

Abstract
Mitochondrial uncoupling protein 1 (UCP1) is responsible for nonshivering thermogenesis in brown adipose tissue (BAT). Upon activation by long-chain fatty acids (LCFAs), UCP1 increases the conductance of the inner mitochondrial membrane (IMM) to make BAT mitochondria generate heat rather than ATP. Despite being a member of the family of mitochondrial anion carriers (SLC25), UCP1 is believed to transport H+ by an unusual mechanism that has long remained unresolved. Here, we achieved direct patch-clamp measurements of UCP1 currents from the IMM of BAT mitochondria. We show that UCP1 is an LCFA anion/H+ symporter. However, the LCFA anions cannot dissociate from UCP1 due to hydrophobic interactions established by their hydrophobic tails, and UCP1 effectively operates as an H+ carrier activated by LCFA. A similar LCFA-dependent mechanism of transmembrane H+ transport may be employed by other SLC25 members and be responsible for mitochondrial uncoupling and regulation of metabolic efficiency in various tissues.
INTRODUCTION
Brown adipose tissue (BAT) specializes in burning fat and is responsible for adaptive, nonshivering thermogenesis in mammals (Cannon and Nedergaard, 2004; Enerbäck et al., 1997). The thermogenic ability of BAT is conferred by uncoupling protein 1 (UCP1), a BAT-specific transport protein of the inner mitochondrial membrane (IMM) (Aquila et al., 1985; Bouillaud et al., 1986; Heaton et al., 1978; Ridley et al., 1986). UCP1 increases IMM conductance for H+ to dissipate the mitochondrial H+ gradient and convert the energy of substrate oxidation into heat (Nicholls and Locke, 1984). UCP1 is activated by long-chain fatty acids (LCFAs) that are produced within brown adipocytes by the lipolysis of cytoplasmic lipid droplets upon adrenergic stimulation of BAT (Cannon and Nedergaard, 2004). Despite the importance of UCP1 for the maintenance of core body temperature and the control of energy intake and expenditure balance, the LCFA-dependent mechanism of UCP1 operation remains elusive (Divakaruni and Brand, 2011; Klingenberg, 2010).
Several mechanisms of LCFA-dependent UCP1 operation have been proposed (Figure S1A available online): (1) that of an H+ uniporter (channel) activated by the allosteric binding of LCFAs (Cannon and Nedergaard, 2004; Rial and González-Barroso, 2001); (2) that of an OH– uniporter (channel) activated by the allosteric binding of LCFAs (Nicholls, 2006); (3) the “H+ buffering” model of UCP1 as an H+ channel in which LCFAs bind to the pore and provide their carboxylic groups to complete the H+ translocation pathway along with the titratable amino acid residues of UCP1 (Klingenberg and Huang, 1999); and (4) the “fatty-acid cycling” model in which UCP1 is an LCFA anion carrier that transports H+ indirectly: UCP1 carries LCFA anions outside the mitochondria where they bind H+ and, in protonated form, “flip-flop” back across the IMM to release the H+ into the mitochondrial matrix (Garlid et al., 1998). Furthermore, UCP1 is inhibited by cytosolic purine nucleotides, and the mechanism by which LCFAs overcome this inhibition also remains controversial (Klingenberg, 2010; Nicholls, 2006; Nicholls and Locke, 1984; Shabalina et al., 2004).
The main difficulty in determining the mechanism of UCP1 operation has been the lack of a direct method to study UCP1 transport activity in its native membrane environment. Here, we use the patch-clamp technique to directly measure UCP1 currents in the native IMM of BAT and provide a detailed analysis of the LCFA-dependent mechanism of UCP1 operation.
RESULTS
Identification and Biophysical Properties of UCP1 Current
To identify UCP1 currents, we applied the whole-cell patch-clamp technique to mitoplasts (Kirichok et al., 2004), vesicles of whole native IMM isolated from mouse BAT (Figure 1A). In the whole-mitoplast mode, the voltage step from 0 to –160 mV, followed by a voltage ramp to +80 mV, elicited a large-amplitude current that was strongly inhibited by the classic UCP1 inhibitor GDP (Figure 1B) and other purine nucleotides such as ATP, GTP, and ADP (data not shown). LCFAs, classic activators of UCP1, strongly potentiated this current (Figures 1C and S1B). Fatty-acid-free bovine serum albumin (BSA) and alpha-cyclodextrin (αCD), which bind LCFAs and remove them from the membrane, strongly inhibited the observed current (Figures 1C and S1C), suggesting that endogenous membrane-associated LCFAs are essential for the current. The original current observed upon breaking-in into mitoplasts and the additional current induced by LCFAs were not present in UCP1–/– mitoplasts (n = 15; Figures 1D and S1H). Therefore, we concluded that the LCFA-dependent, purine nucleotide-sensitive current was mediated by UCP1. It is important to note that the density of the UCP1 current is one of the highest among H+ currents across biological membranes.
Figure 1. Electrophysiological Properties of UCP1 Current.
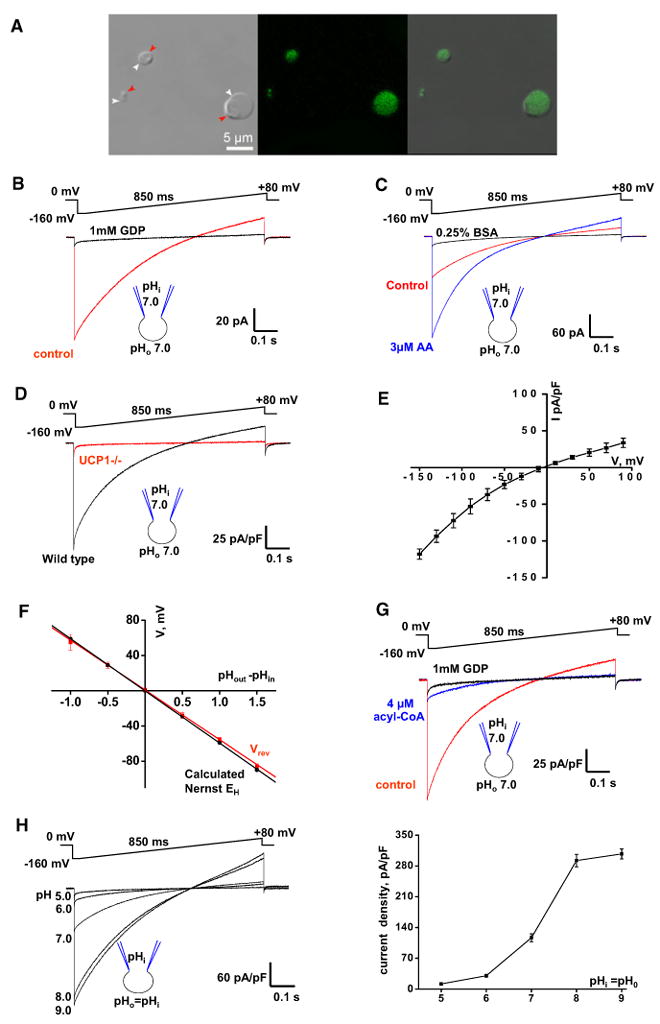
(A) Transmitted, fluorescent, and superimposed images (left to right) of BAT mitoplasts isolated from mice expressing CFP in the mitochondrial matrix (false green color). White arrows, IMM; red arrows, remnants of outer membrane.
(B) Whole-mitoplast putative UCP1 current before (control, red) and after (black) the addition of 1 mM GDP to the bath solution. The voltage protocol is indicated at the top. The pipette-mitoplast diagram indicates the recording conditions. The mitoplast membrane capacitance was 1.1 pF.
(C) Putative UCP1 current (control, red) is potentiated by 3 μM arachidonic acid (AA, blue) and inhibited by 0.25% BSA (black). The mitoplast membrane capacitance was 1.0 pF.
(D) Representative whole-mitoplast currents recorded from wild-type (black) and UCP1–/– (red) mitoplasts.
(E) Current-voltage dependence of IUCP1. Amplitudes were measured at the beginning of the voltage steps shown in Figure S1D; n = 5.
(F) IUCP1 reversal potentials (Vrev) compared to H+ equilibrium potentials (EH) predicted by the Nernst equation. The red line indicates the linear fitting of IUCP1 reversal potentials versus ΔpH; n = 3–10. The black line indicates EH calculated by the Nernst equation at 24°C.
(G) Whole-mitoplast IUCP1 before (control, red), after the addition of 4 μM oleoyl-CoA to the bath solution (blue), and after the subsequent application of 1 mM GDP (black).
(H) Left panel: IUCP1 at different symmetrical pH values. Representative traces recorded from different mitoplasts are shown. Right panel: Mean IUCP1 densities in different symmetrical pH values; n = 4–12. Amplitudes were measured upon stepping from 0 to [C0]160 mV as in the left panel.
Error bars represent standard error of the mean (SEM). See also Figure S1.
We further characterized the biophysical properties of the UCP1 current (IUCP1). At lower membrane potentials (from –90 to +90 mV), when the amplitude of IUCP1 was small, it was virtually time independent (Figure S1D). However, as IUCP1 increased at high negative potentials (beyond –90 mV), its amplitude declined during the voltage step (Figure S1D), likely due to the saturation of the proton buffer inside the mitoplast (see Experimental Procedures). When IUCP1 was recorded from small membrane patches excised from mitoplasts (inside-out mode), no such time dependence was observed, as in this mode, saturation of the buffer on the matrix side of the IMM is less likely (Figure S1E). Although the inside-out IUCP1 was robust, we did not resolve any single-channel openings, suggesting that the amplitude of the UCP1 unitary current is very small.
IUCP1 reversal potentials closely corresponded to the calculated H+ Nernst potentials (Figures 1F, S1F, and S1G), confirming that IUCP1 carries H+. The UCP1 current-voltage (I/V) relationship had mild inward rectification (Figures 1E and S1D), making UCP1 uncoupling more effective at physiologically relevant negative potentials.
Although in the past UCP1 was often postulated to operate as an H+ channel, IUCP1 was the largest at pH 9.0 and reduced dramatically when pH was symmetrically (equally in bath and pipette solutions) decreased to 5.0 (Figure 1H). Based on this pH dependence, we conclude that UCP1 is unlikely to operate as a simple H+ channel, for which we would expect higher current amplitudes at lower pH values. In the experiments presented below, we seek to clarify the mechanism of LCFA-dependent UCP1 operation.
UCP1 Has No Constitutive Activity, and LCFAs Activate UCP1
Besides LCFAs, other UCP1 activators have been proposed, most notably long-chain acyl-CoA (Cannon et al., 1977), coenzyme Q (ubiquinone, CoQ) (Echtay et al., 2000), and 4-hydroxy 2-nonenal (Echtay et al., 2003), but their ability to activate UCP1 remains controversial (Jaburek and Garlid, 2003; Nicholls and Locke, 1984; Shabalina et al., 2006). In our experiments, 4 μM oleoyl-CoA strongly inhibited IUCP1 by 92% ± 1% at –160 mV (n = 4; Figure 1G). Coenzyme Q6 and hydroxy 2-nonenal had no effect on IUCP1 at 5 μM and 3 μM, respectively (data not shown). Thus, our data support the general consensus in the field that LCFAs are the most likely physiological activators of UCP1.
Upon breaking-in, we observed robust IUCP1 in the absence of LCFAs in the recording solutions (Figure 1C), presumably due to the presence of “endogenous” LCFAs in the IMM. Although 0.25% BSA added to the bath strongly inhibited this IUCP1, the inhibition was not complete, as 1 mM GDP caused a further IUCP1 reduction (Figure S2A). However, 0.5% BSA in both the bath and the pipette completely inhibited IUCP1, and 1 mM GDP did not cause any further inhibition (Figure 2A). IUCP1 was also completely inhibited when both the bath and the pipette solution contained 15 mM αCD (Figure 2B). These experiments indicate that UCP1 has no constitutive activity.
Figure 2. UCP1 Is Activated by Endogenous Membrane LCFAs.
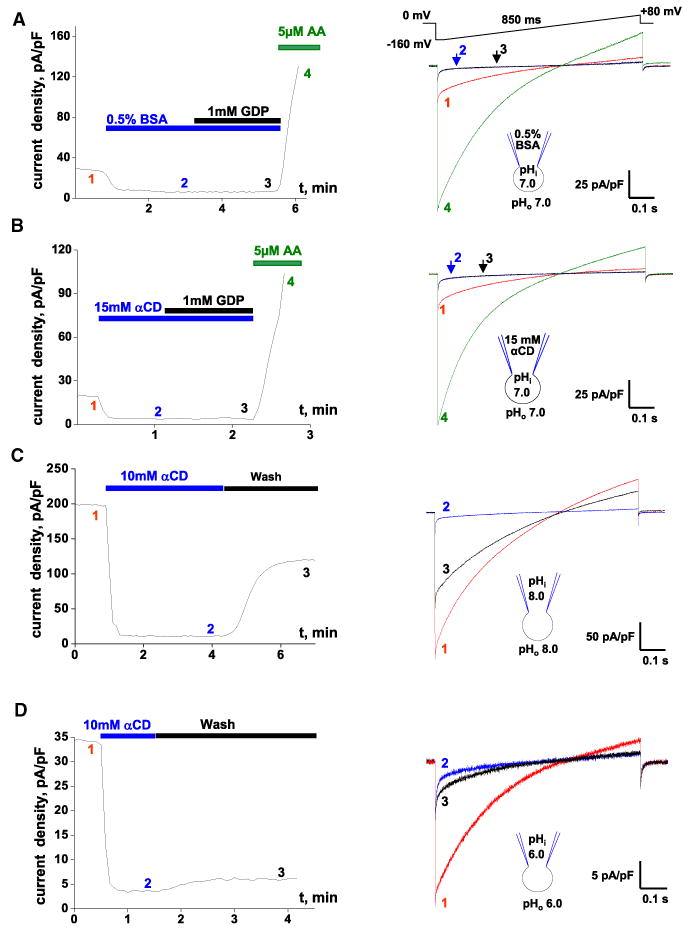
(A) Left panel: Representative time course of IUCP1 amplitude in control (1), upon application of 0.5% BSA (2), and with 0.5% BSA and 1 mM GDP (3). AA (5 μM) was applied at the end to verify that IUCP1 can still be activated (4). Pipette solution contained 0.5% BSA. Amplitudes were measured upon stepping from 0 to –160 mV (see right panel). Right panel: IUCP1 traces recorded at times 1, 2, 3, and 4 as indicated in the left panel.
(B) The same experiment as in (A) but performed with 15 mM αCD in the bath and pipette solutions.
(C) Left panel: Representative time course of IUCP1 amplitude in control (1) and upon the application (2) and subsequent washout (3) of 10 mM αCD at pH 8.0. Right panel: IUCP1 traces recorded at times 1, 2, and 3 as indicated in the left panel.
(D) The same experiment as in (C) but performed at symmetrical pH 6.0.
See also Figure S2.
LCFAs were sufficient to activate UCP1. A mixture of LCFAs with their acceptor methyl-β-cyclodextrin (MβCD) can be used for fast delivery of LCFAs into the membrane (Brunaldi et al., 2010). MβCD simultaneously extracts endogenous fatty activators of UCP1 originally present in the IMM. After deactivation of UCP1 with MβCD, robust IUCP1 was activated by the application of 2 mM oleic acid (OA) mixed with 10 mM MβCD (Figure S2B). Together, these experiments demonstrate that UCP1 has no constitutive activity, and that LCFAs are the likely physiological activators.
LCFAs Are Generated within the IMM
Following the deactivation of IUCP1 by the extraction of endogenous LCFAs using fatty-acid acceptors, a significant fraction of the IUCP1, recovered as bath αCD (Figure 2C) or BSA (Figure S2C), was removed. This IUCP1 recovery was strongly pH dependent, with only a small fraction of the original current recovered at pH 6.0 (Figure 2D) compared to almost one-half at pH 8.0 (Figures 2C and S2D). These results suggest that an UCP1 activator, most likely LCFAs, is produced within the IMM. The smaller IUCP1 recovery at acidic pH correlates with decreased activity of mitochondrial PLA2 at a lower pH (Kinsey et al., 2007; Zurini et al., 1981). Because Ca2+ in our solutions was in the low nM range, the putative PLA2 involved in UCP1 activation must be Ca2+ independent. At low pH, the low PLA2 activity (Figure S2D) may contribute to low IUCP1 amplitudes (Figure 1H).
To further probe the involvement of PLA2 in UCP1 activation, we tested the effect of lysophospholipids (PLA2 produces LCFAs and lysophospholipids) on IUCP1. Given that phosphatidylcholine and phosphatidylethanolamine are the two most abundant phospholipids of the IMM (Osman et al., 2011), lysophosphatidylcholine (lysoPC) and lysophosphatidylethanolamine (lysoPE) should suppress IUCP1 activated as a result of PLA2 activity. Indeed, 4 μM oleoyl-lysoPC or oleoyl-lysoPE inhibited the original IUCP1 by 66% ± 1% (n = 3; Figure 3A) and 66% ± 5% (n = 3; data not shown), respectively. Neither oleoyl-lysoPC nor oleoyl-lysoPE inhibited UCP1 directly: after the removal of endogenous LCFAs with αCD, IUCP1 reactivated with exogenous 1 μM OA was insensitive to 4 μM oleoyl-lysoPC (n = 3; Figure 3B) or oleoyl-lysoPE (n = 3; data not shown). These experiments support the conclusion that endogenous membrane LCFAs generated by PLA2 activate UCP1.
Figure 3. Regulation of IUCP1 by Lysophospolipids.
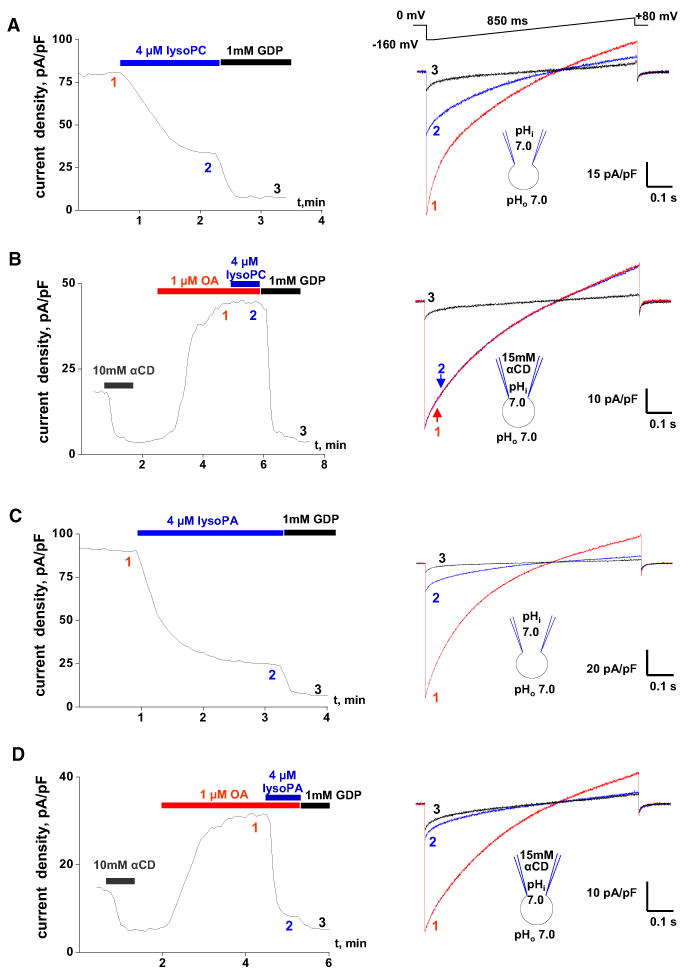
(A) Left panel: Representative time course of the IUCP1 amplitude in control (1), upon the application of 4 mM oleoyl-lysoPC (2), and the subsequent application of 1 mM GDP (3). IUCP1 amplitudes were measured upon stepping from 0 to [C0]160 mV (see right panel). Right panel: IUCP1 traces recorded at times 1, 2, and 3 as indicated in the left panel.
(B) Left panel: Representative time course of the IUCP1 amplitude after the extraction of endogenous LCFAs with 10 mM aCD, reactivation of IUCP1 with 1 mM OA (1), the subsequent addition of 4 mM oleoyl-lysoPC (2), and the application of 1 mM GDP (3). The pipette solution contained 15 mM aCD to extract endogenous membrane LCFAs. Right panel: IUCP1 traces recorded at times 1, 2, and 3 as indicated in the left panel.
(C) The same experiment as in (A) but performed with 4 mM oleoyl-lysoPA instead of oleoyl-lysoPC.
(D) The same experiment as in (B) but performed with 4 mM oleoyl-lysoPA instead of oleoyl-lysoPC.
See also Figure S3.
In contrast to lysoPC and lysoPE, which are neutral overall, the negatively charged lysophosphatidic acid (lysoPA) inhibited both the original IUCP1 (76% ± 2% inhibition, n = 4; Figure 3C) and IUCP1 activated by OA (80% ± 1% inhibition, n = 4; Figure 3D). Thus, lysoPA inhibits UCP1 directly, likely by competing with structurally related LCFAs for UCP1 binding.
The lipid signaling that controls UCP1 activity in intact cells is likely more complex than during the whole-mitoplast patch-clamp. In particular, although PLA2 primarily generates unsaturated LCFAs, mitochondrial phospholipase A1 activity or the hydrolysis of cytoplasmic lipid droplets can generate saturated LCFAs in intact cells. Saturated LCFAs interact with lipid membranes much more strongly than do unsaturated LCFAs (Anel et al., 1993) and can be “stuck” within the IMM bilayer unless extracted with BSA or cyclodextrins. These unsaturated LCFAs can explain the component of the original IUCP1 that never recovers after the removal of BSA and cyclodextrins (Figures 2C and 2D). In contrast, the amplitude of the recovered IUCP1 should be determined only by the equilibrium between the PLA2-dependent production of unsaturated LCFAs and the washout of these LCFAs from membrane. As the recovered IUCP1 completely depends on the PLA2 activity, 4 μM lysoPC inhibited the recovered IUCP1 more strongly than the original IUCP1, by 88% ± 2% (n = 3; Figure S3A). The almost complete inhibition of the recovered IUCP1 by bath lysophospholipids also suggests that the PLA2 must be located on the cytoplasmic face of the IMM (lysophospholipids cannot flip-flop across the membrane; Bhamidipati and Hamilton, 1995).
We further attempted pharmacological identification of the Ca2+-independent mitochondrial PLA2 involved in UCP1 activation. This PLA2 does not belong to the iPLA2 subfamily, as a selective iPLA2 inhibitor bromoenol lactone (BEL) added into the bath and/or pipette solution at 50 μM did not affect IUCP1 (n = 4; data not shown). Two newly identified members of the PLA2 family, cPLA2β and cPLA2γ, can also associate with mitochondrial membranes in a Ca2+-independent manner (Murakami et al., 2011). Pyrrophenone and a related compound RSC-3388 inhibit cPLAβ and cPLA2γ at micromolar concentrations (Ghomashchi et al., 2010). Methyl arachidonyl fluorophosphate (MAFP), a classic inhibitor of Ca2+-dependent cPLA2α, also inhibits cPLA2γ (Stewart et al., 2002). Although 50 μM pyrrophenone inhibited the original IUCP1 by 65% ± 6% (n = 5; Figure S3B), it acted upon UCP1 directly. Indeed, IUCP1 reactivated with 1 μM of exogenous OA was inhibited 77% ± 3% by 50 μM pyrrophenone (n = 3; Figure S3C). Similarly, 50 μM RSC-3388 inhibited the original IUCP1 by 68% ± 4% (n = 3), and IUCP1 activated by 1 μM OA by 73% ± 3%, (n = 3; data not shown). Finally, 50 μM MAFP inhibited the original IUCP1 by 47% ± 3% (n = 3) and the IUCP1 activated by 1 μM OA by 58% ± 6% (n = 3; data not shown). Thus, although pyrrophenone, RSC-3388, and MAFP inhibit IUCP1, they act upon UCP1 directly. The identity of the PLA2 isoform(s) involved in UCP1 activation remains to be established.
LCFA production within the IMM may serve as a physiological mechanism of UCP1 regulation, along with the generation of LCFAs by the lipolysis of cytoplasmic lipid droplets.
Fatty-Acid Analogs Are UCP1 Transport Substrates
Because LCFAs may serve as H+ carriers in the UCP1 uncoupling mechanism (as in the H+ buffering or the fatty-acid cycling models), we compared IUCP1 induced by regular LCFAs that can bind H+ at physiological pH and by low-pKa LCFA analogs that cannot (Jezek and Garlid, 1990; Rial et al., 2004). In solution, regular LCFAs have a pKa around 4.8, but their pKa after incorporation into the phospholipid membrane can increase to 7.5 or higher, depending on their local membrane (or membrane protein) environment (Hamilton, 1998). Thus, the majority of membrane-bound LCFAs are protonated at physiological pH. In contrast, alkylsulfonates, low-pKa LCFA analogs with pKa ≈–2 in solution (Guthrie, 1978), are unprotonated at physiological pH even after incorporation into the membrane.
Before activating UCP1 with exogenous fatty acids and their low-pKa analogs, exogenous membrane LCFAs were extracted with 10 mM αCD. Experiments were performed in symmetrical pH 6.0, as at this pH, endogenous LCFAs recover very poorly after extraction with αCD (Figures 2D and S2D). Under these conditions, 40 μM lauric acid (C11-carboxylate) activated a steady H+IUCP1 in response to the voltage-step protocol (Figure S4A, right panel), as expected for a regular LCFA. In a striking contrast, 100 μM undecanesulfonate (C11-sulfonate, a low-pK analog of lauric acid) induced transient currents in response to the same voltage-step protocol (Figure S4A, left panel and Figure 4B, left panel). These transient currents were carried by UCP1, as they were blocked by GDP (Figure 4B, left panel) and disappeared in mitoplasts from UCP1–/–/ mice (Figure S4B).
Figure 4. Alkylsulfonates Are UCP1 Transport Substrates.
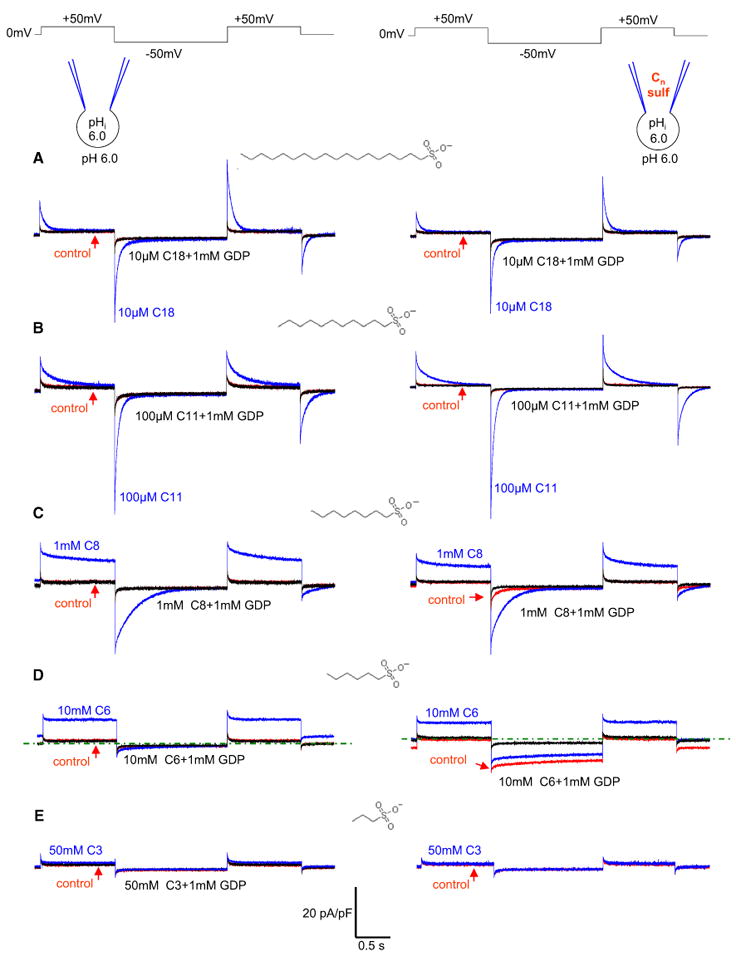
Left panels: Representative IUCP1 recorded after the extraction of endogenous membrane LCFAs with αCD (control, red), after subsequent application of the indicated concentration of Cn-sulfonate (blue), and upon adding 1 mM GDP (black) at symmetrical pH 6.0. The structure of the activating Cn-sulfonate is shown near the currents induced. Right panels: Same as the left panels, except that the pipette solution contained the same concentration of Cn-sulfonate as applied to the bath (10 μM C18 in A, 100 μM C11 in B, 1 mM C8 in C, 10 mM C6 in D, and 50 mM C3 in E). The calibration bar relates to all traces. A zero current level is indicated by the green dotted line in (C) and (D). See also Figure S4.
We next investigated how the length of the hydrophobic tail affects the IUCP1 induced by alkylsulfonates. Similar to C11-sulfonate, octadecanesulfonate (C18-sulfonate) induced a GDP-sensitive transient IUCP1 (Figure 4A, left panel). In contrast, octanesulfonate (C8-sulfonate) induced a GDP-sensitive IUCP1 with different kinetics, with a time-independent outward current and a transient inward current (Figure 4C, left panel). Hexanesulfonate (C6-sulfonate) induced primarily a time-independent outward GDP-sensitive IUCP1 with almost no inward current (Figure 4D, left panel). The currents induced by C8- and C6-sulfonates completely disappeared in UCP1–/– mitoplasts (Figures S4C and S4D). Generally, the shorter the hydrophobic carbon tail, the higher the concentration of alkylsulfonate required to induce IUCP1, with only 10 μM sufficient for C18-sulfonate and 10 mM required for C6-sulfonate (Figures 4A–4D, left panels). C3-sulfonate failed to induce significant IUCP1, even at 50 mM (Figure 4E, left panel).
IUCP1 induced by alkylsulfonates was not carried by H+, but by alkylsulfonates. In particular, the steady current induced by C6-sulfonate (Figure 4D, left panels) always reversed at the C6-sulfonate Nernst potentials (Figures 5A and S5), clearly demonstrating that it was carried by C6-sulfonate and not by H+.
Figure 5. H+ Transport by UCP1 Is Coupled to Transport of LCFA Anions.
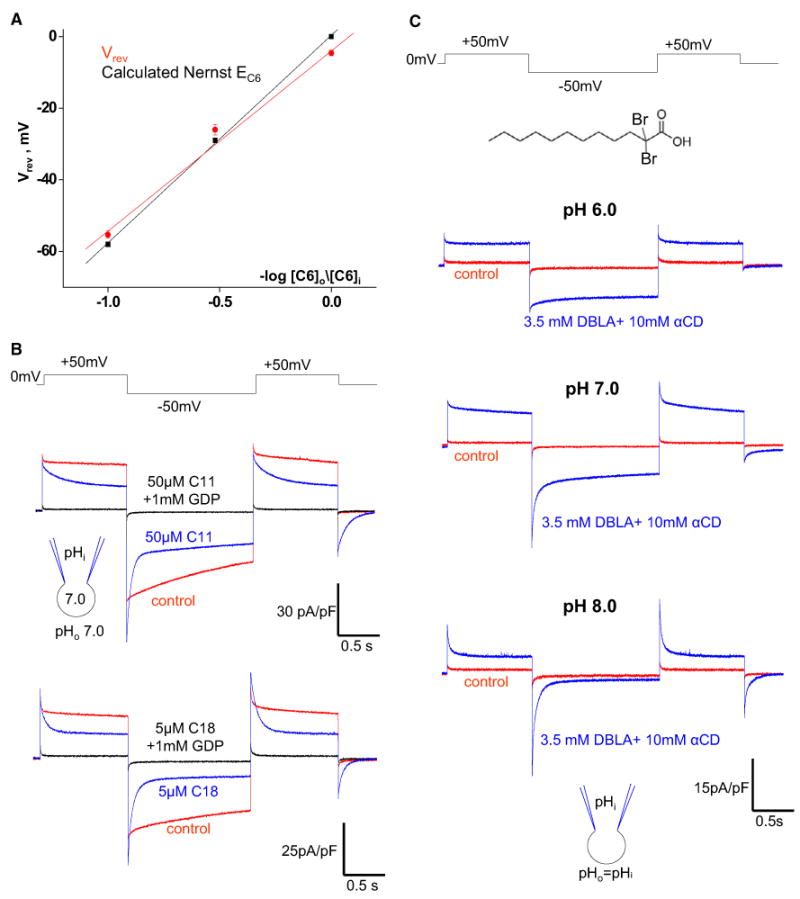
(A) Comparison of reversal potentials (Vrev) of the IUCP1 induced by C6-sulfonate, with C6-sulfonate equilibrium potentials (EC6) predicted by the Nernst equation. The red line indicates the linear fitting of IUCP1 reversal potentials; Vrev versus –log [C6]o/[C6]i, n = 3–6. The black line indicates EC6 calculated by the Nernst equation at 24°C.
(B) Upper panel: The IUCP1 activated by endogenous membrane LCFAs before (control, red) and after the application of 50 μM C11-sulfonate either alone (blue) or in combination with 1 mM GDP (black). Lower panel: The same experiment as in the upper panel but with 5 μM C18-sulfonate.
(C) IUCP1 in 10 mM αCD (control, red) and in 10 mM αCD plus 3.5 mM DBLA (blue) at symmetrical pH 6.0 (upper panel), pH 7.0 (middle panel), and pH 8.0 (lower panel).
Error bars represent SEM. See also Figure S5.
Similarly, the transient IUCP1 induced by C11- and C18-sulfonates (Figures 4A and 4B, left panels) should also be carried by them. C11- and C18-sulfonates differ from C6-sulfonate only by their much longer carbon tails that can establish strong hydrophobic interactions with UCP1 (or the membrane). Thus, although C11- and C18-sulfonates are transported by UCP1, they cannot leave the membrane due to the hydrophobic interactions, resulting in transient currents in response to voltage steps. Indeed, for the C18-sulfonate current (Figure 4A), the net charge translocated upon stepping from +50 to –50 mV was nearly identical to that carried back upon stepping from –50 to +50 mV (the area under the outward transient was only 4% ± 6% larger than under the inward transient; n = 4). For C11-sulfonate, this difference was only 6% ± 3% (n = 4). Although transport of C18- and C11-sulfonates is confined to the membrane, as expected for permeant species, they inhibited the steady H+IUCP1 activated by endogenous membrane LCFAs and replaced it with a transient IUCP1, characteristic of C18- and C11-sulfonates (Figure 5B). C18-sulfonate, with its longer hydrophobic tail, was a stronger inhibitor than C11-sulfonate (Figure 5B). Thus, the UCP1 currents carried by C18- and C11-sulfonates are similar to the gating currents of voltage-gated ion channels (Bezanilla, 2000) or to pre-steady-state currents observed in transporters in the presence of only one out of the two transported charged substrates (Lester et al., 1996; Peres et al., 2004).
C8-sulfonate was a “borderline” case between the short-chain C6-sulfonate and the long-chain C11- and C18-sulfonates. The hydrophobic interactions established by C8-sulfonate are not strong enough to prevent its steady transmembrane flux (as indicated by the steady outward current; Figure 4C, left panel) but are sufficient to keep some C8-sulfonate associated with UCP1 (or the membrane). This trapped C8-sulfonate is expelled by application of –50 mV into the mitoplast, causing the transient inward current (Figure 4C, left panel).
The transient IUCP1 induced by long-chain alkylsulfonates was not a peculiar property of alkylsulfonates, as the same transient IUCP1 could be caused by other low-pKa LCFA analogs independent of whether pKa-changing modifications were introduced into the head group or the hydrophobic tail. Perfluorotridecanoic acid (tail modification; Figure S4E) with a pKa ∼0 (Goss, 2008) and dodecyl sulfate (head modification; Figure S4F) with a pKa ∼–3.4 (Guthrie, 1978) caused transient IUCP1.
Thus, similar to other members of the SLC25 superfamily, UCP1 transports anions. The fact that alkylsulfonates with shorter hydrophobic tails require higher concentrations for permeation (Figure 4, left panels) and inhibition of the steady H+IUCP1 (Figure 5B) suggests that binding to the fatty-acid anion translocation site of UCP1 is improved by hydrophobic interactions.
Because various closely related negatively charged LCFA analogs (including those with carboxylic heads) are transported by UCP1, regular LCFA anions should also be UCP1 transport substrates. Given that LCFAs are required for H+ transport by UCP1, H+ transport should be coupled to the transport of LCFA anions. This conclusion is supported by the fact that long-chain alkylsulfonates inhibit the LCFA-dependent H+ current mediated by UCP1 (Figure 5B) in a competitive manner.
As LCFA anions serve as UCP1 transport substrates, the purely allosteric UCP1 models (1 and 2 in Figure S1A) cannot explain the mechanism of UCP1 operation. Our data also do not support the H+ buffering model (3 in Figure S1A), which does not explain the transport of fatty-acid anions.
H+ Transport by UCP1 Depends on the pKa of the Activating LCFA
Because LCFA anions are UCP1 transport substrates, H+ transport by UCP1 is likely to occur as the result of LCFA transport. Furthermore, H+ transport seems to depend on the ability of the transported LCFA to bind H+, as in the physiological pH range, regular LCFAs always activate steady H+ currents, whereas low-pKa LCFAs are transported alone. If this hypothesis is correct, 1-dibromolauric acid (DBLA), which has a pKa of ∼1.3 (Bruce, 2001; Lide, 1998), exactly in-between those of C11-sulfonate and lauric acid, should be able to activate both steady H+ and transient DBLA anion currents within the physiological pH range.
As we studied IUCP1 induced by DBLA in a broad pH range (6.0–8.0), we applied DBLA on a background of αCD (3.5 mM DBLA mixed with 10 mM αCD) to extract the endogenous membrane LCFAs generated rapidly at high pH and to limit their effect on UCP1. At pH 6.0, DBLA induced steady IUCP1 (Figure 5C, upper panel). At pH 7.0, the steady component of IUCP1 was still large, but a clear transient current appeared (Figure 5C, middle panel). At pH 8.0, almost all IUCP1 induced by DBLA was transient (Figure 5C, lower panel). This experiment links two different phenomena, the steady H+IUCP1 activated by regular LCFAs and the transient IUCP1 mediated by the low-pKa LCFA analogs, and shows that both can be observed with a single LCFA analog. Thus, H+ transport depends on the ability of the activating LCFA to bind H+: the lower the pKa of the activating LCFA, the higher the H+ concentration required to observe H+ transport.
The simplest explanation of the observed dependence of H+ transport on the pKa of the activating LCFA is that LCFA anions directly bind and carry H+ as they are transported by UCP1. When UCP1 transports LCFA anions, they either move within UCP1 or through UCP1 from one IMM leaflet to another. Accordingly, there can be two simple alternative models of LCFA-dependent H+ transport. In the first model, H+ transport occurs outside UCP1 by transbilayer LCFA flip-flop, and UCP1 operates in accordance with the fatty-acid cycling model (Figure S1A). In the second model, H+ is transported through UCP1 by an LCFA anion shuttling within UCP1 (Figure 7A). We will call the latter model the “LCFA-shuttling” model to distinguish it from the fatty-acid cycling model. Further experiments demonstrate that UCP1 is unlikely to operate by the fatty-acid cycling model.
Figure 7. LCFA-Shuttling Model of UCP1 Operation.
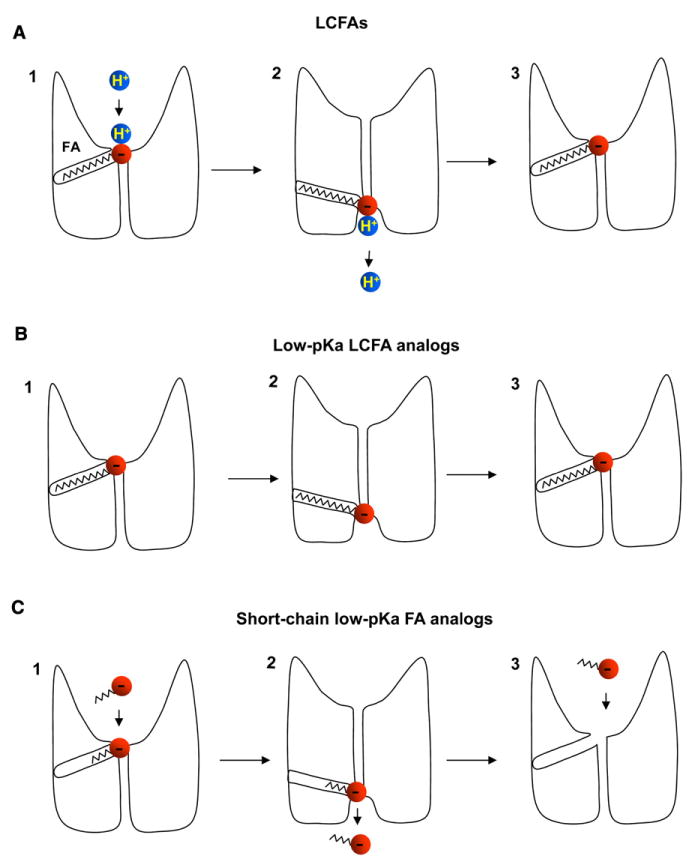
(A) The simplest mechanism of steady H+IUCP1 induced by LCFAs. UCP1 operates as a symporter that transports one LCFA and one H+ per the transport cycle. First, the LCFA anion binds to UCP1 on the cytosolic side at the bottom of a hypothetical cavity (1). H+ binding to UCP1 occurs only after the LCFA anion binds to UCP1(1). The H+ and the LCFA are translocated by UCP1 upon conformational change, and H+ is released on the opposite side of the IMM, whereas the LCFA anion stays associated with UCP1 due to the hydrophobic interactions established by its carbon tail (2). The LCFA anion then returns to initiate another H+ translocation cycle (3). Charge is translocated only in step 3 when the LCFA anion returns without the H+.
(B) The mechanism of transient IUCP1 induced by low-pKa LCFA analogs. A low-pKa LCFA analog can be translocated by UCP1 similar to an LCFA anion. However, the low pKa of the LCFA analog prevents the binding of H+ to UCP1. The negatively charged low-pKa LCFA analog shuttles within the UCP1 translocation pathway in response to the transmembrane voltage, producing transient currents.
(C) The mechanism of steady IUCP1 induced by short-chain low-pKa fatty-acid analogs. The hydrophobic tail is too short to anchor the fatty analog to UCP1, and the analog is translocated through UCP1, producing a steady current. In contrast to LCFAs, the short-chain low-pKa fatty-acid analogs can bind to UCP1 on both sides of the IMM.
The LCFA-Binding Site Is on the Cytoplasmic Side of UCP1
As long-chain alkylsulfonates cannot flip-flop across the membrane, we used them to probe the ability of UCP1 to bind LCFA anions on different faces of the IMM. Pipette C18- and C11-sulfonates failed to induce any measurable IUCP1 (Figures 4A and 4B, right panels, red traces); however, as expected, the subsequent addition of C18- and C11-sulfonates to the bath solution caused robust transient currents (Figures 4A and 4B, right panels, blue traces). Only when the pipette concentration of C11-sulfonate was elevated to 2 mM (below the critical micelle concentration, 20 mM; Annunziata et al., 1999) did C11-sulfonate induce a very small GDP-sensitive transient IUCP1 (Figure 6A, red trace). The subsequent addition of only 100 μM C11-sulfonate to the bath led to a much larger IUCP1 (Figure 6A, blue trace). Thus, UCP1 has a dramatic preference for accepting long-chain alkylsulfonates on the cytosolic side of the IMM. We used C11-sulfonate to further investigate this peculiar asymmetry of UCP1.
Figure 6. Asymmetry of LCFA Binding to UCP1.
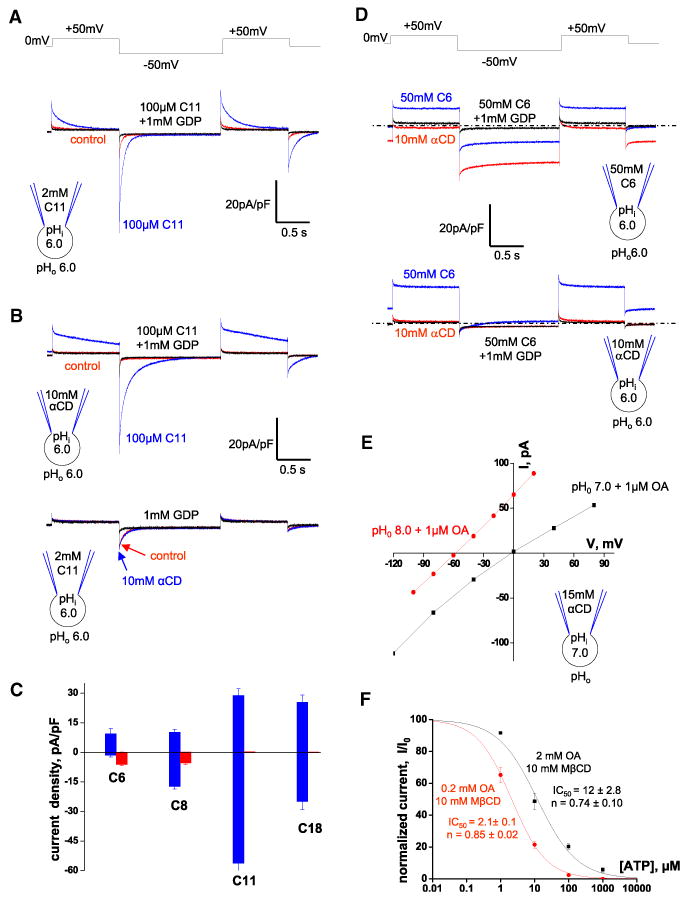
(A) IUCP1 recorded with 2 mM C11-sulfonate in the pipette. Representative IUCP1 recorded upon extraction of endogenous membrane LCFAs with αCD (control, red), in 100 μM C11-sulfonate (blue), and after the addition of 1 mM GDP (black).
(B) Upper panel: IUCP1 recorded with 10 mM αCD in the pipette. Representative IUCP1 recorded upon extraction of endogenous membrane LCFAs with αCD (control, red), in 100 μM C11-sulfonate (blue), and after the addition of 1 mM GDP (black). Lower panel: IUCP1 recorded with 2 mM C11-sulfonate in the pipette. Representative IUCP1 recorded upon extraction of endogenous membrane LCFAs with αCD (control, red), in 10 mM αCD (blue), and after the addition of 1 mM GDP (black).
(C) Amplitudes of the inward (negative) and outward (positive) UCP1 currents induced by various alkylsulfonates added to the bath (blue) or pipette (red) solutions. IUCP1 was recorded with the same alkylsulfonate concentrations using the same voltage protocol as in Figures 4A–4D. IUCP1 was measured in the beginning of the second (–50 mV, inward IUCP1) and third (+50 mV, outward IUCP1) voltage steps. The leak current remaining after application of 1 mM GDP was subtracted.
(D) IUCP1 recorded with 50 mM C6-sulfonate (upper panel) and 10 mM αCD (lower panel) in the pipette solution. Representative IUCP1 in 10 mM αCD (red), in 50 mM C6-sulfonate (blue), and after the addition of 1 mM GDP (black). The zero current level is indicated by the dotted line.
(E) I/V curves of IUCP1 induced by1 mM OA at pH 7.0 (black) and pH 8.0 (red). The pipette solution contained 15 mM αCD, pH 7.0. The IUCP1 amplitudes were measured as indicated in Figure S6B.
(F) The dose dependence of IUCP1 inhibition by ATP at two different concentrations of activating OA.IUCP1 was activated either with 0.2 mM OA mixed with 10 mM MβCD (red curve) or with 2 mM OA mixed with 10 mM MβCD (black curve). Amplitudes were measured upon stepping from 0 to –160 mV as in Figures S6C and S6D.
Error bars represent SEM. See also Figure S6.
Adding 10 mM αCD into the pipette significantly altered the transient IUCP1 induced by 100 μM bath C11-sulfonate (Figure 4B, left panel), making the outward current nearly steady and slowing down the kinetics of the inward current (Figure 6B, upper panel). Obviously, C11-sulfonate tended to abandon the hydrophobic interactions that anchored it within UCP1 or the membrane and to move into the pipette to establish hydrophobic interactions with αCD. In contrast, the small transient IUCP1 induced by 2 mM pipette C11-sulfonate was unaffected by the addition of 10 mM αCD to the bath (Figure 6B, lower panel). These experiments serve as additional evidence for asymmetry in UCP1 substrate binding and show that the permeation of matrix C11-sulfonate into UCP1 is very limited and unassisted by cytosolic αCD.
The access of the matrix C11-sulfonate into UCP1 was not improved by the presence of a cytosolic substrate (as in the obligatory exchange mechanism), as the application of cytosolic lauric acid (C11-carboxylate) did not increase the amplitude of the transient IUCP1 induced by 2 mM matrix C11-sulfonate but simply activated steady currents characteristic of lauric acid (data not shown). Moreover, cytosolic C11- and C18-sulfonates caused the same amplitude of IUCP1 regardless of the presence of the same alkylsulfonate on the matrix side of the IMM (Figures 4A, 4B, and 6A), suggesting that the matrix substrate has no effect on UCP1 function. This supports the conclusion that UCP1 accepts LCFA anions only on the cytosolic side.
Interestingly, in contrast to long-chain alkylsulfonates, C8- and C6-sulfonates induced IUCP1 when applied on either side of the IMM (Figures 4 and 6C). In particular, the application of 50 mM C6-sulfonate on either the matrix (Figure 6D, upper panel, red trace) or cytosolic sides (Figure 6D, lower panel, blue trace) caused about the same amplitude of IUCP1 (see Figure S6A for statistics). Transport of C6-sulfonate did not occur by the obligatory exchange mechanism, as C6-sulfonate currents were recorded in the presence of 10 mM αCD on the opposite side of the IMM to remove all substrates including endogenous membrane LCFAs (Figure 6D). On the contrary, when C6-sulfonate was applied on both sides of the IMM (Figure 6D, upper panel, blue trace), the amplitudes of the inward and outward IUCP1 were smaller than those induced by C6-sulfonate only on one side (Figures 6D and S6A). Thus, C6-sulfonate permeates through UCP1 from both sides by a channel-like mechanism.
These experiments with long-tail and short-tail alkylsulfonates suggest that the long hydrophobic tail prevents the penetration of matrix alkylsulfonates through UCP1. The small IUCP1 observed with unphysiologically high concentrations of matrix C11-sulfonate (Figure 6B, lower panel) indicates that at these high concentrations, the head group can be forced into the UCP1 translocation pathway from the matrix side; however, the long hydrophobic tail cannot enter UCP1 from the matrix side, severely limiting the translocation of the head group and the current.
The exclusive binding of long-chain substrates to UCP1 from the cytosolic side argues strongly against the fatty-acid cycling model because, per this model, UCP1 has to bind and export matrix LCFAs in order to import H+ into mitochondria (Figure S1A).
More evidence against the fatty-acid cycling model comes from measurements of IUCP1 reversal potentials. According to this model, UCP1 is an LCFA anion carrier, and therefore IUCP1 reversal potentials must depend on the transmembrane concentration gradient of LCFA anions. To establish such a gradient, we added 15 mM αCD to the pipette solution to deplete LCFA anions from the inner IMM leaflet and 1 μM OA to the bath solution to saturate the outer leaflet with OA anions. Under these conditions, if UCP1 is an LCFA anion carrier, the IUCP1 reversal potential should be negative, and the IUCP1 amplitude should be significantly larger in the outward direction. However, the IUCP1 reversal potential was ∼0 mV (which corresponds to the Nernst equilibrium potential for H+ but not for LCFA anions), and IUCP1 had equal amplitudes in both directions (Figures 6E and S6B, left panel). In the same experiment, subsequent elevation of the extracellular pH from 7.0 to 8.0 resulted in a dramatic –60 mV shift in the IUCP1 reversal potential, as expected for an H+ current (Figures 6E and S6B, right panel). This result confirms that UCP1 does not operate in accordance with the fatty-acid cycling model.
Given that the cytosolic LCFA-binding site of UCP1 and the strict H+ selectivity are inconsistent with the fatty-acid cycling model, the simplest model of UCP1 operation that fits all our experimental data is the LCFA-shuttling model proposed here. In this model, LCFA anions are UCP1 transport substrates, but they cannot dissociate from UCP1 due to hydrophobic interactions and serve as permanently attached substrates that shuttle within UCP1 and help transport H+ (Figure 7A).
LCFAs Can Overcome Purine Nucleotide Inhibition of UCP1
To activate BAT thermogenesis, LCFAs have to overcome UCP1 inhibition by cytosolic Mg2+-free purine nucleotides (primarily ATP). However, whether LCFAs remove purine nucleotide inhibition by direct competition or a different mechanism has remained controversial (Huang, 2003; Klingenberg, 2010; Nicholls, 2006; Rial et al., 1983; Shabalina et al., 2004; Winkler and Klingenberg, 1994).
To address a possibility of direct competition between LCFAs and purine nucleotides, we compared the inhibition of IUCP1 by ATP at two OA concentrations. To limit the effect of endogenous membrane LCFAs, OA was applied on a background of 10 mM MβCD. IUCP1 activated by 0.2 mM OA/10 mM MβCD was completely inhibited by 100 μM ATP, and 1 mM ATP did not cause additional inhibition (Figure S6C). In contrast, when IUCP1 was activated by 2 mM OA/10 mM MβCD, 100 μM ATP induced only moderate inhibition, and 1 mM ATP further suppressed IUCP1 (Figure S6D). We also compared the dose response of UCP1 inhibition by ATP under these two OA concentrations. IUCP1 activated by 0.2 mM OA/10 mM MβCD was inhibited by ATP with an IC50 of 2.1 ± 0.1 μM, whereas in a 10-fold higher concentration of OA (2 mM OA/10 mM MβCD), ATP inhibited IUCP1 with an IC50 of 12.0 ± 2.8 μM (Figure 6F).
Thus, LCFAs have a double effect on UCP1 activation: they serve as UCP1 transport substrates but can also remove purine nucleotide inhibition. Because purine nucleotides bind on the cytosolic side of UCP1, similar to LCFAs, LCFA anions are likely to directly compete with purine nucleotides.
Cl– Conductance of UCP1 Is Negligible as Compared to Its H+ Conductance
Because UCP1 was also proposed to conduct Cl– (Nicholls, 2006), we compared the amplitudes of Cl– and H+ currents through UCP1 to determine whether Cl– significantly contributes to mitochondrial uncoupling.
As compared to the IUCP1 recorded under Cl–-free conditions (Figure 1B, red trace), the current recorded in symmetrical 150 mM Cl– had a larger outward component observed at the positive voltages (Figure S7A, red trace). This additional outward current was mediated by large-conductance channels (∼100 pS, not shown), as judged by significant step-like fluctuations of the current observed in the positive range of potentials (Figure S7A, red trace). To smooth out these fluctuations and simplify the amplitude analysis, all traces were recorded 30–40 times and averaged to obtain the final traces as presented in Figure S7B.
Addition of 1 mM GDP to the bath led to a dramatic reduction in the inward current observed at negative membrane potentials, but the outward current at positive potentials was largely unaffected (Figures S7A and S7B), suggesting that a significant portion of the outward current was not UCP1 dependent. Indeed, recording from UCP1–/– mitoplasts in symmetrical 150 mM Cl– revealed a UCP1-independent outward current at positive membrane potentials (Figures S7C and S7D). This outward Cl– current was likely mediated by the inner membrane anion channel (IMAC, also called the 108-pS anion channel) (Borecký et al., 1997; Jezek et al., 1989) based on its Cl– permeability, outward rectification (conducting current primarily in the outward direction), and similar single-channel amplitude (around 100 pS).
When we used Mg2+-containing bath and pipette solutions, the UCP1-independent Cl– current was inhibited (Figure S7E, red trace) as expected for IMAC (Jezek et al., 1989), and we could study the Cl– current through UCP1 only. Under these conditions, IUCP1 was not significantly affected by the elevation of Cl– in the bath solution from 2 mM to 75 mM (Figure S7E). However, the reversal potential was slightly shifted by –7 mV, as determined by the UCP1 I/V relationship (Figure S7F). Based on this shift, we calculated that the UCP1 PCl–/PH+ permeability ratio is 4 × 10–6, and UCP1 appears to be highly selective for H+ over Cl–.
DISCUSSION
LCFAs Are Essential for UCP1 Uncoupling
Despite the agreement that LCFAs activate UCP1, there has been a controversy as to whether LCFAs are absolutely required for UCP1 uncoupling or whether UCP1 has some constitutive, LCFA-independent activity (Garlid et al., 1998; Klingenberg, 2010; Nicholls, 2006). Here we demonstrate under well-controlled conditions that native UCP1 requires LCFAs for its H+ transport activity, and that LCFAs serve as permanently attached UCP1 substrates that help to carry H+ through UCP1. We also show that LCFAs are produced within the IMM by a putative PLA2 associated with the IMM. The “constitutive” LCFA-independent UCP1 uncoupling observed in ion flux studies in mitochondria in the presence of albumin (Nicholls, 2006) was likely due to incomplete extraction of LCFAs produced by mitochondrial PLA2.
The Transport Mechanism of UCP1
The simplest model of UCP1 operation that explains all our experimental data is that of an LCFA–/H+ symporter, which simultaneously transports one LCFA anion and one H+ (the LCFA-shuttling model, Figure 7A). This model assumes that H+ transport is coupled to LCFA– transport but does not necessarily assume direct binding of H+ to the LCFA anion. Furthermore, although both LCFA– and H+ are UCP1 transport substrates, LCFA– cannot dissociate from UCP1, and UCP1 operates as a virtual H+ carrier (Figure 7A). Although in accordance with this model, H+ is transported across the membrane, the symport of H+ in association with LCFA– is electroneutral. The charge translocation (the UCP1 current) is produced by the head group of the LCFA anion when, after releasing H+, it returns to the opposite side of the membrane (Figure 7A). Thus, although the steady currents carried by UCP1 are termed “H+ currents” due to the fact that they reverse at the Nernst potentials for H+ (Figure 1F), in accordance with our model, these currents are actually carried by the shuttling LCFA anions. Although the model presented in Figure 7A shows H+ transport only in one direction, that transport is fully reversible: H+ can bind to UCP1 on both sides of the IMM, and UCP1 can transport H+ in both directions. The LCFA-shuttling model correlates well with the prediction (based on symmetry analysis of SLC25 members) that UCP1 transports carboxylic or keto acids in symport with H+ (Robinson et al., 2008).
LCFA anions appear to act as the “principal” UCP1 substrate, as they can be transported by UCP1 independent of H+ transport. H+ appears to be a “secondary” UCP1 substrate because H+ transport by UCP1 depends on LCFAs and their pKa.
Low-pKa LCFA analogs are transported by UCP1 alone, without H+. They cannot dissociate from UCP1 due to hydrophobic interactions and shuttle back and forth within UCP1 as the membrane potential changes, resulting in transient UCP1 currents (Figure 7B). In contrast, short-chain low-pKa fatty-acid analogs do not establish strong hydrophobic interactions and can dissociate from UCP1, producing actual transmembrane currents (Figure 7C).
The LCFA-shuttling model can explain the peculiar pH dependence of IUCP1 (Figure 1H). According to the LCFA-shuttling model (Figure 7A), the H+ current carried by UCP1 will be small either at very high symmetrical pH or very low symmetrical pH. At high pH, there are no H+ to be transported, and at low pH, H+ cannot dissociate from UCP1, and both the LCFA anion and H+ remain associated with UCP1, resulting in electroneutral transport. Thus, the pH dependence of IUCP1 should be bell-shaped, with the maximum around the pKa of the H+-binding site of UCP1. This pKa for H+ transport depends on the pKa of the cotransported LCFA anion. For the IUCP1 activated by regular LCFAs, the pKa for H+ transport appears to be around 8.0–9.0, and therefore, at our experimental pH (5.0–9.0), we observed only the left wing of the bell-shaped pH dependence of the H+ current (Figure 1H). However, for IUCP1 activated by DBLA (pKa is 3.5 units lower than that of regular LCFAs), we likely observed the right wing of the pH dependence, as at pH 8.0, UCP1 primarily carried transient currents mediated by LCFA anions and very little steady H+ current (Figure 5C). In addition to the pH dependence of the UCP1-binding site for H+ transport, the pH dependence of LCFA production within the IMM (Figure S2D) should also contribute to the overall pH dependence of IUCP1.
UCP1 is an anion carrier; however, based on our data, the transported anion must possess a hydrophobic tail, and as the length of this tail decreases, the affinity of the alkyl anion for the substrate-binding site of UCP drops significantly (Figure 4A). The importance of the hydrophobic tail for anion permeation through UCP1 has been previously suggested (Jezek and Garlid, 1990). Therefore, although UCP1 can in principle transport Cl– (Nicholls, 2006), we showed that the Cl– current through UCP1 is small in comparison with the currents produced by LCFA analogs/H+, and that Cl– is unlikely to contribute significantly to UCP1 uncoupling.
The LCFA-shuttling model bears similarities to the recently proposed mechanism of operation of CLC H+/Cl– exchangers (Feng et al., 2010). In these proteins, the transport of H+ is mediated by the carboxylic group of a glutamate residue (the so-called gating glutamate) that shuttles along the channel-like anion translocation pathway.
It is important to understand that the transport mechanism in Figure 7 represents the simplest model that fits our data. Further experiments are required to clarify the exact stoichiometry of transport, binding sites of for H+ and LCFAs, and the conformational change that leads to the translocation of LCFA– and H+.
The Role of LCFAs in the Removal of Purine Nucleotide Inhibition of UCP1
In living cells, UCP1 is tonically inhibited by purine nucleotides, primarily ATP. Purine nucleotides bind on the cytosolic side of UCP1 and seem to occlude the UCP1 translocation pathway (Klingenberg, 2010). The identity of the molecule that overcomes ATP inhibition and helps to activate UCP1 uncoupling in BAT has remained elusive. Two primary candidates, LCFAs and long-chain acyl-CoA, have been suggested but remain controversial (Huang, 2003; Rial et al., 1983; Shabalina et al., 2004; Winkler and Klingenberg, 1994). In our experiments, LCFAs were able to overcome the inhibition of UCP1 by ATP4– (Figure 6F), whereas long-chain acyl-CoA (similar to related purine nucleotides) was a potent UCP1 inhibitor (Figure 1G).
LCFA anions are permeable species and in principle can directly compete with ATP4– that binds near or within the translocation pathway. Due to the significant structural differences between LCFA anions and ATP4–, their binding sites on UCP1 cannot be identical but may partially overlap, so that the electrostatic repulsion between the two negatively charged ligands results in competition. Although LCFAs can overcome ATP inhibition in patch-clamp experiments, the validity of this mechanism for the removal of UCP1 inhibition by ATP in intact brown adipocytes remains to be established.
EXPERIMENTAL PROCEDURES
Isolation of BAT Mitochondria and Mitoplasts
Mice (3–4 weeks old) were sacrificed by CO2 asphyxiation followed by cervical dislocation. Interscapular BAT was isolated, and mitochondria were isolated as in Kirichok et al. (2004). Mitoplasts (vesicles of the whole native IMM) were produced from mitochondria using a French press at 2,000 psi to rupture the outer membrane, which allows for gentle and pure mechanical isolation of mitoplasts (Decker and Greenawalt, 1977).
Immediately before the electrophysiological experiments, 15–50 μl of the mitoplast suspension was added to 500 μl solution containing 150 mM KCl, 10 mM HEPES, and 1 mM EGTA (pH adjusted to 7.2 with KOH) and plated on 5 mm coverslips. For whole-mitoplast experiments, coverslips were pretreated with 0.1% gelatin to reduce adhesion, whereas untreated coverslips were used for outside-out experiments. Only mitoplasts that had a dense, opaque matrix were selected for experiments, as this indicated the integrity of their matrix and thus their membranes.
Patch-Clamp Recordings
Mitoplasts used for patch-clamp experiments were 3–5 μm in diameter and typically had membrane capacitances of 0.5–1.2 pF. Gigaohm seals were formed in the bath solution containing 150 mM KCl, 10 mM HEPES, and 1 mM EGTA, pH 7.2 (adjusted with KOH). A 3 M KCl agar salt bridge was used as the bath reference electrode. Voltage steps of 250–800 mV and 1–50 ms were applied to obtain the whole-mitoplast configuration, as monitored by the appearance of capacitance transients. The access resistance and membrane capacitance of mitoplasts were determined with the Membrane Test tool of pClamp 10 (Molecular Devices). Mitoplasts were stimulated every 5 s.
At very negative membrane potentials, whole-mitoplast IUCP1 showed a time-dependent reduction in amplitude (see, for example, Figure S1D) due to saturation of the H+ buffer inside the mitoplast. To reduce experimental errors associated with such saturation, we measured amplitudes of whole-mitoplast IUCP1 at the beginning of the voltage steps and set the holding voltage at the H+ reversal potential whenever appropriate.
For the experiments shown in Figures 1, 2, 3, 4A–4D, 5, 6A–6C, 6D (lower panel), 6F, S1, S2, S3, S4, S5, and S6B–S6D, pipettes were filled with 150 mM tetramethylammonium hydroxide (TMA), 1.5 mM EGTA, 1–2 mM magnesium gluconate, 150 mM HEPES (MES or CHES), and 2 mM Tris-Cl (pH adjusted to 5.0–9.0 with D-gluconic acid, tonicity adjusted to ∼450 mmol/kg with sucrose). In experiments that required 50 mM hexanesulfonate (C6-sulfonate) or 50 mM propanesulfonate (C3-sulfonate) on the matrix side of the IMM as shown in Figures 4E, 6D (upper panel), and S6A, we used the following pipette solution: 100 mM TMA, 1.5 mM EGTA, 1 mM magnesium gluconate, 100 mM MES, 50 mM sodium hexanesulfonate (or propanesulfonate), and 2 mM Tris-Cl (pH adjusted to 6.0 with D-gluconic acid, tonicity adjusted to ∼450 mmol/kg with sucrose). Typically, pipettes had resistances of 25–40 MΩ, and the access resistance was 40–75 MΩ. The calculated voltage-clamp error associated with the access resistance did not exceed 10 mV.
Whole-mitoplast IUCP1 were recorded in the bath solution containing 150 mM HEPES (or MES or CHES) and 1 mM EGTA (pH adjusted to 5.0–9.0 with D-gluconic acid or Tris base, tonicity adjusted to ∼300 mmol/kg with sucrose). In some instances, 2 mM magnesium gluconate was added to the bath solution to improve membrane stability, which did not change IUCP1 behavior. In experiments requiring 50 mM hexanesulfonate (C6-sulfonate) or 50 mM propanesulfonate (C3-sulfonate) on the cytosolic side of the IMM as shown in Figures 4E, 5A, 6C, 6D, S5, and S6A, we used the following bath solution: 100 mM MES, 50 mM sodium hexanesulfonate (or propanesulfonate), and 1 mM EGTA (pH adjusted to 5.0–9.0 with D-gluconic acid or Tris base, tonicity adjusted to ∼300 mmol/kg with sucrose). All experiments were performed under continuous perfusion of the bath solution.
To record whole-mitoplast Cl– currents (Figures S7A–S7D), we used a pipette solution containing 150 mM CsCl, 150 mM HEPES, and 1 mM EGTA (pH adjusted to 7.0 with Tris base, tonicity adjusted to 450 mmol/kg). The bath solution in these experiments contained 150 mM Tris-Cl, 20 mM HEPES, and 1 mM EGTA (pH adjusted to 7.0 with Tris base, tonicity 300 mmol/kg with sucrose). In experiments investigating the relative selectivity of UCP1 for Cl– versus H+ (Figures S7E and S7D), we used symmetrical bath/pipette solutions that contained 100 mM CsOH, 1 mM Mg gluconate, 1.5 mM EGTA, 150 mM HEPES, and 2 mM Tris-Cl (pH adjusted to 7.0 with D-gluconic acid, tonicity adjusted to 300 mmol/kg with sucrose). The 75 mM Cl– bath solution in these experiments contained 75 mM HCl, 100 mM CsOH, 1 mM Mg gluconate, 1.5 mM EGTA, 150 mM HEPES, and 2 mM Tris-Cl (pH adjusted to 7.0 with Tris base, tonicity adjusted to 300 mmol/kg with sucrose). Membrane potentials were corrected for the liquid junction potential in experiments probing the relative selectivity of UCP1 for Cl– versus H+.
Data acquisition and analysis were performed using PClamp 10 (Molecular Devices) and Origin 7.5 (OriginLab). All electrophysiological data presented were acquired at 10 kHz and filtered at 1 kHz. Statistical data are presented as the mean ± standard error of the mean (SEM).
All general chemicals including fatty acids and their low-pKa analogs were acquired from Sigma (St. Louis, MO, USA). Phospholipase A2 inhibitors and 1-dibromolauric acid were from Cayman Chemical Company (Ann Arbor, MI, USA). Lysophospholipids and acyl-CoA were from Avanti Polar Lipids and Sigma. DBLA was synthesized by Cayman Chemical Company.
Supplementary Material
Acknowledgments
The authors are grateful to Drs. David Nicholls, Eduardo Rial, Robert Farese, Frederick Sachs, and Francesca Fieni for advice and to Dr. Leslie Kozak for the UCP1–/– mice. This work was funded by the NIH Director’s New Innovator Award DP2OD004656, the UCSF Program for Breakthrough Biomedical Research, and a Larry L. Hillblom Foundation Start-up Award. A.F. and Y.K. conceived the project and designed the experiments. A.F. performed most of the experiments. Y.K. and P.V.L. helped with pilot experiments for the project. All authors discussed the results and commented on the manuscript.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes seven figures and can be found with this article online at http://dx.doi.org/10.1016/j.cell.2012.09.010.
References
- Anel A, Richieri GV, Kleinfeld AM. Membrane partition of fatty acids and inhibition of T cell function. Biochemistry. 1993;32:530–536. doi: 10.1021/bi00053a018. [DOI] [PubMed] [Google Scholar]
- Annunziata O, Costantino L, D’Errico G, Paduano L, Vitagliano VV. Transport Properties for Aqueous Sodium Sulfonate Surfactants. J Colloid Interface Sci. 1999;216:16–24. doi: 10.1006/jcis.1999.6269. [DOI] [PubMed] [Google Scholar]
- Aquila H, Link TA, Klingenberg M. The uncoupling protein from brown fat mitochondria is related to the mitochondrial ADP/ATP carrier. Analysis of sequence homologies and of folding of the protein in the membrane. EMBO J. 1985;4:2369–2376. doi: 10.1002/j.1460-2075.1985.tb03941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bezanilla F. The voltage sensor in voltage-dependent ion channels. Physiol Rev. 2000;80:555–592. doi: 10.1152/physrev.2000.80.2.555. [DOI] [PubMed] [Google Scholar]
- Bhamidipati SP, Hamilton JA. Interactions of lyso 1-palmitoylphosphatidylcholine with phospholipids: a 13C and 31P NMR study. Biochemistry. 1995;34:5666–5677. doi: 10.1021/bi00016a043. [DOI] [PubMed] [Google Scholar]
- Borecký J, Jezek P, Siemen D. 108-pS channel in brown fat mitochondria might Be identical to the inner membrane anion channel. J Biol Chem. 1997;272:19282–19289. [PubMed] [Google Scholar]
- Bouillaud F, Weissenbach J, Ricquier D. Complete cDNA-derived amino acid sequence of rat brown fat uncoupling protein. J Biol Chem. 1986;261:1487–1490. [PubMed] [Google Scholar]
- Bruce PY. Organic Chemistry. Upper Saddle River, NJ: Pearson Education; 2001. [Google Scholar]
- Brunaldi K, Huang N, Hamilton JA. Fatty acids are rapidly delivered to and extracted from membranes by methyl-beta-cyclodextrin. J Lipid Res. 2010;51:120–131. doi: 10.1194/jlr.M900200-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- Cannon B, Sundin U, Romert L. Palmitoyl coenzyme A: a possible physiological regulator of nucleotide binding to brown adipose tissue mitochondria. FEBS Lett. 1977;74:43–46. doi: 10.1016/0014-5793(77)80748-5. [DOI] [PubMed] [Google Scholar]
- Decker GL, Greenawalt JW. Ultrastructural and biochemical studies of mitoplasts and outer membranes derived from French-pressed mitochondria. Advances in mitochondrial subfractionation. J Ultrastruct Res. 1977;59:44–56. doi: 10.1016/s0022-5320(77)80027-0. [DOI] [PubMed] [Google Scholar]
- Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 2011;26:192–205. doi: 10.1152/physiol.00046.2010. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Winkler E, Klingenberg M. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature. 2000;408:609–613. doi: 10.1038/35046114. [DOI] [PubMed] [Google Scholar]
- Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ, Portero-Otín M, Pamplona R, Vidal-Puig AJ, Wang S, Roebuck SJ, Brand MD. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J. 2003;22:4103–4110. doi: 10.1093/emboj/cdg412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enerbäck S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, Harper ME, Kozak LP. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature. 1997;387:90–94. doi: 10.1038/387090a0. [DOI] [PubMed] [Google Scholar]
- Feng L, Campbell EB, Hsiung Y, MacKinnon R. Structure of a eukaryotic CLC transporter defines an intermediate state in the transport cycle. Science. 2010;330:635–641. doi: 10.1126/science.1195230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garlid KD, Jabürek M, Jezek P. The mechanism of proton transport mediated by mitochondrial uncoupling proteins. FEBS Lett. 1998;438:10–14. doi: 10.1016/s0014-5793(98)01246-0. [DOI] [PubMed] [Google Scholar]
- Ghomashchi F, Naika GS, Bollinger JG, Aloulou A, Lehr M, Leslie CC, Gelb MH. Interfacial kinetic and binding properties of mammalian group IVB phospholipase A2 (cPLA2beta) and comparison with the other cPLA2 isoforms. J Biol Chem. 2010;285:36100–36111. doi: 10.1074/jbc.M110.165647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goss KU. The pKa values of PFOA and other highly fluorinated carboxylic acids. Environ Sci Technol. 2008;42:456–458. doi: 10.1021/es702192c. [DOI] [PubMed] [Google Scholar]
- Guthrie JP. Hydrolysis of esters of oxy acids: pKa values for strong acids; Brønsted relationship for attack of water at methyl; free energies of hydrolysis of esters of oxy acids; and a linear relationship between free energy of hydrolysis and pKa holding over a range of 20pK units. Can J Chem. 1978;56:2342–2354. [Google Scholar]
- Hamilton JA. Fatty acid transport: difficult or easy? J Lipid Res. 1998;39:467–481. [PubMed] [Google Scholar]
- Heaton GM, Wagenvoord RJ, Kemp A, Jr, Nicholls DG. Brown-adipose-tissue mitochondria: photoaffinity labelling of the regulatory site of energy dissipation. Eur J Biochem. 1978;82:515–521. doi: 10.1111/j.1432-1033.1978.tb12045.x. [DOI] [PubMed] [Google Scholar]
- Huang SG. Binding of fatty acids to the uncoupling protein from brown adipose tissue mitochondria. Arch Biochem Biophys. 2003;412:142–146. doi: 10.1016/s0003-9861(03)00031-6. [DOI] [PubMed] [Google Scholar]
- Jaburek M, Garlid KD. Reconstitution of recombinant uncoupling proteins: UCP1, -2, and -3 have similar affinities for ATP and are unaffected by coenzyme Q10. J Biol Chem. 2003;278:25825–25831. doi: 10.1074/jbc.M302126200. [DOI] [PubMed] [Google Scholar]
- Jezek P, Garlid KD. New substrates and competitive inhibitors of the Cl- translocating pathway of the uncoupling protein of brown adipose tissue mitochondria. J Biol Chem. 1990;265:19303–19311. [PubMed] [Google Scholar]
- Jezek P, Beavis AD, Diresta DJ, Cousino RN, Garlid KD. Evidence for two distinct chloride uniport pathways in brown adipose tissue mitochondria. Am J Physiol. 1989;257:C1142–C1148. doi: 10.1152/ajpcell.1989.257.6.C1142. [DOI] [PubMed] [Google Scholar]
- Kinsey GR, McHowat J, Beckett CS, Schnellmann RG. Identification of calcium-independent phospholipase A2γ in mitochondria and its role in mitochondrial oxidative stress. Am J Physiol Renal Physiol. 2007;292:F853–F860. doi: 10.1152/ajprenal.00318.2006. [DOI] [PubMed] [Google Scholar]
- Kirichok Y, Krapivinsky G, Clapham DE. The mitochondrial calcium uniporter is a highly selective ion channel. Nature. 2004;427:360–364. doi: 10.1038/nature02246. [DOI] [PubMed] [Google Scholar]
- Klingenberg M. Wanderings in bioenergetics and biomembranes. Biochim Biophys Acta. 2010;1797:579–594. doi: 10.1016/j.bbabio.2010.02.012. [DOI] [PubMed] [Google Scholar]
- Klingenberg M, Huang SG. Structure and function of the uncoupling protein from brown adipose tissue. Biochim Biophys Acta. 1999;1415:271–296. doi: 10.1016/s0005-2736(98)00232-6. [DOI] [PubMed] [Google Scholar]
- Lester HA, Cao Y, Mager S. Listening to neurotransmitter transporters. Neuron. 1996;17:807–810. doi: 10.1016/s0896-6273(00)80213-5. [DOI] [PubMed] [Google Scholar]
- Lide DR. Handbook of Chemistry and Physics. 87. Boca Raton, FL: CRC Press; 1998. [Google Scholar]
- Murakami M, Taketomi Y, Miki Y, Sato H, Hirabayashi T, Yamamoto K. Recent progress in phospholipase A2 research: from cells to animals to humans. Prog Lipid Res. 2011;50:152–192. doi: 10.1016/j.plipres.2010.12.001. [DOI] [PubMed] [Google Scholar]
- Nicholls DG. The physiological regulation of uncoupling proteins. Biochim Biophys Acta. 2006;1757:459–466. doi: 10.1016/j.bbabio.2006.02.005. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Locke RM. Thermogenic mechanisms in brown fat. Physiol Rev. 1984;64:1–64. doi: 10.1152/physrev.1984.64.1.1. [DOI] [PubMed] [Google Scholar]
- Osman C, Voelker DR, Langer T. Making heads or tails of phospholipids in mitochondria. J Cell Biol. 2011;192:7–16. doi: 10.1083/jcb.201006159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peres A, Giovannardi S, Bossi E, Fesce R. Electrophysiological insights into the mechanism of ion-coupled cotransporters. News Physiol Sci. 2004;19:80–84. doi: 10.1152/nips.01504.2003. [DOI] [PubMed] [Google Scholar]
- Rial E, González-Barroso MM. Physiological regulation of the transport activity in the uncoupling proteins UCP1 and UCP2. Biochim Biophys Acta. 2001;1504:70–81. doi: 10.1016/s0005-2728(00)00240-1. [DOI] [PubMed] [Google Scholar]
- Rial E, Poustie A, Nicholls DG. Brown-adipose-tissue mitochondria: the regulation of the 32000-Mr uncoupling protein by fatty acids and purine nucleotides. Eur J Biochem. 1983;137:197–203. doi: 10.1111/j.1432-1033.1983.tb07815.x. [DOI] [PubMed] [Google Scholar]
- Rial E, Aguirregoitia E, Jiménez-Jiménez J, Ledesma A. Alkylsulfonates activate the uncoupling protein UCP1: implications for the transport mechanism. Biochim Biophys Acta. 2004;1608:122–130. doi: 10.1016/j.bbabio.2003.11.001. [DOI] [PubMed] [Google Scholar]
- Ridley RG, Patel HV, Gerber GE, Morton RC, Freeman KB. Complete nucleotide and derived amino acid sequence of cDNA encoding the mitochondrial uncoupling protein of rat brown adipose tissue: lack of a mitochondrial targeting presequence. Nucleic Acids Res. 1986;14:4025–4035. doi: 10.1093/nar/14.10.4025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson AJ, Overy C, Kunji ER. The mechanism of transport by mitochondrial carriers based on analysis of symmetry. Proc Natl Acad Sci USA. 2008;105:17766–17771. doi: 10.1073/pnas.0809580105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina IG, Jacobsson A, Cannon B, Nedergaard J. Native UCP1 displays simple competitive kinetics between the regulators purine nucleotides and fatty acids. J Biol Chem. 2004;279:38236–38248. doi: 10.1074/jbc.M402375200. [DOI] [PubMed] [Google Scholar]
- Shabalina IG, Petrovic N, Kramarova TV, Hoeks J, Cannon B, Nedergaard J. UCP1 and defense against oxidative stress 4-Hydroxy-2-nonenal effects on brown fat mitochondria are uncoupling protein 1-independent. J Biol Chem. 2006;281:13882–13893. doi: 10.1074/jbc.M601387200. [DOI] [PubMed] [Google Scholar]
- Stewart A, Ghosh M, Spencer DM, Leslie CC. Enzymatic properties of human cytosolic phospholipase A(2)γ. J Biol Chem. 2002;277:29526–29536. doi: 10.1074/jbc.M204856200. [DOI] [PubMed] [Google Scholar]
- Winkler E, Klingenberg M. Effect of fatty acids on H+ transport activity of the reconstituted uncoupling protein. J Biol Chem. 1994;269:2508– 2515. [PubMed] [Google Scholar]
- Zurini M, Hugentobler G, Gazzotti P. Activity of phospholipase A2 in the inner membrane of rat-liver mitochondria. Eur J Biochem. 1981;119:517–521. doi: 10.1111/j.1432-1033.1981.tb05638.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.