

Abstract
A resin-assisted enrichment method has been developed for specific isolation of protein N-terminal peptides to facilitate LC-MS/MS characterization of proteolytic processing, a major form of posttranslational modifications. In this method, protein thiols are blocked by reduction and alkylation, and protein lysine residues are converted to homoarginines. Protein N-termini are selectively converted to reactive thiol groups, and the thiol-containing N-terminal peptides are then captured by a thiol-affinity resin with high specificity (>97%). The efficiencies of these sequential reactions were demonstrated to be nearly quantitative. The resin-assisted N-terminal peptide enrichment approach was initially applied to a cell lysate of the filamentous fungus Aspergillus niger. Subsequent C-MS/MS analyses resulted in the identification of 1672 unique protein N-termini or proteolytic cleavage sites from 690 unique proteins.
Keywords: proteolytic processing, N-terminal peptides, enrichment, Aspergillus niger
INTRODUCTION
Proteolytic processing, the irreversible hydrolysis of peptide bonds by proteases, is an important posttranslational modification (PTM) for regulating protein functions in living organisms.1 Due to the inherent complexity of the proteome, the identification of proteolytic cleavage sites typically requires selective enrichment of protein N-terminal peptides prior to liquid chromatography-tandem mass spectrometry (LC-MS/MS)-based analysis.2–3 Several negative and positive selection methods have been reported for enriching the N-terminal peptides. For negative selection, non-N-terminal peptides can be removed using amine-reactive scavengers2, 4–6 or via chromatographic separations; 3, 7–8 however, drawbacks of this approach include multiple scavenging reactions4, 6 or extensive fractionation steps.3, 7–8 Furthermore, negative selection using scavenger materials may suffer from non-specific binding of non-N-terminal peptides and non-specific loss of N-terminal peptides on the resin due to the relatively large amount of amine-reactive scavenging materials required to remove all non-N-terminal peptides.
Conceptually, positive selection strategies in which N-terminal peptides are directly captured using a chemical derivatization strategy have the potential to achieve high enrichment specificity. This advantage of high specificity compensates one limitation of the positive selection strategies; that is, in vivo modified N-terminal peptides will not be enriched.9 In positive selection strategies, a biotin affinity tag is often introduced to the protein N-termini through either enzymatic10–12 or chemical labeling.9, 13 The biotin tag enables selective capture of protein N-terminal peptides by immobilized avidin beads;9–13 however, a major disadvantage of avidin-biotin enrichment is the non-covalent nature of the binding, which can lead to nonspecific binding and relatively poor enrichment specificity.14–16 To address this issue, a number of studies have utilized solid-phase or resin-assisted covalent enrichment approaches that offer simpler, more efficient ways of capturing cysteine-containing peptides16–17 and other PTMs, such as S-nitrosylation,14, 18–19 phosphorylation,15 and tyrosine nitration.20 For example, we demonstrated the utility of resin-assisted covalent enrichment for proteomics applications, using thiol-affinity resins to capture cysteine-containing peptides via disulfide-exchange.17
In this work, we introduce a chemical strategy that enables resin-assisted enrichment of N-terminal peptides from complex mixtures. The strategy involves several blocking and chemical derivatization steps that selectively convert protein N-termini into reactive thiol groups amenable to capture by thiol-affinity resins. All chemical reactions demonstrate nearly quantitative yields. Initial application of our N-terminal peptide strategy to a whole cell lysate of Aspergillus niger followed by LC-MS/MS revealed 1672 unique protein N-termini or proteolytic cleavage sites from 690 unique proteins, indicating a high enrichment efficiency.
EXPERIMENTAL SECTION
Culture conditions
Aspergillus niger (NRRL3122) was grown and harvested as previously described.21 Briefly, A. niger was grown on potato dextrose agar plates at 30°C for 4 days for culture maintenance and spore production. Spores were harvested by washing with sterile 0.8% Tween 80 and counted using a hemocytometer.
Protein extraction
To extract proteins from whole cells, the mycelia were separated from the medium by filtering through two layers of sterile miracloth (EMD Chemicals Inc., Gibbstown, NJ). Approximately 5 g of frozen mycelia were ground into fine powder in liquid nitrogen using a chilled mortar and pestle. The sample was then re-suspended in 10 mL of 8 M urea buffer containing 0.5 % dithiothreitol (DTT) and 0.5% protease inhibitor cocktail (for use with fungal and yeast extracts; #P8215, Sigma-Aldrich, St. Louis, MO). After incubating at room temperature for 1 h with intermittent vortexing, insoluble material was removed by centrifugation (15,000 × g for 15 min). The supernatant was transferred to a new tube and centrifuged again (15,000 × g for 15 min). The collected protein sample in the supernatant was subjected to acetone precipitation by adding four volumes of cold acetone (−20 °C) and the sample was placed at −20 °C overnight. After centrifugation, the protein pellet was re-dissolved in 6 M guanidine hydrochloride containing 250 mM triethylammonium bicarbonate (TEAB, Sigma-Aldrich) buffer (pH 8.2) and stored at −20° C until further use.
Alkylation of protein thiols
Protein samples (100–150 μg) dissolved in 6 M guanidine hydrochloride containing 250 mM TEAB were denatured and reduced in 10 mM DTT for 1h at 37° C followed by alkylation of cysteine residues with 40 mM iodoacetamide (IAA) for 1 h at 37° C in the dark. The TEAB buffer was selected for amine derivatization because it does not react with amine-reactive reagents and is easy to remove.
Chemical derivatizations of amino groups
The pH of alkylated protein samples was adjusted to 10.5 by the addition of 5N NaOH solution. The samples were mixed with o-methylisourea solution (pH 10.5) to reach a final concentration of 1 M o-methylisourea. After brief vortexing and centrifugation, the samples were incubated for >24 h at 4°C with constant shaking. The pH of the samples was checked again after the first 12h incubation and readjusted to 10.5 with 5N NaOH if necessary. 20% trifluoroacetic acid was added to drop the pH of the protein samples to ~8.0. The samples were then labeled using N-succinimidyl S-acetylthioacetate (SATA, Pierce, Rockford, IL) at a SATA/protein ratio of 50 nmol/μg without additional desalting or buffer exchange. The samples were incubated for 1 h at room temperature. The SATA-labeling step was repeated by adding a new aliquot of SATA reagent into the sample. Subsequently, a 10-fold excess of hydroxylamine to SATA was added to the sample, which was then incubated for 2 h at room temperature to quench any unreacted SATA and to release the thiol groups from the protein N-termini. Following chemical derivatization, the protein samples were desalted by buffer exchange into 50 mM ammonium bicarbonate buffer, using Amicon Ultra centrifugal filters with 10K molecular weight cut off (Millipore, Billerica, MA).
Protein digestion
After determining protein concentration using a BCA protein assay (Pierce), acetonitrile was added to the desalted protein samples to reach a final concentration of 20% (v/v) in order to increase protein solubility and digestion efficiency. Samples were digested overnight at 37°C, using sequencing-grade modified-trypsin (Promega, Madison, WI) with a 1:50 enzyme to protein ratio (w/w). Digested samples were lyophilized to remove acetonitrile and buffer solution in preparation for resin-assisted enrichment of N-terminal peptides.
Capture of N-terminal peptides using thiol-reactive resin
A slightly modified version of our cysteinyl peptide enrichment protocol17 was used to capture the derivatized N-terminal peptides. The lyophilized peptides were resuspended in 150 μL of 50 mM Tris-HCl/20 mM EDTA buffer (pH 7.5) and 1.0 μL of 100 mM DTT. After incubating for 1 h at 37 °C, the samples were subjected to pre-washed thiol-reactive resin in a spin column (Pierce) that contained 35 mg of dried thiopropyl Sepharose 6B (GE Healthcare, Pittsburgh, PA) and had a final volume of ~100 μL after hydration. The mixture was incubated for 1 h at room temperature to allow sufficient time for the resin to capture thiol-containing N-terminal peptides. Non-N-terminal peptides were removed by washing the resin five times with each of the following solutions: 2M urea, 2 M NaCl, and 80% acetonitrile in 0.1% trifluoroacetic acid. The disulfide-based covalently captured N-terminal peptides were eluted with 2 × 100 μL of 20 mM DTT and then alkylated using 100 μL of 80 mM IAA. The samples were desalted using C18 SPE prior to LC-MS/MS analysis.
LC-MS/MS analysis
The N-terminal peptide samples were analyzed using a fully automated nano-LC system22 coupled on-line with an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific) modified with an in-house manufactured electrospray ionization interface. The reversed phase capillary column (75 μm i.d. × 65 cm long) column was slurry packed using 3 μm Jupiter C18 particles (Phenomenex). The column was connected via a Valco 100 μm i.d. stainless steel union to a chemically etched fused silica emitter (20 μm i.d.). Mobile phases consisted of 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). An exponential gradient that started with 100% A and gradually increased to 60% B over the course of 100 min at a flow-rate of ~500 nL/min was utilized for the LC separation. The LTQ-Orbitrap was operated in data-dependent mode with an m/z range of 400–2000. The ten most abundant ions determined by MS were selected for MS/MS, which used a normalized collision energy setting of 35%. A dynamic exclusion of 1 min was applied to reduce repetitive analysis of the same abundant precursor ion.
Data analysis
LC-MS/MS raw data were converted into .dta files using Extract_MSn (version 3.0) in Bioworks Cluster 3.2 (Thermo Fisher Scientific, Cambridge, MA). The SEQUEST algorithm23 (version 27, revision 12) was used to search all MS/MS spectra against the Aspergillus niger JGI data base (version 2006 2006–04–04; consisting of 11200 protein entries) using a parameter file with static cysteine alkylation (+57.022 Da), static lysine guanidination (+42.022 Da), and dynamic alkylated SATA modification (+131.004 Da) on the N-terminal residue of a peptide. Peptides were filtered based on the MS-generating function (MSGF) score24–25 value. The final peptide identifications have <1% false discovery rate (FDR) at the unique peptide level. Only fully tryptic and semi-tryptic peptides were considered.
RESULTS AND DISCUSSION
Resin-assisted enrichment strategy
Our strategy for enriching N-terminal peptides is depicted in Fig. 1a. In this strategy, the protein cysteine residues are first blocked through reduction by DTT and alkylation by IAA. Protein lysine ε-amines are then converted into homoarginine by a guanidination reaction using o-methylisourea (Fig. 1b).13, 26 After blocking all lysine ε-amines, a protected thiol-ester group is introduced to the protein N-termini by SATA, an amine reactive reagent. The protected thiol-ester group is then readily converted to free thiols by treating the samples with hydroxylamine (Fig. 1c). If the reactions of these blocking and derivatization steps are achieved with high efficiencies only the protein N-termini contain free thiols. These thiol-containing N-terminal peptides can be enriched either at the protein level or at the peptide level.19 For protein-level enrichment, on-resin trypsin digestion can be performed and all non-N-terminal peptides can be removed prior to the elution of N-terminal peptides. For peptide level enrichment, the thiol-containing N-terminal peptides are captured after in-solution trypsin digestion by the thiol-affinity resin as previously described.17, 19 In this work, the peptide-level enrichment protocol was utilized to take advantage of the better binding capacity that the resin has for peptides. All derivatization steps were performed in guanidine HCl to avoid urea-related N-terminal carbamylation, a potential artifact for N-terminal peptides.
Figure 1.
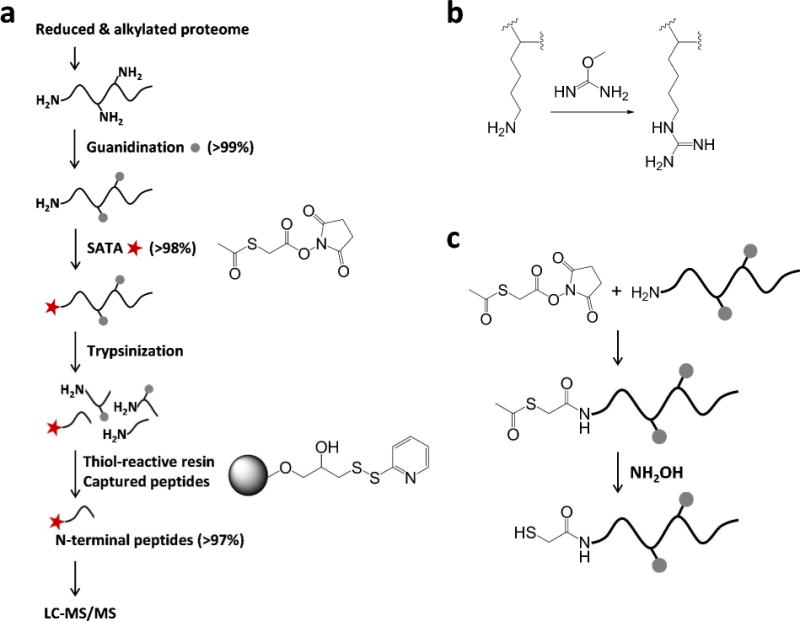
Schematic diagrams of thiol affinity resin-assisted enrichment of N-terminal peptides. (a) Overall enrichment strategy. The reaction or capture efficiency is indicated in parentheses. (b) Guanidination reaction. (b) SATA-labeling.
Assessment of derivatization reaction efficiencies
Effective blocking of protein thiols is an essential first step in achieving the enrichment specificity necessary to capture N-terminal peptides using a thiol-affinity resin. Mouse plasma was used as a test sample (Supplementary Table 1) and spectral counting as a semi-quantitative measure.27–28 Our data showed that the conditions (10 mM DTT and 40 mM IAA) used to reduce/alkylate cysteines at the protein level resulted in nearly complete alkylation (99.6%). This finding was further confirmed by MS/MS spectra obtained for cell lysate samples of A niger, i.e., ~3100 spectra were identified as N-terminal peptides while only 15 spectra were identified as cysteine-containing non-N-terminal peptides (Supplementary Table 2). These results demonstrated that the small percentage (~0.5%) of unalkylated cysteine-containing peptides had only a negligible effect on the final enrichment specificity for N-terminal peptides.
Another factor critical to the success of this approach is the efficiency and selectivity of converting lysine ε-amines to homoarginines by the guanidination reaction at the protein level (Fig. 1b). If guanidination efficiency is low, then unblocked lysine residues compromise the enrichment efficiency of N-terminal peptides. Conversely, over-guanidination of N-terminal amines can occur in some reaction conditions, e.g., high temperature or poorly controlled pH.29–30 Over-guanidination results in a loss of N-terminal peptides, as blocked N-terminal amines will not be converted to reactive thiols. The selectivity of guanidination on lysine ε-amines rather than N-terminal α-amines has been largely attributed to the steric protection of α-amines given by the peptide bond and the amino side chains.29, 31 Indeed, guanidination on α-amines was often pnly observed on N-terminal glycine due to the reduced steric hindrance of glycine.29, 31 Another potentially important factor contributing to the selectivity is that the lysine ε-amines is a much better nucleophile than the N-terminal α-amine due to the electron-withdrawing nature of the adjacent carbonyl group. After exploring a range of conditions, we observed that the most effective reaction condition was incubation at 4° C and pH 10.5 for 24 h, as shown by the spectral count data (Table 1). Figure 2a further shows the specificity of guanidination for lysine amines, which was found to be ~ 99% based on the extracted ion chromatogram (XIC) peak areas. In contrast, incubation at 37 °C for 4 h resulted in a ~ 5% increase in undesirable N-terminal over-guanidination (Fig. 2b). We note that in both cases the guanidination efficiency was high as only guanidinated peptides were detected.
Table 1.
Assessment of guanidination efficiency of lysine ε-amines under different conditions (Spectral count of identified peptides was used for the estimation).
| Samples | Incubation conditions | Identified peptides | Lysine-containing peptides | Guanidinated lysine-peptides | Guanidination efficiency |
|---|---|---|---|---|---|
| Bovine serum albumin | 4° C, 24 h | 473 | 262 | 260 | 99.2% |
| Bovine serum albumin | 37° C, 4 h | 310 | 148 | 145 | 98.0% |
| Bovine serum albumin | 65° C, 20 min | 839 | 440 | 207 | 47% |
| Aspergillus nigerlysate | 4° C, 24 h | 2,725 | 1,300 | 1,290 | 99.2% |
All reactions were performed at pH 10.5. The number of identified peptides indicates all identified MS/MS spectra with FDR <0.5%.
Figure 2.
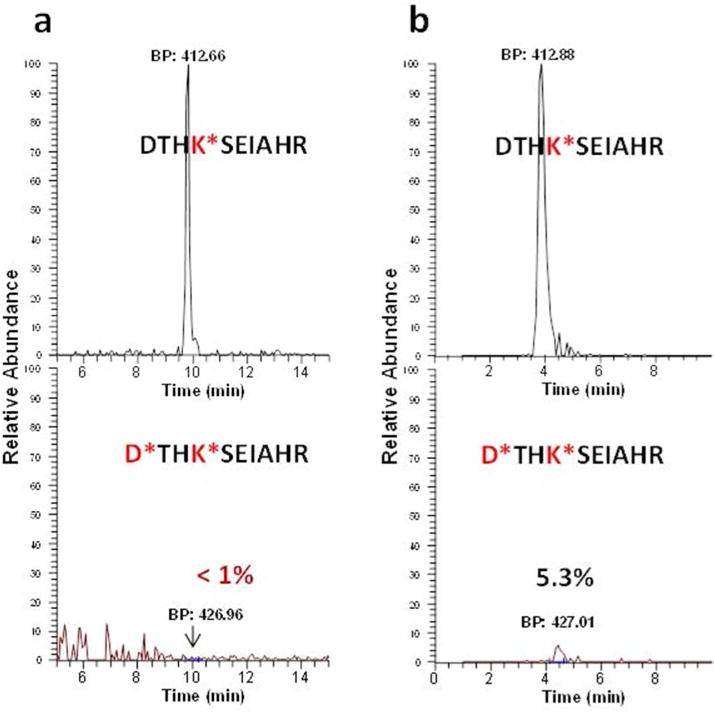
Specificity of guanidination on lysine amines. Extracted ion chromatographs of N-terminal peptides (DTHKSEIAHR) of bovine serum albumin from two different incubation conditions: (a) at 4 °C for 24 h, and (b) at 37 °C for 4 h. The undesirably guanidinated peptide on protein N-terminal amine was display in the bottom. Note that there was an ~5 min shift of retention times with a 100-min separation for the observed peptides between two different runs. This was due to the relative poor reproducibility in retention times for LC separations operated under a constant pressure.
Next, we tested the reaction efficiency of introducing a protected thiol group to primary amines. Due to the limited number of N-terminal peptides available when using a standard protein or standard protein mixture, lysine amines were utilized to examine the reaction efficiency, as N-terminal and lysine amines should exhibit similar reaction efficiencies when using amine-reactive reagents. Three reagents (dimethyl 3,3′-dithiobispropionimidate, N-succinimidyl 3–(2-pyridyldithio)propionate, and SATA) that contain a thiol group were initially explored using bovine serum albumin (BSA). The N-succinimidyl 3–(2–pyridyldithio)propionate reagent encountered a solubility issue in aqueous buffer and a substantial portion of organic solvent (~40% acetonitrile) was necessary to dissolve it in a coupling buffer, which caused protein precipitation during the course of the reaction. The protein labeling of BSA using dimethyl 3,3′-dithiobispropionimidate reagent was observed to have a low labeling efficiency (~45%) based on MS/MS spectral count data. The SATA reagent was observed to be the best for this step based on its high labeling efficiency at the protein level, as well as its good solubility in buffer solution. Based on the MS/MS spectral count data, ~93% of lysine-containing peptides from BSA were labeled with SATA reagent. Moreover, ~98% reaction efficiency was achieved based on the XICs of major peaks (Supplementary Figure 1). The observed high reaction efficiency was consistent with our previous work, which used the SATA reagent for a similar reaction and demonstrated that the release of free thiol groups from SATA labeled-peptides using hydroxylamine was close to 100%.20
In our strategy, alkylation, guanidination, SATA-labeling, and thioester deprotection reactions are performed sequentially in one sample vial. This was done without additional sample clean-up in order to minimize potential protein loss (note that pH of the sample was adjusted for each reaction as necessary). We observed that this sequential reaction strategy did not compromise the efficiency of any chemical reaction based on our final enriched N-terminal peptide identification data (Supplementary Table 2). Following buffer exchange to remove all reagents and trypsin digestion, the peptide mixture was subjected to resin-assisted enrichment of thiol-containing peptides. In general, the procedure involved a buffer exchange step and a single SPE clean-up step for all chemical reactions, including resin-assisted capturing.
Application to an A. niger cell lysate
Following optimization, we applied the strategy to profile proteolytic cleavage sites of proteins in A. niger, a filamentous fungus of interest for biomass degradation due to its ability to produce many extracellular hydrolytic enzymes.32 Based on the results from LC-MS/MS analysis of two process replicates of enriched samples, 1672 uniquely labeled N-terminal peptides from 690 proteins were identified with FDR <0.5% (Supplementary Tables 2–5). Furthermore, ~98% of the MS/MS spectra were identified as labeled thiol-containing N-terminal peptides, demonstrating the high specificity of the enrichment strategy. Among the non-specific peptides, 0.5% was from unblocked cysteine-containing peptides and 1.2% from non-specific bound peptides (Supplementary Table 2).
An interesting observation related to the identified N-terminal peptide sequences is that 423 of the identified N-terminal peptides ended with a homoarginine residue (Supplementary Table 3). This result suggest that the homoarginine residue, which is slightly longer but structurally similar to arginine (Fig. 1b), remains susceptible to trypsin cleavage, although the catalytic kinetics may be slower than those of either lysine or arginine residues. Figure 3 shows a representative MS/MS spectrum of a homoarginine-ending N-terminal peptide. The ability of trypsin to cleave at the converted homoarginine residue is an advantage of our method as it allows more N-terminal peptides ending with a lysine residue (>30%) to be identified. If the arginine residue was the only enzymatic cleavage site available after lysine residues are blocked, these peptides most likely would be too large (e.g., >5000 Da) to be identified by LC-MS/MS. For example, the long arginine-terminal peptide (54 amino acid residues and >6000 Da) shown in Figure 3 could only be identified because of cleavage on the homoarginine residue. It should be noted that the use of multiple enzymes such as trypsin and Glu-C is an alternative strategy for facilitating identification of large peptides by providing additional cleavage sites.13
Figure 3.
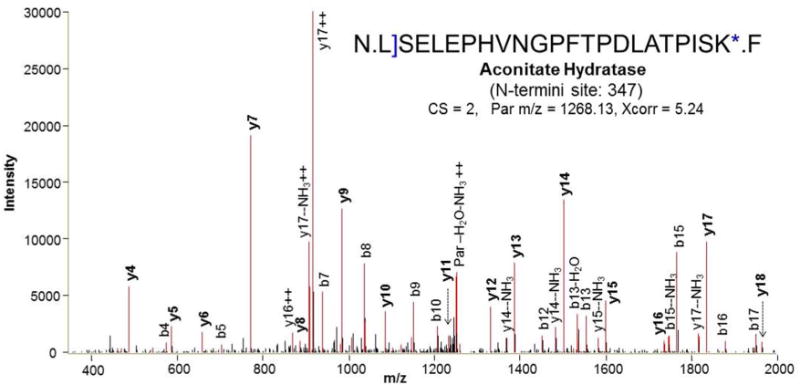
MS/MS spectrum of N-terminal peptide of aconitate hydratase with guanidinated lysine C-terminus. The single bracket and asterisk indicate SATA-labeled and guanidinated residues, respectively.
The identified N-terminal peptides were further examined for known annotations. Figure 4a shows that only 5.6% were annotated as known or predicted sites that comprised intact N-termini, sites with methionine initiator removed, and sites with a signal peptide or a mitochondrial targeting peptide removed (Supplementary Table 4). Our data confirmed 27 N-terminal cleavage sites predicted to remove either signal or mitochondrial targeting peptides (from SignalP33–34 and TargetP33 prediction tools), as well as 62 original protein N-termini–among which are proteins with methionine initiator removed. These results lend support to the validity of our method and also the accuracy of the predictions. Six other sites that were predicted to remove mitochondrial targeting peptides were either one or two residues off the observed cleavage sites (Supplementary Table 4), which raises the possibility that the predictions may be inaccurate.
Figure 4.
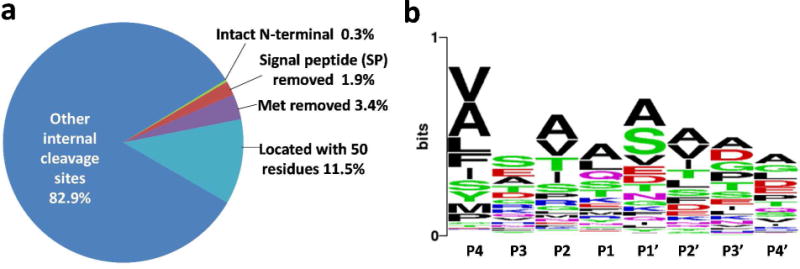
(a) The distribution of the identified N-terminal sites. (b) Sequence motif of N-terminal cleavage sites; potential sites of signal-peptide removal, which were located within first 50 amino acid residues.
A motif analysis was performed for the non-annotated proteolytic cleavage sites that were located within the first 50 amino acid residues (Fig. 4b). The alanine-rich and relatively serine-rich sequence motifs of these peptides provide only weak evidence that some of the sites may correspond to signal peptides being removed. Overall, the majority of the identified N-terminal sites originated from internal protein cleavage sites with no apparent specific sequence motifs, which suggests a complexity of protease activities in A. niger. This observation is consistent with the finding that A. niger produces a variety of proteases.35
CONCLUSIONS
Resin-assisted enrichment of N-terminal peptides coupled with LC-MS/MS provides an effective approach for proteome-wide profiling of proteolytic cleavage sites. The enrichment protocol requires relatively small amounts of starting material (100–150 μg total proteins) to yield 5–10 μg total N-terminal peptides for LC-MS/MS. Furthermore, resin-assisted thiol-affinity capture can be performed at either the peptide-level as in this study or at the protein-level, depending on the applications. Protein-level capture is detergent-friendly in that all reagents can be readily removed after capturing thiol-containing proteins on the resin.
The strategy also is amenable to quantitative studies (e.g., for identifying protease substrates or novel functional proteins) by using an 18O-labeled “universal” reference,36–37 stable isotope labeling by amino acids in cell cultures,38 or label-free quantitative approaches. In addition to facilitating the discovery of specific functional proteolytic isoforms in quantitative studies, the N-termini profiling may provide a resource for other applications, such as characterization of intact protein isoforms using top-down proteomics.39
Supplementary Material
Acknowledgments
Portions of this work were supported by the U. S. Department of Energy (DOE) Early Career Research Award, NIH grants DP2OD006668 and P41GM103493, and the DOE Office of Biological and Environmental Research Genome Sciences Program under the Pan-omics project. Experimental work was performed in the Environmental Molecular Sciences Laboratory, a DOE/BER national scientific user facility at Pacific Northwest National Laboratory (PNNL) in Richland, Washington. PNNL is operated by Battelle for the DOE under contract DE-AC05-76RLO-1830.
Footnotes
Supporting Information Available
Supplemental information includes Supplementary Tables 1 to 5 and Supplementary figure 1. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Lopez-Otin C, Overall CM. Nat Rev Mol Cell Biol. 2002;3:509–19. doi: 10.1038/nrm858. [DOI] [PubMed] [Google Scholar]
- 2.McDonald L, Robertson DH, Hurst JL, Beynon RJ. Nat Methods. 2005;2:955–957. doi: 10.1038/nmeth811. [DOI] [PubMed] [Google Scholar]
- 3.Gevaert K, Goethals M, Martens L, Van Damme J, Staes A, Thomas GR, Vandekerckhove J. Nat Biotechnol. 2003;21:566–569. doi: 10.1038/nbt810. [DOI] [PubMed] [Google Scholar]
- 4.McDonald L, Beynon RJ. Nat Protoc. 2006;1:1790–8. doi: 10.1038/nprot.2006.317. [DOI] [PubMed] [Google Scholar]
- 5.Kleifeld O, Doucet A, auf dem Keller U, Prudova A, Schilling O, Kainthan RK, Starr AE, Foster LJ, Kizhakkedathu JN, Overall CM. Nat Biotechnol. 2010;28:281–8. doi: 10.1038/nbt.1611. [DOI] [PubMed] [Google Scholar]
- 6.Zhang X, Ye J, Engholm-Keller K, Hojrup P. Proteomics. 2011;11:81–93. doi: 10.1002/pmic.201000453. [DOI] [PubMed] [Google Scholar]
- 7.Van Damme P, Martens L, Van Damme J, Hugelier K, Staes A, Vandekerckhove J, Gevaert K. Nat Methods. 2005;2:771–7. doi: 10.1038/nmeth792. [DOI] [PubMed] [Google Scholar]
- 8.Impens F, Colaert N, Helsens K, Ghesquiere B, Timmerman E, De Bock PJ, Chain BM, Vandekerckhove J, Gevaert K. Mol Cell Proteomics. 2010;9:2327–33. doi: 10.1074/mcp.M110.001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Xu G, Shin SB, Jaffrey SR. Proc Natl Acad Sci U S A. 2009;106:19310–5. doi: 10.1073/pnas.0908958106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mahrus S, Trinidad JC, Barkan DT, Sali A, Burlingame AL, Wells JA. Cell. 2008;134:866–876. doi: 10.1016/j.cell.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wildes D, Wells JA. Proc Natl Acad Sci U S A. 2010;107:4561–6. doi: 10.1073/pnas.0914495107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agard NJ, Maltby D, Wells JA. Mol Cell Proteomics. 2010;9:880–93. doi: 10.1074/mcp.M900528-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Timmer JC, Enoksson M, Wildfang E, Zhu W, Igarashi Y, Denault JB, Ma Y, Dummitt B, Chang YH, Mast AE, Eroshkin A, Smith JW, Tao WA, Salvesen GS. Biochem J. 2007;407:41–48. doi: 10.1042/BJ20070775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Forrester MT, Thompson JW, Foster MW, Nogueira L, Moseley MA, Stamler JS. Nat Biotechnol. 2009;27:557–559. doi: 10.1038/nbt.1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qian WJ, Goshe MB, Camp DG, 2nd, Yu LR, Tang K, Smith RD. Anal Chem. 2003;75:5441–50. doi: 10.1021/ac0342774. [DOI] [PubMed] [Google Scholar]
- 16.Zhou H, Ranish JA, Watts JD, Aebersold R. Nat Biotechnol. 2002;20:512–5. doi: 10.1038/nbt0502-512. [DOI] [PubMed] [Google Scholar]
- 17.Liu T, Qian WJ, Strittmatter EF, Camp DG, Anderson GA, Thrall BD, Smith RD. Analytical Chemistry. 2004;76:5345–5353. doi: 10.1021/ac049485q. [DOI] [PubMed] [Google Scholar]
- 18.Liu M, Hou J, Huang L, Huang X, Heibeck TH, Zhao R, Pasa-Tolic L, Smith RD, Li Y, Fu K, Zhang Z, Hinrichs SH, Ding SJ. Analytical Chemistry. 2010;82:7160–8. doi: 10.1021/ac100569d. [DOI] [PubMed] [Google Scholar]
- 19.Su D, Shukla AK, Chen B, Kim JS, Nakayasu E, Qu Y, Aryal U, Weitz K, Clauss TR, Monroe ME, Camp li DG, Bigelow DJ, Smith RD, Kulkarni RN, Qian WJ. Free Radic Biol Med. 2012;57C:68–78. doi: 10.1016/j.freeradbiomed.2012.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Q, Qian WJ, Knyushko TV, Clauss TR, Purvine SO, Moore RJ, Sacksteder CA, Chin MH, Smith DJ, Camp DG, 2nd, Bigelow DJ, Smith RD. J Proteome Res. 2007;6:2257–68. doi: 10.1021/pr0606934. [DOI] [PubMed] [Google Scholar]
- 21.Wang L, Aryal UK, Dai Z, Mason AC, Monroe ME, Tian ZX, Zhou JY, Su D, Weitz KK, Liu T, Camp DG, 2nd, Smith RD, Baker SE, Qian WJ. Journal of Proteome Research. 2012;11:143–56. doi: 10.1021/pr200916k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Livesay EA, Tang K, Taylor BK, Buschbach MA, Hopkins DF, LaMarche BL, Zhao R, Shen Y, Orton DJ, Moore RJ, Kelly RT, Udseth HR, Smith RD. Anal Chem. 2008;80:294–302. doi: 10.1021/ac701727r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Eng JK, McCormack AL, Yates JR. Journal of The American Society for Mass Spectrometry. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 24.Kim S, Gupta N, Pevzner PA. J Proteome Res. 2008;7:3354–3363. doi: 10.1021/pr8001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim S, Mischerikow N, Bandeira N, Navarro JD, Wich L, Mohammed S, Heck AJ, Pevzner PA. Mol Cell Proteomics. 2010;9:2840–52. doi: 10.1074/mcp.M110.003731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brancia FL, Montgomery H, Tanaka K, Kumashiro S. Analytical Chemistry. 2004;76:2748–55. doi: 10.1021/ac030421+. [DOI] [PubMed] [Google Scholar]
- 27.Liu H, Sadygov RG, Yates JR., 3rd Anal Chem. 2004;76:4193–201. doi: 10.1021/ac0498563. [DOI] [PubMed] [Google Scholar]
- 28.Qian WJ, Jacobs JM, Camp DG, II, Monroe ME, Moore RJ, Gritsenko MA, Calvano SE, Lowry SF, Xiao W, Moldawer LL, Davis RW, Tompkins RG, Smith RD. Proteomics. 2005;5:572–584. doi: 10.1002/pmic.200400942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beardsley RL, Reilly JP. Anal Chem. 2002;74:1884–90. doi: 10.1021/ac015613o. [DOI] [PubMed] [Google Scholar]
- 30.Kim JS, Song JS, Kim Y, Park SB, Kim HJ. Analytical and bioanalytical chemistry. 2012;402:1911–9. doi: 10.1007/s00216-011-5642-7. [DOI] [PubMed] [Google Scholar]
- 31.Beardsley RL, Karty JA, Reilly JP. Rapid Commun Mass Spectrom. 2000;14:2147–53. doi: 10.1002/1097-0231(20001215)14:23<2147::AID-RCM145>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- 32.Andersen MR, Salazar MP, Schaap PJ, van de Vondervoort PJ, Culley D, Thykaer J, Frisvad JC, Nielsen KF, Albang R, Albermann K, Berka RM, Braus GH, Braus-Stromeyer SA, Corrochano LM, Dai Z, van Dijck PW, Hofmann G, Lasure LL, Magnuson JK, Menke H, Meijer M, Meijer SL, Nielsen JB, Nielsen ML, van Ooyen AJ, Pel HJ, Poulsen L, Samson RA, Stam H, Tsang A, van den Brink JM, Atkins A, Aerts A, Shapiro H, Pangilinan J, Salamov A, Lou Y, Lindquist E, Lucas S, Grimwood J, Grigoriev IV, Kubicek CP, Martinez D, van Peij NN, Roubos JA, Nielsen J, Baker SE. Genome Res. 2011;21:885–97. doi: 10.1101/gr.112169.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Emanuelsson O, Brunak S, von Heijne G, Nielsen H. Nature protocols. 2007;2:953–71. doi: 10.1038/nprot.2007.131. [DOI] [PubMed] [Google Scholar]
- 34.Petersen TN, Brunak S, von Heijne G, Nielsen H. Nat Methods. 2011;8:785–6. doi: 10.1038/nmeth.1701. [DOI] [PubMed] [Google Scholar]
- 35.Schaal R, Kupfahl C, Buchheidt D, Neumaier M, Findeisen P. J Microbiol Methods. 2007;71:93–100. doi: 10.1016/j.mimet.2007.05.010. [DOI] [PubMed] [Google Scholar]
- 36.Qian WJ, Liu T, Petyuk VA, Gritsenko MA, Petritis BO, Polpitiya AD, Kaushal A, Xiao W, Finnerty CC, Jeschke MG, Jaitly N, Monroe ME, Moore RJ, Moldawer LL, Davis RW, Tompkins RG, Herndon DN, Camp DG, Smith RD. J Proteome Res. 2009;8:290–9. doi: 10.1021/pr800467r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim JS, Fillmore TL, Liu T, Robinson E, Hossain M, Champion BL, Moore RJ, Camp DG, 2nd, Smith RD, Qian WJ. Mol Cell Proteomics. 2011;10:M110007302. doi: 10.1074/mcp.M110.007302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Mol Cell Proteomics. 2002;1:376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 39.Tran JC, Zamdborg L, Ahlf DR, Lee JE, Catherman AD, Durbin KR, Tipton JD, Vellaichamy A, Kellie JF, Li M, Wu C, Sweet SM, Early BP, Siuti N, LeDuc RD, Compton PD, Thomas PM, Kelleher NL. Nature. 2011;480:254–8. doi: 10.1038/nature10575. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.