

Abstract
Mouse-adapted transmissible spongiform encephalopathy (TSE) strains are routinely distinguished based on reproducible disease characteristics in a given mouse line following inoculation via a consistent route. We investigated whether different administration routes (oral, intragastric (i.g.) and intracerebral (i.c.)) can alter the disease characteristics in IM mice after serial dilution of a stabilized mouse-adapted bovine spongiform encephalopathy (BSE) strain (301V). In addition, the infectivity of distal ileum and mesenteric lymph nodes (ln) sampled at three time points (35 days postinoculation (dpi), 70 dpi and terminal disease) after i.g. inoculation of 301V strain was assessed in mice by i.c. challenge. Strain characteristics were assessed according to standard methodology and PrPSc immunohistochemistry deposition patterns. Mean incubation periods were prolonged following oral or i.g. inoculations compared to the i.c. route. Lesion profiles following i.c. challenges were elevated compared to i.g. and oral routes although vacuolation in the dorsal medulla was consistently high irrespective of the route of administration. Nevertheless, the same PrPSc deposition pattern was associated with each route of administration. Distal and mesenteric ln infectivity was detected as early as 35 dpi and displayed consistent lesion profiles and PrPSc deposition patterns. Our data suggest that although 301V retained its properties, some phenotypic parameters were affected by the route of inoculation. We conclude that bioassay data should be interpreted carefully and should be standardized for route of inoculation.
Keywords: 301V, bovine spongiform encephalopathy, mouse bioassay, prion, transmissible spongiform encephalopathy
Prion diseases, or transmissible spongiform encephalopathies (TSEs), are a group of fatal neurodegenerative disorders that affect humans and animals. These diseases include bovine spongiform encephalopathy (BSE) in cattle, scrapie in sheep and goats and Creutzfeldt–Jakob disease (CJD) in humans. Prion diseases share the same pathogenic mechanism, which involves the misfolding and aggregation of the normal host-encoded cellular membrane prion protein (PrPC) into an oligomeric β-sheet-rich pathogenic isoform (PrPSc) (Prusiner 1982). Although prion diseases share similar disease characteristics, which can include spongiform vacuolation, neuronal loss, PrPSc deposition, amyloid plaque formation and gliosis, the disease phenotype can vary between different prion diseases (Gambetti et al. 2003; Gavier-Widén et al. 2005; Konold et al. 2006).
Transmissible spongiform encephalopathy strains are determined by the phenotype of the disease they cause in specific hosts (Bruce et al. 2002) suggesting that in addition to strain specific parameters, host specific parameters can also influence the ability to distinguish individual TSE strains. The mouse bioassay is currently the gold standard for measuring prion infectivity and for discriminating TSE strains. Two different wild type mouse lines are routinely used for strain discrimination studies, as phenotypic characteristics are mouse line specific. Further research indicated that the Prnp gene was the major factor responsible for these properties (Westaway et al. 1987), and as a result, the effect of TSEs on wild type mouse lines can be deduced according to their Prnp (Prnpa or Prnpb) allele. Other genetic factors may also be implicated although their impact is unknown (Manolakou et al. 2001). Research has shown that classical BSE is caused by a single strain of agent which after serial passage in a given mouse line produces a unique and consistent phenotype denoted 301C in Prnpa (usually C57BL/6 or RIII) or 301V in Prnpb (usually VM or IM) mice (Fraser et al. 1992). The BSE agent has been shown to retain its characteristics upon passage in numerous species and subsequent bioassay in RIII mice (Green et al. 2004).
Inoculation of a TSE agent via the i.c. route into a given species, including mice, is widely regarded as the most effective route of transmission. Conversely, peripheral routes of infection are notably less effective and reveal prolonged incubation periods and lower attack rates (Kimberlin & Walker 1989; Maignien et al. 1999; Taylor et al. 2001). However, such routes permit a more natural reproduction of the pathogenesis of the disease as i.c. inoculations bypass many of the processes that occur naturally during the course of the disease (Barlow & Middleton 1990).
Indeed, after i.c. inoculation, the agent is deposited directly to the brain and as a result the incubation period shortens considerably (Mohan et al. 2005). It has been reported that normal gastric secretions (pH 1.3) can provide a temporary barrier against some ovine scrapie strains. C57BL/6 or Tg338 mice challenged i.g. with scrapie respectively showed a marked reduction in positive mice in comparison with mice subjected to increased levels of gastric pH (2.4 and 5.4 respectively) (Martinsen et al. 2002, 2011).
Although the effect of the genetic background of the host on TSE strains is well established, there is restricted information regarding the effect of the inoculation route (Kimberlin & Walker 1986; Thackray et al. 2003; Novakofski et al. 2005; Langevin et al. 2011). A recent study suggested that using a predefined classical scrapie strain at primary passage, different routes of inoculation (peripheral or i.c.) can affect the resulting phenotype. Differences included clinical signs, PrPSc deposition profile and neuropathology (Langevin et al. 2011).
Administration of 301V via the oral route rather than the i.c. route may not only impact on incubation period but also on other phenotypic parameters which are used to define TSE strains such as lesion profiles and PrPSc distribution in the brain. In addition, it may play an important role in elucidating aspects of the pathogenesis of TSEs. It is essential to understand the impact of alternative inoculation routes as there may be TSE sources which are not suitable for inoculation via the i.c. route. This may include contaminated material which resists antibiotic treatment and maybe deemed ethically more suitable for oral administration or materials such as pituitary extracts which may cause adverse reactions if deposited directly to the brain. Furthermore, environmental studies that contain whole soil structures or faeces are more suitable for oral challenge due to the nature of the matrix (Johnson et al. 2007; Safar et al. 2008).
In this study, we use a stabilized mouse line so that the effect of route could be isolated. The first objective of this study was to determine whether different inoculation routes would alter the phenotypic presentation of a stabilized 301V strain in mice. The peripheral dissemination of the agent during the disease course was also evaluated.
Material and methods
Sample preparation and mouse inoculation
Four serial dilutions, 10−1–10−4 (w/v in normal saline), of a reference 301V strain (supplied by Roslin Institute) obtained through serial transmission and subsequent biological cloning were prepared in sterile saline. Each dilution was administered to groups of twenty IM (Prnpb) mice aged six to ten weeks via i.c., intragastric (i.g.) or oral routes. For i.c. inoculations, 20 μl of homogenate was deposited directly in the right hemisphere of each mouse as described (Corda et al. 2012). Mice inoculated orally or via the i.g. route were challenged with 500 μl of inoculum (250 μl in the morning and 250 μl in the afternoon on the same day). Orally inoculated mice were housed singly on the day of inoculation to ensure each individual was exposed to the same dose. An additional group of animals was inoculated i.g. with 250 μl of the 10−1 dilution in the morning only.
To study peripheral pathogenesis, 20 IM mice were challenged i.g. with 10−1 (w/v) IM-passaged 301V (250 μl am and 250 μl pm on the same day). Five of these mice were euthanized at 35 and 70 days postinoculation (dpi), whilst the remaining 10 mice were left until the onset of terminal disease. The brain, mesenteric lymph node (ln) and distal ileum were sampled from each mouse. Distal ileum and mesenteric ln collected at 35 dpi, 70 dpi and onset of terminal disease were each homogenized to 1% (w/v) with sterile saline and administered i.c. into 20 IM mice.
All mice were monitored from 50 days postinoculation for clinical signs and euthanized using carbon dioxide when clinical end point had been reached, which was defined as having a clinically positive score for TSE in two consecutive weeks, or two of three weeks. Euthanasia also occurred if significant deterioration in mobility, or inability to consume food or drink, was observed at any point during the monitoring process.
Ethical approval
All work was carried out in accordance with the Animals (Scientific Procedures) Act 1986 under licence from the home office and approval of the study by the local ethics committee.
Histopathological and immunohistochemical analysis
Brains were removed and immediately fixed in 10% neutral buffered formalin for a minimum of 3 days at room temperature before being cut at four coronal sections to reveal caudal medulla, rostral medulla, midbrain, thalamic and frontal levels required for lesion scoring and profiling as detailed below. Tissues were processed and embedded in paraffin wax. Sections (3 μm thick) were mounted on slides and stained with haematoxylin and eosin (H&E). The slides were assessed blind by a single reader, and the TSE status was diagnosed based on the presence or absence of TSE-specific vacuolation. The severity of the lesions in TSE positive samples was assessed semi-quantitatively on a scale of 0–5 in nine specific neuroanatomical grey matter areas and from 0–3 in three white matter areas of the brain (Figure 1). The scores were plotted against the respective brain areas to produce lesion profiles using an established method as previously described (Fraser & Dickinson 1968). Due to the variation of individual mouse profiles, the mean lesion profile for a given inoculum must be generated from a minimum of five clinically and histopathologically positive mice to be considered reliable. For immunohistochemistry (IHC), sections were labelled with rabbit polyclonal antibody Rb486 which recognizes amino acids from 221 to 233 (YQRESQAYYQRGA) of bovine PrP using a standard method as described previously (Green et al. 2004). Clinically and histopathologically positive mice from each inoculum representing the minimum, median and maximum incubation period were further analysed by IHC.
Figure 1.
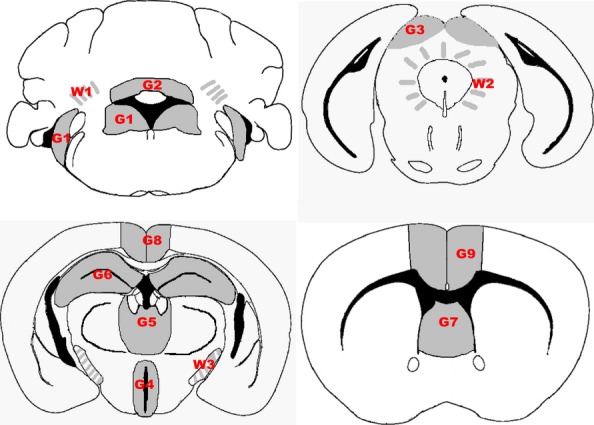
Mandatory scoring areas used to construct a lesion profile. Grey matter areas scored (indicated by grey shading) were G1, dorsal medulla nuclei including cochlear nuclei; G2, granular layer of the cerebellar cortex adjacent to the fourth ventricle; G3, superior colliculus; G4, hypothalamus; G5, thalamus; G6, hippocampus; G7, septal nuclei of the paraterminal body; G8, cerebral cortex (at the level of G4 and G5); and G9, cerebral cortex (at the level of G7). White matter areas scored (indicated by grey shaded lines) were W1, cerebellar white matter; W2, mesencephalic tegmentum; and W3, the cerebral peduncles.
Results
Attack rate and incubation period analysis
The number of positive mice and the range and mean incubation period for each route of administration with 301V in IM mice are presented in Table 1. Regarding the 10−1 dilution the inoculation route did not have any effect on the attack rates which were consistently high (90–100%), although the incubation period of i.c. challenged mice was 50% shorter compared to the other routes of inoculation. For the i.c. challenges, the attack rates were 100% and the incubation periods increased in relation to the dilution. Oral dose attack rates remained high for the 10−1 and 10−2 dilution, but a sharp decline was observed for the 10−3 dilution and the agent was unable to transmit at 10−4 dilution. A moderately decreased attack rate and slightly prolonged incubation period were observed in animals challenged with 250 μl i.g. compared to those which received 500 μl inoculum of the same strength and via the same route. The least successful serial titration in terms of attack rates was i.g. challenge; however, transmission occurred with all dilutions.
Table 1.
Transmission data of 301V titrations from brain material via three different inoculation routes
| Inoculation route (dose) | Dilution | Clinically +ve and pathologically +ve | Clinically −ve and pathologically +ve | ||
|---|---|---|---|---|---|
| n* | Mean (range) dpi | n | Mean (range) dpi | ||
| i.c. (20 μl) | 10−1 | 20 | 123 (118–127) | 0 | (−) |
| 10−2 | 20 | 128 (126–130) | 0 | (−) | |
| 10−3 | 19 | 135 (134–136) | 1 | (115) | |
| 10−4 | 20 | 151 (147–163) | 0 | (−) | |
| Oral (500 μl) | 10−1 | 18 | 264 (226–331) | 0 | (−) |
| 10−2 | 14 | 274 (251–300) | 0 | (−) | |
| 10−3 | 4 | 279 (254–321) | 0 | (−) | |
| 10−4 | 0 | (−) | 0 | (−) | |
| i.g. (500 μl) | 10−1 | 18 | 244 (173–278) | 1 | (221) |
| 10−2 | 4 | 423 (278–589) | 1 | (267) | |
| 10−3 | 3 | 310 (293–320) | 0 | (−) | |
| 10−4 | 1 | (299) | 0 | (−) | |
| i.g. (250 μl) | 10−1 | 14 | 263 (190–419) | 0 | (−) |
n represents number of positive mice by histopathological examination.
Each dilution (tissue w/v in normal saline) was inoculated into 20 mice.
Orally and i.c. challenged mice showed a gradual increase in mean incubation period in relation to an increase in serial dilution. For the oral and i.g. inoculation routes, the range of incubation periods overlapped between successive dilutions, whilst in i.c. mice, there was a minor overlap between the 10−1 and 10−2 dilutions.
Mice challenged i.g. with 10−2 dilution showed a marked increase in incubation period which decreased in subsequent titration. The most effective inoculation route was i.c. challenge due to consistently high attack rates and short incubation periods although a rise in incubation periods occurred throughout the dilution series.
The transmission data from mice challenged i.c. with distal ileum or mesenteric ln derived from mice inoculated i.g. with 301V are presented in Table 2. The attack rates and incubation periods indicate a maximum infectivity point of PrPSc in the distal ileum soon after 35 dpi as there was only a small variation in these two parameters compared to 70 dpi and terminal disease. In contrast, mice inoculated with mesenteric ln at 35 dpi displayed less than a third of the attack rate compared to distal ileum, suggesting that the mesenteric ln infection appears secondary to the distal ileum. However, it is likely that PrPSc levels in both of these tissues peaked as early as 70 dpi demonstrated by attack rates and incubation periods that were similar to those observed in mice inoculated with the same tissues collected at terminal disease. Even at terminal disease, these peripheral tissues seem to be at least 10−4 less infectious compared to i.c. challenged brain.
Table 2.
Transmission data of IM mice inoculated i.c. with peripheral tissues collected at specified points from mice challenged i.g. with 301V
| Tissue | Collection time | Clinically +ve and pathologically +ve | Clinically −ve and pathologically +ve | ||
|---|---|---|---|---|---|
| n* | Mean (range) dpi | n | Mean (range) dpi | ||
| Distal ileum | 35 dpi | 16 | 198 (151–421) | 0 | (−) |
| 70 dpi | 18 | 174 (156–222) | 0 | (−) | |
| Terminal disease | 18 | 177 (164–213) | 1 | (207) | |
| Mesenteric ln | 35 dpi | 5 | 240 (197–307) | 0 | (−) |
| 70 dpi | 18 | 160 (152–166) | 0 | (−) | |
| Terminal disease | 15 | 158 (150–168) | 5 | 154 (146–156) | |
n indicates number of positive mice by histopathological examination.
Each source was inoculated into 20 mice; dpi indicates days postinoculation.
Lesion profile analysis
Figure 2a shows the lesion profiles derived after inoculation of 10−1 dilution through various administration routes. The lesion profile at 10−1 from i.c. challenge was elevated compared to i.g. and oral routes. However, mandatory area G1 (medulla) was scored consistently high irrespective of the route of administration.
Figure 2.
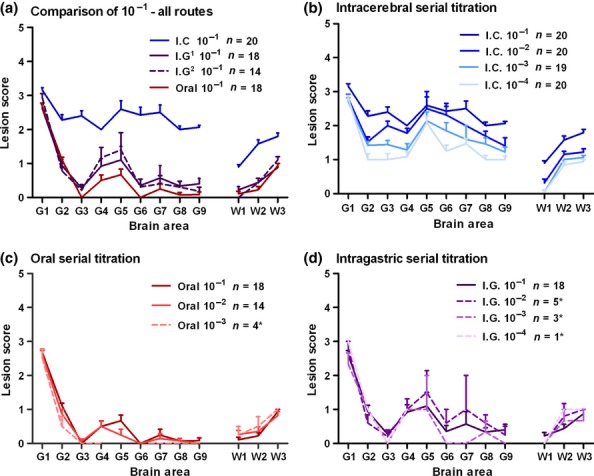
Lesion profiles at 10−1 dilution for each route of inoculation (a) and lesion profiles from each titration series (c–d). Lesion profiles were constructed using five or more clinically and histopathologically positive mice unless stated (*). Error bars indicate standard error of the mean; n indicates the number of mice which contributed to the lesion profile; i.c., intracerebral inoculation (20 μl dose); oral, oral inoculation (500 μl dose); i.g.1, intragastric inoculation (500 μl dose); i.g.2, intragastric inoculation (250 μl dose).
Lesion profiles consistently showed peaks at G1 and G5 and to a lesser extent in area G7 irrespective of administration route or dilution (Figure 2a–d). Mice challenged i.c. showed a drop in vacuolation intensity along the lesion profile with increasing dilutions (Figure 2b) which also corresponded with an increase in the mean incubation period (Table 1). In addition, i.c challenges produced the most intense lesion profiles when compared to the other inoculation routes (Figure 2b). However, oral dosing appeared to produce the most stable and reproducible lesion profile across the titration series (Figure 2c). Inoculation via the i.g. route gave rise to a lesion profile contour similar to oral dose which was also relatively consistent across the dilution series (Figure 2d).
Figure 3 shows lesion profiles from mice challenged i.c. with mesenteric ln or distal ileum collected at different time points. Mesenteric ln challenged animals displayed a small degree of variation in brain areas G3 and G7 but this did not alter the contour of the profile which remained consistent irrespective of the collection time point of the tissue (Figure 3a). Lesion profiles produced from mice challenged with distal ileum were consistent and similar to those derived from mesenteric ln (Figure 3b).
Figure 3.
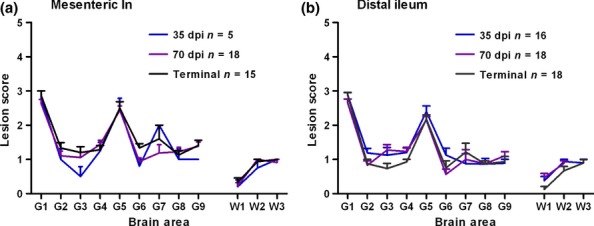
Lesion profiles derived from mice challenged i.c. with mesenteric ln (a) or distal ileum (b) derived from mice inoculated i.g. with 301V and collected at three different time points during the development of the disease. Lesion profiles were constructed using five or more clinically and histopathologically positive mice. Error bars indicate standard error of the mean; n indicates the number of mice which contributed to the lesion profile.
Immunohistochemistry analysis
All analysed mice showed the same characteristic PrPSc pattern distribution irrespective of the route of administration. Hallmarks of this pattern include the absence of significant deposition in the periaqueductal grey matter and the solitary tract, in addition to stellate-type deposition across the molecular layer of the dentate gyrus in the hippocampus (Figure 4) as previously described in Prnpb mice (Spiropoulos et al. 2011; Corda et al. 2012). These features were also observed in mice challenged with distal ileum and mesenteric ln from 301V infected mice at all time points (Figure 4). Although qualitatively the patterns were indistinguishable, some difference was observed in the intensity of the labelling in the dentate gyrus of the hippocampus relating to different administration routes (Figure 4) and inocula prepared from peripheral tissues at different time points (Figure 5). The most intense labelling was observed in the i.c. inoculated mice (Figure 4c) followed by i.g. (Figure 4g) and oral (Figure 4e) inoculations. The intensity across the periaqueductal grey and other brain areas was similar irrespective of inoculation route or dilution (Figure 3). In mice challenged i.c. with peripheral tissues, the most intense labelling in the hippocampal area was associated with terminal disease, whilst the weakest labelling was associated with tissues collected at 35 dpi (Figure 5). No marked differences were observed in the intensity of immunolabelling in the periaqueductal grey or any other brain areas that were examined.
Figure 4.
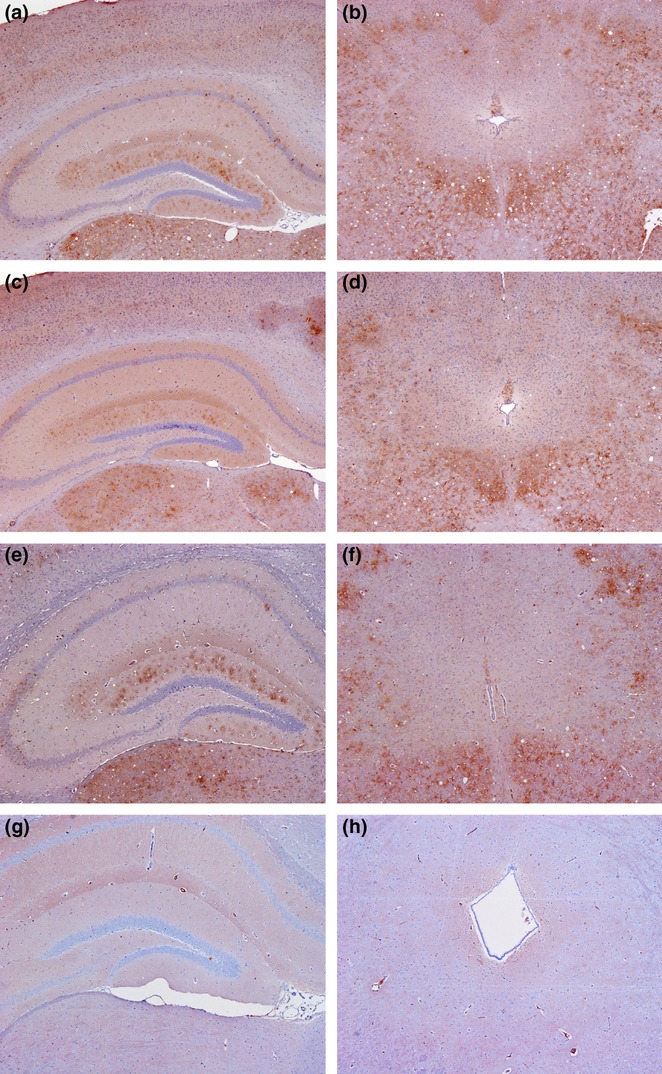
PrPSc labelling in hippocampus and midbrain of each inoculation route. Stellate-type immunolabelling in i.c. (a) oral (c) and i.g. (e) inoculated mice in the dentate gyrus of the hippocampus. The periaquaductal grey is encased by a surrounding layer of granular deposition in i.c. (b) oral (d) and i.g. (f) administered mice. (g) and (h) are negative controls; i.c., intracerebral inoculation (20 μl dose); oral, oral inoculation (500 μl dose); i.g.1, intragastric inoculation (500 μl dose).
Figure 5.
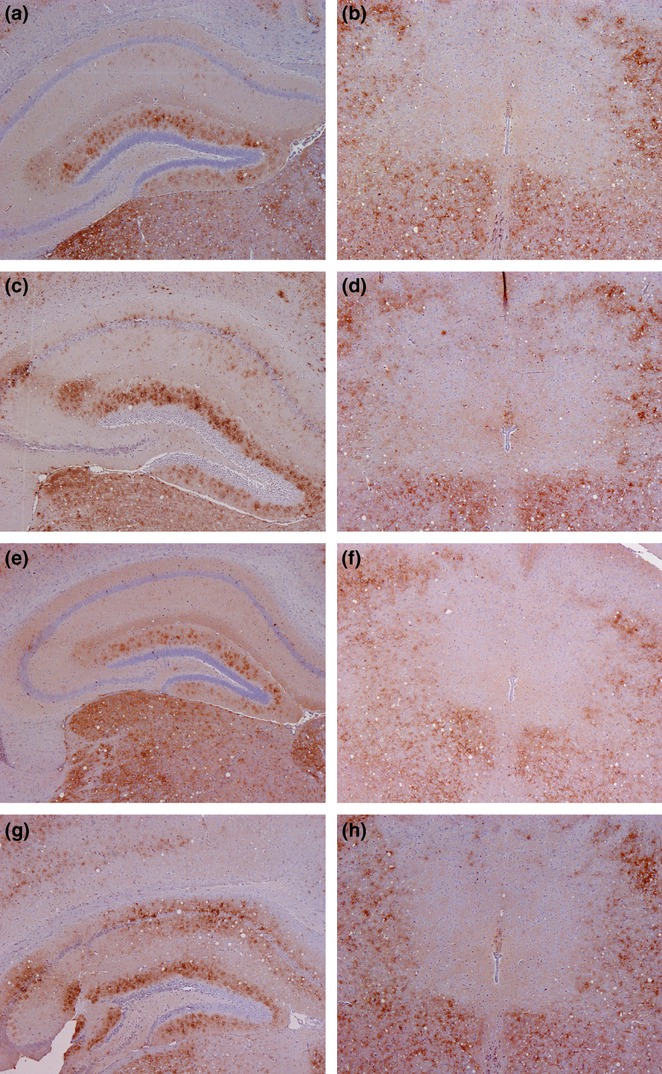
PrPSc labelling in hippocampus and midbrain in recipient mice challenged i.c. with mesenteric ln or distal ileum. A marked difference in PrPSc was observed at 35 dpi (mesenteric ln (a) and distal ileum (e)) compared to terminal disease (mesenteric ln (c) and distal ileum (g)) in the dentate gyrus of the hippocampus. However, no difference in intensity was shown in the midbrain between 35 dpi (mesenteric ln (b) and distal ileum (f)) and terminal disease (mesenteric ln (d) and distal ileum (h)).
Discussion
The results presented here illustrate that although different administration routes affected certain phenotypic parameters, notably lesion profile intensity and incubation periods, the strain remained stable based on PrPSc deposition pattern. The strain used in this study, 301V, is characterized by short incubation periods of approximately 120 dpi after i.c. inoculation (Bruce et al. 2002). This phenotypic parameter was elevated after i.g. or oral challenges which may be attributed to the time required for the agent to colonize and multiply in the peripheral lymphoid tissues prior to transportation to the brain via nerves of the autonomic nervous system (Prusiner 1982; Hoffmann et al. 2007; Kratzel et al. 2007; Van Keulen et al. 2008; Wemheuer et al. 2011). However, the mean incubation period following oral challenge correlates with previously published results using IM mice (González et al. 2005). A previous study (Martinsen et al. 2002) had suggested that normal gastric secretions may provide a temporary barrier against some scrapie strains. However, in our study, the distal ileum and mesenteric ln were diagnosed positive after 35 dpi, suggesting that gastric acid had little or no effect in mice inoculated i.g. with murine adapted BSE.
The general outline of the lesion profiles appeared to remain stable across each dilution series although the intensity of the lesion profiles of i.g. and oral challenges was notably decreased compared to those following i.c. challenges. A comparable drop in the lesion profile intensity was also observed for i.c. inoculations as the dilution of the inoculum increased. This reduction in the lesion intensity with increasing dilution in the i.c. inoculations has not been reported in TSE bioassays in other species or indeed in any other mouse lines. The appearance of this phenomenon in this data set cannot be explained as, except for the titre of the inocula, all other parameters such as route and site of inoculation, mouse genetic background, clinical monitoring, euthanasia at terminal stage disease, postmortem procedures and subsequent handling of the brain samples were consistent among the different dilutions. However, if dilutions had been prepared in brain homogenate instead of normal saline, the concentration of exogenous brain components would have remained constant. Under these circumstances, after i.c. inoculation, the host might have reacted more vigorously during prion pathogenesis reducing the observed differences. Nonetheless, i.c. inoculation of brain homogenate from healthy TSE free animals does not produce any form of disease or pathology in the recipients (J Spiropoulos, unpublished data).
Immunohistochemistry observations mirror those of the lesion profiles. Specifically, the route of inoculation did not have any effect on the qualitative traits of the PrPSc pattern induced by 301V in this mouse line although the intensity of the labelling was decreased in the hippocampus of mice that were challenged i.g. or orally.
These findings are difficult to explain as mice were analysed at terminal disease stage and the observed lesions reflected the full extent of the pathology inflicted in the host by this specific strain. In this study, only one characterized TSE strain was used, and differences in the PrPSc signal intensity were mainly observed in the hippocampal area. This could be attributed to a relatively slower propagation of the agent in these areas as the titre of the agent decreased. Irrespective of titre or administration route, the vacuolation score in the medulla was consistently high. A possible explanation for this may be that the medulla represents a clinical target area in this model (Kimberlin & Walker 1986; Brandner 2003). The medulla oblongata controls autonomic functions such as breathing, digestion, heart function, swallowing and sneezing (Hadjikoutis et al. 2000). Evidence suggests that the medulla is comprised of numerous regulatory centres of the autonomic nervous system and is regarded as the first point of contact with trafficking prions from the spinal cord or the vagus nerve (Hadjikoutis et al. 2000; Mabbott & MacPherson 2006; Wemheuer et al. 2011). If the target areas responsible for clinical signs are affected more by the agent, this potentially has an effect on the level of vacuolation and PrPSc intensity in the brain of the mouse (Ayers et al. 2009). Therefore, it is feasible that when the lesion reached a threshold point in this area, the mice succumbed to clinical disease irrespective of the severity of the pathology in other brain areas.
These data suggest that various phenotypic characteristics that contribute to strain characterization retained their qualitative properties although they may have shown quantitative differences. Taking this into account, the PrPSc deposition patterns and to a lesser extent lesion profiles indicate that the strain characteristics remained stable and were unaffected by the inoculation route or the infectious titre. In a similar study, in addition to the quantitative changes, there were also differences in the regional distribution of spongiform changes and PrPSc between i.c. and intraperitoneally (i.p.) challenged mice that were attributable to neuroanatomical areas which were affected in i.c., but not in i.p. inoculated mice. This could be because the duration of the replication phase, as estimated by the time elapsed between the first moment of PrPSc detection and the terminal stage of disease, in the brain of i.p. challenged mice was the shortest recorded among other representative TSE rodent models, whereas after i.c. inoculation, this duration remained average (Langevin et al. 2011). Consistent with this view, after neuroinvasion of the brainstem via the autonomic nervous system following incubation period inoculation, the animal would die in a relatively short period of time as the agent accumulates early in neuronal centres that are vital for sustaining life, prior to extensive PrPSc accumulation in other brain areas. These apparently qualitative differences may not indicate a novel phenotype but a variant phenotype of the specific strain which is a function of the combination of inoculation route, TSE strain and host. Indeed, the characteristic phenotype of the strain was recovered after i.c. inoculation of brains recovered from the i.p. inoculated animals (Langevin et al. 2011). We did not observe such a wide phenotypic diversity in this study probably because in the TSE/host model, we used the replication phase in i.p. inoculated mice is relatively prolonged allowing the infection to spread beyond the brain stem areas to the rest of the brain before the involvement of vital neuronal centres leads to animal death.
Results from this study show that following i.g. challenge PrPSc infectivity can be detected in the ileum and in mesenteric ln at a very early stage of the disease (35 dpi) and was shown to persist until the terminal stage of the infection. A point of maximum infectivity was observed at 70 dpi in both tissues as incubation periods were not notably reduced beyond this time point and attack rates remained similar to mice inoculated with such tissues collected at terminal disease. Interestingly the lesion profiles from distal ileum and mesenteric ln at terminal disease mimicked the lesion profiles generated after i.c. challenge of 10−4 brain inoculum, suggesting that the oral or i.g. inoculations correspond to lower titre i.c. challenges. In addition, the mean incubation period for mesenteric ln collected at terminal disease was only 7 days longer than the mean incubation period of mice challenged i.c. with a 10−4 dilution indicating titre effect although the characteristics of the strain remained. However, despite these results, it is not recommended to directly compare data derived from different inoculation routes or tissues as they may obscure interpretation particularly on uncharacterized strains. As a result, any route of inoculation could be used to identify TSE strains as long as the comparisons are restricted within data derived from a single route. Inoculations via the i.c. route produce the shortest possible incubation periods and the highest attack rates and are therefore from a diagnostic point of view the preferred method particularly if bioassays are used to identify cases integral to policy requirements (Béringue et al. 2008).
Collectively, these data indicate that the decreased intensity in vacuolation observed after i.g. or oral challenge might be attributed to the different routes the agent takes before it reaches the brain. Our data suggest that although 301V retained its properties in this sequence of experiments, some phenotypic parameters can be affected by the route of inoculation. Therefore, it is imperative that bioassay data must be interpreted carefully and should be standardized for each route of inoculation as murine bioassay, either in wild type or more recently in transgenic mice, is still considered the most reliable method for discriminating TSE strains particularly when specific TSEs (for example classical scrapie) are attributed to multiple prion strains (Thackray et al. 2011; Beck et al. 2012; Corda et al. 2012).
Acknowledgments
The authors would like to thank colleagues in histopathology for their skilled technical support. We also thank Ian Dexter and Karen Hartley and their teams for their technical expertise and support.
Competing Interests
The authors declare that they have no competing interests.
Funding source
This study was funded by VLA Seedcorn Project SC0042 and Defra Project SE1849.
References
- Ayers JI, Kincaid AE, Bartz JC. Prion strain targeting independent of strain-specific neuronal tropism. J. Virol. 2009;83:81–87. doi: 10.1128/JVI.01745-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow RM, Middleton DJ. Dietary transmission of bovine spongiform encephalopathy to mice. Vet. Rec. 1990;126:111–112. [PubMed] [Google Scholar]
- Beck KE, Sallis RE, Lockey R, et al. Use of murine bioassay to resolve ovine transmissible spongiform encephalopathy cases showing a bovine spongiform encephalopathy molecular profile. Brain Pathol. 2012;22:265–279. doi: 10.1111/j.1750-3639.2011.00526.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Béringue V, Vilotte J-L, Laude H. Prion agent diversity and species barrier. Vet. Res. 2008;39:47. doi: 10.1051/vetres:2008024. [DOI] [PubMed] [Google Scholar]
- Brandner S. CNS pathogenesis of prion diseases. Br. Med. Bull. 2003;66:131–139. doi: 10.1093/bmb/66.1.131. [DOI] [PubMed] [Google Scholar]
- Bruce ME, Boyle A, Cousens S, et al. Strain characterization of natural sheep scrapie and comparison with BSE. J. Gen. Virol. 2002;83:695–704. doi: 10.1099/0022-1317-83-3-695. [DOI] [PubMed] [Google Scholar]
- Corda E, Beck KE, Sallis RE, et al. The interpretation of disease phenotypes to identify TSE strains in mice: characterisation of BSE using PrPSc distribution patterns in the brain. Vet. Res. 2012;43:86. doi: 10.1186/1297-9716-43-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser H, Bruce ME, Chree A, McConnell I, Wells GA. Transmission of bovine spongiform encephalopathy and scrapie to mice. J. Gen. Virol. 1992;73(Pt 8):1891–1897. doi: 10.1099/0022-1317-73-8-1891. [DOI] [PubMed] [Google Scholar]
- Fraser H, Dickinson AG. The sequential development of the brain lesion of scrapie in three strains of mice. J. Comp. Pathol. 1968;78:301–311. doi: 10.1016/0021-9975(68)90006-6. [DOI] [PubMed] [Google Scholar]
- Gambetti P, Kong Q, Zou W, Parchi P, Chen SG. Sporadic and familial CJD: classification and characterisation. Br. Med. Bull. 2003;66:213–239. doi: 10.1093/bmb/66.1.213. [DOI] [PubMed] [Google Scholar]
- Gavier-Widén D, Stack MJ, Baron T, Balachandran A, Simmons M. Diagnosis of transmissible spongiform encephalopathies in animals: a review. J. Vet. Diagn. Invest. 2005;17:509–527. doi: 10.1177/104063870501700601. [DOI] [PubMed] [Google Scholar]
- González L, Terry L, Jeffrey M. Expression of prion protein in the gut of mice infected orally with the 301V murine strain of the bovine spongiform encephalopathy agent. J. Comp. Pathol. 2005;132:273–282. doi: 10.1016/j.jcpa.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Green R, Horrocks C, Wilkinson A, Hawkins SAC, Ryder SJ. Primary isolation of the bovine spongiform encephalopathy agent in mice: agent definition based on a review of 150 transmissions. J. Comp. Pathol. 2004;132:117–131. doi: 10.1016/j.jcpa.2004.08.002. [DOI] [PubMed] [Google Scholar]
- Hadjikoutis S, Pickersgill TP, Dawson K, Wiles CM. Abnormal patterns of breathing during swallowing in neurological disorders. Brain. 2000;123(Pt 9):1863–1873. doi: 10.1093/brain/123.9.1863. [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Ziegler U, Buschmann A, et al. Prions spread via the autonomic nervous system from the gut to the central nervous system in cattle incubating bovine spongiform encephalopathy. J. Gen. Virol. 2007;88:1048–1055. doi: 10.1099/vir.0.82186-0. [DOI] [PubMed] [Google Scholar]
- Johnson CJ, Pedersen JA, Chappell RJ, McKenzie D, Aiken JM. Oral transmissibility of prion disease is enhanced by binding to soil particles. PLoS Pathog. 2007;3:e93. doi: 10.1371/journal.ppat.0030093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimberlin RH, Walker CA. Pathogenesis of scrapie (strain 263K) in hamsters infected intracerebrally, intraperitoneally or intraocularly. J. Gen. Virol. 1986;67(Pt 2):255–263. doi: 10.1099/0022-1317-67-2-255. [DOI] [PubMed] [Google Scholar]
- Kimberlin RH, Walker CA. Pathogenesis of scrapie in mice after intragastric infection. Virus Res. 1989;12:213–220. doi: 10.1016/0168-1702(89)90040-3. [DOI] [PubMed] [Google Scholar]
- Konold T, Lee YH, Stack MJ, et al. Different prion disease phenotypes result from inoculation of cattle with two temporally separated sources of sheep scrapie from Great Britain. BMC Vet. Res. 2006;2:31. doi: 10.1186/1746-6148-2-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kratzel C, Kruger D, Beekes M. Relevance of the regional lymph node in scrapie pathogenesis after peripheral infection of hamsters. BMC Vet. Res. 2007;3:22. doi: 10.1186/1746-6148-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langevin C, Andreoletti O, Le Dur A, Laude H, Beringue V. Marked influence of the route of infection on prion strain apparent phenotype in a scrapie transgenic mouse model. Neurobiol. Dis. 2011;41:219–225. doi: 10.1016/j.nbd.2010.09.010. [DOI] [PubMed] [Google Scholar]
- Mabbott NA, MacPherson GG. Prions and their lethal journey to the brain. Nat. Rev. Microbiol. 2006;4:201–211. doi: 10.1038/nrmicro1346. [DOI] [PubMed] [Google Scholar]
- Maignien T, Lasmezas CI, Beringue V, Dormont D, Deslys JP. Pathogenesis of the oral route of infection of mice with scrapie and bovine spongiform encephalopathy agents. J. Gen. Virol. 1999;80(Pt 11):3035–3042. doi: 10.1099/0022-1317-80-11-3035. [DOI] [PubMed] [Google Scholar]
- Manolakou K, Beaton J, McConnell I, et al. Genetic and environmental factors modify bovine spongiform encephalopathy incubation period in mice. Proc. Natl Acad. Sci. USA. 2001;98:7402–7407. doi: 10.1073/pnas.121172098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinsen TC, Taylor DM, Johnsen R, Waldum HL. Gastric acidity protects mice against prion infection? Scand. J. Gastroenterol. 2002;37:497–500. doi: 10.1080/00365520252903017. [DOI] [PubMed] [Google Scholar]
- Martinsen TC, Benestad SL, Moldal T, Waldum HL. Inhibitors of gastric acid secretion increase the risk of prion infection in mice. Scand. J. Gastroenterol. 2011;46:1418–1422. doi: 10.3109/00365521.2011.619277. [DOI] [PubMed] [Google Scholar]
- Mohan J, Bruce ME, Mabbott NA. Neuroinvasion by scrapie following inoculation via the skin is independent of migratory Langerhans cells. J. Virol. 2005;79:1888–1897. doi: 10.1128/JVI.79.3.1888-1897.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novakofski J, Brewer MS, Mateus-Pinilla N, Killefer J, McCusker RH. Prion biology relevant to bovine spongiform encephalopathy. J. Anim. Sci. 2005;83:1455–1476. doi: 10.2527/2005.8361455x. [DOI] [PubMed] [Google Scholar]
- Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science (New York, NY.) 1982;216:136–144. doi: 10.1126/science.6801762. [DOI] [PubMed] [Google Scholar]
- Safar JG, Lessard P, Tamgüney G, et al. Transmission and detection of prions in feces. J. Infect. Dis. 2008;198:81–89. doi: 10.1086/588193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiropoulos J, Lockey R, Sallis RE, et al. Isolation of prion with BSE properties from farmed goat. Emerg. Infect. Dis. 2011;17:2253–2261. doi: 10.3201/eid1712.110333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor DM, Fernie K, Steele PJ, Somerville RA. Relative efficiency of transmitting bovine spongiform encephalopathy to RIII mice by the oral route. Vet Rec. 2001;148:345–346. doi: 10.1136/vr.148.11.345. [DOI] [PubMed] [Google Scholar]
- Thackray AM, Klein MA, Bujdoso R. Subclinical prion disease induced by oral inoculation. J. Virol. 2003;77:7991–7998. doi: 10.1128/JVI.77.14.7991-7998.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackray AM, Hopkins L, Lockey R, Spiropoulos J, Bujdoso R. Emergence of multiple prion strains from single isolates of ovine scrapie. J. Gen. Virol. 2011;92:1482–1491. doi: 10.1099/vir.0.028886-0. [DOI] [PubMed] [Google Scholar]
- Van Keulen LJM, Bossers A, Van Zijderveld F. TSE pathogenesis in cattle and sheep. Vet. Res. 2008;39:24. doi: 10.1051/vetres:2007061. [DOI] [PubMed] [Google Scholar]
- Wemheuer WM, Benestad SL, Wrede A, et al. PrPSc spreading patterns in the brain of sheep linked to different prion types. Vet. Res. 2011;42:32. doi: 10.1186/1297-9716-42-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westaway D, Goodman PA, Mirenda CA, McKinley MP, Carlson GA, Prusiner SB. Distinct prion proteins in short and long scrapie incubation period mice. Cell. 1987;51:651–662. doi: 10.1016/0092-8674(87)90134-6. [DOI] [PubMed] [Google Scholar]