

Abstract
The history of stem cell gene therapy is strongly linked to the development of gene therapy for severe combined immunodeficiencies (SCID) and especially adenosine deaminase (ADA)-deficient SCID. Here we discuss the developments achieved in over two decades of clinical and laboratory research that led to the establishment of a protocol for the autologous transplant of retroviral vector-mediated gene-modified hematopoietic stem cells, which has proved to be both successful and, to date, safe. Patients in trials in three different countries have shown long-term immunological and metabolic correction. Nevertheless, improvements to the safety profile of viral vectors are underway and will undoubtedly reinforce the position of stem cell gene therapy as a treatment option for ADA-SCID.
Keywords: adenosine deaminase, severe combined immunodeficiency, gene therapy, hematopoietic stem cell, retrovirus, clinical trial
Adenosine deaminase deficiency
Severe combined immunodeficiencies (SCID) are a set of diseases caused by monogenic disorders that impair T-cell development and, depending on the gene implicated, can be associated with faults in the development of other hematopoietic lineages. At least ten different molecular defects that cause SCID have been described1 and ADA deficiency accounts for ~10% to 20% of the total number of cases.
Consistent with the clinical and immunological profile of SCID, ADA-deficient patients present with severe lymphopenia, absence of cellular- and humoral-mediated immunity, recurrence of opportunistic infections and failure to thrive. In addition to the immunological manifestations and because ADA is a ubiquitously expressed protein, patients also present with skeletal, lung, liver, gastrointestinal tract and neural abnormalities. In its most severe form, ADA-SCID is fatal within the first year of life.
Adenosine deaminase (ADA, EC3.5.4.4) is an enzyme of the purine salvage pathway, expressed at different levels in all tissues of the body, with the highest levels detected in the thymus.2 ADA catalyzes the conversion of deoxydenosine (dAdo) and adenosine (Ado) to deoxyinosine and inosine, respectively (Figure 1).2 In the absence of ADA activity, dAdo accumulates in extracellular compartments and within cells, where it is converted by the enzyme deoxycytidine kinase (dCydK) to deoxyadenosine trisphosphate (dATP). The build-up of both dAdo and dATP is the main cause of lymphotoxicity and the immunological alterations. dATP inhibits ribonucleotide reductase, an enzyme that participates in DNA replication and repair,3 induces apoptosis in immature thymocytes4 and interferes with terminal deoxynucleotidyl transferase (TdT) activity, thus limiting V(D)J recombination and antigen receptor diversity.5 dAdo accumulation inactivates the enzyme S-adenosylhomocysteine hydrolase (SAHH)6 and results in inhibition of transmethylation reactions necessary for effective lymphocyte activation. Elevated levels of Ado, acting through cell surface G protein coupled receptors, may contribute to immune dysfunction7,8 and pulmonary inflammation associated with ADA deficiency.9
Figure 1.

Pathogenesis of ADA deficiency
Abbreviations: ADA, adenosine deaminase; ADCY adenylyl cyclise; Ado, adenosine; AdoHCy, S-adenosylhomocysteine; AdoK, adenosine kinase; AdoR, adenosine receptors; c-AMP, cyclic adenosine monophosphate; d-Ado, 2-deoxyadenosine; d-AMP, deoxyadenosine monophosphate; d-ATP, deoxyadenosine triphosphate; dCydK, deoxycytidine kinase; d-Ino, deoxyinosine; G, g-proteins; Ino, inosine; SAHH, S-adenosylhomocysteine hydrolase.
ADA-SCID treatment
The pathogenesis of ADA-SCID arises from the accumulation of toxic metabolites in intra- and extracellular compartments. The nucleosides Ado and dAdo are transported to the plasma once the catalytic activity of dCydK is saturated whereas dATP is trapped inside the cells. In principle, any form of treatment that aids the clearance of Ado and dAdo from the plasma will shift the dynamic equilibrium of metabolites from within cells and then achieve detoxification, ultimately promoting immune recovery. Thus delivery of ADA enzyme, whether exogenously (eg, enzyme replacement therapy) or packaged within cells (allogeneic stem cell transplant or gene therapy), is the goal for correction of the disease phenotype.
Hematopoietic stem cell transplant
Allogeneic hematopoietic stem cell transplant (HSCT) has been the mainstay of patients with ADA-SCID. In terms of survival, the success rate of the transplant depends heavily on the bone marrow donor: according to the latest data,10 transplants from fully human leukocyte antigen (HLA)-matched sibling and related donors are highly successful with survival rates of ~90%. However, survival rates following transplants from matched unrelated and mismatched related donors (haplo-identical) are less good with the latest data showing ~67% and 43% survival respectively. In the case of matched unrelated and HLA-haploidentical transplant, it is necessary to administer chemotherapeutic agents in order to improve the chances of donor cell engraftment and the morbidity and mortality associated with these transplants may in part be attributable to the toxicity of the cytoreductive regime.
HSCT confers good immune recovery as measured by lymphocyte counts and function and antibody production. Metabolic correction is also achieved, with dATP levels significantly reduced from pre-transplant levels, although they remain higher than in normal individuals11 or patients on PEG-ADA.12 It is possible that HSCT ameliorates the symptoms manifested in the lungs or the liver; in the animal model for ADA-SCID, the transplant of ADA+/+ bone marrow in neonatal ADA−/− mice was able to prevent the inflammation and thickening of the lung tissue, as well as promoting the absence of liver toxicity.13 However, HSCT does not appear to correct the central nervous system (CNS)-related features of the disease, as ADA-SCID patients treated with HSCT have ongoing cognitive and behavioral abnormalities,14–16 despite effective metabolic detoxification and immune recovery.
Enzyme replacement therapy
Intramuscular injections of Polyethylene glycol (PEG)ylated bovine ADA, or enzyme replacement therapy (ERT) offers an alternative treatment for ADA-SCID. By maintaining PEG-ADA levels about 100-fold above the normal, it is possible to bring down the circulation levels of Ado and dAdo, but this requires weekly or biweekly administration. PEG-ADA is often preferentially used when no suitable donor is available for an HSCT transplant or to stabilize a patient in preparation for HSCT.
Immune recovery becomes evident at about 2 to 4 months after starting therapy with initial B and NK cell recovery followed by T-cell reconstitution. Long-term administration seems to be well tolerated, the probability of surviving 20 years while on ERT is about 80%.17 Although these patients are clinically well (ie, free of opportunistic infections), there is evidence of a decline in immune function parameters over time. In a retrospective study by Chan and colleagues all nine ADA-deficient patients on long-term PEG-ADA (5 to 12 years) showed lymphocyte counts below normal and decreasing over time, as well as a decline in mitogen-induced proliferative response.18 Malacarne and colleagues also showed a decrease in thymic function in patients on long-term enzyme replacement therapy.19
As for the effects of PEG-ADA on the nonimmunological symptoms of the disease, there is no formal study in long-term patients; however some information can be taken from case reports and studies in ADA-SCID mice. A neonate ADA-SCID patient presenting with hepatitis, hyperbilirrubinemia and immune deficiency experienced recovery from all symptoms after receiving PEG-ADA.20
In addition, the pulmonary insufficiency observed as one of the prominent features in the mouse model for ADA-SCID can be prevented by injecting the pups with a low dose of PEG-ADA (100 to 500 units/kg body weight) from birth.21 The plasma levels attained with the low dose protocol were below that which is considered therapeutic in humans, and therefore failed to reduce the Ado, dAdo, and dATP levels in high-turnover tissues like the thymus and the spleen. A high-dose protocol (1000 to 5000 units/kg body weight) is required to reach therapeutic levels in plasma, reduce metabolite levels in thymus and spleen and ultimately achieve immune recovery.
As for the CNS abnormalities, there is no study with regard to the effect of PEG-ADA, as no single center has enough patients on long-term ERT to compare with the outcome of HSCT patients.
ADA-SCID as a good candidate for gene therapy
Although the existing treatment options give hope to patients with an otherwise lethal disease, there are certain disadvantages associated with both options. As discussed patients undergoing HSCT have a high probability of survival only if a fully matched related donor is available; haploidentical transplants have a low success rate in ADA-SCID patients and should only be undertaken when there is no other option available.17 PEG-ADA promotes rapid detoxification of metabolites, is able to improve the clinical well-being of the patient and achieve some immune recovery. Perhaps the most important limitations for its use are its high cost (£150,000 to £200,000 per patient per year), the need for frequent administration and poor immune recovery over a long term and the lack of availability in certain countries.
Over 20 years ago, the need to provide an alternative option for those patients for whom a suitable bone marrow donor could not be found or who were not able to start on PEG-ADA, was identified.
Stem cell gene therapy may provide that option for a number of reasons. The use of gene-modified autologous stem cells avoids the risks of graft versus host disease and also the need for full cytoreductive conditioning regimes and their concomitant risks. In addition to this and with ADA being an enzyme, tight gene expression regulation is not crucial and overexpression is not likely to cause detrimental effects.
On the other hand it may not be necessary to reach ADA normal levels to obtain a beneficial outcome. ADA mutation carriers are usually normal despite having ~20% to 50% of the normal activity and according to Hirschhorn and others, it is possible to see normal immune function in individuals with as low as 5% of the normal ADA.22,23
In addition, gene-corrected cells are expected to have a selective advantage over their deficient counterparts in a toxic environment. Such a conclusion comes from rare cases of somatic mosaicism, where patients experience immune recovery after emergence of a clone that has reverted the mutation in the ADA gene to normal.24
Pre-clinical studies towards the development of gene therapy
The gene encoding ADA was cloned and made available as a cDNA in the early 1980s25–27 and it was the first gene to be associated with a form of SCID;28 therefore it became the focus of some of the first attempts to cure diseases with gene therapy.
By the mid-1980s, the advent of retroviral vector technology made it possible to deliver therapeutic genes into cells in a stable and efficient manner, since these vectors are able to integrate into the genome of the host cell. Valerio and colleagues and Friedman and colleagues reported the construction of the first retroviral vectors with the human ADA cDNA under the control of the viral long terminal repeat (LTR), and effectively transduced and expressed the transgene in NIH3T3 mouse fibroblasts29 and the mouse T-cell lymphoma BL/VL3,30 respectively. Using a similar retroviral vector, Kantoff and colleagues transduced T and B ADA-deficient cells derived from patients and were able to express sufficient ADA to correct their hypersensitivity to 2′ deoxyadenosine.31
It was not until Joyner and colleagues reported the first transduction of mouse hematopoietic stem cells with retrovirus in vitro,32 followed by the demonstration that retrovirus-infected mouse bone marrow cells were capable of contributing to the reconstitution of lethally-irradiated recipients and retained the ability to transmit the transgene to their daughter cells in vivo33,34 that the prospect of modifying human stem cells in the same way could be envisaged. Belmont and colleagues then published the successful transduction of mouse hematopoietic progenitors with an ADA-neoR retroviral vector.35 Viruses expressing human ADA were used in transplant experiments in mice and followed up to 6 months after transplant36,37 Expression of ADA could be detected in all hematopoietic lineages, with an activity comparable or even superior to the endogenous mouse ADA activity37
Following the promising results observed in the murine models, the first attempts were made at reconstituting large, nonhuman primate models with bone marrow cells that had been subjected to the retrovirus transduction procedure.38–40 It was soon evident that primate stem cells were not as easily transduced with retroviruses as were mouse stem cells; the maximum proportion of peripheral blood cells containing the provirus in these models was 1%.
Retroviruses require host cell division to accomplish genome integration and in primates, a larger proportion of stem cells are in the quiescent (G0) status, therefore the efficiency of transduction is reduced. Stimulation of proliferation with combinations of cytokines and growth factors, particularly interleukin-3 (IL-3) and IL-6 improved the transduction efficiency in human progenitor cells.41
An important piece of information obtained from these early studies was the absence of adverse events attributed to the recombinant retroviral vector. T-cell lymphomas developed in irradiated rhesus monkey recipients after transplantation with stem cells infected with a retrovirus vector preparation, but this was due to contamination with replication-competent retrovirus (RCR)42,43 These observations underlined the importance of screening for RCR in the viral supernatants used in clinical studies.
Whilst a number of researchers were focusing their efforts on overcoming the barriers to achieve efficient stem cell transduction, others were testing the transduction of peripheral blood lymphocytes (PBLs) as alternative targets. PBLs obtained from patients under PEG-ADA treatment were transduced with a human ADA retrovirus and then injected into immunodeficient mice. Human T-cells were recovered from the spleen and peritoneum of vector-positive recipients but not from mock-infected PBL recipients. The recovered T-cells presented normal antigen-specific immune responses44 and so in this model, by expression of ADA in ADA- T-cells, it was possible to rescue immune function. In a rhesus monkey model, PBLs were recovered from donor animals, transduced with retrovirus, selected for transgene expression and then re-infused in the donor animals. Gene-marked cells were detected for up to 2 years45 suggesting that long term therapy could be provided through transduction of peripheral cells. In light of these promising results, the first clinical trials for ADA-SCID were initiated in the early 1990s.
Early clinical studies
Table 1 lists the gene therapy trials performed between 1990 and 1996. The first ever clinical gene therapy study was started at the NIH for ADA-SCID in 1990, enrolling 2 patients who had been treated with PEG-ADA for a minimum of 9 months and had not achieved immune reconstitution.46 Lymphocytes were recovered from their blood by leukapheresis and stimulated with OKT3 monoclonal antibody (10 ng/mL) and recombinant IL-2 (rIL-2) (1000 U/mL) before infecting them with supernatant containing the LASN retrovirus, a vector that included both the ADA and the neoR genes. The cells were then expanded without any selective pressure and later re-infused into the patient. The procedure was repeated eleven times over a period of two years and the children continued to receive PEG-ADA, albeit at a lower dose.
Table 1.
Early ADA-SCID gene therapy clinical trials
| Study | Retroviral vector | Envelope | Cell type | Number of patients |
|---|---|---|---|---|
| Blaese NIH46 | LASN | Ampho | PBL | 2 |
| Bordignon Milan48 | DCAm, DCAI | Ampho | BM and PBL | 2 |
| Kohn LA49 | LASN | Ampho | UCB CD34+ | 3 |
| Hoogerbrugge Netherlands50 | LgAL | Ampho | BM CD34+ | 3 |
| Onodera Japan47 | LASN | Ampho | PBL | 1 |
Abbreviations: Ampho, amphotrophic; PBL, peripheral blood lymphocytes; BM, bone marrow; UCB, umbilical cord blood.
Gene marking in CD3+ cells was as high as 50% in the first patient, but only 1% in the second. Although, some improvement in antigen-dependent responses, isohemagglutinin titers and lymphocyte ADA levels were observed after gene therapy, the clinical benefit of the therapy could not be clearly evaluated as PEG-ADA was administered alongside the gene-modified T-cells. The trials did demonstrate however that gene-modified T-cells were able to persist in the circulation for as long as ten years after treatment, an unexpected finding. The same vector and protocol were used to treat a patient in Japan47 and similar results to those observed in the first patient of the NIH trial were obtained.
A second trial transduced both T-cells and bone marrow progenitors, each with a different version of the same retroviral vector that carried different restriction enzyme patterns.48 This allowed investigators to study the contribution of transduced lymphocytes or marrow progenitors to the development of peripheral blood lymphocytes. Analysis of the PBLs showed that up to one year after treatment, the transduced cells were mainly derived from gene-modified PBLs and after this time point cells arising from BM progenitors were evident. Again it was difficult to discern the clinical benefit of the therapy alone, as PEG-ADA continued to be administered. Immune function parameters that had declined while on ERT recovered after gene therapy (GT) treatment, suggesting that improvement was associated with the reconstitution of gene-modified cells.
Contemporary to this trial was the study of Blaese and colleagues in three neonatal ADA-SCID patients treated with transduced umbilical cord blood (UCB) stem cells.49 The three patients had been diagnosed for ADA-SCID in utero and their cord blood was recovered at the time of delivery and the CD34+ population isolated and transduced using a cell-free supernatant containing the LASN retrovirus, in the presence of IL-3, IL-6, and SCF. Bone marrow samples were taken 1 year after infusion and the proportion of vector-positive cells within the CD34+ population was determined to be 1%. Despite this, the frequency of vector-positive leukocytes in the peripheral blood was only between 0.001% to 0.03%. This result was attributed to the fact that all patients were maintained on PEG-ADA which may have blunted the survival advantage conferred to gene-modified cells and had allowed the recovery of endogenous untransduced T-cells through effective metabolic detoxification. Importantly these latter two studies demonstrated that gene modification of bone marrow progenitors could result in the development of long-lived lymphoid populations.
The results of a multicenter European trial using CD34+ cells as the target for gene modification was published in 1996 by Hoogerbrugge and colleagues.50 The CD34+ population was isolated from the bone marrow of 3 patients and transduced by co-cultivation with a packaging cell line in the presence of IL-3. Although some vector-positive clonable progenitors were detected in the infused samples, the vector was detectable only at very low levels in the peripheral blood or the bone marrow of the patients and no clinical benefit was observed.
Development to improve gene therapy protocols
The lack of efficacy in these early studies prompted investigators to reconsider the requirements for successful gene therapy in ADA-SCID. Two major issues were identified as being of great importance. Firstly all patients in these studies were not subject to conditioning prior to infusion of the gene-modified stem cells. The role of conditioning may allow the engraftment of a greater gene-modified marrow progenitors thereby allowing more robust lymphoid development. The other major issue was the use of PEG-ADA alongside gene therapy. It was argued that the removal of PEG-ADA would allow a greater survival or growth advantage to gene-modified cells. Evidence for this latter assertion came from a report published in 2002 by Aiuti and colleagues.51 A group of 6 patients enrolled in the Milan study were treated with multiple infusions of autologous transduced PBLs whilst maintained on ERT. In one of these patients, ERT was withdrawn due to complications. After PEG-ADA withdrawal, the number of gene marked PBLs rose from 10% to almost 100%. The ADA activity in PBLs increased an average of 3-fold the value before ERT discontinuation. The gene corrected T-cell population that developed after ERT withdrawal was functional with normal anti CD3 responses and specific responses to vaccination. These two major developments, explicitly the use of a conditioning regime and the withdrawal or avoidance of ERT led to the first studies showing successful gene therapy for ADA-SCID.
Two other laboratory developments helped to pave the way for the second-generation clinical studies; namely the improved cytokine cocktails and the co-localization of stem cells with virus on fibronectin. The infection of human stem cells had been shown in in vitro studies to be most efficient when cells were maintained over stromal layer cultures. However, co-cultivation was not clinically acceptable and it was therefore necessary to improve the cell-free systems in order to achieve the same level of transduction without loss of multipotency and self-renewal ability of the cells. Several cytokine combinations were tested on the CD34+ Thy-1+ Lin- subset, a population thought to contain primitive hematopoietic stem cells.52 The combination of SCF, Flt3-ligand and TPO, all cytokines derived from stroma and involved in stem cell proliferation, proved optimal in stimulating cell division without loss of CD34+ or Thy-1 expression.53 This combination minus TPO and addition of IL-3, was shown to be effective in clinical trials of gene therapy for the X-linked form of SCID.54
Meanwhile, investigators also developed a way of enhancing the interaction between target cells and retroviral particles. The protein fibronectin participates in the adhesion of stem cells to the extracellular matrix of the bone marrow; Hanenberg and colleagues showed that target cells and retrovirus can adhere to neighboring regions of the same fragment in fibronectin-covered plates, thus increasing the likelihood of cell/virus contact and consequently of infection.55 The CH-296 fibronectin fragments were later made commercially available as Retronectin®.
Successful clinical trials of gene therapy for ADA-SCID
The combination of these improvements and the experience gained from the previous clinical trials led the group of Aiuti and colleagues to embark on a new clinical trial in 2000, enrolling 2 patients that lacked an HLA-matched sibling and for whom there was no PEG-ADA available.56 Bone marrow CD34+ cells were isolated from the patients, pre-stimulated with SCF, Flt3-ligand TPO, and IL-3 and subjected to three rounds of infection with an amphotrophic envelope retrovirus on Retronectin®-coated and virus pre-loaded bags. The patients underwent mild conditioning with busulfan (4 mg/kg) prior to the infusion of gene-modified CD34+ cells. This dose was approximately one quarter of the dose that would be used for a conventional HSCT After an initial period of neutropenia, blood cell counts increased progressively, thymopoiesis as measured by the numbers of naïve T-cells produced and the presence of T-cell receptor excision circles (TRECs) in CD3+ cells was restored and normal production of CD4+ and CD8+ T-cell subsets, as well as an heterogeneous repertoire of T-cell receptor variable region β chain and normal proliferative responses to antigens were present. B-cell functions seemed to be improving at least in one patient, permitting the discontinuation of immunoglobulin therapy. The vector was detected in most blood lineages, confirming that multipotent transduced-stem cells had engrafted.
With these positive results, the trial recruited thirteen more patients over the last 7 years, with the inclusion criteria extending to patients who had failed to respond to haplo-BMT or PEG-ADA. A summary of the results for the first 10 patients was published recently57 One year after gene therapy, on average, the proportion of gene-modified cells in the bone marrow was about 5.1% of the CD34+. In the peripheral blood the marking was at least one order of magnitude higher in the lymphocyte population (T, B, and NK cells) than in the bone marrow stem cell compartment, and comparable to the latter in the granulocyte population. This means that, in the presence of a cytotoxic environment, there is preferential differentiation of the corrected progenitors and/or selective proliferation of the gene-modified cells in the periphery. The median ADA levels in peripheral blood mononuclear cells and erythrocytes corresponded to 33.6% and 1.9% of the levels in normal controls, respectively. The presence of ADA in red cells led to a substantial decrease of toxic metabolites in this lineage. Immune reconstitution was observed in 9 out of 10 patients. Despite the fact that less than half of them reached normal blood cell counts, this was sufficient to protect patients from infection and helped them to lead a normal life free of social restriction. Only two patients required the administration of PEG-ADA after gene therapy. There were no adverse events that could be attributed to the gene-modified cells. Although an analysis of the vector integration sites revealed insertions near proto-oncogenic58 there was no evidence for clonal selection or proliferation.
In 2003, our group started a clinical trial recruiting patients for whom no haplo-identical bone marrow transplant was available and who were failing on ERT. The vector used in this trial was a gammaretrovirus, encoding only the ADA gene and pseudotyped with the GALv (Gibbon-ape leukemia virus) envelope. PEG-ADA treatment was stopped 1 month before gene therapy, CD34+ cells were isolated from the bone marrow and transduced using a similar protocol as that followed in the Italian trial. The patients were conditioned with a single dose of melphalan. The results for the first patient enrolled in this study, 2 years after starting treatment, were published in 2006.59 Again, a rise in T, B, and NK cell counts was observed in this patient, together with the acquisition of proliferative responses to PHA and CD3 stimulation. Recovery of thymopoiesis, assessed by the detection of TRECs in CD4+ and CD8+ populations, was confirmed as well as the presence of a normal heterogeneous T-cell repertoire. Metabolic detoxification was achieved with dATP levels falling to those found in patients after HSCT and red blood cell ADA activity was found to be at 10% of the normal level. The gene marking ranged from 50% in T and NK cells to >0.1% in myeloid cells.
Five more patients have been treated to date (unpublished data). Similar to that reported for patient 1, two more patients have experienced good immune recovery and metabolic detoxification, and in a fourth, gene-modified cells make dominant contributions to immune recovery even though PEG-ADA was re-started. Failure of gene therapy has been observed in two individuals due to a poor stem cell harvest in the first, and low transduction efficiency in the second. No adverse events attributed to the procedure have been observed to date, although integrations near proto-oncogenes have been detected (unpublished data).
A two-center trial has been conducted at the National Institute of Health and Children’s Hospital of Los Angeles, and the results confirm the importance of conditioning and myelosuppression for successful engraftment of gene-modified cells. In the initial phase of this study (reported at the ASGT meeting 2008, Sokolic and colleagues), 4 patients were treated in the absence of pre-conditioning; 2 of these patients had low levels of vector-positive cells, resulting in no sustained immune recovery. In the second trial phase after substantial protocol amendment, the use of pre-conditioning as well as ERT withdrawal has led to better immune recovery and metabolic detoxification (F Candotti, unpublished data).
If the results from all three studies in Italy, UK, and US are combined, over 20 patients have been treated and followed for at least 1 year after gene therapy. All treated patients have survived the procedure and approximately two-thirds have shown near full or partial immune recovery. Certainly, in terms of safety, GT appears to be superior to allogeneic HSCT from MUD and haplo-identical donors. Efficacy in terms of immune recovery is better than that following ERT especially for thymopoiesis and is comparable to that following HSCT. GT therefore provides an effective alternative for those patients for whom no well matched family donor is available. For the nonimmunological abnormalities of ADA-deficiency, it still remains to be seen whether gene therapy can ameliorate some of the symptoms previously mentioned and especially the cognitive and neurological complications.
Fortunately, and in contrast to what has been observed in the clinical trials for SCID-X1 to date, no major adverse events have been observed in any of the clinical trials for ADA-SCID. Five out of 20 patients undergoing SCID-X1 gene therapy developed T-cell leukemia between 2 and 6 years post-treatment using gammaretroviral vectors in 2 trials performed in France60 and London.61 In all cases, insertion of the vector near a proto-oncogene resulting in its subsequent aberrant expression was an initiating event in leukemogenesis. Although insertions in similar proto-oncogenes have been observed in patients treated for ADA-SCID, clonal proliferation has not been seen in any patients. The reasons for this are not clear but may relate in part to the fact that ADA is a housekeeping enzyme and not directly involved in signaling pathways that control cell proliferation. It is also important to note that the kinetics of T-cell recovery and turnover in gene therapy for ADA-SCID patients was slower than in gene therapy for SCID-X1 patients and thus the accumulation of other genetic events required to cause transformation may not manifest within a similar time frame. Therefore, it is possible that there is still a latent risk and close follow-up is necessary to fully assess the safety of this therapy.
The future
The retroviral vectors tested in the clinical trials discussed above contain strong enhancer elements in their viral LTRs, thus having the inherent risk of causing insertional mutagenesis; a phenomenon by which the inserted provirus is capable of activating the expression of a neighboring proto-oncogene, leading to malignant transformation (Figure 2). Initially it was thought that the integration profile of retroviruses was random, hence making the risk of insertional mutagenesis low. Following the development of leukemia in SCID-X1 patients, it was demonstrated that retroviruses have a preference for integrating within the regulatory regions of actively transcribed genes,62 therefore increasing the chance of causing gene deregulation.
Figure 2.
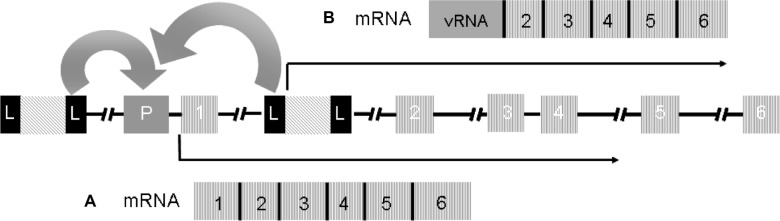
Mechanisms of insertional mutagenesis.
Schematic representation of two copies of a provirus integrated upstream the regulatory regions and between exons 1 and 2 of a proto-oncogene. The full-length LTRs in the provirus are represented by black boxes L; P represents the promoter region of the gene; the numbered boxes represent the exons; the arrows represent the orientation of transcription. A) as reported in some of the patients of the SCID-X1 trials, the enhancer elements of the LTR can activate the expression of the proto-oncogene or B) as reported by Bokhoven et al in vitro,80 fusion transcripts of viral RNA and cellular RNA are generated due to aberrant splicing between the splicing donor site downstream of the 5′ LTR and a splicing acceptor site upstream of exon 2 of the proto-oncogene.
The development of new vectors with better safety profiles is important to improve the risk-benefit profile of all hematopoietic stem cell gene therapies. The integration profile of lentiviral vectors differs from that of retroviral vectors in that insertions occur preferentially within transcriptional units but not immediately surrounding the transcriptional start site,62 reducing the probability of gene deregulation. A recent study in a tumor-prone animal model compared lentiviruses and gamma retroviruses with matched LTRs and concluded that lentiviruses provide a 10-fold reduction in the risk of oncogenesis.63 Moreover, what makes lentiviral vectors a more attractive option than retroviral vectors is their capacity for transducing non-dividing cells. Human bone marrow CD34+ cells only need a 24-hours pre-stimulation period before lentivirus infection,64 a reduced ex vivo incubation period would mean a better recovery of modified stem cells after transduction and consequently superior engraftment.
Further, by deleting the enhancer elements within the LTR, and driving gene expression from an internal promoter, a layer of safety can be added to both retroviral and lentiviral vectors. Such vectors, known as self-inactivating viruses (SIN) have been shown to be less genotoxic than conventional LTR-based vectors in both in vitro65,66 and in vivo67,63 models of transformation and are anticipated to be used in the next generation of clinical trials (Figure 3). A SIN lentiviral vector for ADA-SCID treatment has already been tested at the pre-clinical level13 and it will not be long until it enters the clinical trial phase.
Figure 3.
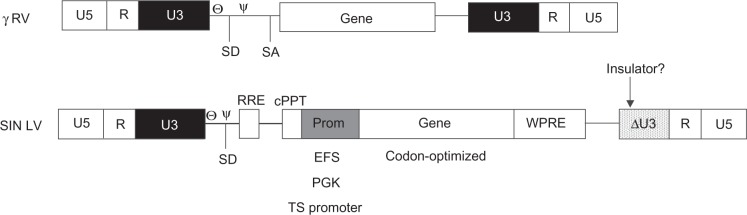
Comparing the architecture of the γ retroviruses tested in SCID clinical trials and the design of the future generations of vectors.
The new generations of vectors for HSC gene therapy will be lentiviruses without enhancer regions in the LTRs and cellular promoters placed downstream of the major splicing donor site. Codon-optimized genes will be useful in models where overexpression is not a limit. The introduction of insulator sequences in the LTRs may provide another layer of safety particularly when the internal regulatory region includes enhancer elements. The WPRE region improves RNA processing and boosts SIN vector titers.
Abbreviations: U5, unique region 5; R, repeat region; U3, unique region 3; ,Θ primer binding site; Ψ, packaging signal; SD, splicing donor site; SA, splicing acceptor site; RRE, Rev-responsive element; cPPT central polypurine tract; Prom, internal promoter; EFS, elongation factor 1 α short promoter; PGK, phosphoglycerate kinase promoter; TS promoter, tissue specific promoter; WPRE, woodchuck hepatitis virus post-transcriptional regulatory element; SIN LV, self-inactivating lentivirus.
To date, the most popular choice of internal promoter for constitutive transgene expression is the intron-less version of the elongation factor 1 α promoter (EFS). Compared to the spleen focus forming virus U3 region (SFFV), EFS is 2- to 3-fold less active in hematopoietic cells,68 but provides a reduction of 10-fold or more in the risk of genotoxicity in vitro.66
Another attractive choice for use as an internal regulatory element is the ubiquitously acting chromatin opening element (UCOE) from the human HNRPA2B1-CBX3 locus. This element is able to efficiently drive gene expression whilst resisting insertion-site position effects and does not include enhancer elements that could increase the risk of insertional mutagenesis.69
An additional modification that could further improve the safety profile of future generations of vectors is the inclusion of insulator elements in the 3′ LTR that results in the gene expression cassette being flanked at both ends in the integrated provirus. Insulators are “DNA elements that define the boundaries of co-ordinately regulated chromatin domains”.70 Insulator elements can act as heterochromatin barriers, sheltering the chromatin domain from silencing; or as enhancer-blockers, protecting the regulatory region within the domain from the effects of a distal enhancer. Some insulators, like the chicken β-globin hypersensitive site 4 (cHS4) can have both functions.71 The cHS4 element was first characterized as a 1.2 kb-long fragment,72 about 50% of the insulator activity was later mapped to a 250 bp-long core element that contains a binding site for the CTCF zinc finger protein.73 It was observed that 2 copies of the core element placed in tandem can provide the insulator function of the 1.2 kb element.73
Inclusion of the 1.2 kb fragment into a gammaretroviral vector with intact LTRs reduces its capacity for dysregulating gene expression in the surroundings of the insertion site by at least 3-fold, and its ability to cause malignant transformation by 6-fold in an in vivo model of tumor formation, but it does not completely eliminate either.74 These results are in agreement with what was observed in a system recreating the retroviral insertion in the vicinity of the LMO2 proto-oncogene -as found in some of the patients in the SCID-X1 trials-, whereby the presence of the insulator was not enough to block the activity of the intact LTR.75 This would mean that extra effort needs to be put into isolating more powerful insulator sequences than the cHS4, or that is better to use less potent enhancer-promoter combinations, as in a SIN design with internal cellular regulatory regions. Alas, the inclusion of the 1.2 kb element, or indeed any long sequence, in the LTR of a SIN lentivector results in low vector titers due to interference with the reverse transcription process and increased frequency of homologous recombination.76 Substituting the 1.2 kb fragment with a doublet of the 250 bp element is not a viable option either, as it has been shown that one copy of the core can be deleted at high frequency during viral replication.77
Since most of the enhancer-blocking activity of insulators can be mapped to CTCF binding sites, an obvious strategy would be to construct artificial insulator elements that combine a few of these sites, with enough variability between them to avoid the possibility of recombination. Such a strategy was explored by Ramezani and colleagues, whereby a combination of the CTCF binding sites from the cHS4 and the BEAD-1 insulators was capable of reducing the genotoxic potential of a SIN lentivirus carrying the murine stem cell virus (MSCV) LTR enhancer-promoter as an internal regulatory region.78 Other combinations are being tested at the moment79 and hopefully they can contribute to the generation of a safer vector.
Summary
The joint efforts of many research groups over twenty years have led to the development of stem cell gene therapy for severe combined immunodeficiencies. ADA-SCID patients particularly benefit from this new option, as fully-matched donor bone marrow transplants are not always available and PEG-ADA treatment is costly and not always effective long term. The data gathered from the successful gene therapy clinical trials indicate that this form of therapy is effective at providing immune recovery and to date, safe. The development of new viral vectors that improve the safety profile of stem cell gene therapy will be crucial to make it a first line of treatment option.
Footnotes
Disclosures
The authors have no conflicts of interest that are directly relevant to the content of this review.
References
- 1.Fischer A, Le DF, Hacein-Bey-Abina S, et al. Severe combined immunodeficiency. A model disease for molecular immunology and therapy. Immunol Rev. 2005;203:98–109. doi: 10.1111/j.0105-2896.2005.00223.x. [DOI] [PubMed] [Google Scholar]
- 2.Hershfield MS, Mitchell BS. Immunodeficiency diseases caused by adenosine deaminase deficiency and purine nucleoside phosphorylase deficiency. In: Scriver CR, Beaudet AL, Sly WS, Valle D, editors. The Metabolic and Molecular Basis of Inherited Disease. New York, NY: McGraw-Hill; 1995. pp. 1725–1768. [Google Scholar]
- 3.Lee N, Russell N, Ganeshaguru K, et al. Mechanisms of deoxyadenosine toxicity in human lymphoid cells in vitro: relevance to the therapeutic use of inhibitors of adenosine deaminase. Br J Haematol. 1984;56(1):107–119. doi: 10.1111/j.1365-2141.1984.tb01276.x. [DOI] [PubMed] [Google Scholar]
- 4.Apasov SG, Blackburn MR, Kellems RE, Smith PT, Sitkovsky MV. Adenosine deaminase deficiency increases thymic apoptosis and causes defective T cell receptor signaling. J Clin Invest. 2001;108(1):131–141. doi: 10.1172/JCI10360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gangi-Peterson L, Sorscher DH, Reynolds JW, Kepler TB, Mitchell BS. Nucleotide pool imbalance and adenosine deaminase deficiency induce alterations of N-region insertions during V(D)J recombination. J Clin Invest. 1999;103(6):833–841. doi: 10.1172/JCI4320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benveniste P, Zhu W, Cohen A. Interference with thymocyte differentiation by an inhibitor of S-adenosylhomocysteine hydrolase. J Immunol. 1995;155(2):536–544. [PubMed] [Google Scholar]
- 7.Hershfield MS. New insights into adenosine-receptor-mediated immunosuppression and the role of adenosine in causing the immunodeficiency associated with adenosine deaminase deficiency. Eur J Immunol. 2005;35(1):25–30. doi: 10.1002/eji.200425738. [DOI] [PubMed] [Google Scholar]
- 8.Cassani B, Mirolo M, Cattaneo F, et al. Altered intracellular and extracellular signaling leads to impaired T-cell functions in ADA-SCID patients. Blood. 2008;111(8):4209–4219. doi: 10.1182/blood-2007-05-092429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Blackburn MR, Kellems RE. Adenosine deaminase deficiency: metabolic basis of immune deficiency and pulmonary inflammation. Adv Immunol. 2005;86:1–41. doi: 10.1016/S0065-2776(04)86001-2. [DOI] [PubMed] [Google Scholar]
- 10.Booth C, Hershfield M, Notarangelo L, et al. Management options for adenosine deaminase deficiency; proceedings of the EBMT satellite workshop (Hamburg, March 2006) Clin Immunol. 2007;123(2):139–147. doi: 10.1016/j.clim.2006.12.009. [DOI] [PubMed] [Google Scholar]
- 11.Hirschhorn R, Roegner-Maniscalco V, Kuritsky L, Rosen FS. Bone marrow transplantation only partially restores purine metabolites to normal in adenosine deaminase-deficient patients. J Clin Invest. 1981;68(6):1387–1393. doi: 10.1172/JCI110389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Booth C, Gaspar HB. Pegademase bovine (PEG-ADA) for the treatment of infants and children with severe combined immunodeficiency (SCID) Biologics. 2009;3:349–358. [PMC free article] [PubMed] [Google Scholar]
- 13.Mortellaro A, Hernandez RJ, Guerrini MM, et al. Ex vivo gene therapy with lentiviral vectors rescues adenosine deaminase (ADA)-deficient mice and corrects their immune and metabolic defects. Blood. 2006;108(9):2979–2988. doi: 10.1182/blood-2006-05-023507. [DOI] [PubMed] [Google Scholar]
- 14.Rogers MH, Lwin R, Fairbanks L, Gerritsen B, Gaspar HB. Cognitive and behavioral abnormalities in adenosine deaminase deficient severe combined immunodeficiency. J Pediatr. 2001;139(1):44–50. doi: 10.1067/mpd.2001.115023. [DOI] [PubMed] [Google Scholar]
- 15.Honig M, Albert MH, Schulz A, et al. Patients with adenosine deaminase deficiency surviving after hematopoietic stem cell transplantation are at high risk of CNS complications. Blood. 2007;109(8):3595–3602. doi: 10.1182/blood-2006-07-034678. [DOI] [PubMed] [Google Scholar]
- 16.Titman P, Pink E, Skucek E, et al. Cognitive and behavioral abnormalities in children after hematopoietic stem cell transplantation for severe congenital immunodeficiencies. Blood. 2008;112(9):3907–3913. doi: 10.1182/blood-2008-04-151332. [DOI] [PubMed] [Google Scholar]
- 17.Gaspar HB, Aiuti A, Porta F, Candotti F, Hershfield MS, Notarangelo LD. How I treat ADA deficiency. Blood. 2009;114(17):3524–3532. doi: 10.1182/blood-2009-06-189209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chan B, Wara D, Bastian J, et al. Long-term efficacy of enzyme replacement therapy for adenosine deaminase (ADA)-deficient severe combined immunodeficiency (SCID) Clin Immunol. 2005;117(2):133–143. doi: 10.1016/j.clim.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 19.Malacarne F, Benicchi T, Notarangelo LD, et al. Reduced thymic output, increased spontaneous apoptosis and oligoclonal B cells in polyethylene glycol-adenosine deaminase-treated patients. Eur J Immunol. 2005;35(11):3376–3386. doi: 10.1002/eji.200526248. [DOI] [PubMed] [Google Scholar]
- 20.Bollinger ME, Arredondo-Vega FX, Santisteban I, Schwarz K, Hershfield MS, Lederman HM. Brief report: hepatic dysfunction as a complication of adenosine deaminase deficiency. N Engl J Med. 1996;334(21):1367–1371. doi: 10.1056/NEJM199605233342104. [DOI] [PubMed] [Google Scholar]
- 21.Blackburn MR, Aldrich M, Volmer JB, et al. The use of enzyme therapy to regulate the metabolic and phenotypic consequences of adenosine deaminase deficiency in mice. Differential impact on pulmonary and immunologic abnormalities. J Biol Chem. 2000;275(41):32114–32121. doi: 10.1074/jbc.M005153200. [DOI] [PubMed] [Google Scholar]
- 22.Hirschhorn R. Immunodeficiency due to adenosine deaminase. In: Ochs HD, Smith CIE, Puck JM, editors. Primary Immunodeficiency Diseases: A Molecular and Genetic Approach. New York, NY: Oxford University Press Inc, USA; 1998. [Google Scholar]
- 23.Daddona PE, Mitchell BS, Meuwissen HJ, Davidson BL, Wilson JM, Koller CA. Adenosine deaminase deficiency with normal immune function. An acidic enzyme mutation. J Clin Invest. 1983;72(2):483–492. doi: 10.1172/JCI110996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hirschhorn R. In vivo reversion to normal of inherited mutations in humans. J Med Genet. 2003;40(10):721–728. doi: 10.1136/jmg.40.10.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wiginton DA, Adrian GS, Friedman RL, Suttle DP, Hutton JJ. Cloning of cDNA sequences of human adenosine deaminase. Proc Natl Acad Sci U S A. 1983;80(24):7481–7485. doi: 10.1073/pnas.80.24.7481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Valerio D, McIvor RS, Williams SR, et al. Cloning of human adenosine deaminase cDNA and expression in mouse cells. Gene. 1984;31(1–3):147–153. doi: 10.1016/0378-1119(84)90205-1. [DOI] [PubMed] [Google Scholar]
- 27.Daddona PE, Shewach DS, Kelley WN, Argos P, Markham AF, Orkin SH. Human adenosine deaminase. cDNA and complete primary amino acid sequence. J Biol Chem. 1984;259(19):12101–12106. [PubMed] [Google Scholar]
- 28.Giblett ER, Anderson JE, Cohen F, Pollara B, Meuwissen HJ. Adenosinedeaminase deficiency in two patients with severely impaired cellular immunity. Lancet. 1972;2(7786):1067–1069. doi: 10.1016/s0140-6736(72)92345-8. [DOI] [PubMed] [Google Scholar]
- 29.Valerio D, Duyvesteyn MG, van der Eb AJ. Introduction of sequences encoding functional human adenosine deaminase into mouse cells using a retroviral shuttle system. Gene. 1985;34(2–3):163–168. doi: 10.1016/0378-1119(85)90124-6. [DOI] [PubMed] [Google Scholar]
- 30.Friedman RL. Expression of human adenosine deaminase using a transmissable murine retrovirus vector system. Proc Natl Acad Sci U S A. 1985;82(3):703–707. doi: 10.1073/pnas.82.3.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kantoff PW, Kohn DB, Mitsuya H, et al. Correction of adenosine deaminase deficiency in cultured human T and B cells by retrovirus-mediated gene transfer. Proc Natl Acad Sci U S A. 1986;83(17):6563–6567. doi: 10.1073/pnas.83.17.6563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Joyner A, Keller G, Phillips RA, Bernstein A. Retrovirus transfer of a bacterial gene into mouse haematopoietic progenitor cells. Nature. 1983;305(5934):556–558. doi: 10.1038/305556a0. [DOI] [PubMed] [Google Scholar]
- 33.Keller G, Paige C, Gilboa E, Wagner EF. Expression of a foreign gene in myeloid and lymphoid cells derived from multipotent haematopoietic precursors. Nature. 1985;318(6042):149–154. doi: 10.1038/318149a0. [DOI] [PubMed] [Google Scholar]
- 34.Eglitis MA, Kantoff P, Gilboa E, Anderson WF. Gene expression in mice after high efficiency retroviral-mediated gene transfer. Science. 1985;230(4732):1395–1398. doi: 10.1126/science.2999985. [DOI] [PubMed] [Google Scholar]
- 35.Belmont JW, Henkel-Tigges J, Chang SM, et al. Expression of human adenosine deaminase in murine haematopoietic progenitor cells following retroviral transfer. Nature. 1986;322(6077):385–387. doi: 10.1038/322385a0. [DOI] [PubMed] [Google Scholar]
- 36.Lim B, Apperley JF, Orkin SH, Williams DA. Long-term expression of human adenosine deaminase in mice transplanted with retrovirus-infected hematopoietic stem cells. Proc Natl Acad Sci U S A. 1989;86(22):8892–8896. doi: 10.1073/pnas.86.22.8892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wilson JM, Danos O, Grossman M, Raulet DH, Mulligan RC. Expression of human adenosine deaminase in mice reconstituted with retrovirus-transduced hematopoietic stem cells. Proc Natl Acad Sci U S A. 1990;87(1):439–443. doi: 10.1073/pnas.87.1.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kantoff PW, Gillio AP, McLachlin JR, et al. Expression of human adenosine deaminase in nonhuman primates after retrovirus-mediated gene transfer. J Exp Med. 1987;166(1):219–234. doi: 10.1084/jem.166.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Bodine DM, McDonagh KT, Brandt SJ, et al. Development of a high-titer retrovirus producer cell line capable of gene transfer into rhesus monkey hematopoietic stem cells. Proc Natl Acad Sci U S A. 1990;87(10):3738–3742. doi: 10.1073/pnas.87.10.3738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.van BV, Kukler A, Heidt PJ, Valerio D. Long-term expression of human adenosine deaminase in rhesus monkeys transplanted with retrovirus-infected bone-marrow cells. Proc Natl Acad Sci U S A. 1992;89(16):7640–7644. doi: 10.1073/pnas.89.16.7640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nolta JA, Kohn DB. Comparison of the effects of growth factors on retroviral vector-mediated gene transfer and the proliferative status of human hematopoietic progenitor cells. Hum Gene Ther. 1990;1(3):257–268. doi: 10.1089/hum.1990.1.3-257. [DOI] [PubMed] [Google Scholar]
- 42.Donahue RE, Kessler SW, Bodine D, et al. Helper virus induced T-cell lymphoma in nonhuman primates after retroviral mediated gene transfer. J Exp Med. 1992;176(4):1125–1135. doi: 10.1084/jem.176.4.1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vanin EF, Kaloss M, Broscius C, Nienhuis AW. Characterization of replication-competent retroviruses from nonhuman primates with virus-induced T-cell lymphomas and observations regarding the mechanism of oncogenesis. J Virol. 1994;68(7):4241–4250. doi: 10.1128/jvi.68.7.4241-4250.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ferrari G, Rossini S, Giavazzi R, et al. An in vivo model of somatic cell gene therapy for human severe combined immunodeficiency. Science. 1991;251(4999):1363–1366. doi: 10.1126/science.1848369. [DOI] [PubMed] [Google Scholar]
- 45.Culver KW, Morgan RA, Osborne WR, et al. In vivo expression and survival of gene-modified T lymphocytes in rhesus monkeys. Hum Gene Ther. 1990;1(4):399–410. doi: 10.1089/hum.1990.1.4-399. [DOI] [PubMed] [Google Scholar]
- 46.Blaese RM, Culver KW, Miller AD, et al. T lymphocyte-directed gene therapy for ADA-SCID: initial trial results after 4 years. Science. 1995;270(5235):475–480. doi: 10.1126/science.270.5235.475. [DOI] [PubMed] [Google Scholar]
- 47.Onodera M, Ariga T, Kawamura N, et al. Successful peripheral T-lymphocyte-directed gene transfer for a patient with severe combined immune deficiency caused by adenosine deaminase deficiency. Blood. 1998;91(1):30–36. [PubMed] [Google Scholar]
- 48.Bordignon C, Notarangelo LD, Nobili N, et al. Gene therapy in peripheral blood lymphocytes and bone marrow for ADA- immunodeficient patients. Science. 1995;270(5235):470–475. doi: 10.1126/science.270.5235.470. [DOI] [PubMed] [Google Scholar]
- 49.Kohn DB, Weinberg KI, Nolta JA, et al. Engraftment of gene-modified umbilical cord blood cells in neonates with adenosine deaminase deficiency. Nat Med. 1995;1(10):1017–1023. doi: 10.1038/nm1095-1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hoogerbrugge PM, van BV, Fischer A, et al. Bone marrow gene transfer in three patients with adenosine deaminase deficiency. Gene Ther. 1996;3(2):179–183. [PubMed] [Google Scholar]
- 51.Aiuti A, Vai S, Mortellaro A, et al. Immune reconstitution in ADA-SCID after PBL gene therapy and discontinuation of enzyme replacement. Nat Med. 2002;8(5):423–425. doi: 10.1038/nm0502-423. [DOI] [PubMed] [Google Scholar]
- 52.Baum CM, Weissman IL, Tsukamoto AS, Buckle AM, Peault B. Isolation of a candidate human hematopoietic stem-cell population. Proc Natl Acad Sci U S A. 1992;89(7):2804–2808. doi: 10.1073/pnas.89.7.2804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Luens KM, Travis MA, Chen BP, Hill BL, Scollay R, Murray LJ. Thrombopoietin, kit ligand, and flk2/flt3 ligand together induce increased numbers of primitive hematopoietic progenitors from human CD34+Thy-1+Lin- cells with preserved ability to engraft SCID-hu bone. Blood. 1998;91(4):1206–1215. [PubMed] [Google Scholar]
- 54.Cavazzana-Calvo M, Hacein-Bey S, de Saint BG, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science. 2000;288(5466):669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 55.Hanenberg H, Hashino K, Konishi H, Hock RA, Kato I, Williams DA. Optimization of fibronectin-assisted retroviral gene transfer into human CD34+ hematopoietic cells. Hum Gene Ther. 1997;8(18):2193–2206. doi: 10.1089/hum.1997.8.18-2193. [DOI] [PubMed] [Google Scholar]
- 56.Aiuti A, Slavin S, Aker M, et al. Correction of ADA-SCID by stem cell gene therapy combined with nonmyeloablative conditioning. Science. 2002;296(5577):2410–2413. doi: 10.1126/science.1070104. [DOI] [PubMed] [Google Scholar]
- 57.Aiuti A, Cattaneo F, Galimberti S, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360(5):447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 58.Aiuti A, Cassani B, Andolf G, et al. Multilineage hematopoietic reconstitution without clonal selection in ADA-SCID patients treated with stem cell gene therapy. J Clin Invest. 2007;117(8):2233–2240. doi: 10.1172/JCI31666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaspar HB, Bjorkegren E, Parsley K, et al. Successful reconstitution of immunity in ADA-SCID by stem cell gene therapy following cessation of PEG-ADA and use of mild preconditioning. Mol Ther. 2006;14(4):505–513. doi: 10.1016/j.ymthe.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 60.Hacein-Bey-Abina S, Garrigue A, Wang GP, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118(9):3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Howe SJ, Mansour MR, Schwarzwaelder K, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118(9):3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300(5626):1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- 63.Montini E, Cesana D, Schmidt M, et al. The genotoxic potential of retroviral vectors is strongly modulated by vector design and integration site selection in a mouse model of HSC gene therapy. J Clin Invest. 2009;119(4):964–975. doi: 10.1172/JCI37630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Charrier S, Dupre L, Scaramuzza S, et al. Lentiviral vectors targeting WASp expression to hematopoietic cells, efficiently transduce and correct cells from WAS patients. Gene Ther. 2007;14(5):415–428. doi: 10.1038/sj.gt.3302863. [DOI] [PubMed] [Google Scholar]
- 65.Modlich U, Bohne J, Schmidt M, et al. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood. 2006;108(8):2545–2553. doi: 10.1182/blood-2005-08-024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zychlinski D, Schambach A, Modlich U, et al. Physiological Promoters reduce the genotoxic risk of integrating gene vectors. Mol Ther. 2008;16(4):718–725. doi: 10.1038/mt.2008.5. [DOI] [PubMed] [Google Scholar]
- 67.Montini E, Cesana D, Schmidt M, et al. Hematopoietic stem cell gene transfer in a tumor-prone mouse model uncovers low genotoxicity of lentiviral vector integration. Nat Biotechnol. 2006;24(6):687–696. doi: 10.1038/nbt1216. [DOI] [PubMed] [Google Scholar]
- 68.Schambach A, Bohne J, Chandra S, et al. Equal potency of gammaretroviral and lentiviral SIN vectors for expression of O6-methylguanine-DNA methyltransferase in hematopoietic cells. Mol Ther. 2006;13(2):391–400. doi: 10.1016/j.ymthe.2005.08.012. [DOI] [PubMed] [Google Scholar]
- 69.Zhang F, Thornhill SI, Howe SJ, et al. Lentiviral vectors containing an enhancer-less ubiquitously acting chromatin opening element (UCOE) provide highly reproducible and stable transgene expression in hematopoietic cells. Blood. 2007;110(5):1448–1457. doi: 10.1182/blood-2006-12-060814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Strachan T, Read AP. Human Molecular Genetics 2. 2nd ed. Wilmington, DE: Wiley-Liss; 1999. [Google Scholar]
- 71.Gaszner M, Felsenfeld G. Insulators: exploiting transcriptional and epigenetic mechanisms. Nat Rev Genet. 2006;7(9):703–713. doi: 10.1038/nrg1925. [DOI] [PubMed] [Google Scholar]
- 72.Chung JH, Whiteley M, Felsenfeld G. A 5’ element of the chicken beta-globin domain serves as an insulator in human erythroid cells and protects against position effect in Drosophila. Cell. 1993;74(3):505–514. doi: 10.1016/0092-8674(93)80052-g. [DOI] [PubMed] [Google Scholar]
- 73.Chung JH, Bell AC, Felsenfeld G. Characterization of the chicken beta-globin insulator. Proc Natl Acad Sci U S A. 1997;94(2):575–580. doi: 10.1073/pnas.94.2.575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Li CL, Xiong D, Stamatoyannopoulos G, Emery DW. Genomic and functional assays demonstrate reduced gammaretroviral vector genotoxicity associated with use of the cHS4 chromatin insulator. Mol Ther. 2009;17(4):716–724. doi: 10.1038/mt.2009.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ryu BY, Evans-Galea MV, Gray JT, Bodine DM, Persons DA, Nienhuis AW. An experimental system for the evaluation of retroviral vector design to diminish the risk for proto-oncogene activation. Blood. 2008;111(4):1866–1875. doi: 10.1182/blood-2007-04-085506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Urbinati F, Arumugam P, Higashimoto T, et al. Mechanism of reduction in titers from lentivirus vectors carrying large inserts in the 3’LTR. Mol Ther. 2009;17(9):1527–1536. doi: 10.1038/mt.2009.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ramezani A, Hawley TS, Hawley RG. Stable gammaretroviral vector expression during embryonic stem cell-derived in vitro hematopoietic development. Mol Ther. 2006;14(2):245–254. doi: 10.1016/j.ymthe.2006.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ramezani A, Hawley TS, Hawley RG. Combinatorial incorporation of enhancer-blocking components of the chicken beta-globin 5’HS4 and human T-cell receptor alpha/delta BEAD-1 insulators in self-inactivating retroviral vectors reduces their genotoxic potential. Stem Cells. 2008;26(12):3257–3266. doi: 10.1634/stemcells.2008-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mitra K, Lenz J. A combinatorial insulator approach confers effective protection against insertional activation of oncogenes in a stringent tumorigenesis model. Mol Ther. 2009;27(17(Suppl 1)):S142. [Google Scholar]
- 80.Bokhoven M, Stephen SL, Knight S, et al. Insertional gene activation by lentiviral and gammaretroviral vectors. J Virol. 2009;83(1):283–294. doi: 10.1128/JVI.01865-08. [DOI] [PMC free article] [PubMed] [Google Scholar]