

Abstract
Background
We have recently demonstrated that treatment with suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor, significantly improves survival in a rodent model of lipopolysaccharide (LPS)-induced endotoxic shock. However, the precise mechanisms have not been well defined. The aim of this study was to investigate the impact of SAHA treatment on gene expression profiles at an early stage of shock.
Methods
Male C57B1/6J mice were treated with or without SAHA (50 mg/kg, i.p), followed by a lethal dose of LPS (20 mg/kg, i.p) and a second dose of SAHA. Lungs of the animals (LPS and SAHA+LPS groups, n=3/group) were harvested 3 hours post-LPS insult. Sham mice (no LPS and no SAHA) served as controls. RNA was isolated from the tissues and gene expression was analyzed using Affymatrix microarray (23,000 genes). A lower confidence bound (LCB) of fold change was determined for comparison of LPS vs. SAHA+LPS, and genes with LCB >2 were considered to be differentially expressed. RT-PCR, western blotting and tissue staining were performed to verify the key changes. Network graphs were used to determine gene interaction, and biological relevance.
Results
The expression of many genes known to be involved in septic pathophysiology changed after the LPS insult. Interestingly, a number of genes not previously implicated in the septic response were also altered. SAHA treatment attenuated expression of several key genes involved in inflammation. It also reduced neutrophil infiltration in lungs and histological evidence of acute lung injury. Further analysis confirmed genes engaged in the cellular and humoral arms of innate immune system that were specifically inhibited by SAHA. Gene network analysis identified numerous molecules for the potential development of targeted therapies.
Conclusions
Administration of SAHA in a rodent model of LPS shock rapidly modulates gene transcription, with an attenuation of inflammatory mediators derived from both arms (cellular and humoral) of the innate immune system. This may be a novel mechanism responsible for the survival advantage seen with SAHA treatment.
Keywords: Endotoxic shock, microarray, immune, inflammation, suberoylanilide hydroxamic acid, lung
INTRODUCTION
Sepsis continues to be the leading cause of morbidity and mortality in intensive care units and is responsible for more than 200,000 deaths each year 1, 2. Sepsis manifests as a systemic inflammatory response syndrome (SIRS) that is caused by infection 3. At the molecular level, this inflammatory response is characterized acutely by an upregulation of innate immunological mediators that increase endothelial permeability, neutrophil infiltration, platelet aggregation, and reduce blood flow. Inflammation and tissue hypoxia further derange host immunity leading to aberrant leukocyte trafficking and increased ischemic insult4. Unless treated effectively, it can progress to hemodynamic instability (septic shock) and organ injury, which often begins in the lungs but can progress to multi-organ failure.
Despite aggressive research efforts early recognition of sepsis remains challenging, and in the absence of effective targeted therapies outcomes continue to be poor 1. Changes in gene expression patterns constitute a key regulatory step in the innate immune response and therefore in the acute pathology of sepsis. Two opposing enzyme families, histone deacetylases (HDAC) and histone acetyl transferases (HAT), control the balance of acetylation in a cell, and can be therapeutically targeted by a class of drugs called histone deacetylase inhibitors (HDACI). Acetylation of lysine residues on histones neutralizes the attraction between positively charged histones and negatively charged nucleic acids, which increases the accessibility of DNA to transcription machinery, thereby facilitating gene expression. In gram negative sepsis, bacterial outer membrane component lipoylpolysaccharide (LPS) binds to the host toll like receptor 4 (TLR 4) to alter gene expression, which promotes an acute phase immune response. Although it lacks the complexity of poly-microbial infections, LPS administration recapitulates much of the cellular and systemic manifestations of sepsis in a highly reproducible fashion, making it an attractive experimental agent5, 6.
Innate immune system is the first line of defense against pathogen invasion. It has two components (cellular and humoral) that complement each other. Toll-like receptors (TLR), involved in the cellular arm, represent a family of related pathogens associated molecular patterns (PAMPs) receptors that recognize a variety of conserved microbial structures such as lipopolysaccharide (TLR4), lipoproteins and lipoarabinomannans (TLR2) and flagellin (TLR5) 7. Recognition of these structures not only causes activation and differentiation of cells directly involved in innate immunity, but also maturation of professional antigen-presenting cells, which initiate and shape the adaptive immune response 8. Components of the humoral arm include soluble pattern recognition molecules (PRMs) that recognize PAMPs and initiate the immune response in coordination with the cellular arm, therefore acting as functional ancestors of antibodies.
As a key component of humoral arm of innate immunity, pentraxin 3 (PTX3) has antibody-like functions: binding pathogens, activating and regulating complement cascade, agglutination and neutralization, facilitation of recognition via cellular receptors (opsonization), and regulation of inflammation. Macrophages, neutrophils, dendritic cells and endothelial cells are major producers of PTX3 in response to Toll-like receptor (TLR) engagement such as binding of LPS to TLR4 9 and inflammatory cytokines such as TNF-α and IL-1β 10. To date, effects of HDACI treatment on the PTX3 have not been studied in the setting of LPS-induced inflammation.
We have recently demonstrated that treatment with suberoylanilide hydroxamic acid (SAHA), a histone deacetylase inhibitor (HDACI), significantly improves survival in a rodent model of LPS-induced shock 11. However, the precise mechanisms underlying SAHA’s anti-inflammatory and pro-survival effects in the sepsis have not been well defined. Since the initiation and progression of septic shock are highly complex, multiple gene products and pathways are involved 12. Likewise, SAHA’s protective effects are likely the result of integrated influences on gene and protein expression involving multiple pathways in cellular and humoral immune responses. Hence, the aim of the current study was to use high-throughput methods to investigate the multidimensional impact of SAHA treatment on gene expression profiles at an early stage of LPS-induced shock.
MATERIALS AND METHODS
Materials
LPS (from Salmonella typhosa) and dimethyl sulfoxide (DMSO) were purchased from Sigma (St. Louis, MO). Suberoylanilide hydroxamic acid was purchased from Biomol International (Plymouth Meeting, PA). Primary antibody against PTX3 was purchased from R & D Systems (Minneapolis, MN). Anti-actin antibody was purchased from Sigma Chemical, Co. Anti-mouse and anti-rabbit IgG secondary antibodies were purchased from Amersham Biosciences (Piscataway, NJ). Protease Inhibitor Cocktail II was purchased from Calbiochem (San Diego, CA). All other chemicals in this study were of analytical grade and obtained from Sigma if not mentioned otherwise.
Animals
All the research was conducted in compliance with the Animal Welfare Act and was approved by the Institutional Animal Care and Use Committee. Male C57BL/6J mice (6 - 8 weeks; 20–25 g weight; Jackson Labs, Bar Harbor, ME) were housed in plastic cages, with free access to chow and water, and kept at room temperature (24 ± 2 °C) with alternative 12-h cycles of light and darkness.
Model
Male C57BL/6J mice were divided into three groups (1) single dose of LPS (20 mg/kg, dissolved in isotonic saline) was given intraperitoneal (i.p.) to induce lethal shock; (2) a single does of LPS (20 mg/kg i.p.) with SAHA (50 mg/kg, dissolved in DMSO) i.p. pre-treatment (2 hours prior to LPS) and post-treatment (2 hours after LPS); and (3) sham (no LPS, no SAHA).
DNA Microarrays
DNA microarrays were performed at the Genomics Core at Beth Israel Deaconess Medical Center. Total RNA was prepared from mouse lung tissue in RNAlater solution using an RNeasy Mini kit (Qiagen, Valencia, CA). Amounts, purity, and integrity of RNA were evaluated by UV spectrophotometry and an RNA-nano Bioanalyzer (Agilent, Palo Alto, CA). One μg of total RNA from each sample was used to robotically prepare antisense biotinylated RNA on the Affymetrix GeneChip Array Station (Affymetrix, Santa Clara, CA). Synthesis of single-stranded cDNA was done with the use of T7-oligo (dT) primer using the GeneChip HT One-Cycle cDNA Synthesis Kit (Affymetrix). The GeneChip HT IVT Labeling Kit was used to convert cDNA into cRNA. The biotinylated cRNAs were subsequently fragmented and mixed with hybridization cocktails, and hybridized onto the Affymetrix HT-mouse 430 A & B GeneChip (a high throughput platform array in a 24-well format) at 48 °C over 16 h. Array washing and staining were done using the Affymetrix GeneChip Array Station (Caliper Life Science, Mountain View, CA) following a robotic protocol according to the manufacturer’s instructions (Affymetrix). Arrays were then scanned by the GeneChip HT Scanner (Affymetrix), an automated epi-fluorescence imaging scanner, and signal intensity for each transcript (background-subtracted and adjusted for noise) was determined using Genechip operating software 2.0. The scanned array images were analyzed by dChip 13.
Gene Expression Analysis
Gene data analysis was performed using dChip software 11. CEL files (primary Affymetrix array data files) were loaded and normalized at the probe cell level by the Invariate Set Normalization method 14. A lower confidence bound (LCB) of fold change was determined for the comparison of LPS vs. SAHA+LPS, and genes with LCB > 1.5 were considered to be differentially expressed. The ANOVA test was carried out using a P value < 0.05 in order to define a set of significantly up- or down-regulated genes. The resulting genes were filtered for gene presence calls of >20 in 50% of samples. Hierarchical clustering analysis was performed on the genes that met the above criteria.
Real-time Polymerase Chain Reaction (Real-Time PCR)
RNA was converted into cDNA with High-Capacity cDNA Reverse Transcription kit, following the manufacturer’s protocol (Applied Biosystems, Foster City, CA). Equal amounts of cDNA were submitted to the PCR in the presence of SYBR green Master Mix, forward and reverse primers, and the ABI PRISM 7300 Real Time PCR detection machine. Primers were designed for specific genes using primer3 software, and are shown below. PCR was performed with 40 cycles of 15 seconds at 95 °C, and 1 minute at 60 °C. GAPDH was used as an internal control. Each sample was run in triplicates. Relative mRNA expression was calculated using the parameter threshold cycle (CT) values. The ΔCT was the difference in the CT values derived from the specific gene being assayed and the GAPDH mRNA. ΔΔCT represented the difference between the paired samples, as calculated by the formula ΔCT of a sample − ΔCT of reference (the average ΔCT of sham samples). The amount of target, normalized to GAPDH and reference, was calculated as 2−ΔΔCT.
Primers of selected genes for real-time PCR.
| Gene name | Forward primer (5′ – 3′) | Reverse primer (5′ – 3′) |
|---|---|---|
| TRAF6 | TTGCACATTCAGTGTTTTTGG | TGCAAGTGTCGTGCCAAG |
| PTX3 | AGGGTGGACTACAGATTGG | CCGATCCCAGATATTGAAGCC |
| TLR2 | GCCACCATTTCCACGGAC | GGCTTCCTCTTGGCCTGG |
| MyD88 | GGAGTCCGAGAAGCCTTTAC | GGATCATCTCCTGCACAAAC |
| TNFR-1 | CCGGGCCACCTGGTCCG | CAAGTAGGTTCCTTTGTG |
| TNFR-2 | GTCGCGCTGGTCTTCGAACTG | GGTATACATGCTTGCCTCACAGTC |
| CCL3 | ACTGCCTGCTGCTTCTCCTACA | AGGAAAATGACACCTGGCTGG |
| GAPDH | GGAGCGAGACCCCACTAACA | ACATACTCAGCACCGGCCTC |
Network Analysis
Network of selected genes was analyzed with VisAnt software that is freely available at http://visant.bu.edu 15.
Histology
Lungs were harvested 24 hours after LPS injection, fixed in 10% formalin, and embedded in paraffin. Tissues sections were stained with hematoxylin and eosin (H&E), and analyzed by a pathologist who was blinded to the group allocation of the samples.
Measurement of Myeloperoxidase (MPO) Activity
To estimate the extent of neutrophil infiltration into the lung, MPO was quantified according to the manufacturer’s instruction (Mouse MPO ELISA kit; Cell Sciences, Canton, MA). Mouse lungs (50 mg wet weight) from sham, LPS, and SAHA + LPS groups were removed and homogenized in 1 mL of a lysis buffer (200 mM NaCl, 5 mM EDTA, 10 mM Tris, 10% glycine [vol/vol], 1 mM phenylmethylsulfonyl fluoride, 1 2g/mL leupeptide, 28 2g/mL aprotinin). The samples were centrifuged twice at 1,500 g at 4 °C for 15 min, and supernatants were analyzed for MPO levels by sandwich enzyme-linked immunosorbent assay (ELISA).
Evaluation of LPS-induced Serum PTX3 in Vivo
Quantitative determination in the blood of mice was made using Quantikine ELISA Kit (R&D Systems, Minneapolis, MN) for PTX3 according to manufacturer’s instruction. The concentration of cytokine was measured by optical densitometry at 450 nm in a SpectramaxPlus 384 microplate reader (Molecular Devices, Sunnyvale, CA). All of the analyses were performed in triplicates.
Western Blot Analysis
Proteins (~100 μg per lane) were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) on 12% polyacrylamide gels and transferred onto nitrocellulose membranes (Bio-Rad Laboratories, Hercules, CA). The membranes were blocked in 0.05% PBS-Tween (PBST) containing 5% milk (Bio-Rad Laboratories) and then incubated with the primary antibody at 4 °C overnight. The primary antibody was detected by incubation with horseradish peroxidase-coupled second antibody (1:3,000 in PBST with 5% milk) at room temperature for 2 h. The chemiluminescence detection was performed by using Western Lighting Chemiluminescence Reagent Plus (PerkinElmer LAS, Inc., Boston, MA). Films were developed using a standard photographic procedure and quantitative analysis of detected bands was carried out by densitometer scanning using VersaDoc Imaging System (BioRad Laboratories).
Statistical Analysis
Statistical differences were determined by Student t tests for two-group and analysis of variance (ANOVA) for multiple-group comparisons, respectively using statistical software package (SSPS, Chicago, IL). Differences were considered to be statistically significant when p values were less than 0.05.
RESULTS
LPS and LPS+SAHA Treatment Differentially Regulate Gene Expression Acutely in Endotoxic Shock
mRNA levels of 208/22,626 genes were found to be differentially regulated in the LPS animals compared to the LPS and SAHA treated animals. Some of these genes that are well-known to be involved in septic pathophysiology (e.g. TNF-α, IL-1β, IL-10) changed after the LPS insult; however, a number of genes not previously implicated in the septic response, but known to be involved in innate immune and inflammatory responses, were also found to be altered. SAHA treatment attenuated most of these gene expression changes (Fig. 1).
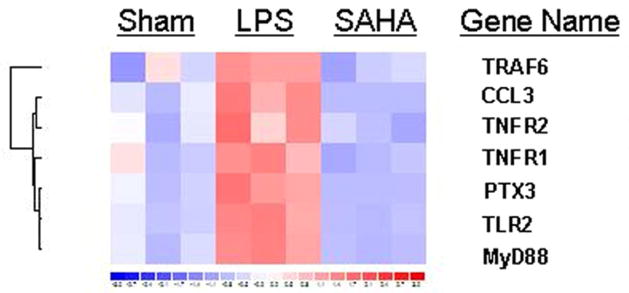
Figure 1. Hierarchical clustering of inflammatory response genes in LPS insult and SAHA treatment.

Each row represents a cDNA on the Affymetrix chip and each column represents an individual lung mRNA sample. Red represents over-expressed genes, and blue represents under-expressed genes. Sham: no LPS and no SAHA treated animals; LPS: lipopolysaccharide (LPS) treated animals; SAHA: suberoylanilide hydroxamic acid (SAHA) plus LPS treated animals.
Validation of Selected Genes with Real-Time PCR
dChiP analysis identified 12 key pathways that were specifically modulated by SAHA. A pro-inflammatory pathway involving tumor necrosis factor receptor 1 and 2 (TNFR1 and 2), tumor necrosis factor receptor associated factor 6 (TRAF6), toll like receptor 2 (TLR2), PTX3, myeloid differentiation primary response gene 88 (MyD88) and chemokine ligand 3 (CCL3) was up-regulated following LPS insult but down regulated by SAHA treatment (Fig 2A). These seven inflammatory response genes that were modulated by SAHA and identified by the microarray studies were verified using real-time PCR. In the microarray studies, TNFR-1 was down-regulated by 1.8-fold, TNFR-2 by 4-fold, TRAF6 gene by 1.84-fold, TLR2 by 9.5-fold, PTX3 by 110.7-fold, MyD88 by 9-fold, and CCL3 by 44.8-fold, in SAHA treated group compared with LPS group. Real-time PCR confirmed the down-regulation of all these genes (TNFR-1: 9.37-fold, p = 0.01; TNFR-2: 5.45-fold, p = 0.006; TRAF6: 1.93-fold, p = 0.024; TLR2: 18.27-fold, p = 0.0005; MyD88: 10.9-fold, p = 0.002; CCL3: 10.4-fold, p = 0.0001; PTX3: 32.04-fold, p = 0.03. Fig. 2B).
Figure 2. Real-time PCR evaluations of genes identified as differentially expressed between LPS insult and SAHA + LPS groups.

(A) A heat map showing seven of the genes in nine lung tissue samples are differentially expressed between LPS-induced septic shock and SAHA treatment groups. (B) Real-time PCR showing expression of TNFR1, TNFR2, TLR2, PTX3, MyD88, TRAF6 and CCL3 genes is significantly different between LPS-induced septic shock and SAHA treatment groups. The symbol * indicates that a value significantly (p<0.05) differs from LPS group. RQ, real-time quantitative PCR for relative mRNA expression levels (fold changes from sham).
SAHA Decreases Pulmonary Inflammatory Cell Infiltration
In the sham group, light microscopy showed minimal inflammatory cell infiltration in the lungs. In contrast, after LPS insult, there was development of overt tissue edema and infiltration of inflammatory cells. SAHA treatment markedly reduced the edema and cell infiltration (Fig. 3A). Furthermore, MPO activity as an index of neutrophils infiltration into the lung was examined. LPS-induced lung MPO activity was markedly increased compared with normal control. SAHA treatment significantly decreased the activity of MPO by 5-fold after LPS insult (P < 0.05) (Fig. 3B).
Figure 3. SAHA inhibits inflammatory infiltration into lungs of mice during the progression of LPS-induced septic shock.
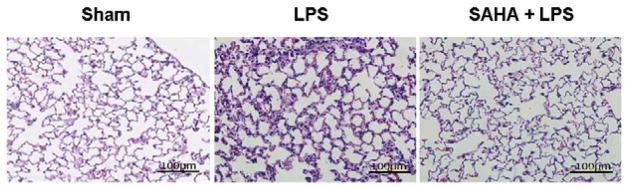
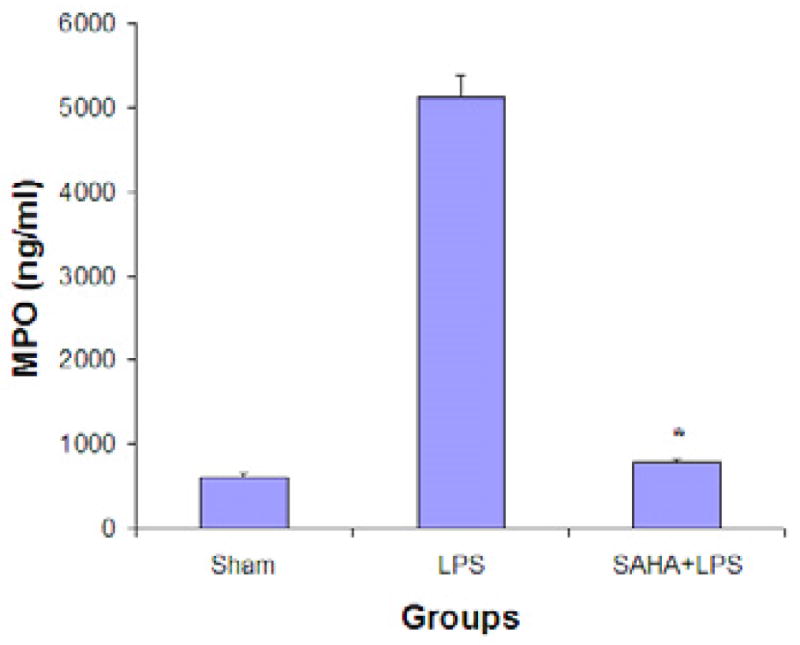
(A). Representative slides of lung sections (n = 3) from animal groups of sham, LPS, and SAHA + LPS. The pathologic changes were examined by hematoxylin and eosin (H&E) staining and light microscopy in the lung tissues of mice treated with or without SAHA at 24 h after LPS injection. The markedly increased infiltration of inflammatory cells such as neutrophils was observed in the lungs of mice from the LPS group, which was attenuated by SAHA treatment. Scale bars represent 100 μm. (B). Myeloperoxidase (MPO) activity was assessed for neutrophil infiltration in a lung of mice treated with or without SAHA in LPS insult. The activity was analyzed by quantifying MPO levels in lung tissues. Values represent the means ± SD (n=3). The symbol * indicates that a value significantly (p<0.05) differs from LPS group.
SAHA Decreases Protein Levels of PTX3 in Lung and Blood
PTX3 has recently been reported to accelerate lung injury in model of high tidal volume ventilation in mice 16. At the same time we have previously reported attenuation of lung injury with HDACI treatment. However, it was unclear if this benefit was through the modulation of the PTX3 levels. Therefore, we decided to look in more detail at the changes in PTX3 profile with SAHA treatment. In normal condition and at 1 h after LPS insult or SAHA treatment, PTX3 protein in circulation and the lung tissues was hardly detectable. LPS insult increased levels of the PTX3 in a time-dependent manner. At 24 h, the levels of PTX3 reached maximum among the designed time course. Protein levels in circulation and lung correlated with each other. SAHA treatment significantly reduced levels of PTX3 protein in lungs (p < 0.002 - 0.005) (Fig 4) as well as blood (p < 0.001) (Fig 5).
Figure 4. SAHA decreases expression of PTX3 in lungs of mice during the progression of LPS-induced endotoxic shock.

Whole tissue lysate of mouse lung from sham, LPS and LPS+SAHA groups at different time points after LPS insult was subjected to western blotting with anti-PTX3 and anti-actin antibodies. Specific bands were quantified by densitometry and expressed as means±SD (n=3). The symbol * indicates that a value significantly (p < 0.002) differs from the LPS group. Note: all time points (hours) in this study use LPS injection as the reference point (time zero).
Figure 5. SAHA decreases protein levels of PTX3 in circulation during the progression of LPS-induced endotoxic shock.
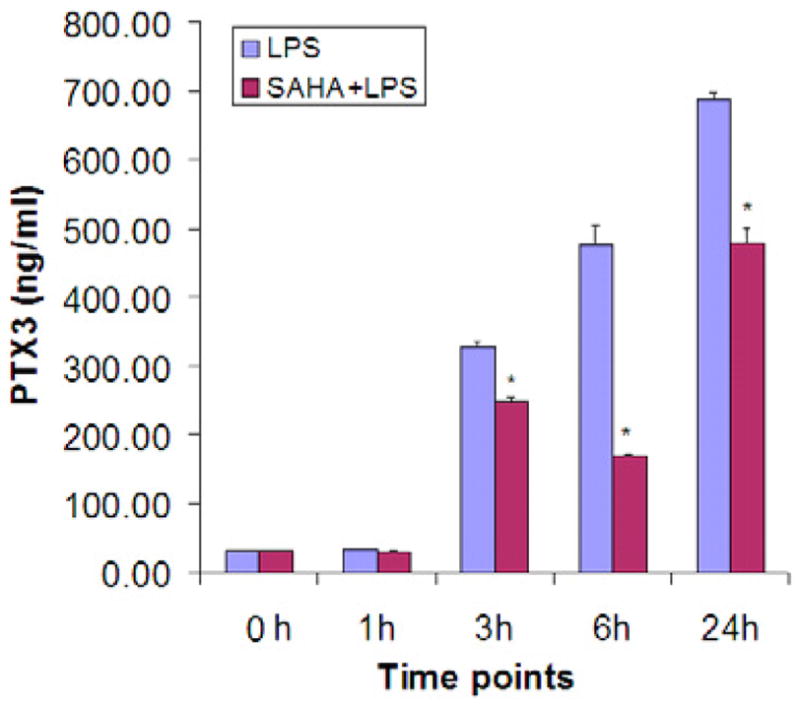
Serum levels of PTX3 protein were analyzed by ELISA (described in materials and methods). All analyses were performed in triplicate. The error bar indicates standard errors of means (n=3). The symbol * indicates that a value significantly (p < 0.001) differs from the LPS group.
Network Analysis: SAHA Attenuates Expression of Pro-Inflammatory Genes
Recently, VisAnt has been used for integrating biomolecular data into a cohesive, graphical interface 17. To analyze whether the seven selected genes could interact with each other in the context of LPS insult and SAHA treatment, we entered these seven genes into the VisAnt program and examined the computed results. As shown in Figure 6, there are some direct interactions between TLR2-MyD88 and MyD88-TRAF6, consistent with previous reports. Additionally, there are numerous indirect interactions among these genes, several of which have not been reported in the context of severe inflammation and pharmacologic treatment. This figure indicates that LPS insult may stimulate pathologic, pro-inflammatory gene-gene interactions, and that SAHA treatment likely attenuates the expression of these genes and their interactions.
Figure 6. Protein interaction map of the selected differentially expressed genes.

The VisAnt tool was utilized for mining and visualizing possible protein-protein interaction of the seven selected inflammatory response genes in septic shock and SAHA treatment. Green nodes represent genes that were differentially expressed between the LPS and SAHA treatment groups. Pink nodes represent genes that are possibly involved in the interaction. Symbol – in green nodes indicates that gene expression is inhibited by SAHA treatment.
DISCUSSION
We have previously demonstrated that SAHA treatment significantly improves survival in a lethal model of LPS-induced shock in mice 11. However, the precise anti-inflammatory mechanisms that are activated by this treatment remain poorly defined. In this study, high-throughput analysis identified numerous genes involved in inflammatory pathways that were rapidly upregulated by LPS exposure but attenuated by SAHA treatment. A number of pro-inflammatory genes noted in this study (i.e. coding for TLR4, TNF-α, IL-1β) are well known to be associated with LPS-induced (endotoxic) shock. But we also identified several other pro-inflammatory genes from humoral innate immune system that are “perceived as a world apart” 18 and not typically implicated in the pathophysiology of endotoxic shock. Regulation of these pro-inflammatory genes, in addition to those from a cellular arm of innate immunity, represents a critical mechanism underlying HDACIs’ pro-survival effect. Our current investigation has demonstrated for the first time that treatment of LPS shock by SAHA modulates the expression of genes controlling both the cellular and humoral arms of innate immune response. Further analyses indicated that PTX3 might be an important target gene involved in the LPS-induced inflammation, acute lung injury and endotoxic shock.
It is already known that TNFR1 and 2, TRAF6, TLR2, MyD88, and CCL3 (involved in cellular innate immunity) play key roles in the balance between infection clearance and injurious inflammation. TLRs are a class of membrane-bound, pathogen and danger associated molecular pattern receptors (PPRs and DPRs) that initiate and perpetuate the innate immune response in the acute phase of septic shock. TLR4 is specifically activated by LPS 19 while TLR2 is stimulated by early phase cytokines and endogenous “alarmins” including components released from necrotic cells, late phase cytokines like high mobility group box protein 1 (HMGB-1), and heat shock proteins 20. Both TLR2 and TLR4 are known to be up-regulated in septic patients compared to healthy controls 21. In this model of LPS-shock, TLR4 is likely responsible for initiating the acute endotoxic response while TLR2 and 4 may contribute to the progression of endotoxic pathology along with other PPRs and DPRs. Myeloid differentiation protein 88 (MyD88) is a central adaptor protein for many TLRs, including TLR2 22, 23. Extracellular activation TLR2 leads to intracellular recruitment of MyD88. MyD88 in turn recruits IL-1 receptor associated kinases (IRAK), and activates TNF receptor associated factor 6 (TRAF6) 24. Active TRAF6 is necessary for proteasome-independent activation of the inhibitor of IκB kinase (IKK), which frees NF-κB from its inhibitor protein (IκB) and enables it to enter the nucleus and promote transcription of pro-inflammatory genes 25, including tumor necrosis factor α (TNFα). TNFα is the earliest response cytokine in response to LPS insult 26 and serves as a central mediator in septic shock. TNFα is well-known to be upregulated in response to LPS exposure and is produced by resident and circulating mononuclear phagocytes 27. TNFα interacts with two receptors, tumor necrosis factor receptor 1 and 2 (TNFR1 and TNFR2), and triggers a downstream signaling cascade that activates transcription factor nuclear factor kappa B (NF-κB). Activated NF-κB binds targeted sites on host DNA and increases the production of other early phase cytokines such as interleukin-1β (IL-1β) and interleukin-6 (IL-6), and alters the expression profile of leukocyte attractants 27.
Importantly PTX3, a fluid-phase pattern-recognition molecule, was also found to be involved in the present model of endotoxic shock and SAHA treatment. Normally, PTX3 plasma levels are very low but increase in several pathological conditions, including infections 28. High concentrations of circulating PTX3 reflect release of the long pentraxin from degranulating neutrophils as well as gene expression-dependent production 27. Deban and colleagues recently have reported that PTX3, released from activated leukocytes, functions locally to dampen neutrophil recruitment at the site of inflammation 29. However, it remains undefined whether LPS-induced PTX3 could systemically inhibit neutrophil infiltration in lung when LPS is administered intraperitoneally (i.p.). Results of our current experiments demonstrate that i.p. injection of LPS can dramatically increase PTX expression and pulmonary neutrophil infiltration, which are attenuated by SAHA treatment.
We have demonstrated that SAHA treatment significantly attenuates PTX3 levels both in the lung and peripheral blood, but the reduction profile is somewhat different. In the lung, the levels of PTX3 protein peaked at 6 h, and then declined whereas in peripheral blood the highest levels of PTX3 occurred at 24 h. In addition to differences in techniques of measurement, this discrepancy could have also resulted from potentially different sources of PTX3 protein production. It has been reported that macrophages, neutrophils, dendritic cells and endothelial cells are major producers of PTX3 in response to Toll-like receptor engagement 30 and inflammatory cytokines 10. In the lung, Han and colleagues found that lung epithelial cells appear to be a major local source for PTX3 production, which could be induced in vivo from these cells by LPS or other inflammatory stimuli, and may be an important mediator for host defense and lung tissue damage 31.
We discovered that seven genes were significantly up-regulated following exposure to LPS. This early response (3 hours) at the mRNA level may have played a role in the later (24 hrs) development of pulmonary infiltration. In contrast, SAHA treated animals exhibited attenuated transcription of these genes and reduced pulmonary inflammation and MPO activity. These results underscore the importance of maintaining the tenuous equilibrium between protective and pathologic immunity and among genes related to cellular and humoral arms of innate immune response. Modulation of expression of these genes appears to be a potential mechanism through which SAHA could reduce NF-κB-mediated pro-inflammatory gene expression, attenuate pulmonary neutrophil infiltration, prevent acute lung injury, and promote survival. Moreover, preliminary evidence suggests that targeting the pathogen associated molecular pattern receptors (both cellular and humoral) might be a promising way to modulate the inflammatory response after trauma, surgery, or infection, and limit the secondary pathological progression to septic shock without compromising the innate immune system’s infection fighting function.
Networks are a natural paradigm for describing many biological systems such as gene regulation, protein interactions, biological pathways and others. VisANT is a Java-based web tool that can be used for viewing gene interactions, combined and overlaid with annotation from Gene Ontology, GenBank, Kyoto Encyclopedia of Genes and Genomes (KEGG), and SwissProt 17. In the present study we introduced VisAnt for the first time into our data analysis. Although the data are not absolutely comprehensive, VisAnt tools can help us to understand complex biological systems and guide future research. Using gene network analysis to identify key pathways in septic shock (Fig. 6), we selected seven genes—TNFR1 and 2, TRAF6, TLR2, PTX3, MyD88, and CCL3 and confirmed changes in their mRNA expression by real time-PCR. Each of these gene products is integrally involved in the onset and progressive derangement of the immune and inflammatory response to endogenous and exogenous stress signals.
This study has limitations that must be acknowledged. LPS challenge, although well established, does not completely replicate all aspects of septic shock. We are currently validating these findings in a more realistic model of cecal ligation and puncture. Also, for logistical reasons we could not measure many other genes and pathways that might be involved. Another limitation is that the animals were treated with SAHA prior to receiving LPS, which is obviously not clinically realistic. We have subsequently shown that survival is significantly improved (from 0 to 80%) even when SAHA is administered 2 hrs after LPS insult 32. Therefore, while this high-throughput study serves a hypothesis generating function, future studies exploring additional time points, multiple organs, different dosing schemes, polymicrobial models of sepsis, and more detailed biochemical assays are needed to fully explore modulation of gene expression in the treatment of septic shock.
In conclusion, treatment with SAHA attenuates inflammation and decreases neutrophil infiltration in the lungs. In this study we have demonstrated that a key mechanism appears to be rapid modulation of transcription of genes in both cellular and humoral arms of innate immunity, with an attenuation of inflammatory mediators including TNFR1 and 2, TRAF6, TLR2, PTX3, MyD88, and CCL3. The present data suggest that protein acetylation is a key mechanism that regulates the expression of genes involved in the host response to microbial components (e.g., LPS). The broad anti-inflammatory and immune-modulation properties of SAHA might be beneficial in the treatment of septic shock. In addition, profiling of gene expression in response to a pan-HDAC inhibitor (SAHA) will serve as reference for future studies with isoform-specific inhibitors of HDAC. Moreover, our results suggest that PTX3 should be investigated further as a potential target for specific pharmacological interventions.
Acknowledgments
Supported by NIH RO1 GM084127 (to HBA) and a generous research endowment by the Polsky family. Data presented at the 7th Annual Academic Surgical Congress in Las Vegas, NV (February, 2012).
ABBREVIATIONS
- CCL3
Chemokine ligand 3
- ELISA
Enzyme-linked immunosorbent assay
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- HAT
Histone Acetyltransferase
- HDAC
Histone deacetylase
- HDACI
Histone deacetylase inhibitor
- IL
Interleukin
- IRAK
IL-1 receptor associated kinases
- LCB
Lower confidence bound
- LPS
Lipopolysaccharide
- MPO
Myeloperoxidase
- MyD88
Myeloid differentiation primary response gene 88
- PAMP
Pathogens associated molecular pattern
- PCR
Polymerase chain reaction
- PRM
Pattern recognition molecule
- PTX3
Pentraxin 3
- SAHA
Suberoylanilide hydroxamic acid
- TLR
Toll-like receptor
- TNFR
Tumor necrosis factor receptor
- TRAF6
Tumor necrosis factor receptor associated factor 6
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Angus DC, Linde-Zwirble W, Lidicker JM, Clermont G, Carcillo J, Pinsky M. Epidemiology of severe sepsis in the United States: Analysis of incidence, outcome, and associated costs of care. Crit Care Med. 2001;29(7):1303–1310. doi: 10.1097/00003246-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Alberti C, Brun-Buisson C, Goodman SV, Guidici D, Granton J, Moreno R, Smithies M, Thomas O, Artigas A, Le Gall JR. Influence of systemic inflammatory response syndrome and sepsis on outcome of critically ill infected patients. Am J Respir Crit Care Med. 2003;168:77–78. doi: 10.1164/rccm.200208-785OC. [DOI] [PubMed] [Google Scholar]
- 3.Russell JA. Management of sepsis. N Engl J Med. 2006;355:1699–1713. doi: 10.1056/NEJMra043632. [DOI] [PubMed] [Google Scholar]
- 4.Cinel I, Dellinger RP. Advances in pathogenesis and management of sepsis. Curr Opin Infect Dis. 2007;20:345–352. doi: 10.1097/QCO.0b013e32818be70a. [DOI] [PubMed] [Google Scholar]
- 5.Ianaro A, Tersigni M, D’Acquisto F. New Insight in LPS Antagonist. Mini Rev Med Chem. 2009;9(3):306–317. doi: 10.2174/1389557510909030306. [DOI] [PubMed] [Google Scholar]
- 6.Coimbra R, Melbostad H, Loomis W, Porcides RD, Wolf P, Tobar M, Hoyt DB. LPS-induced acute lung injury is attenuated by phosphodiesterase inhibition: effects on proinflammatory mediators, metalloproteinases, NFkappaB, and ICAM-1 expression. J Trauma. 2006;60:115–125. doi: 10.1097/01.ta.0000200075.12489.74. [DOI] [PubMed] [Google Scholar]
- 7.Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- 8.Janeway CA, Jr, Medzhito R. Innate immune recognition. Annu Rev Immunol. 2002;20:197–216. doi: 10.1146/annurev.immunol.20.083001.084359. [DOI] [PubMed] [Google Scholar]
- 9.Jeannin P, Bottazzi B, Sironi M, Doni A, Rusnati M, Presta M, Maina V, Maqistrelli G, Haeuw JF, Hoeffel G, Thieblemont N, Corvaia N, Garlanda C, Delneste Y, Mantovani A. Complexity and complementarity of outer membrane protein A recognition by cellular and humoral innate immunity receptors. Immunity. 2005;22:551–560. doi: 10.1016/j.immuni.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 10.Doni A, Michela M, Bottazzi B, Peri G, Valentino S, Polentarutti N, Garlanda C, Mantovani A. Regulation of PTX3, a key component of humoral innate immunity in human dendritic cells: stimulation by IL-10 and inhibition by IFN-γ. J Leukoc Biol. 2006;79:797–802. doi: 10.1189/jlb.0905493. [DOI] [PubMed] [Google Scholar]
- 11.Li Y, Liu B, Zhao H, Sailhamer EA, Zhang X, Kheirbek T, Finkelstein R, Velmahos G, deMoya M, Hales CA, Alam HB. Protective Effect of Suberoylanilide Hydroxamic Acid against LPS-induced Septic Shock in Rodents. Shock. 2009;32(5):517–523. doi: 10.1097/SHK.0b013e3181a44c79. [DOI] [PubMed] [Google Scholar]
- 12.Wurfel MM. Microarray-based analysis of ventilator-induced lung injury. Proc Am Thorac Soc. 2007;4(1):77–84. doi: 10.1513/pats.200608-149JG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li C. Automating dChip: toward reproducible sharing of microarray data analysis. BMC Bioinformatics. 2008;9:231. doi: 10.1186/1471-2105-9-231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li C, Hung Wong W. Model-based analysis of oligonucleotide arrays: model validation, design issues and standard error application. Genome Biol. 2001;2(8):research0032. doi: 10.1186/gb-2001-2-8-research0032. Epub 2001 Aug 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hu Z, Mellor J, Wu J, Yamada T, Holloway D, Delisi C. VisANT: data-integrating xisual framework for biological networks and modules. Nucleic Acids Res. 2005;33:353–357. doi: 10.1093/nar/gki431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Real JM, Spilborghs GM, Morato-Marques M, de Moura RP, Negri EM, Camargo AA, Deheinzelin D, Dias AA. Pentraxin 3 accelerates lung injury in high tidal volume ventilation in mice. Mol Immunol. 2012;51:82–90. doi: 10.1016/j.molimm.2012.02.113. [DOI] [PubMed] [Google Scholar]
- 17.Hu Z, Mellor J, Wu J, Delisi C. VisAnt: an online visualization and analysis tool for biological interaction data. BMC Bioinformatics. 2004;5:17. doi: 10.1186/1471-2105-5-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bottazzi B, Doni A, Garlanda C, Mantovani A. An integrated view of humoral innate immunity: pentraxins as a paradigm. Annu Rev Immunol. 2010;28:157–183. doi: 10.1146/annurev-immunol-030409-101305. [DOI] [PubMed] [Google Scholar]
- 19.Medzhitov R, Preston-Hurlburt P, Janeway CA., Jr A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 1997;388:394–397. doi: 10.1038/41131. [DOI] [PubMed] [Google Scholar]
- 20.Bianchi ME. DAMPs, PAMPs and Alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 21.Armstrong L, Medford AR, Hunter KJ, Uppington KM, Millar AB. Differential expression of Toll-like receptor (TLR)-2 and TLR-4 on monocytes in human sepsis. Clin Exp Immunol. 2004;136:312–319. doi: 10.1111/j.1365-2249.2004.02433.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kawai T, Adachi O, Ogawa T, Takeda K, Akira S. Unresponsiveness of MyD88-deficient mice to endotoxin. Immunity. 1999;11:115–122. doi: 10.1016/s1074-7613(00)80086-2. [DOI] [PubMed] [Google Scholar]
- 23.Janssens S, Burns K, Vercammen E, Tsay HC, Beyaert R. MyD88S, a splice variant of MyD88, differentially modulates NF-kappaB- and AP-1-dependent gene expression. FEBS Lett. 2003;548:103–107. doi: 10.1016/s0014-5793(03)00747-6. [DOI] [PubMed] [Google Scholar]
- 24.Li X, Qin J. Modulation of Toll-interleukin 1 receptor mediated signaling. J Mol Med. 2005;83:258–266. doi: 10.1007/s00109-004-0622-4. [DOI] [PubMed] [Google Scholar]
- 25.Medzhitov R, Preston-Hurlburt P, Kopp E, Stadlen A, Chen C, Ghosh S, Janeway CA. MyD88 is an adaptor protein in the hToll/IL-1 receptor family signaling pathways. Mol Cell. 1998;2:253–258. doi: 10.1016/s1097-2765(00)80136-7. [DOI] [PubMed] [Google Scholar]
- 26.Cinel I, Opal S. Molecular biology of inflammation and sepsis: a primer. Crit Care Med. 2009;37 (1):291–305. doi: 10.1097/CCM.0b013e31819267fb. [DOI] [PubMed] [Google Scholar]
- 27.Cohen J. The immunopathogenesis of sepsis. Nature. 2002;420:885–891. doi: 10.1038/nature01326. [DOI] [PubMed] [Google Scholar]
- 28.Huttunen R, Hurme M, Ajttoniemi J, Huhtala H, Vuento R, Laine J, Jylhava J, Syrjanen J. High plasma level of long pentraxin 3 (PTX3) is associated with fatal disease in bacteremic patients: a prospective cohort study. PLoS One. 2011;6(3):e17653. doi: 10.1371/journal.pone.0017653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V, Cuccovillo I, Bastone A, Gobbi M, Valentino S, Doni A, Garland C, Danese S, Salvatori G, Sassano M, Evangelista V, Rossi B, Zenaro E, Constantin G, Laudanna C, Bottazzi B, Mantovani A. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol. 2010;11(4):328–334. doi: 10.1038/ni.1854. [DOI] [PubMed] [Google Scholar]
- 30.Jeannin P, Bottazzi B, Sironi M, Doni A, Rusnati M, Presta M, Maina V, Magistrelli G, Haeuw JF, Hoeffel G, Thieblemont N, Corvaia N, Garlanda C, Delneste Y, Mantovani A. Complexity and complementarity of outer membrane protein A recognition by cellular and humoral innate immunity receptors. Immunity. 2005;22(5):551–560. doi: 10.1016/j.immuni.2005.03.008. [DOI] [PubMed] [Google Scholar]
- 31.Han B, Mura M, Andrade CF, Okutani D, Lodyga M, dos Santos CC, Keshavjee S, Matthay M, Liu M. TNFalpha-induced long pentraxin PTX3 expression in human lung epithelial cells via JNK. J Immunol. 2005;175(12):8303–8311. doi: 10.4049/jimmunol.175.12.8303. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Liu B, Fukudome EY, Kochanek AR, Finkelstein R, Chong W, Jin G, Lu J, deMoya M, Velmahos GC, Alam HB. Surviving lethal septic shock without fluid resuscitation in a rodent model. Surgery. 2010;48(2):246–254. doi: 10.1016/j.surg.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]