

Abstract
Respiratory syncytial virus (RSV) is the most common cause of childhood viral bronchiolitis and lung injury. Inflammatory responses significantly contribute to lung pathologies during RSV infections and bronchiolitis but the exact mechanisms have not been completely defined. The double-stranded RNA-activated protein kinase (PKR) functions to inhibit viral replication and participates in several signaling pathways associated with innate inflammatory immune responses. Using a functionally defective PKR (PKR−/−) mouse model, we investigated the role of this kinase in early events of RSV-induced inflammation. Our data showed that bronchoalveolar lavage (BAL) fluid from infected PKR−/− mice had significantly lower levels of several innate inflammatory cytokines and chemokines. Histological examinations revealed that there was less lung injury in infected PKR−/− mice as compared to the wild type. A genome-wide analysis showed that several early antiviral and immune regulatory genes were affected by PKR activation. These data suggest that PKR is a signaling molecule for immune responses during RSV infections.
Introduction
Respiratory syncytial virus (RSV) accounts for the majority of virus-associated respiratory illnesses in early infancy (Hall and others 1976; McIntosh and others 1978). Although RSV infections are well tolerated in healthy individuals, resulting in a mild respiratory infection, amongst the very young, the elderly, or immunocompromised patients, severe respiratory disease leading to hospitalization and death can occur (Moore and Peebles 2006). Several studies have revealed that during RSV infections lung injury is mediated both by direct viral damage and by the inflammatory responses elicited against the virus (Tripp and others 2000; Durbin and others 2002; Rutigliano and Graham 2004). Thus far, the molecular mechanisms of RSV-induced inflammation and lung injury are not yet completely understood.
Studies by Tripp et al. and Estripeaut et al. using murine models showed that chemokines and cytokines MIP-1α, MCP-1, IP-10, and IFN-γ were induced within 24 h by RSV infections (Tripp and others 2000; Estripeaut and others 2008). Interestingly, the study by Tripp et al. showed that chemokine induction was maximally induced at 8 h post-infection, which was subsequently reduced by 18 h post-infection (Tripp and others 2000). In addition, both in vivo and in vitro studies have shown that in response to RSV infection, several cytokines including TNF-α, IL-1, IL-6, IFN-β are potently induced (Midulla and others 1993; Noah and Becker 1993; Arnold and others 1994; Matsuda and others 1995; Meusel and Imani 2003; McNamara and others 2004).
While there is evidence that these cytokines and chemokines are important for immune responses against viral infections, they can also be deleterious by contributing to tissue injury. TNF-α, in particular, has been shown to lead to lung injury and exacerbate RSV-associated pathologies in mice (Neuzil and others 1996; Zhao and others 2000; Hussell and others 2001; Rutigliano and Graham 2004). In addition to cytokines, chemokines have also been linked to the severity of RSV-associated disease. In vitro exposure of both primary human airway epithelial cells and human airway epithelial cell lines to RSV resulted in the production of several chemokines (Olszewska-Pazdrak and others 1998; Zhang and others 2001), and high levels of chemokines were found to be associated with severe inflammation in RSV-infected infants and young children (Sheeran and others 1999; Welliver and others 2002).
We hypothesized that the double-stranded RNA-activated protein kinase (PKR) may be important in the early events during RSV infection and may likely be involved in recruitment of inflammatory cells into the lungs. The role of PKR in anti-RSV responses in vivo has not been previously reported. PKR is a cytoplasmic serine/threonine kinase that is activated early during virus infections with a key role in antiviral defense. It is induced in an inactive form by type I interferons and it requires interaction specifically with dsRNA for activation (Krust and others 1984; Galabru and others 1989). It has a well-characterized role in the inhibition of translation through phosphorylation of eukaryotic initiation factor 2α (eIF-2α) (Lebleu and others 1976; Samuel 1979; de Haro and others 1996), which leads to reduction in viral replication. In addition to the antiviral effects, activated PKR participates in regulating several signaling pathways involved in immune responses such as NF-κB, MAPK, STAT-1, NF-AT, and IRFs pathways (Kumar and others 1994; Langland and others 1999; Zamanian-Daryoush and others 2000; Gil and others 2001; Goh and others 2000; Kehoe and others 2001; García and others 2006).
Despite the studies demonstrating that mice lacking expression of a functional PKR are more susceptible to virus infections (Balachandran and others 2000; Stojdl and others 2000; Guidotti and others 2002; Carr and others 2006), there has been little on the role of PKR in RSV infection or RSV-induced lung inflammation, lung injury, or RSV-induced early gene expression. In the current study, our data showed that although replication of RSV was enhanced in the absence of a functional PKR, there was significantly less lung inflammation and injury in RSV-infected PKR−/− mice as compared to the WT control mice. In addition, a genome-wide gene expression analysis revealed several early antiviral and immune response genes that are regulated by PKR.
Materials and Methods
Mice, virus, and infection
The 6- to 10-week-old male WT (B6.129PF1/J) (The Jackson Labs, Bar Harbor, ME) and PKR−/− (Yang and others 1995) mice were used in all experiments. Mice were housed and maintained in specific pathogen-free conditions and used in accordance with the regulations of the National Institutes of Health. The human long strain of RSV-A2 (originally from Dr. B.S. Graham, NIH) was propagated in HEp-2 cells (American Type Culture Collection). Viral stocks were prepared by harvesting HEp-2 cells in fresh medium and 3 quick freeze thaw cycles followed by high-speed centrifugation (10,000g) to remove cellular debris. The viral titer was then determined by plaque assay as previously described (Graham and others 1988). For in vivo infections, anesthetized mice were made to aspirate, intratracheally, RSV at 1×107 pfu/mouse or vehicle (virus-free medium). For in vitro infection of mouse cells, lungs were digested in collagenase, passed through a cell strainer, and then infected with RSV at a multiplicity of infection (MOI) of 2.5 pfu/cell.
RNA extraction and RT-PCR
Total RNA was extracted from lung tissue by homogenization in TRIzol (Invitrogen, Carlsbad, CA). cDNA was synthesized with SuperScript reverse transcriptase (Invitrogen) and amplified with a SYBR Green (Bio-Rad, Hercules, CA) master mix containing primers to either GAPDH, RSV-G, or RSV-N. The sequences are as follows: GAPDH, forward TTC ACC ACC ATG GAG AAG GC, reverse GGC ATG GAC TGT GGT CAT GA; RSV-G, forward AAG TCA ACC CTG CAA TCC AC, reverse GCA TAT GCT GCA GGG TAC AA; and RSV-N, forward AGG ATT GTT TAT GAA TGC CTA TGG T, reverse GCT TTT GGG TTG TTC AAT ATA TGG TAG. Data were collected and analyzed with MyiQ software (Bio-Rad, Hercules, CA).
Western blot assays
Lung samples were first homogenized in 1×Laemmli sample buffer containing 1% SDS and 2-mercaptoethanol without additional protease or phosphatase inhibitors (Bio-Rad Laboratories, Hercules, CA) and the chromosomal DNA was sheared by passing the samples several times through a 26-gauge needle. Proteins were separated by SDS-PAGE, transferred onto nitrocellulose, and probed with antibodies to β-actin (Sigma-Aldrich, St. Louis, MO), RSV (Chemicon, Temecula, CA), phospho-JNK, JNK, phospho-p38 MAPK, p38 MAPK, phospho-ERK, and ERK (Cell Signaling Technologies, Danvers, MA). The immunoblotted proteins were then visualized by the enhanced chemiluminescence reagents (GE Healthcare Bio-Sciences, Piscataway, NJ).
Electrophoretic mobility shift assay
Extracts were prepared by homogenizing lung tissues in buffer containing (50 mM HEPES pH 7.0, 250 mM NaCl, 5 mM EDTA, 1 mM dithiothreitol, and 0.1% NP-40). Clarified protein extracts were then incubated with 32P-labeled NF-κB-specific oligonucleotide (5′ CAA CGG CAG GGG AAT TCC CCT CTC CTT 3′) (Boothby and others 1997) in a reaction mixture containing, 2 μg poly (dI–dC), 10 μg BSA, 20 mM HEPES pH 7.9, 5% glycerol, 1 mM EDTA, 1% NP-40, and 5 mM dithiothreitol. The mixture was incubated at room temperature for 15 min. The resultant protein–DNA complexes were resolved on a native 4% polyacrylamide gel and were visualized by autoradiography.
Measurement of protein, albumin, and cytokine levels in the BAL fluid
At 24 h post-infection, bronchoalveolar lavage (BAL) was performed using phosphate-buffered saline. Total protein levels within the BAL fluid were measured by Bradford assay (Bio-Rad Laboratories, Hercules, CA). ELISA was used to measure mouse albumin (Bethyl Laboratories, Montgomery, TX), IP-10 (R&D Systems, Minneapolis, MN), and IFN-β (PBL biomedical Lab, Piscataway, NJ). Bio-Plex Cytokine Assay (Bio-Rad Laboratories) was used to determine the levels of all other cytokines and chemokines.
Histology and differential analysis
A hemocytometer was used to determine total cell number in the BAL fluid. For differentials, cells were affixed to slides, and were stained using the HEMA-3 stain (Fisher Scientific, Pittsburg, PA). The stained cells were counted and differentiated based on color and morphology. For histological examinations, lungs (9 mice per group) were fixed with 10% buffered formalin. Paraffin-embedded tissues were cut into 5 μM sections, affixed to glass slides, and then stained with hematoxylin and eosin. A pathologist blinded to animal genotype and treatment evaluated the slides using a semiquantitative histological score method. The scoring system, which grades (0=none, 1=minimal, 2=mild, 3=moderate, 4=severe) the degree of inflammatory cells infiltration in the perivascular, bronchial, and alveolar region, in addition to changes in bronchial walls and alveolar macrophage accumulation, has been previously described (Stapleton and others 2005; Jaradat and others 2006). Data were averaged from 2 separate experiments and 2-tailed Student's t-test was used to determine statistical significance.
Microarray, linear amplification, labeling protocol, and feature extraction
Gene expression analysis was conducted using Agilent Whole Mouse Genome 4x44 multiplex format oligo arrays (014868) (Agilent Technologies, Palo Alto, CA) following the Agilent 1-color microarray-based gene expression analysis protocol. Starting with 500 ng of total RNA, Cy3-labeled cRNA was produced according to manufacturer's protocol. For each sample, 1.65 μg of Cy3-labeled cRNAs were fragmented and hybridized for 17 h in a rotating hybridization oven. Slides were washed and then scanned with an Agilent Scanner. Data was obtained using the Agilent Feature Extraction software (v9.5), using the 1-color defaults for all parameters. The Agilent Feature Extraction Software performed error modeling and adjusting for additive and multiplicative noise. The resulting data were processed using the RosettaResolver® system (version 7.2) (Rosetta Biosoftware, Kirkland, WA).
The Ingenuity Pathway Analysis (IPA; Ingenuity Systems, www.ingenuity.com) was used to identify the biological functions and/or diseases that were most significant to the data sets. Genes from the data sets that met the normalized cutoff of 2-fold were then analyzed for biological functions and/or disease relevance in the Ingenuity Pathways Knowledge Base (Ingenuity Systems, www.ingenuity.com). Fischer's exact test was then used to calculate P values.
Results
PKR regulates RSV replication
Since it is reported that replication of several viruses is increased in PKR−/− mice (Balachandran and others 2000; Stojdl and others 2000; Carr and others 2006), we first examined mRNA accumulation in the mice. Mice were infected with RSV (1×107 pfu/mice) and after 1, 5, 14, and 21 days, RSV presence in infected lungs was determined by RT-PCR. As shown in Figure 1A, the level of RSV-G mRNA was significantly increased in the lungs of PKR−/− mice as compared to the WT. At late times during infection (21 days), the level of RSV was equally reduced in both mice strains, likely due to adaptive immune responses. Since our experiments were aimed at early events, we have focused on 1 day post-infection (Fig. 1B, RSV-G and RSV-N RT-PCR). In addition, Western blot of total lung proteins extracted at day 1 post-infection showed greater amounts of RSV proteins in infected PKR−/− mice as compared to the WT (Fig. 1C). This suggests that as expected RSV accumulates to higher levels in PKR−/− mice.
FIG. 1.

Viral load is increased in the lungs of double-stranded RNA-activated protein kinase (PKR)-deficient mice. (A) Mice (6 mice per group) were infected with 1×107 pfu of respiratory syncytial virus (RSV). At indicated times, lungs were isolated and total lung RNA was extracted for RT-PCR using respiratory syncytial virus (RSV)-G-specific primers. (B) To further investigate early events, RNA from another 24 h post-infection time point was used in RT-PCR using specific primers to both RSV-G (left panel) and RSV-N (right panel) (6 mice per group, each represented by a dot). RT-PCR data are expressed as relative fold induction over mock normalized against GAPDH. (C) Viral protein expression in mouse lungs, 24 h after inoculation, was determined in lung lysates with an anti-RSV polyclonal antibody. Data are from lungs of 2 separate mice; arrows indicate RSV proteins. (D) To assess differences in replication or infectivity, total lung cells were infected and at time 0, 6, and 24 h post-infection, total RNA was extracted and used in RT-PCR for the detection of RSV-G mRNA levels (n=3).
To confirm that the difference was due to replication and not to differences in initial infectivity, total lung cells were isolated and infected in vitro. Despite similar levels of RSV-G mRNA at 6 h post-infection, the RSV-G mRNA level in PKR−/− lung cells at 24 h post-infection was significantly greater than that detected in cells from WT mice (Fig. 1D). These data suggest that the observed increase in RSV load in vivo is likely due to enhanced replication and not increases in initial infectivity.
PKR is necessary for maximal expression of inflammatory cytokines and chemokines
RSV infections of both mouse and human have been shown to induce the expression of a variety of cytokines and chemokines (Arnold and others 1994; Franke and others 1994; Meusel and Imani 2003; Jafri and others 2004). Therefore, we examined the effects of PKR in inflammatory response during RSV infection. Mice were infected with 1×107 pfu/mouse and after 24 h BAL fluid and lung tissue samples were collected. Analysis of BAL fluid revealed that infected WT mice had approximately 3- to 30-fold higher levels of the inflammatory cytokines and chemokines, IFN-β, IL-1β, IL-6, TNF-α, CCL2/Monocyte chemotactic protein-1 (MCP-1), CCL5/RANTES, CXCL10/IP-10 (IFN-inducible protein 10), and IL-8 as compared to RSV-infected PKR−/− mice (Fig. 2). It is noteworthy that of all cytokines and chemokines tested, TNF-α was the most potently affected by the absence of PKR (∼30-fold). Taken together, these data indicate that PKR is necessary for maximal expression of inflammatory cytokines and chemokines during RSV infection in vivo.
FIG. 2.

Double-stranded RNA-activated protein kinase (PKR) regulates induction of cytokines and chemokines. PKR−/− or wild-type (WT) mice were infected with respiratory syncytial virus (RSV; 1×107 pfu/mouse) and after 24 h, the mice were anesthetized and BAL fluid was collected. The levels of cytokines and chemokines were measured as described in Materials and Methods. The error bars represent standard error of the mean (SEM) where averages were generated from 3 separate mice in each group.
Several of the cytokines and chemokines shown in Figure 2 are attractants of inflammatory cell influx into the lungs. To assess the influx of inflammatory cells, total cell number was determined in BAL fluid. The data showed a 2-fold increase in cells within the BAL fluid collected from RSV-infected WT mice as compared to PKR−/− mice (Fig. 3A). Differential analysis of the cellular infiltrates revealed that the majority of the cells that migrated into the lungs were neutrophils and a significantly greater percentage of neutrophils migrated into the lungs of the WT mice as compared to PKR−/− mice (Fig. 3B).
FIG. 3.

Inflammatory cell influx into lungs is affected by double-stranded RNA-activated protein kinase (PKR). (A) The total number of viable cells present within the bronchoalveolar lavage (BAL) was determined by trypan blue exclusion using a hemocytometer (n=2, 3 mice/group). (B) Identity of cells within the BAL was determined by fixing the cells onto slides, followed by hemotoxylin–eosin staining and differential counting based on size and morphology. The data represent 2 separate experiments with 6 mice in each group.
RSV-induced lung injury is reduced in the absence of PKR
A critical consequence of respiratory viral infections is lung injury, which is also associated with inflammatory cell transmigration into the lungs and increased vascular permeability. Therefore, to assess lung injury, we first examined lung permeability by measuring the level of serum albumin in BAL fluid at 24 h post-infection. Data in Figure 4A and 4B showed that the level of total protein and albumin was ∼5-fold higher within the BAL fluid of the RSV-infected WT lungs as compared to mock-infected WT. In contrast, albumin levels within the BAL fluid of RSV-infected PKR−/− did not change (Fig. 4B). This suggests that PKR is required for lung damage, which allows the extravasation of serum protein into the lung milieu.
FIG. 4.
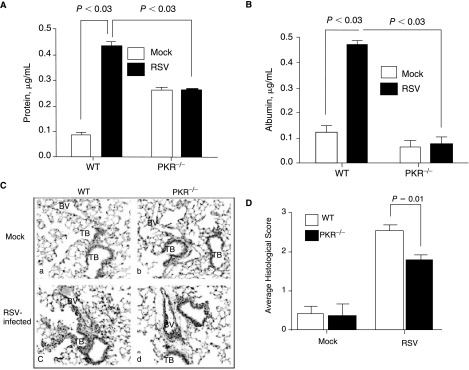
Respiratory syncytial virus (RSV)-induced albumin extravasation and lung injury are reduced in the absence of double-stranded RNA-activated protein kinase (PKR). WT and PKR−/− mice were mock treated or were infected with RSV. After 24 h, the level of total protein (A) and albumin (B) was determined in the bronchoalveolar lavage (BAL) fluid. The error bars represent SEM; data are representative of 2 separate experiments (3 mice in each experiment). Inflammation and lung injury in the infected WT and PKR−/− mice were determined at 24 h post-infection. (C) Lung sections were prepared as described in Materials and Methods and stained with hematoxylin and eosin. (D) Lung inflammation was quantified by a pathologist as described in Materials and Methods. Data are an average of 2 separate experiments. Abbreviations: TB, terminal bronchiole; BV, blood vessel.
To further access the level of lung injury, we performed a histological examination of the lungs. The quantitative unbiased histological score showed that the degree of inflammation and lung damage in WT mice was significantly higher than in PKR−/− mice (Fig. 4C and 4D). In addition, the histological examination of RSV-infected mice lungs showed a significantly higher perivascular/peribronchiolar infiltrates of polymorphonulcear (PMN) cells, lymphocytes, and eosinophils in WT than in PKR−/− mice (Fig. 4C and 4D). These data provide further evidence that RSV-induced lung injury requires PKR.
PKR is required for RSV-induced activation of MAPK and NF-κB pathways
PKR has been reported to be necessary for activation of the MAPK and NF-κB signaling pathways, which are essential for inflammatory responses (Kumar and others 1994; Langland and others 1999; Goh and others 2000; Zamanian-Daryoush and others 2000; Gil and others 2001). Furthermore, RSV infection of human bronchial epithelial cells has previously been shown to activate both ERK1/2 and p38 MAPK (Monick and others 2001; Pazdrak and others 2002; Meusel and Imani 2003; Kong and others 2004). We, therefore, determined whether there was any alteration in activation of p38 MAPK, JNK, and ERK in lungs of RSV-infected PKR−/− mice. After 24 h of infection, total lung protein extracts were prepared and were used in Western blots. Data in Figure 5A showed that RSV infection robustly activated the MAPK pathways in WT mouse lungs. However, there was little increase in activation of these pathways in extracts prepared from PKR−/− mice (Fig. 5A).
FIG. 5.

Activation of MAPK and NF-κB pathways is attenuated in the absence of double-stranded RNA-activated protein kinase (PKR). (A) Whole cell extracts were prepared from lungs of mock- or RSV-infected mice at 24 h post-infection. The levels of phosphorylated and total JNK, p38 MAPK, and ERK proteins were determined by Western blot. Each lane represents extracts prepared from a single animal. The data are representative of 3 independent experiments each with similar results. (B) At 24 h post-infection, total protein was extracted from mock- or RSV-infected mouse lungs (n=3 per group) and analyzed by EMSA using an NF-κB-specific probe. The positions of p65/p50 heterodimer and p50/p50 homodimer are indicated by the arrows. A β-actin Western blot was performed to confirm that equal amounts of proteins were used. (C) The changes in activation of NF-κB were quantified by excising the shifted bands from the gel represented in panel B and measuring radioactivity in a scintillation counter. Data represent average of counts from 3 separate lanes.
Next, using EMSA, we examined the activation state of NF-κB within the RSV-infected lungs. The data in Figure 5B showed that activation state of NF-κB was higher in infected WT lungs than in PKR−/− lungs (Fig. 5B). Quantitative analysis of the shifted bands confirmed that there was a significant difference between NF-κB activation in RSV-infected lungs (Fig. 5C). These data demonstrated that PKR activation affects several signaling pathways in vivo.
A genome-wide analysis of PKR-regulated early innate immune genes
We next examined the role of PKR in global expression of RSV-induced early genes by microarray analysis. PKR−/− and WT mice were infected with RSV and after 24 h, total RNA was extracted from lungs and was used with Agilent mouse whole genome 4x44 gene expression arrays. At a minimum of 2-fold increase, a high stringency analysis of the up-regulated genes showed that RSV infection induced 142 genes in WT and 99 genes in PKR−/− mice (see Supplementary data at the NCBI Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/projects/geo). The comparison of the gene array data showed that from the genes that were not up-regulated in PKR−/− mice, 29 of the genes were associated with interferon and immune responses (Table 1). These included PKR, TLR-3, ADAR, RNA helicase, and several members of major histocompatibility complex (MHC) genes. This suggests that PKR plays a pivotal role in early immune responses during RSV infections and may also affect adaptive immunity by enhancing antigen presentation.
Table 1.
Analysis of Double-Stranded RNA-Activated Protein Kinase-Dependent Genes
| Gene | Description | Fold change | P value |
|---|---|---|---|
| Ifi44 | Interferon-induced protein 44 | 83.77 | 0.00062 |
| H28 | Histocompatibility 28 | 81.34 | 0.0003 |
| Apod | Apolipoprotein D | 51.28 | 0.00063 |
| Ifit2 | Interferon-induced protein with tetratricopeptide repeats 2 | 48.21 | 0.00039 |
| Mpa21 | Macrophage activation 2 like | 25.24 | 0.00061 |
| Tgtp | T-cell-specific GTPase | 23.76 | 0.00046 |
| Igtp | Interferon gamma-induced GTPase | 19.30 | 0.00025 |
| Trim30 | Tripartite motif-containing 30 | 15.66 | 0.00072 |
| Ifi204 | Interferon-activated gene 204 | 12.44 | 0.00078 |
| ligp2 | Interferon-inducible GTPase 2 | 11.93 | 0.00014 |
| Isg20 | Interferon-stimulated protein | 11.00 | 0.00089 |
| H2-T24 | Histocompatibility 2. T region locus 24 | 10.86 | 0.00002 |
| Cc18 | Chemokine (C–C motif) ligand 8 | 9.18 | 0.00079 |
| Eif2ak2 | PKR, eukaryotic translation initiation factor 2-alpha kinase | 8.69 | 0.00049 |
| Stat2 | Signal transducer and activator of transcription 2 | 7.83 | 0.0008 |
| Adar | Adenosine deaminase. RNA-specific | 7.50 | 0.0004 |
| Ifi35 | Interferon-induced protein 35 | 5.83 | 0.00043 |
| H2-M2 | Histocompatibility 2, M regon locus 2 | 5.23 | 0.00044 |
| H2-Q1 | Histocompatibility 2, Q region locus 1 | 5.09 | 0.00097 |
| Irf9 | Interferon regulatory factor 9 | 5.00 | 0.00055 |
| Ifitm7 | Interferon-induced transmembrane protein 7 | 4.97 | 0.00032 |
| Ly6a | Lymphocyte antigen 6 complex, locus A | 4.92 | 0.00062 |
| Tapbp | TAP-binding protein | 4.29 | 0.00066 |
| Nmi | N-myc (and STAT) interactor | 3.98 | 0.00027 |
| Tlr3 | Toll-like receptor 3 | 3.97 | 0.0004 |
| Trim21 | Tripartite motif-containing 21 | 3.77 | 0.00066 |
| Tafsf10 | Tumor necrosis factor (ligand) superfamily, member 10 | 3.02 | 0.0008 |
| H2-K1 | Histocompatibility 2, K1, K region | 2.65 | 0.00088 |
| Trim25 | Tripartite motif-containing 25 | 2.43 | 0.00098 |
Fold changes of PKR-dependent genes that are involved in immune and interferon pathways with highest significance are represented. For complete gene list, refer to Supplementary data at the NCBI Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/projects/geo.
We next used Ingenuity Pathway Analysis (IPA) to categorize PKR-dependent genes in relation to disease and disorders. As expected, the data revealed that PKR-dependent genes were most likely associated with the inflammatory and immunologic disorders (Fig. 6). In addition, cancer, connective tissue, and skeletal/muscular disorders were also significantly associated with PKR activation.
FIG. 6.

Functional analysis of differentially expressed genes. The Ingenuity Pathway Analysis (IPA) of disease and functional relevance for double-stranded RNA-activated protein kinase (PKR)-dependent genes are represented. The −log (significance) indicates an increase in confidence for each category.
Discussion
RSV is the most common cause of viral respiratory infections in infants, which can lead to severe bronchiolitis, hospitalization, and mortality. In murine models, RSV infections leads to induction of TNF-α, IL-6, IL-8, MCP-1, RANTES, and CXCL10 (IP-10) (Graham and others 2000; Tripp and others 2000; Jafri and others 2004; Miller and others 2004; Estripeaut and others 2008). In the early steps, the presence of viral infections leads to an innate immune response and there is a rapid (8–24 h) and transient rise in the expression of chemokines and cytokines (Tripp and others 2000; Estripeaut and others 2008). Therefore, we hypothesized that PKR, a dsRNA-specific kinase, which is activated early during viral infections could play a role in RSV-induced immunity. To date, there has been no report on the role of PKR in RSV infection, immune responses, and lung injury.
PKR is activated by dsRNA, which is present during the life cycle of most viruses (Jacobs and Langland 1996). Once activated, PKR phosphorylates eIF-2α, leading to a reduction in translation and thus to an attenuation of viral replication. Several groups using the PKR−/− mouse have demonstrated that PKR is a key host factor for resistance to viral replication (Balachandran and others 2000; Stojdl and others 2000; Guidotti and others 2002; Carr and others 2006). Consistent with the role of PKR in viral replication, our data showed that unlike WT mice, PKR−/− mice (Yang and others 1995; Baltzis and others 2002) had a greater load of RSV at 24 h post-infection (Fig. 1A and 1B). Based on an in vitro kinetic analysis, we showed that the difference was due to viral replication and not initial infectivity (Fig. 1D).
In addition to participating in antiviral responses, PKR also functions as a signaling intermediate in several pathways including MAPK and NF-κB (Kumar and others 1994; Goh and others 2000b; Zamanian-Daryoush and others 2000; Zhou and others 2003). We previously reported that RSV infection of human epithelial cells led to activation of MAPK and subsequently inflammatory cytokine expression (Meusel and Imani 2003). Also, RSV infection of airway epithelial cells was shown to induce activation of ERK and p38 MAPK (Mastronarde and others 1996; Pazdrak and others 2002). In the present study, we report that while activation of the MAPK and NF-κB pathways occurred in WT lung tissue during RSV infection, there was considerably less activation of these factors in the lungs of PKR−/− mice (Fig. 5). This suggests that in response to RSV, PKR is required for maximal activation of these signaling pathways in vivo.
Several in vitro studies using the PKR activator, dsRNA, have provided evidence for the involvement of PKR in the development of inflammatory responses. We previously reported that dsRNA activation of PKR was required for induction of inflammatory cytokines in human bronchial epithelial cells (Meusel and others 2002). Also, treatment of cells with dsRNA led to activation of PKR and increased production of IL-8 and RANTES (Gern and others 2003). In our experiments, induction of cytokines and chemokines was increased dramatically within the BAL fluid of RSV-infected WT mice as compared to PKR−/− mice. The induction of cytokines and chemokines was accompanied with a corresponding increase in cellular influx into the lungs (Fig. 3B).
Previously, we reported that during intracerebral infection using mouse-adapted polio virus, both PKR−/− and transgenic mice expressing a trans-dominant negative mutant form of PKR, displayed a less severe central nervous system inflammation and tissue damage than their WT cohort (Scheuner and others 2003). Based on these reports and our current data, we believe that PKR is required for both control of RSV replication, and mounting a robust inflammatory immune response.
It is reported that in addition to direct viral pathologies, inflammatory responses can participate in tissue damage during viral infections (Tripp 2004). Wang et al. reported that the presence of neutrophils enhanced RSV-induced cell damage and detachment from culture dishes, suggesting that neutrophilic infiltrate into the lungs may contribute to the pathogenesis of RSV airway disease (Basler and others 2000). In this report, we investigated RSV-induced lung injury by comparing the levels of neutrophils, total protein, albumin, and histological score (Figs. 3 and 4). Our data showed that the levels of neutrophil influx, total protein, and albumin were greater in BAL fluid collected from RSV-infected WT mice than from PKR−/− mice. Consistent with these observations, the histological score of WT mice was significantly greater than PKR−/− mice. These data suggest that PKR is a key factor in RSV-induced lung injury and liquid extravasations into the lungs.
A high stringency comparison of the genes that were induced by RSV in WT and PKR−/− mice showed that PKR activation was required for induction of 101 genes, 29 of which were associated with early immune responses (See supplementary data at the NCBI Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/projects/geo). Expectedly several interferon-induced genes were affected by PKR activation (Table 1). Also, major histocompatibility complex (MHC) genes such as H2-M2 and H2-Q1 were up-regulated only in WT mice (Table 1). This finding is interesting because all of these MHC molecules are considered as the non-classical MHC class-I molecules with low polymorphism. Since members of the non-classical MHC molecules are involved in activation of natural killer (NK) cells (Sambrook and Beck 2007), it is tempting to speculate that PKR plays a role in viral recognition by these cells. Additional evidence for association of PKR with inflammation and immune responses was revealed in functional analysis of the PKR-regulated genes, which showed that this pathway was associated with inflammatory and immunological disorders (Fig. 6).
In addition to the MHC molecules, several of the antiviral molecules such as PKR, TLR-3, adenosine deaminase acting on RNA (ADAR), and interferon regulatory factor-9 (IRF-9) were up-regulated in WT and not in PKR−/− mice. This further suggests that PKR is critical for antiviral responses during RSV infections.
An interesting observation in our experiments was the induction of p65/p50 heterodimerization and p50/p50 homodimerization of NF-κB complexes (Fig. 5B). While activation of the p65/p50 heterodimeric form of NF-κB is required for expression of many genes, the p50 subunit has been shown to be essential for expression of IgE (Delphin and Stavnezer 1995; Kuchroo and others 1995). In as much as IgE is a central molecule in allergic responses, these data suggest that PKR may play a role in RSV induction of IgE expression. This is consistent with our previous in vitro data, which demonstrated that PKR activation regulated IgE class switching in human B lymphocytes (Rager and others 1998). Previous reports have also shown that IgE levels are higher in RSV-infected mice exposed to ragweed (Leibovitz and others 1988) and RSV-specific IgE was higher in RSV-infected infants (Welliver and others 1981; Welliver and Duffy 1993).
Collectively, our data establishes a dual role for PKR in RSV infections. First, it is necessary for controlling replication and second, it participates in signal transduction, leading to innate inflammatory responses. These responses, while important for the overall immunity to RSV, likely contribute to lung injury.
Acknowledgments
We would like to thank Ms. Kimwa Walker and the NIEHS Histology and Micro array core facilities for their assistance. This research was supported entirely by the Intramural Research Program at National Institute of Environmental Health Sciences/NIH.
References
- Arnold R. Humbert B. Werchau H. Gallati H. Konig W. Interleukin-8, interleukin-6, and soluble tumour necrosis factor receptor type I release from a human pulmonary epithelial cell line (A549) exposed to respiratory syncytial virus. Immunology. 1994;82(1):126–133. [PMC free article] [PubMed] [Google Scholar]
- Balachandran S. Roberts PC. Brown LE. Truong H. Pattnaik AK. Archer DR. Barber GN. Essential role for the dsRNA-dependent protein kinase PKR in innate immunity to viral infection. Immunity. 2000;13(1):129–141. doi: 10.1016/s1074-7613(00)00014-5. [DOI] [PubMed] [Google Scholar]
- Baltzis D. Li S. Koromilas AE. Functional characterization of pkr gene products expressed in cells from mice with a targeted deletion of the N terminus or C terminus domain of PKR. J Biol Chem. 2002;277(41):38364–38372. doi: 10.1074/jbc.M203564200. [DOI] [PubMed] [Google Scholar]
- Basler CF. Wang X. Muhlberger E. Volchkov V. Paragas J. Klenk HD. Garcia-Sastre A. Palese P. The Ebola virus VP35 protein functions as a type I IFN antagonist. Proc Natl Acad Sci USA. 2000;97(22):12289–12294. doi: 10.1073/pnas.220398297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boothby MR. Mora AL. Scherer DC. Brockman JA. Ballard DW. Perturbation of the T lymphocyte lineage in transgenic mice expressing a constitutive repressor of nuclear factor (NF)-kappaB. J Exp Med. 1997;185(11):1897–1907. doi: 10.1084/jem.185.11.1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carr DJ. Wuest T. Tomanek L. Silverman RH. Williams BR. The lack of RNA-dependent protein kinase enhances susceptibility of mice to genital herpes simplex virus type 2 infection. Immunology. 2006;118(4):520–526. doi: 10.1111/j.1365-2567.2006.02403.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Haro C. Mendez R. Santoyo J. The eIF-2alpha kinases and the control of protein synthesis. FASEB J. 1996;10(12):1378–1387. doi: 10.1096/fasebj.10.12.8903508. [DOI] [PubMed] [Google Scholar]
- Delphin S. Stavnezer J. Characterization of an interleukin 4 (IL-4) responsive region in the immunoglobulin heavy chain germline epsilon promoter: regulation by NF-IL-4, a C/EBP family member and NF-kappa B/p50. J Exp Med. 1995;181(1):181–192. doi: 10.1084/jem.181.1.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durbin JE. Johnson TR. Durbin RK. Mertz SE. Morotti RA. Peebles RS. Graham BS. The role of IFN in respiratory syncytial virus pathogenesis. J Immunol. 2002;168(6):2944–2952. doi: 10.4049/jimmunol.168.6.2944. [DOI] [PubMed] [Google Scholar]
- Estripeaut D. Torres JP. Somers CS. Tagliabue C. Khokhar S. Bhoj VG. Grube SM. Wozniakowski A. Gomez AM. Ramilo O. Jafri HS. Mejias A. Respiratory syncytial virus persistence in the lungs correlates with airway hyperreactivity in the mouse model. J Infect Dis. 2008;198(10):1435–1443. doi: 10.1086/592714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franke G. Freihorst J. Steinmuller C. Verhagen W. Hockertz S. Lohmann-Matthes ML. Interaction of alveolar macrophages and respiratory syncytial virus. J Immunol Methods. 1994;174(1–2):173–184. doi: 10.1016/0022-1759(94)90020-5. [DOI] [PubMed] [Google Scholar]
- Galabru J. Katze MG. Robert N. Hovanessian AG. The binding of double-stranded RNA and adenovirus VAI RNA to the interferon-induced protein kinase. Eur J Biochem. 1989;178(3):581–589. doi: 10.1111/j.1432-1033.1989.tb14485.x. [DOI] [PubMed] [Google Scholar]
- García MA. Gil J. Ventoso I. Guerra S. Domingo E. Rivas C. Esteban M. Impact of protein kinase PKR in cell biology: from antiviral to antiproliferative action. Microbiol Mol Biol Rev. 2006;70(4):1032–1060. doi: 10.1128/MMBR.00027-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gern JE. French DA. Grindle KA. Brockman-Schneider RA. Konno S. Busse WW. Double-stranded RNA induces the synthesis of specific chemokines by bronchial epithelial cells. Am J Respir Cell Mol Biol. 2003;28(6):731–737. doi: 10.1165/rcmb.2002-0055OC. [DOI] [PubMed] [Google Scholar]
- Gil J. Rullas J. Garcia MA. Alcami J. Esteban M. The catalytic activity of dsRNA-dependent protein kinase, PKR, is required for NF-kappaB activation. Oncogene. 2001;20(3):385–394. doi: 10.1038/sj.onc.1204109. [DOI] [PubMed] [Google Scholar]
- Goh KC. deVeer MJ. Williams BR. The protein kinase PKR is required for p38 MAPK activation and the innate immune response to bacterial endotoxin. EMBO J. 2000;19(16):4292–4297. doi: 10.1093/emboj/19.16.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham BS. Johnson TR. Peebles RS. Immune-mediated disease pathogenesis in respiratory syncytial virus infection. Immunopharmacology. 2000;48(3):237–247. doi: 10.1016/s0162-3109(00)00233-2. [DOI] [PubMed] [Google Scholar]
- Graham BS. Perkins MD. Wright PF. Karzon DT. Primary respiratory syncytial virus infection in mice. J Med Virol. 1988;26(2):153–162. doi: 10.1002/jmv.1890260207. [DOI] [PubMed] [Google Scholar]
- Guidotti LG. Morris A. Mendez H. Koch R. Silverman RH. Williams BR. Chisari FV. Interferon-regulated pathways that control hepatitis B virus replication in transgenic mice. J Virol. 2002;76(6):2617–2621. doi: 10.1128/JVI.76.6.2617-2621.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall CB. Douglas RG., Jr. Geiman JM. Respiratory syncytial virus infections in infants: quantitation and duration of shedding. J Pediatr. 1976;89(1):11–15. doi: 10.1016/s0022-3476(76)80918-3. [DOI] [PubMed] [Google Scholar]
- Hussell T. Pennycook A. Openshaw PJ. Inhibition of tumor necrosis factor reduces the severity of virus-specific lung immunopathology. Eur J Immunol. 2001;31(9):2566–2573. doi: 10.1002/1521-4141(200109)31:9<2566::aid-immu2566>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Jacobs BL. Langland JO. When two strands are better than one: the mediators and modulators of the cellular responses to double-stranded RNA. Virology. 1996;219(2):339–349. doi: 10.1006/viro.1996.0259. [DOI] [PubMed] [Google Scholar]
- Jafri HS. Chavez-Bueno S. Mejias A. Gomez AM. Rios AM. Nassi SS. Yusuf M. Kapur P. Hardy RD. Hatfield J. Rogers BB. Krisher K. Ramilo O. Respiratory syncytial virus induces pneumonia, cytokine response, airway obstruction, and chronic inflammatory infiltrates associated with long-term airway hyperresponsiveness in mice. J Infect Dis. 2004;189(10):1856–1865. doi: 10.1086/386372. [DOI] [PubMed] [Google Scholar]
- Jaradat M. Stapleton C. Tilley SL. Dixon D. Erikson CJ. McCaskill JG. Kang HS. Angers M. Liao G. Collins J. Grissom S. Jetten AM. Modulatory role for retinoid-related orphan receptor alpha in allergen-induced lung inflammation. Am J Respir Crit Care Med. 2006;174(12):1299–1309. doi: 10.1164/rccm.200510-1672OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehoe KE. Brown MA. Imani F. Double-stranded RNA regulates IL-4 expression. J Immunol. 2001;167(5):2496–2501. doi: 10.4049/jimmunol.167.5.2496. [DOI] [PubMed] [Google Scholar]
- Kong X. San Juan H. Behera A. Peeples ME. Wu J. Lockey RF. Mohapatra SS. ERK-1/2 activity is required for efficient RSV infection. FEBS Lett. 2004;559(1–3):33–38. doi: 10.1016/S0014-5793(04)00002-X. [DOI] [PubMed] [Google Scholar]
- Krust B. Galabru J. Hovanessian AG. Further characterization of the protein kinase activity mediated by interferon in mouse and human cells. J Biol Chem. 1984;259(13):8494–8498. [PubMed] [Google Scholar]
- Kuchroo VK. Das MP. Brown JA. Ranger AM. Zamvil SS. Sobel RA. Weiner HL. Nabavi N. Glimcher LH. B7-1 and B7-2 costimulatory molecules activate differentially the Th1/Th2 developmental pathways: application to autoimmune disease therapy. Cell. 1995;80(5):707–718. doi: 10.1016/0092-8674(95)90349-6. [DOI] [PubMed] [Google Scholar]
- Kumar A. Haque J. Lacoste J. Hiscott J. Williams BR. Double-stranded RNA-dependent protein kinase activates transcription factor NF-kappa B by phosphorylating I kappa B. Proc Natl Acad Sci USA. 1994;91(14):6288–6292. doi: 10.1073/pnas.91.14.6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langland JO. Kao PN. Jacobs BL. Nuclear factor-90 of activated T-cells: A double-stranded RNA-binding protein and substrate for the double-stranded RNA-dependent protein kinase, PKR. Biochemistry. 1999;38(19):6361–6368. doi: 10.1021/bi982410u. [DOI] [PubMed] [Google Scholar]
- Lebleu B. Sen GC. Shaila S. Cabrer B. Lengyel P. Interferon, double-stranded RNA, and protein phosphorylation. Proc Natl Acad Sci USA. 1976;73(9):3107–3111. doi: 10.1073/pnas.73.9.3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibovitz E. Freihorst J. Piedra PA. Ogra PL. Modulation of systemic and mucosal immune responses to inhaled ragweed antigen in experimentally induced infection with respiratory syncytial virus implication in virally induced allergy. Int Arch Allergy Appl Immunol. 1988;86(1):112–116. doi: 10.1159/000234615. [DOI] [PubMed] [Google Scholar]
- Mastronarde JG. He B. Monick MM. Mukaida N. Matsushima K. Hunninghake GW. Induction of interleukin (IL)-8 gene expression by respiratory syncytial virus involves activation of nuclear factor (NF)-kappa B and NF-IL-6. J Infect Dis. 1996;174(2):262–267. doi: 10.1093/infdis/174.2.262. [DOI] [PubMed] [Google Scholar]
- Matsuda K. Tsutsumi H. Okamoto Y. Chiba C. Development of interleukin 6 and tumor necrosis factor alpha activity in nasopharyngeal secretions of infants and children during infection with respiratory syncytial virus. Clin Diagn Lab Immunol. 1995;2(3):322–324. doi: 10.1128/cdli.2.3.322-324.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntosh K. Masters HB. Orr I. Chao RK. Barkin RM. The immunologic response to infection with respiratory syncytial virus in infants. J Infect Dis. 1978;138(1):24–32. doi: 10.1093/infdis/138.1.24. [DOI] [PubMed] [Google Scholar]
- McNamara PS. Flanagan BF. Selby AM. Hart CA. Smyth RL. Pro- and anti-inflammatory responses in respiratory syncytial virus bronchiolitis. Eur Respir J. 2004;23(1):106–112. doi: 10.1183/09031936.03.00048103. [DOI] [PubMed] [Google Scholar]
- Meusel TR. Imani F. Viral induction of inflammatory cytokines in human epithelial cells follows a p38 mitogen-activated protein kinase-dependent but NF-kappaB-independent pathway. J Immunol. 2003;171(7):3768–3774. doi: 10.4049/jimmunol.171.7.3768. [DOI] [PubMed] [Google Scholar]
- Meusel TR. Kehoe KE. Imani F. Protein kinase R regulates double-stranded RNA induction of TNF-alpha but not IL-1 beta mRNA in human epithelial cells. J Immunol. 2002;168(12):6429–6435. doi: 10.4049/jimmunol.168.12.6429. [DOI] [PubMed] [Google Scholar]
- Midulla F. Villani A. Panuska JR. Dab I. Kolls JK. Merolla R. Ronchetti R. Respiratory syncytial virus lung infection in infants: immunoregulatory role of infected alveolar macrophages. J Infect Dis. 1993;168(6):1515–1519. doi: 10.1093/infdis/168.6.1515. [DOI] [PubMed] [Google Scholar]
- Miller AL. Bowlin TL. Lukacs NW. Respiratory syncytial virus-induced chemokine production: linking viral replication to chemokine production in vitro and in vivo. J Infect Dis. 2004;189(8):1419–1430. doi: 10.1086/382958. [DOI] [PubMed] [Google Scholar]
- Monick M. Staber J. Thomas K. Hunninghake G. Respiratory syncytial virus infection results in activation of multiple protein kinase C isoforms leading to activation of mitogen-activated protein kinase. J Immunol. 2001;166(4):2681–2687. doi: 10.4049/jimmunol.166.4.2681. [DOI] [PubMed] [Google Scholar]
- Moore ML. Peebles RS., Jr. Respiratory syncytial virus disease mechanisms implicated by human, animal model, and in vitro data facilitate vaccine strategies and new therapeutics. Pharmacol Ther. 2006;112(2):405–424. doi: 10.1016/j.pharmthera.2006.04.008. [DOI] [PubMed] [Google Scholar]
- Neuzil KM. Tang YW. Graham BS. Protective Role of TNF-alpha in respiratory syncytial virus infection in vitro and in vivo. Am J Med Sci. 1996;311(5):201–204. doi: 10.1097/00000441-199605000-00001. [DOI] [PubMed] [Google Scholar]
- Noah TL. Becker S. Respiratory syncytial virus-induced cytokine production by a human bronchial epithelial cell line. Am J Physiol. 1993;265(5 Pt 1):L472–L478. doi: 10.1152/ajplung.1993.265.5.L472. [DOI] [PubMed] [Google Scholar]
- Olszewska-Pazdrak B. Casola A. Saito T. Alam R. Crowe SE. Mei F. Ogra PL. Garofalo RP. Cell-specific expression of RANTES, MCP-1, and MIP-1alpha by lower airway epithelial cells and eosinophils infected with respiratory syncytial virus. J Virol. 1998;72(6):4756–4764. doi: 10.1128/jvi.72.6.4756-4764.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pazdrak K. Olszewska-Pazdrak B. Liu T. Takizawa R. Brasier AR. Garofalo RP. Casola A. MAPK activation is involved in post-transcriptional regulation of RSV-induced RANTES gene expression. Am J Physiol Lung Cell Mol Physiol. 2002;283(2):L364–L372. doi: 10.1152/ajplung.00331.2001. [DOI] [PubMed] [Google Scholar]
- Rager KJ. Langland JO. Jacobs BL. Proud D. Marsh DG. Imani F. Activation of antiviral protein kinase leads to immunoglobulin E class switching in human B cells. J Virol. 1998;72(2):1171–1176. doi: 10.1128/jvi.72.2.1171-1176.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutigliano JA. Graham BS. Prolonged production of TNF-alpha exacerbates illness during respiratory syncytial virus infection. J Immunol. 2004;173(5):3408–3417. doi: 10.4049/jimmunol.173.5.3408. [DOI] [PubMed] [Google Scholar]
- Sambrook JG. Beck S. Evolutionary vignettes of natural killer cell receptors. Curr Opin Immunol. 2007;19(5):553–560. doi: 10.1016/j.coi.2007.08.002. [DOI] [PubMed] [Google Scholar]
- Samuel CE. Mechanism of interferon action: phosphorylation of protein synthesis initiation factor eIF-2 in interferon-treated human cells by a ribosome-associated kinase processing site specificity similar to hemin-regulated rabbit reticulocyte kinase. Proc Natl Acad Sci USA. 1979;76(2):600–604. doi: 10.1073/pnas.76.2.600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheuner D. Gromeier M. Davies MV. Dorner AJ. Song B. Patel RV. Wimmer EJ. McLendon RE. Kaufman RJ. The double-stranded RNA-activated protein kinase mediates viral-induced encephalitis. Virology. 2003;317(2):263–274. doi: 10.1016/j.virol.2003.08.010. [DOI] [PubMed] [Google Scholar]
- Sheeran P. Jafri H. Carubelli C. Saavedra J. Johnson C. Krisher K. Sanchez PJ. Ramilo O. Elevated cytokine concentrations in the nasopharyngeal and tracheal secretions of children with respiratory syncytial virus disease. Pediatr Infect Dis J. 1999;18(2):115–122. doi: 10.1097/00006454-199902000-00007. [DOI] [PubMed] [Google Scholar]
- Stapleton CM. Jaradat M. Dixon D. Kang HS. Kim SC. Liao G. Carey MA. Cristiano J. Moorman MP. Jetten AM. Enhanced susceptibility of staggerer (RORalphasg/sg) mice to lipopolysaccharide-induced lung inflammation. Am J Physiol Lung Cell Mol Physiol. 2005;289(1):L144–L152. doi: 10.1152/ajplung.00348.2004. [DOI] [PubMed] [Google Scholar]
- Stojdl DF. Abraham N. Knowles S. Marius R. Brasey A. Lichty BD. Brown EG. Sonenberg N. Bell JC. The murine double-stranded RNA-dependent protein kinase PKR is required for resistance to vesicular stomatitis virus. J Virol. 2000;74(20):9580–9585. doi: 10.1128/jvi.74.20.9580-9585.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripp RA. Pathogenesis of respiratory syncytial virus infection. Viral Immunol. 2004;17(2):165–181. doi: 10.1089/0882824041310513. [DOI] [PubMed] [Google Scholar]
- Tripp RA. Jones L. Anderson LJ. Respiratory syncytial virus G and/or SH glycoproteins modify CC and CXC chemokine mRNA expression in the BALB/c mouse. J Virol. 2000;74(13):6227–6229. doi: 10.1128/jvi.74.13.6227-6229.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welliver RC. Duffy L. The relationship of RSV-specific immunoglobulin E antibody responses in infancy, recurrent wheezing, and pulmonary function at age 7–8 years. Pediatr Pulmonol. 1993;15(1):19–27. doi: 10.1002/ppul.1950150104. [DOI] [PubMed] [Google Scholar]
- Welliver RC. Garofalo RP. Ogra PL. Beta-chemokines, but neither T helper type 1 nor T helper type 2 cytokines, correlate with severity of illness during respiratory syncytial virus infection. Pediatr Infect Dis J. 2002;21(5):457–461. doi: 10.1097/00006454-200205000-00033. [DOI] [PubMed] [Google Scholar]
- Welliver RC. Wong DT. Sun M. Middleton E., Jr. Vaughan RS. Ogra PL. The development of respiratory syncytial virus-specific IgE and the release of histamine in nasopharyngeal secretions after infection. N Engl J Med. 1981;305(15):841–846. doi: 10.1056/NEJM198110083051501. [DOI] [PubMed] [Google Scholar]
- Yang YL. Reis LF. Pavlovic J. Aguzzi A. Schafer R. Kumar A. Williams BR. Aguet M. Weissmann C. Deficient signaling in mice devoid of double-stranded RNA-dependent protein kinase. EMBO J. 1995;14(24):6095–6106. doi: 10.1002/j.1460-2075.1995.tb00300.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamanian-Daryoush M. Mogensen TH. DiDonato JA. Williams BR. NF-kappaB activation by double-stranded-RNA-activated protein kinase (PKR) is mediated through NF-kappaB-inducing kinase and IkappaB kinase. Mol Cell Biol. 2000;20(4):1278–1290. doi: 10.1128/mcb.20.4.1278-1290.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y. Luxon BA. Casola A. Garofalo RP. Jamaluddin M. Brasier AR. Expression of respiratory syncytial virus-induced chemokine gene networks in lower airway epithelial cells revealed by cDNA microarrays. J Virol. 2001;75(19):9044–9058. doi: 10.1128/JVI.75.19.9044-9058.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao MQ. Stoler MH. Liu AN. Wei B. Soguero C. Hahn YS. Enelow RI. Alveolar epithelial cell chemokine expression triggered by antigen-specific cytolytic CD8(+) T cell recognition. J Clin Invest. 2000;106(6):R49–R58. doi: 10.1172/JCI9786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou HR. Lau AS. Pestka JJ. Role of double-stranded RNA-activated protein kinase R (PKR) in deoxynivalenol-induced ribotoxic stress response. Toxicol Sci. 2003;74(2):335–344. doi: 10.1093/toxsci/kfg148. [DOI] [PubMed] [Google Scholar]