., Drobac E., Crowder M.E., Kong L., Alvarez F.J., Sumner C.J., O'Donovan M.J. Early functional impairment of sensory-motor connectivity in a mouse model of spinal muscular atrophy. Neuron. 2011;10:453–467. doi: 10.1016/j.neuron.2010.12.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Le T.T., McGovern V.L., Alwine I.E., Wang X., Massoni-Laporte A., Rich M.M., Burghes A.H. Temporal requirement for high SMN expression in SMA mice. Hum. Mol. Genet. 2011;20:3578–3591. doi: 10.1093/hmg/ddr275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lutz C.M., Kariya S., Patruni S., Osborne M.A., Liu D., Henderson C.E., Li D.K., Pellizzoni L., Rojas J., Valenzuela D.M., et al. Postsymptomatic restoration of SMN rescues the disease phenotype in a mouse model of severe spinal muscular atrophy. J. Clin. Invest. 2011;121:3029–3041. doi: 10.1172/JCI57291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gogliotti R.G., Quinlan K.A., Barlow C.B., Heier C.R., Heckman C.J., Didonato C.J. Motor neuron rescue in spinal muscular atrophy mice demonstrates that sensory-motor defects are a consequence, not a cause, of motor neuron dysfunction. J. Neurosci. 2012;32:3818–3829. doi: 10.1523/JNEUROSCI.5775-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martinez T.L., Kong L., Wang X., Osborne M.A., Crowder M.E., Van Meerbeke J.P., Xu X., Davis C., Wooley J., Goldhamer D.J., et al. Survival motor neuron protein in motor neurons determines synaptic integrity in spinal muscular atrophy. J. Neurosci. 2012;32:8703–8715. doi: 10.1523/JNEUROSCI.0204-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lee A.J., Awano T., Park G.H., Monani U.R. Limited phenotypic effects of selectively augmenting the SMN protein in the neurons of a mouse model of severe spinal muscular atrophy. PLoS ONE. 2012;7:e46353. doi: 10.1371/journal.pone.0046353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sleigh J.N., Gillingwater T.H., Talbot K. The contribution of mouse models to understanding the pathogenesis of spinal muscular atrophy. Dis. Model Mech. 2011;4:457–467. doi: 10.1242/dmm.007245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hua Y., Sahashi K., Rigo F., Hung G., Horev G., Bennett C.F., Krainer A.R. Peripheral SMN restoration is essential for long-term rescue of a severe spinal muscular atrophy mouse model. Nature. 2011;478:123–126. doi: 10.1038/nature10485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Foust K.D., Wang X., McGovern V.L., Braun L., Bevan A.K., Haidet A.M., Le T.T., Morales P.R., Rich M.M., Burghes A.H., et al. Rescue of the spinal muscular atrophy phenotype in a mouse model by early postnatal delivery of SMN. Nat. Biotechnol. 2010;28:271–274. doi: 10.1038/nbt.1610. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 25.Passini M.A., Bu J., Roskelley E.M., Richards A.M., Sardi S.P., O'Riordan C.R., Klinger K.W., Shihabuddin L.S., Cheng S.H. CNS-targeted gene therapy improves survival and motor function in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2010;120:1253–1264. doi: 10.1172/JCI41615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Dominguez E., Marais T., Chatauret N., Benkhelifa-Ziyyat S., Duque S., Ravassard P., Carcenac R., Astord S., Pereira de Moura A., Voit T., et al. Intravenous scAAV9 delivery of a codon-optimized SMN1 sequence rescues SMA mice. Hum. Mol. Genet. 2011;20:681–693. doi: 10.1093/hmg/ddq514. [DOI] [PubMed] [Google Scholar]
- 27.Passini M.A., Bu J., Richards A.M., Kinnecom C., Sardi S.P., Stanek L.M., Hua Y., Rigo F., Matson J., Hung G., et al. Antisense oligonucleotides delivered to the mouse CNS ameliorate symptoms of severe spinal muscular atrophy. Sci. Transl. Med. 2011;3:72ra18. doi: 10.1126/scitranslmed.3001777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Porensky P.N., Mitrpant C., McGovern V.L., Bevan A.K., Foust K.D., Kaspar B.K., Wilton S.D., Burghes A.H. A single administration of morpholino antisense oligomer rescues spinal muscular atrophy in mouse. Hum. Mol. Genet. 2011;21:1625–1638. doi: 10.1093/hmg/ddr600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Jarecki J., Chen X., Bernardino A., Coovert D.D., Whitney M., Burghes A., Stack J., Pollok B.A. Diverse small-molecule modulators of SMN expression found by high-throughput compound screening: early leads towards a therapeutic for spinal muscular atrophy. Hum. Mol. Genet. 2005;14:2003–2018. doi: 10.1093/hmg/ddi205. [DOI] [PubMed] [Google Scholar]
- 30.Thurmond J., Butchbach M.E., Palomo M., Pease B., Rao M., Bedell L., Keyvan M., Pai G., Mishra R., Haraldsson M., et al. Synthesis and biological evaluation of novel 2,4-diaminoquinazoline derivatives as SMN2 promoter activators for the potential treatment of spinal muscular atrophy. J. Med. Chem. 2008;51:449–469. doi: 10.1021/jm061475p. [DOI] [PubMed] [Google Scholar]
- 31.Butchbach M.E., Singh J., Thorsteinsdottir M., Saieva L., Slominski E., Thurmond J., Andresson T., Zhang J., Edwards J.D., Simard L.R., et al. Effects of 2,4-diaminoquinazoline derivatives on SMN expression and phenotype in a mouse model for spinal muscular atrophy. Hum. Mol. Genet. 2010;19:454–467. doi: 10.1093/hmg/ddp510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Singh J., Salcius M., Liu S.W., Staker B.L., Mishra R., Thurmond J., Michaud G., Mattoon D.R., Printen J., Christensen J., et al. DcpS as a therapeutic target for spinal muscular atrophy. ACS Chem. Biol. 2008;3:711–722. doi: 10.1021/cb800120t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Liu H., Rodgers N.D., Jiao X., Kiledjian M. The scavenger mRNA decapping enzyme DcpS is a member of the HIT family of pyrophosphatases. EMBO J. 2002;21:4699–4708. doi: 10.1093/emboj/cdf448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jodelka F.M., Ebert A.D., Duelli D.M., Hastings M.L. A feedback loop regulates splicing of the spinal muscular atrophy-modifying gene, SMN2. Hum. Mol. Genet. 2010;19:4906–4917. doi: 10.1093/hmg/ddq425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pellizzoni L., Yong J., Dreyfuss G. Essential role for the SMN complex in the specificity of snRNP assembly. Science. 2002;298:1775–1779. doi: 10.1126/science.1074962. [DOI] [PubMed] [Google Scholar]
- 36.Gabanella F., Butchbach M.E., Saieva L., Carissimi C., Burghes A.H., Pellizzoni L. Ribonucleoprotein assembly defects correlate with spinal muscular atrophy severity and preferentially affect a subset of spliceosomal snRNPs. PLoS ONE. 2007;2:e921. doi: 10.1371/journal.pone.0000921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ling K.K., Gibbs R.M., Feng Z., Ko C.P. Severe neuromuscular denervation of clinically relevant muscles in a mouse model of spinal muscular atrophy. Hum. Mol. Genet. 2012;21:185–195. doi: 10.1093/hmg/ddr453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martinez-Hernandez R., Bernal S., Also-Rallo E., Alias L., Barcelo M., Hereu M., Esquerda J.E., Tizzano E.F. Synaptic defects in type I spinal muscular atrophy in human development. J. Pathol. 2012;229:49–61. doi: 10.1002/path.4080. [DOI] [PubMed] [Google Scholar]
- 39.Wadman R.I., Vrancken A.F., van den Berg L.H., van der Pol W.L. Dysfunction of the neuromuscular junction in spinal muscular atrophy types 2 and 3. Neurology. 2012;79:2050–2055. doi: 10.1212/WNL.0b013e3182749eca. [DOI] [PubMed] [Google Scholar]
- 40.Imlach W.L., Beck E.S., Choi B.J., Lotti F., Pellizzoni L., McCabe B.D. SMN is required for sensory-motor circuit function in Drosophila. Cell. 2012;151:427–439. doi: 10.1016/j.cell.2012.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen V., Liu H., Liu S.W., Jiao X., Kiledjian M. DcpS scavenger decapping enzyme can modulate pre-mRNA splicing. RNA. 2008;14:1132–1142. doi: 10.1261/rna.1008208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Z., Lotti F., Dittmar K., Younis I., Wan L., Kasim M., Dreyfuss G. SMN deficiency causes tissue-specific perturbations in the repertoire of snRNAs and widespread defects in splicing. Cell. 2008;133:585–600. doi: 10.1016/j.cell.2008.03.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lotti F., Imlach W.L., Saieva L., Beck E.S., Hao le T., Li D.K., Jiao W., Mentis G.Z., Beattie C.E., McCabe B.D., et al. An SMN-dependent U12 splicing event essential for motor circuit function. Cell. 2012;151:440–454. doi: 10.1016/j.cell.2012.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baumer D., Lee S., Nicholson G., Davies J.L., Parkinson N.J., Murray L.M., Gillingwater T.H., Ansorge O., Davies K.E., Talbot K. Alternative splicing events are a late feature of pathology in a mouse model of spinal muscular atrophy. PLoS Genet. 2009;5:e1000773. doi: 10.1371/journal.pgen.1000773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Avila A.M., Burnett B.G., Taye A.A., Gabanella F., Knight M.A., Hartenstein P., Cizman Z., Di Prospero N.A., Pellizzoni L., Fischbeck K.H., et al. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J. Clin. Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gabanella F., Carissimi C., Usiello A., Pellizzoni L. The activity of the spinal muscular atrophy protein is regulated during development and cellular differentiation. Hum. Mol. Genet. 2005;14:3629–3642. doi: 10.1093/hmg/ddi390. [DOI] [PubMed] [Google Scholar]
- 47.Pellizzoni L., Baccon J., Rappsilber J., Mann M., Dreyfuss G. Purification of native survival of motor neurons complexes and identification of Gemin6 as a novel component. J. Biol. Chem. 2002;277:7540–7545. doi: 10.1074/jbc.M110141200. [DOI] [PubMed] [Google Scholar]
- 48.Ruggiu M., McGovern V.L., Lotti F., Saieva L., Li D.K., Kariya S., Monani U.R., Burghes A.H., Pellizzoni L. A role for SMN exon 7 splicing in the selective vulnerability of motor neurons in spinal muscular atrophy. Mol. Cell. Biol. 2012;32:126–138. doi: 10.1128/MCB.06077-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Del Castillo J., Katz B. Quantal components of the end-plate potential. J. Physiol. 1954;124:560–573. doi: 10.1113/jphysiol.1954.sp005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.