

Abstract
Well-established rules of translational initiation have been used as a cornerstone in molecular biology to understand gene expression and to frame fundamental questions on what proteins a cell synthesizes, how proteins work and to predict the consequences of mutations. For a group of neurological diseases caused by the abnormal expansion of short segments of DNA (e.g. CAG•CTG repeats), mutations within or outside of predicted coding and non-coding regions are thought to cause disease by protein gain- or loss-of-function or RNA gain-of-function mechanisms. In contrast to these predictions, the recent discovery of repeat-associated non-ATG (RAN) translation showed expansion mutations can express homopolymeric expansion proteins in all three reading frames without an AUG start codon. This unanticipated, non-canonical type of protein translation is length-and hairpin-dependent, takes place without frameshifting or RNA editing and occurs across a variety of repeat motifs. To date, RAN proteins have been reported in spinocerebellar ataxia type 8 (SCA8), myotonic dystrophy type 1 (DM1), fragile X tremor ataxia syndrome (FXTAS) and C9ORF72 amyotrophic lateral sclerosis/frontotemporal dementia (ALS/FTD). In this article, we review what is currently known about RAN translation and recent progress toward understanding its contribution to disease.
INTRODUCTION
Repeat-expansion disorders are a class of neurological and neuromuscular diseases caused by the expansion of short repetitive elements within the human genome. The genic location of the expansion has been traditionally used to classify these disorders into coding expansions caused by protein gain-of-function effects, and non-coding expansions caused by either a loss-of-function of the affected gene or RNA gain-of-function effects (1–3). For protein gain-of-function diseases, the expansion mutation is translated as part of a larger open-reading frame (ORF), resulting in the expression of a mutant protein that disrupts cellular function and induces toxicity. For example, in Huntington's disease (HD) the CAG expansion mutation is translated as part of the huntingtin protein, which results in protein aggregation and cellular dysfunction (4). For RNA gain-of-function disorders, non-coding expansion RNAs accumulate as nuclear foci that sequester RNA-binding proteins and lead to a loss of their normal function (5,6). For example, in myotonic dystrophy type 1 (DM1) and type 2 (DM2), CUG or CCUG expansion RNAs sequester MBNL proteins from their normal splicing targets, such that the resulting MBNL loss-of-function leads to alternative splicing dysregulation (7–10). The recent discovery of repeat-associated non-ATG (RAN) translation (11) showed that microsatellite expansions do not follow the canonical rules of translation initiation and can generate a series of unexpected repeat proteins. This finding opens the door to new paradigms in disease mechanisms and cell biology. In this review, we discuss the discovery of RAN translation, what is currently known about its molecular biology and progress toward understanding its contribution to disease.
INITIAL DISCOVERY OF RAN TRANSLATION IN SCA8
RAN translation was initially discovered by Zu et al. (11) while investigating the molecular mechanisms of spinocerebellar ataxia type 8 (SCA8). SCA8 is a dominantly inherited, slowly progressive neurodegenerative disorder caused by a CTG•CAG repeat expansion (12). Both RNA and protein disease mechanisms likely operate in SCA8 as bidirectional transcription produces both a CUG expansion transcript that forms RNA foci (13) and a CAG expansion transcript with an unusual ATG-initiated ORF encoding a nearly pure polyGln expansion protein (Fig. 1A) (14). The first evidence for RAN translation came when Zu et al., trying to separate the RNA and protein gain-of-function effects, found that removing the only ATG initiation codon within an SCA8 minigene did not prevent expression of the polyGln protein (Fig. 1B) (11). Subsequent experiments with epitope-tagged minigenes showed CAG expansions lacking an ATG initiation codon produce distinct homopolymeric protein products in all the three reading frames, polyGln, polyAla and polySer (Fig. 1C). Because these findings were novel, and completely unexpected multiple approaches were used to characterize the transcripts and to establish the identity of these proteins.
Figure 1.
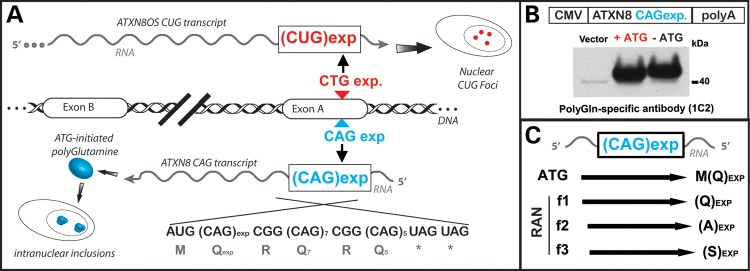
RAN translation in spinocerebellar ataxia type 8 (SCA8). (A) Prior to the discovery of RAN translation, bidirectional transcription at the SCA8 locus was known to produce RNA foci from the CUG expansion transcript and a polyglutamine expansion protein from the CAG expansion transcript (13,14). The CAG expansion transcript has an unusual short ORF with an ATG initiation codon immediately upstream of the CAG expansion and a series of stop codons immediately after the repeat (14). Both RNA and protein gain-of-function effects are evident in SCA8. (B) To separate the effects of the CUGEXP transcript from the polyGln protein, the ATG immediate upstream of the CAG expanded repeat was mutated in an ATXN8 minigene (11). Unexpectedly, this mutation did not prevent the expression of the polyglutamine protein and was the first indication of RAN translation. (C) Schematic diagram showing CAG-repeat expansion expressing both ATG-initiated polyGln and non-ATG initiated polyGln, polyAla, polySer RAN proteins repeats in all the three reading frames.
Analysis of polyribosome-bound transcripts showed no evidence of RNA editing that could have introduced a start codon (11). Immunoprecipitation and analysis of C- and N-terminal epitope-tagged constructs demonstrated that RAN translation does not require frameshifting, and that RAN occurs in all the reading frames even in the presence of an ATG-initiated ORF. Additionally, a combination of epitope tags, tritium labeling and mass spectrometry unequivocally proved that these proteins contain expanded polyAla, polyGln or polySer repeat tracts. For polyalanine, mass spectrometry identified a series of N-terminal peptides containing varying numbers of alanines. No peptides containing N-terminal methionine were identified. These data suggest that translation in the polyAla reading frame begins with an alanine, and that start sites occur at various positions throughout the length of the repeat tract.
Additional experiments on RAN translation (11) demonstrated a number of features. First, immunofluorescence showed RAN proteins expressed from all the three reading frames can accumulate in a single cell, although more frequently only one or two RAN proteins were found. Second, RAN proteins expressed across CAG expansions increase apoptosis, suggesting a potential contribution to disease. Third, RAN proteins are expressed across hairpin-forming CAG but not across non-hairpin-forming CAA repeats in cell culture. These data suggest that structured RNAs may be required for RAN translation. Fourth, RAN translation also occurs across CUG expansion transcripts. Fifth, longer CAG repeat tracts are associated with the simultaneous expression of multiple protein products with a different length threshold required for translation in each frame (Table 1). Taken together, these data demonstrated that CAG and CUG expansion transcripts undergo a novel type of protein translation in which homopolymeric proteins are expressed in all the three reading frames without an ATG-initiation codon.
Table 1.
In vitro characteristics and in vivo detection of RAN translation
| Disorder | Repeat | RAN protein (in vitro) | Threshold (in vitro) | Tissue (in vivo) | Refs. |
|---|---|---|---|---|---|
| SCA8 | CAG | polyGln | >42 repeats | ATG-polyGln, cerebellum and brain stem (14) | (11) |
| polyAla | >73 repeats | Cerebellum | (11) | ||
| polySer | >58 repeats | ND | (11) | ||
| DM1 | CAGa | polyGln | ND | Myoblasts, skeletal muscle, peripheral blood leukocytes | (11) |
| FXTAS | CGG | polyGly | >30 repeats | Frontal cortex, cerebellum, hippocampus | (24) |
| polyAla | >88 repeats | ND | (24) | ||
| polyArg | UD | ND | (24) | ||
| ALS/FTD | GGGGCC | polyGlyPro | >145 repeats | Cerebellum, hippocampus, iPSC-derived neurons, neocortex, medial and lateral geniculate nuclei, testes | (25–27) |
| polyGlyAla | >38 repeats | Cerebellum, hippocampus | (26) | ||
| polyGlyArg | UD | Cerebellum, hippocampus | (26) |
ND, not examined and not determined; UD, examined but undetermined.
aAntisense transcript.
IN VIVO EVIDENCE FOR RAN TRANSLATION IN SCA8
After establishing that RAN translation occurs in transfected cells, Zu et al. looked for evidence that SCA8 RAN proteins are expressed in vivo (11). SCA8 is characterized by severe cerebellar atrophy with Purkinje cell degeneration and loss of granule cells (14). Zu et al. (11) developed antibodies against the unique C-terminal region of the predicted CAG-encoded polyAla RAN protein and showed polyAla-positive immunostaining in cerebellar Purkinje cells from human SCA8 but not control autopsy tissue. These antibodies also detected polyAla RAN proteins in Purkinje cells from an established mouse model of SCA8 (14). SCA8 Purkinje cells are also known to accumulate CUG RNA foci (13) and poly-Gln inclusions (14). Although additional studies are needed to understand the effects of RAN proteins in SCA8, the accumulation of the SCA8 polyAla protein in Purkinje cells suggests that RAN proteins may contribute to disease.
RAN TRANSLATION IN MYOTONIC DYSTROPHY TYPE 1
Additional in vivo evidence for RAN translation was demonstrated in myotonic dystrophy (11). DM1, one of the best examples of an RNA gain-of-function disease (5), is caused by a CTG expansion in the 3′ UTR of the DMPK gene (15–17). Antisense transcripts in the CAG direction have also been reported (11,18). To determine whether RAN translation also occurs for DM1, Zu et al. (11) performed immunostaining with two types of antibodies: (i) a well-established monoclonal antibody that detects expanded polyGln tracts (19,20) and (ii) a novel antibody developed to detect the unique C-terminal region of the predicted CAG encoded poly-Gln RAN protein (11). Positive immunostaining was observed in DM1 myoblasts, skeletal muscle and blood. Similar staining was found in an established DM1 mouse model (21,22), which showed staining of cardiomyocytes and leukocytes (11). Additionally, in both humans and mice polyGln aggregates co-localized with caspase-8, an early indicator of polyGln-induced apoptosis (23). Although RNA gain-of-function effects in DM1 are known to cause specific alternative splicing changes, these results suggest the possibility that RAN translation may also contribute to this disorder.
The discovery of RAN translation combined with growing evidence that many microsatellite expansion mutations are transcribed in both directions (2) suggests that in addition to previously considered gene products, expansion mutations may also express up to six additional RAN proteins (Fig. 2)—each of which may contribute to disease (Fig. 3). Consistent with this prediction, RAN translation has recently been reported in two additional disorders: fragile X-associated tremor ataxia syndrome (FXTAS) (24) and C9ORF72 amyotrophic lateral sclerosis/frontotemporal dementia (ALS/FTD) (25–27).
Figure 2.

Model of RAN translation across repeats in coding and non-coding gene regions. Schematic diagram showing mutations located in intronic or exonic regions with expression of distinct RAN proteins in three frames from sense and antisense directions. For expansions in introns, sense and antisense transcripts (not shown) produce RAN proteins with different repeat motifs and distinct C-terminal regions not corresponding to any endogenous proteins. For repeat-expansion mutations located in ORFs, up to six distinct RAN proteins may be produced from sense and antisense transcripts (see upper inset for antisense RAN proteins). The RAN protein expressed in the ORF is predicted to start at or close to the repeat and contain the same C-terminal region as the protein expressed from the canonical ATG-initiated ORF. Variability of RAN proteins will occur when expressed from: sense or antisense transcripts; different repeat motifs and with variations in C-terminal sequences.
Figure 3.

Potential pathways of pathogenesis of repeat-associated disorders. Bidirectional transcription of an expanded repeat will produced two transcripts (blue = antisense, red = sense), each potentially capable of structure formation and contributions to pathogenesis. In the RNA toxicity model (1st upper and lower panels/light gray), the structures formed by the expanded repeats sequester cellular RNA-binding proteins, thereby interrupting their normal cellular function. The expanded repeats and sequestered proteins may form foci, which may contribute to toxicity or serve a protective function. The proteins sequestered will depend on the structures formed by the RNAs and protein affinity to the structures. For example, expanded CUG transcripts in DM1 sequester the MBNL family of splicing factors which leads to a loss of MBNL function and alternative splicing abnormalities. In the protein gain-/loss-of-function model (second upper panel, medium gray), the ATG-initiated production of expanded proteins may: 1) disrupt or overwhelm cellular pathways (i.e. proteasomes or autophagy) designed to clear aberrant proteins; directly contribute to cellular apoptosis or damage; aggregate or form inclusions that serve a protective function or exacerbate toxicity; or 2) disrupt the normal function of the protein. For example, in huntington's disease, the mutant huntingtin protein disrupts multiple regulatory pathways, including transcription, ubiquitin proteasomal system, autophagy and synaptic transmission. The discovery of RAN translation has added a third potential pathway for disease (upper and lower panels, dark gray). Up to six additional repeat-containing proteins may be produced from the expanded sense and antisense transcripts. RAN proteins may contribute to pathogenesis in a similar or even amplified manner as the protein gain-/loss-of-function pathway. RAN proteins are found within affected patient tissues, suggesting that they contribute to disease.
RAN TRANSLATION AND THE CGG REPEATS OF FRAGILE X TREMOR ATAXIA SYNDROME (FXTAS)
Fragile X-associated tremor ataxia syndrome (FXTAS) is a late-onset disorder that primarily affects the cerebellum and causes coordination deficits and cognitive decline (28–30). This is caused by a specific range of expanded CGG repeats (55–200 repeats) within the 5′ UTR of the FMR1 gene (28), whereas longer repeats (>200 CGGs) are associated clinically distinct Fragile X syndrome (31). In contrast to the transcriptional silencing and loss of protein expression in Fragile X syndrome (32), FXTAS is associated with increased CGG transcripts that accumulate as RNA foci in human autopsy tissue (33). The associated increased mRNA expression, neurodegeneration and CGG-repeat containing neuronal inclusions (33–35) suggested an RNA gain-of-function mechanism. However, not all aspects of disease pathology, such as inclusion size and associated proteins (34,36), are readily explained by this mechanism. Recent work by Todd et al. (24) has shown that RAN translation may explain some of these incongruous aspects of FXTAS pathology.
Initially, Todd et al. (24), noticed aggregates in a fly model designed to express a non-coding CGGEXP mutation upstream of a GFP reporter. This group performed a series of experiments to understand the molecular basis of these aggregates and to test the hypothesis that FXTAS CGG expansion mutations undergo RAN translation. First, they showed evidence from Drosophila, including mass spectrometry, that a high-molecular weight fusion protein is expressed that contains a homopolymeric glycine expansion. Second, in transfected mammalian cells they showed CGG expansions trigger RAN translation in at least two out of three reading frames producing polyGly-GFP and polyAla–GFP fusion proteins. Third, in the polyAla frame, RAN translation is length dependent with polyAla detected using constructs with 88 but not 30 CGG repeats. In contrast, polyGly expression occurred with 88, 50 and 30 CGGs (Table 1). While the poly-Gly protein was produced from constructs containing only 30 repeats, aggregation was only associated with longer repeats tracts. Fourth, these authors performed a number of experiments that indicate translation initiation can begin upstream of the CGG repeat in the polyGly reading frame. Fifth, these authors show evidence that the polyGly RAN protein accumulates as aggregates in several model systems and in human FXTAS brains using several custom C-terminal antibodies. In summary, Todd et al. (24) provide strong evidence that FXTAS CGG expansions undergo RAN translation, and that at least one of the predicted homopolymeric RAN proteins accumulates in FXTAS brains.
RAN TRANSLATION AND C9ORF72 ALS
A large G4C2 hexanucleotide repeat expansion in intron 1 of the C9orf72 gene was recently identified as the most common cause of ALS/FTD (37,38). Repeat tracts in unaffected controls typically contain fewer than 23 G4C2 repeats, while expansions in ALS/FTD patients range from hundreds to more than 1000 repeats (37–39). Initially, haploinsufficiency and RNA gain-of-function were suggested as possible disease mechanisms because the expansion mutation decreases C9ORF72 transcript levels and G4C2 expansion transcripts form RNA foci (37). Two recent studies suggest RAN translation as a third possible mechanism (25,26).
RAN translation of the C9ORF72 G4C2 hexanucleotide expansion mutation is predicted to result in the expression of dipeptide proteins: GlyPro (GP), GlyArg (GR) and GlyAla (GA). Two groups developed antibodies to these predicted dipeptide motifs and used them to examine patient tissues to look for in vivo evidence of RAN translation (25,26). Mori et al. (26) used antibodies to all three predicted dipeptide products, while Ash et al. (25) focused on the GP frame. Both the groups performed a detailed examination of patient tissues and showed that these antibodies recognize inclusions in C9ORF72 ALS/FTD autopsy tissue. In the Mori et al. study(26), the GA antibody, and to a much lesser extent the GP and GR antibodies, detected inclusions in the cerebellum, hippocampus and other brain regions. These inclusions were similar in shape and abundance to typical ALS/FTD inclusions (40) and colocalized with p62 but not phospho-TDP-43 (26). Inclusions that are p62-positive/phospho-TDP43 negative are classic features of ALS/FTD pathology (40–43). In the Ash et al. study (25), the GP antibodies detected widespread neuronal cytoplasmic and intranuclear inclusions throughout the central nervous system. These inclusions were also morphologically similar to the classic ALS inclusions (25). In both the studies, these antibodies did not detect aggregates in C9ORF72-negative disease controls (25,26). More recently, Almeida et al. (27) showed that neurons derived from C9ORF72-positive iPS cells have GP-positive aggregates, elevated p62 levels and an increased sensitivity to cellular stress induced by autophagy inhibitors. Taken together, data from these studies suggest that dipeptide repeat proteins, expressed by RAN translation, contribute to ALS/FTD.
COMMON THEMES IN RAN TRANSLATION
RAN translation has now been reported in four diseases and has been shown to occur across four different types of repeat motifs: CAG, CUG, CGG and GGGGCC. Among this diversity, several common themes are emerging. First, RAN translation is repeat length-dependent with translation more likely with longer expansion mutations. Second, RAN translation in different reading frames have different length thresholds, such that longer repeats are more likely to result in the accumulation of a cocktail of RAN proteins expressed from different reading frames. It is possible that the simultaneous expression of RAN proteins across long repeats may plays a role in anticipation, the earlier onset and increased disease severity associated with longer repeats. Third, all RAN-competent repeat motifs described to date form unusual secondary structures (44–48). Fourth, all disorders in which RAN translation has been reported to date have neurological features.
NEXT STEPS IN RAN TRANSLATION
What are the critical next steps in RAN translation research? From the analysis so far, it is clear that research needs to move beyond the observational and into the mechanistic. For example, what are the precise RNA structural, sequence and protein factor requirements for RAN translation? Answering these questions will yield important clues to the breadth and scope of RAN translation. Future analysis also needs to be extended beyond immunological approaches to more detailed structural and biochemical analyses of RAN translation proteins in disease. Antibody-based techniques are often subject to artifacts and technical problems, which may be particularly problematic for antibodies directed against repeat motifs themselves. Additionally multiple approaches will be necessary to validate results, especially given the possibility of overlap between RAN translation and other cellular processes. For example, the products of RAN translation and frameshifting may appear to be identical when looking at regions only downstream of the repeat motif. Given the discovery of RAN translation, previous reports of frameshifting for disorders such as SCA3 and HD (49–51) warrant re-examination. A more general question is does RAN translation occur across all microsatellite expansion diseases and if so when, where and why? Additional studies will be required to sort out which RAN proteins are toxic and their potential contribution to disease.
CONCLUSIONS
In summary, RAN translation is a novel mechanism that impacts our basic understanding of gene expression, cell biology and disease. Because more than 30 diseases are caused by microsatellite expansion mutations RAN translation may produce an abundant, yet previously unrecognized set of mutant proteins that contribute to a large category of neurological diseases. Additionally, recent evidence from ribosome profiling studies (24,52–58) suggests that translation is more widespread than previously appreciated. Furthermore, because >50% of the human genome consists of repetitive DNA and repetitive, hairpin-forming sequences undergo RAN translation, the discovery of RAN translation could reveal an abundant, yet previously unrecognized category of repeat-containing proteins.
FUNDING
This work was supported by the National Institutes of Health to (P01NS058901 and R01NS040389), Muscular Dystrophy Association, Keck Foundation, CHDI and Target ALS to L.P.W.R., and the Myotonic Dystrophy Foundation to J.D.C. Funding to pay the Open Access publication charges for this article was provided by the Center for NeuroGenetics, College of Medicine, University of Florida.
ACKNOWLEDGEMENT
The authors wish to thank Dr Tao Zu and Ms. Yuanjing Liu for helpful comments and suggestions.
Conflict of Interest statement. L.P.W.R. is named as an inventor on patents for a gene test for SCA8 and on pending patents on RAN translation.
REFERENCES
- 1.Orr H.T., Zoghbi H.Y. Trinucleotide repeat disorders. Annu. Rev. Neurosci. 2007;30:575–621. doi: 10.1146/annurev.neuro.29.051605.113042. doi:10.1146/annurev.neuro.29.051605.113042. [DOI] [PubMed] [Google Scholar]
- 2.Batra R., Charizanis K., Swanson M.S. Partners in crime: bidirectional transcription in unstable microsatellite disease. Hum. Mol. Genet. 2010;19:R77–R82. doi: 10.1093/hmg/ddq132. doi:10.1093/hmg/ddq132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nelson D.L., Orr H.T., Warren S.T. The unstable repeats—three evolving faces of neurological disease. Neuron. 2013;77:825–843. doi: 10.1016/j.neuron.2013.02.022. doi:10.1016/j.neuron.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Labbadia J., Morimoto R.I. Huntington's disease: underlying molecular mechanisms and emerging concepts. Trends Biochem. Sci. 2013;38:378–385. doi: 10.1016/j.tibs.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ranum L.P., Cooper T.A. RNA-mediated neuromuscular disorders. Annu. Rev. Neurosci. 2006;29:259–277. doi: 10.1146/annurev.neuro.29.051605.113014. doi:10.1146/annurev.neuro.29.051605.113014. [DOI] [PubMed] [Google Scholar]
- 6.Echeverria G.V., Cooper T.A. RNA-binding proteins in microsatellite expansion disorders: mediators of RNA toxicity. Brain Res. 2012;1462:100–111. doi: 10.1016/j.brainres.2012.02.030. doi:10.1016/j.brainres.2012.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Charizanis K., Lee K.Y., Batra R., Goodwin M., Zhang C., Yuan Y., Shiue L., Cline M., Scotti M.M., Xia G., et al. Muscleblind-like 2-mediated alternative splicing in the developing brain and dysregulation in myotonic dystrophy. Neuron. 2012;75:437–450. doi: 10.1016/j.neuron.2012.05.029. doi:10.1016/j.neuron.2012.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kanadia R.N., Johnstone K.A., Mankodi A., Lungu C., Thornton C.A., Esson D., Timmers A.M., Hauswirth W.W., Swanson M.S. A muscleblind knockout model for myotonic dystrophy. Science. 2003;302:1978–1980. doi: 10.1126/science.1088583. doi:10.1126/science.1088583. [DOI] [PubMed] [Google Scholar]
- 9.Mankodi A., Urbinati C.R., Yuan Q.P., Moxley R.T., Sansone V., Krym M., Henderson D., Schalling M., Swanson M.S., Thornton C.A. Muscleblind localizes to nuclear foci of aberrant RNA in myotonic dystrophy types 1 and 2. Hum. Mol. Genet. 2001;10:2165–2170. doi: 10.1093/hmg/10.19.2165. doi:10.1093/hmg/10.19.2165. [DOI] [PubMed] [Google Scholar]
- 10.Liquori C.L., Ricker K., Moseley M.L., Jacobsen J.F., Kress W., Naylor S.L., Day J.W., Ranum L.P. Myotonic dystrophy type 2 caused by a CCTG expansion in intron 1 of ZNF9. Science. 2001;293:864–867. doi: 10.1126/science.1062125. doi:10.1126/science.1062125. [DOI] [PubMed] [Google Scholar]
- 11.Zu T., Gibbens B., Doty N.S., Gomes-Pereira M., Huguet A., Stone M.D., Margolis J., Peterson M., Markowski T.W., Ingram M.A., et al. Non-ATG-initiated translation directed by microsatellite expansions. Proc. Natl Acad. Sci. USA. 2011;108:260–265. doi: 10.1073/pnas.1013343108. doi:10.1073/pnas.1013343108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Koob M.D., Moseley M.L., Schut L.J., Benzow K.A., Bird T.D., Day J.W., Ranum L.P. An untranslated CTG expansion causes a novel form of spinocerebellar ataxia (SCA8) Nat. Genet. 1999;21:379–384. doi: 10.1038/7710. doi:10.1038/7710. [DOI] [PubMed] [Google Scholar]
- 13.Daughters R.S., Tuttle D.L., Gao W., Ikeda Y., Moseley M.L., Ebner T.J., Swanson M.S., Ranum L.P. RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genet. 2009;5:e1000600. doi: 10.1371/journal.pgen.1000600. doi:10.1371/journal.pgen.1000600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Moseley M.L., Zu T., Ikeda Y., Gao W., Mosemiller A.K., Daughters R.S., Chen G., Weatherspoon M.R., Clark H.B., Ebner T.J., et al. Bidirectional expression of CUG and CAG expansion transcripts and intranuclear polyglutamine inclusions in spinocerebellar ataxia type 8. Nat. Genet. 2006;38:758–769. doi: 10.1038/ng1827. doi:10.1038/ng1827. [DOI] [PubMed] [Google Scholar]
- 15.Brook J.D., McCurrach M.E., Harley H.G., Buckler A.J., Church D., Aburatani H., Hunter K., Stanton V.P., Thirion J.P., Hudson T., et al. Molecular basis of myotonic dystrophy: expansion of a trinucleotide (CTG) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell. 1992;68:799–808. doi: 10.1016/0092-8674(92)90154-5. doi:10.1016/0092-8674(92)90154-5. [DOI] [PubMed] [Google Scholar]
- 16.Fu Y.H., Pizzuti A., Fenwick R.G., Jr, King J., Rajnarayan S., Dunne P.W., Dubel J., Nasser G.A., Ashizawa T., de Jong P., et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science. 1992;255:1256–1258. doi: 10.1126/science.1546326. doi:10.1126/science.1546326. [DOI] [PubMed] [Google Scholar]
- 17.Mahadevan M., Tsilfidis C., Sabourin L., Shutler G., Amemiya C., Jansen G., Neville C., Narang M., Barcelo J., O'Hoy K., et al. Myotonic dystrophy mutation: an unstable CTG repeat in the 3′ untranslated region of the gene. Science. 1992;255:1253–1255. doi: 10.1126/science.1546325. doi:10.1126/science.1546325. [DOI] [PubMed] [Google Scholar]
- 18.Cho D.H., Thienes C.P., Mahoney S.E., Analau E., Filippova G.N., Tapscott S.J. Antisense transcription and heterochromatin at the DM1 CTG repeats are constrained by CTCF. Mol. Cell. 2005;20:483–489. doi: 10.1016/j.molcel.2005.09.002. doi:10.1016/j.molcel.2005.09.002. [DOI] [PubMed] [Google Scholar]
- 19.Lescure A., Lutz Y., Eberhard D., Jacq X., Krol A., Grummt I., Davidson I., Chambon P., Tora L. The N-terminal domain of the human TATA-binding protein plays a role in transcription from TATA-containing RNA polymerase II and III promoters. EMBO J. 1994;13:1166–1175. doi: 10.1002/j.1460-2075.1994.tb06366.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trottier Y., Devys D., Imbert G., Saudou F., An I., Lutz Y., Weber C., Agid Y., Hirsch E.C., Mandel J.L. Cellular localization of the Huntington's disease protein and discrimination of the normal and mutated form. Nat. Genet. 1995;10:104–110. doi: 10.1038/ng0595-104. doi:10.1038/ng0595-104. [DOI] [PubMed] [Google Scholar]
- 21.Seznec H., Agbulut O., Sergeant N., Savouret C., Ghestem A., Tabti N., Willer J.C., Ourth L., Duros C., Brisson E., et al. Mice transgenic for the human myotonic dystrophy region with expanded CTG repeats display muscular and brain abnormalities. Hum. Mol. Genet. 2001;10:2717–2726. doi: 10.1093/hmg/10.23.2717. doi:10.1093/hmg/10.23.2717. [DOI] [PubMed] [Google Scholar]
- 22.Gomes-Pereira M., Foiry L., Nicole A., Huguet A., Junien C., Munnich A., Gourdon G. CTG Trinucleotide repeat ‘big jumps’: large expansions, small mice. PLoS Genet. 2007;3:e52. doi: 10.1371/journal.pgen.0030052. doi:10.1371/journal.pgen.0030052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.U M., Miyashita T., Ohtsuka Y., Okamura-Oho Y., Shikama Y., Yamada M. Extended polyglutamine selectively interacts with caspase-8 and -10 in nuclear aggregates. Cell Death Differ. 2001;8:377–386. doi: 10.1038/sj.cdd.4400819. doi:10.1038/sj.cdd.4400819. [DOI] [PubMed] [Google Scholar]
- 24.Todd P.K., Oh S.Y., Krans A., He F., Sellier C., Frazer M., Renoux A.J., Chen K.C., Scaglione K.M., Basrur V., et al. CGG repeat-associated translation mediates neurodegeneration in fragile X tremor ataxia syndrome. Neuron. 2013;78:440–455. doi: 10.1016/j.neuron.2013.03.026. doi:10.1016/j.neuron.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ash P.E., Bieniek K.F., Gendron T.F., Caulfield T., Lin W.L., Dejesus-Hernandez M., van Blitterswijk M.M., Jansen-West K., Paul J.W., Rademakers R., et al. Unconventional translation of C9ORF72 GGGGCC expansion generates insoluble polypeptides specific to c9FTD/ALS. Neuron. 2013;77:639–646. doi: 10.1016/j.neuron.2013.02.004. doi:10.1016/j.neuron.2013.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mori K., Weng S.M., Arzberger T., May S., Rentzsch K., Kremmer E., Schmid B., Kretzschmar H.A., Cruts M., Van Broeckhoven C., et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339:1335–1338. doi: 10.1126/science.1232927. [DOI] [PubMed] [Google Scholar]
- 27.Almeida S., Gascon E., Tran H., Chou H.J., Gendron T.F., Degroot S., Tapper A.R., Sellier C., Charlet-Berguerand N., Karydas A., et al. Modeling key pathological features of frontotemporal dementia with C9ORF72 repeat expansion in iPSC-derived human neurons. Acta Neuropathol. 2013 doi: 10.1007/s00401-013-1149-y. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hagerman R.J., Leehey M., Heinrichs W., Tassone F., Wilson R., Hills J., Grigsby J., Gage B., Hagerman P.J. Intention tremor, parkinsonism, and generalized brain atrophy in male carriers of fragile X. Neurology. 2001;57:127–130. doi: 10.1212/wnl.57.1.127. doi:10.1212/WNL.57.1.127. [DOI] [PubMed] [Google Scholar]
- 29.Hagerman P. Fragile X-associated tremor/ataxia syndrome (FXTAS): pathology and mechanisms. Acta Neuropathol. 2013;126:1–19. doi: 10.1007/s00401-013-1138-1. doi:10.1007/s00401-013-1138-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jacquemont S., Hagerman R.J., Leehey M., Grigsby J., Zhang L., Brunberg J.A., Greco C., Des Portes V., Jardini T., Levine R., et al. Fragile X premutation tremor/ataxia syndrome: molecular, clinical, and neuroimaging correlates. Am. J. Hum. Genet. 2003;72:869–878. doi: 10.1086/374321. doi:10.1086/374321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verkerk A.J., Pieretti M., Sutcliffe J.S., Fu Y.H., Kuhl D.P., Pizzuti A., Reiner O., Richards S., Victoria M.F., Zhang F.P., et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile×syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. doi:10.1016/0092-8674(91)90397-H. [DOI] [PubMed] [Google Scholar]
- 32.Pieretti M., Zhang F.P., Fu Y.H., Warren S.T., Oostra B.A., Caskey C.T., Nelson D.L. Absence of expression of the FMR-1 gene in fragile X syndrome. Cell. 1991;66:817–822. doi: 10.1016/0092-8674(91)90125-i. doi:10.1016/0092-8674(91)90125-I. [DOI] [PubMed] [Google Scholar]
- 33.Tassone F., Iwahashi C., Hagerman P.J. FMR1 RNA within the intranuclear inclusions of fragile X-associated tremor/ataxia syndrome (FXTAS) RNA Biol. 2004;1:103–105. doi: 10.4161/rna.1.2.1035. doi:10.4161/rna.1.2.1035. [DOI] [PubMed] [Google Scholar]
- 34.Greco C.M., Berman R.F., Martin R.M., Tassone F., Schwartz P.H., Chang A., Trapp B.D., Iwahashi C., Brunberg J., Grigsby J., et al. Neuropathology of fragile X-associated tremor/ataxia syndrome (FXTAS) Brain. 2006;129:243–255. doi: 10.1093/brain/awh683. doi:10.1093/brain/awh683. [DOI] [PubMed] [Google Scholar]
- 35.Jin P., Zarnescu D.C., Zhang F., Pearson C.E., Lucchesi J.C., Moses K., Warren S.T. RNA-mediated neurodegeneration caused by the fragile X premutation rCGG repeats in Drosophila. Neuron. 2003;39:739–747. doi: 10.1016/s0896-6273(03)00533-6. doi:10.1016/S0896-6273(03)00533-6. [DOI] [PubMed] [Google Scholar]
- 36.Iwahashi C.K., Yasui D.H., An H.J., Greco C.M., Tassone F., Nannen K., Babineau B., Lebrilla C.B., Hagerman R.J., Hagerman P.J. Protein composition of the intranuclear inclusions of FXTAS. Brain. 2006;129:256–271. doi: 10.1093/brain/awh650. doi:10.1093/brain/awh650. [DOI] [PubMed] [Google Scholar]
- 37.DeJesus-Hernandez M., Mackenzie I.R., Boeve B.F., Boxer A.L., Baker M., Rutherford N.J., Nicholson A.M., Finch N.A., Flynn H., Adamson J., et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256. doi: 10.1016/j.neuron.2011.09.011. doi:10.1016/j.neuron.2011.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Renton A.E., Majounie E., Waite A., Simon-Sanchez J., Rollinson S., Gibbs J.R., Schymick J.C., Laaksovirta H., van Swieten J.C., Myllykangas L., et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron. 2011;72:257–268. doi: 10.1016/j.neuron.2011.09.010. doi:10.1016/j.neuron.2011.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Majounie E., Renton A.E., Mok K., Dopper E.G., Waite A., Rollinson S., Chio A., Restagno G., Nicolaou N., Simon-Sanchez J., et al. Frequency of the C9orf72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol. 2012;11:323–330. doi: 10.1016/S1474-4422(12)70043-1. doi:10.1016/S1474-4422(12)70043-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Al-Sarraj S., King A., Troakes C., Smith B., Maekawa S., Bodi I., Rogelj B., Al-Chalabi A., Hortobágyi T., Shaw C.E. P62 positive, TDP-43 negative, neuronal cytoplasmic and intranuclear inclusions in the cerebellum and hippocampus define the pathology of C9orf72-linked FTLD and MND/ALS. Acta Neuropathol. 2011;122:691–702. doi: 10.1007/s00401-011-0911-2. doi:10.1007/s00401-011-0911-2. [DOI] [PubMed] [Google Scholar]
- 41.Nakano T., Nakaso K., Nakashima K., Ohama E. Expression of ubiquitin-binding protein p62 in ubiquitin-immunoreactive intraneuronal inclusions in amyotrophic lateral sclerosis with dementia: analysis of five autopsy cases with broad clinicopathological spectrum. Acta Neuropathol. 2004;107:359–364. doi: 10.1007/s00401-004-0821-7. doi:10.1007/s00401-004-0821-7. [DOI] [PubMed] [Google Scholar]
- 42.Neumann M., Sampathu D.M., Kwong L.K., Truax A.C., Micsenyi M.C., Chou T.T., Bruce J., Schuck T., Grossman M., Clark C.M., et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. doi:10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 43.Pikkarainen M., Hartikainen P., Alafuzoff I. Ubiquitinated p62-positive, TDP-43-negative inclusions in cerebellum in frontotemporal lobar degeneration with TAR DNA binding protein 43. Neuropathology. 2010;30:197–199. doi: 10.1111/j.1440-1789.2009.01043.x. doi:10.1111/j.1440-1789.2009.01043.x. [DOI] [PubMed] [Google Scholar]
- 44.Napierała M., Krzyzosiak W.J. CUG Repeats present in myotonin kinase RNA form metastable ‘slippery’ hairpins. J. Biol. Chem. 1997;272:31079–31085. doi: 10.1074/jbc.272.49.31079. doi:10.1074/jbc.272.49.31079. [DOI] [PubMed] [Google Scholar]
- 45.Shin J., Charizanis K., Swanson M.S. Pathogenic RNAs in microsatellite expansion disease. Neurosci. Lett. 2009;466:99–102. doi: 10.1016/j.neulet.2009.07.079. doi:10.1016/j.neulet.2009.07.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fratta P., Mizielinska S., Nicoll A.J., Zloh M., Fisher E.M., Parkinson G., Isaacs A.M. C9orf72 hexanucleotide repeat associated with amyotrophic lateral sclerosis and frontotemporal dementia forms RNA G-quadruplexes. Sci. Rep. 2012;2:1016. doi: 10.1038/srep01016. doi:10.1038/srep01016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Broda M., Kierzek E., Gdaniec Z., Kulinski T., Kierzek R. Thermodynamic stability of RNA structures formed by CNG trinucleotide repeats. implication for prediction of RNA structure. Biochemistry. 2005;44:10873–10882. doi: 10.1021/bi0502339. doi:10.1021/bi0502339. [DOI] [PubMed] [Google Scholar]
- 48.Reddy K., Zamiri B., Stanley S.Y., Macgregor R.B., Pearson C.E. The disease-associated r(GGGGCC)n repeat from the C9orf72 gene forms tract length-dependent uni- and multimolecular RNA G-quadruplex structures. J. Biol. Chem. 2013;288:9860–9866. doi: 10.1074/jbc.C113.452532. doi:10.1074/jbc.C113.452532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Toulouse A., Au-Yeung F., Gaspar C., Roussel J., Dion P., Rouleau G.A. Ribosomal frameshifting on MJD-1 transcripts with long CAG tracts. Hum. Mol. Genet. 2005;14:2649–2660. doi: 10.1093/hmg/ddi299. doi:10.1093/hmg/ddi299. [DOI] [PubMed] [Google Scholar]
- 50.Davies J.E., Rubinsztein D.C. Polyalanine and polyserine frameshift products in Huntington's disease. J. Med. Genet. 2006;43:893–896. doi: 10.1136/jmg.2006.044222. doi:10.1136/jmg.2006.044222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Stochmanski S.J., Therrien M., Laganière J., Rochefort D., Laurent S., Karemera L., Gaudet R., Vyboh K., Van Meyel D.J., Di Cristo G., et al. Expanded ATXN3 frameshifting events are toxic in Drosophila and mammalian neuron models. Hum. Mol. Genet. 2012;21:2211–2218. doi: 10.1093/hmg/dds036. doi:10.1093/hmg/dds036. [DOI] [PubMed] [Google Scholar]
- 52.Ingolia N.T., Ghaemmaghami S., Newman J.R., Weissman J.S. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324:218–223. doi: 10.1126/science.1168978. doi:10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ingolia N.T. Genome-wide translational profiling by ribosome footprinting. Methods Enzymol. 2010;470:119–142. doi: 10.1016/S0076-6879(10)70006-9. [DOI] [PubMed] [Google Scholar]
- 54.Ingolia N.T., Lareau L.F., Weissman J.S. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 2011;147:789–802. doi: 10.1016/j.cell.2011.10.002. doi:10.1016/j.cell.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ingolia N.T., Brar G.A., Rouskin S., McGeachy A.M., Weissman J.S. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat. Protoc. 2012;7:1534–1550. doi: 10.1038/nprot.2012.086. doi:10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Stern-Ginossar N., Weisburd B., Michalski A., Le V.T., Hein M.Y., Huang S.X., Ma M., Shen B., Qian S.B., Hengel H., et al. Decoding human cytomegalovirus. Science. 2012;338:1088–1093. doi: 10.1126/science.1227919. doi:10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Chew G.L., Pauli A., Rinn J.L., Regev A., Schier A.F., Valen E. Ribosome profiling reveals resemblance between long non-coding RNAs and 5′ leaders of coding RNAs. Development. 2013;140:2828–2834. doi: 10.1242/dev.098343. doi:10.1242/dev.098343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lee S., Liu B., Huang S.X., Shen B., Qian S.B. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc. Natl Acad. Sci. USA. 2012;109:E2424–E2432. doi: 10.1073/pnas.1207846109. doi:10.1073/pnas.1207846109. [DOI] [PMC free article] [PubMed] [Google Scholar]