

Abstract
Medulloblastoma, the most common pediatric malignant brain tumor often arises sporadically; however, in a subgroup of patients, there exist familial conditions such as Fanconi anemia with biallelic BRCA2 mutation that predispose patients to developing medulloblastoma. Biallelic inactivation of BRCA2 in Fanconi anemia has been previously described in only 11 patients with medulloblastoma in the literature to date. Here we report two siblings diagnosed with central nervous system embryonal tumors at an early age in association with biallelic BRCA2 inactivation, including the first reported case of a spinal cord primitive neuroectodermal tumor (PNET) in a BRCA2/FANCD1 kindred.
Keywords: Brain tumor, Primitive Neuroectodermal tumor, Medulloblastoma, Fanconi Anemia, BRCA2 gene mutation
Introduction
Fanconi anemia is an autosomal recessive cancer susceptibility disorder. The complementation group D1 in Fanconi anemia has been linked to a variety of solid tumors, including Wilms tumor and medulloblastoma. Specifically, biallelic inactivation of BRCA2 in Fanconi anemia has been reported in a small number of patients with medulloblastoma. Recently, the breast cancer susceptibility gene, BRCA2, has also been associated with Fanconi anemia complementation group D1 (FANCD1). We present two cases of central nervous system embryonal tumors associated with biallelic BRCA2 inactivation diagnosed in siblings at an early age in addition to a review of the current literature. Interestingly, the current case did not have the typical clinical features of Fanconi anemia, and as a result, the diagnosis was made after extreme chemosensitivity.
Case Report
A 15 month old male (Sibling 1) presented with a 2-month history of decreased bilateral upper-extremity use with anisocoria, right ptosis, and facial weakness. Magnetic resonance imaging (MRI) of the spine revealed an intramedullary lesion extending inferiorly from spinal levels C2 to T2 (Supplemental Figure 1A). The patient underwent a partial resection (80%) of the tumor. Pathological assessment showed a high-grade neuroepithelial tumor (Supplemental Figure 1B). The differential diagnosis included spinal primitive neuroectodermal tumor (PNET) and high-grade glioma, although the former interpretation was preferred on the basis of the cytological features as noted in Supplemental Figure 1B. His treatment protocol included cisplatin, vincristine, etoposide, cyclophosphamide, and methotrexate. Following the first chemotherapy course, complications included myelosuppression, severe mucositis, disseminated fungal infection, and bacteremia. The second course of chemotherapy consisting of vincristine, etoposide and temozolomide, was better tolerated; however, the third chemotherapy course was complicated by prolonged pancytopenia and the development of hepatic venous occlusion resulting in death 3 months after initial diagnosis.
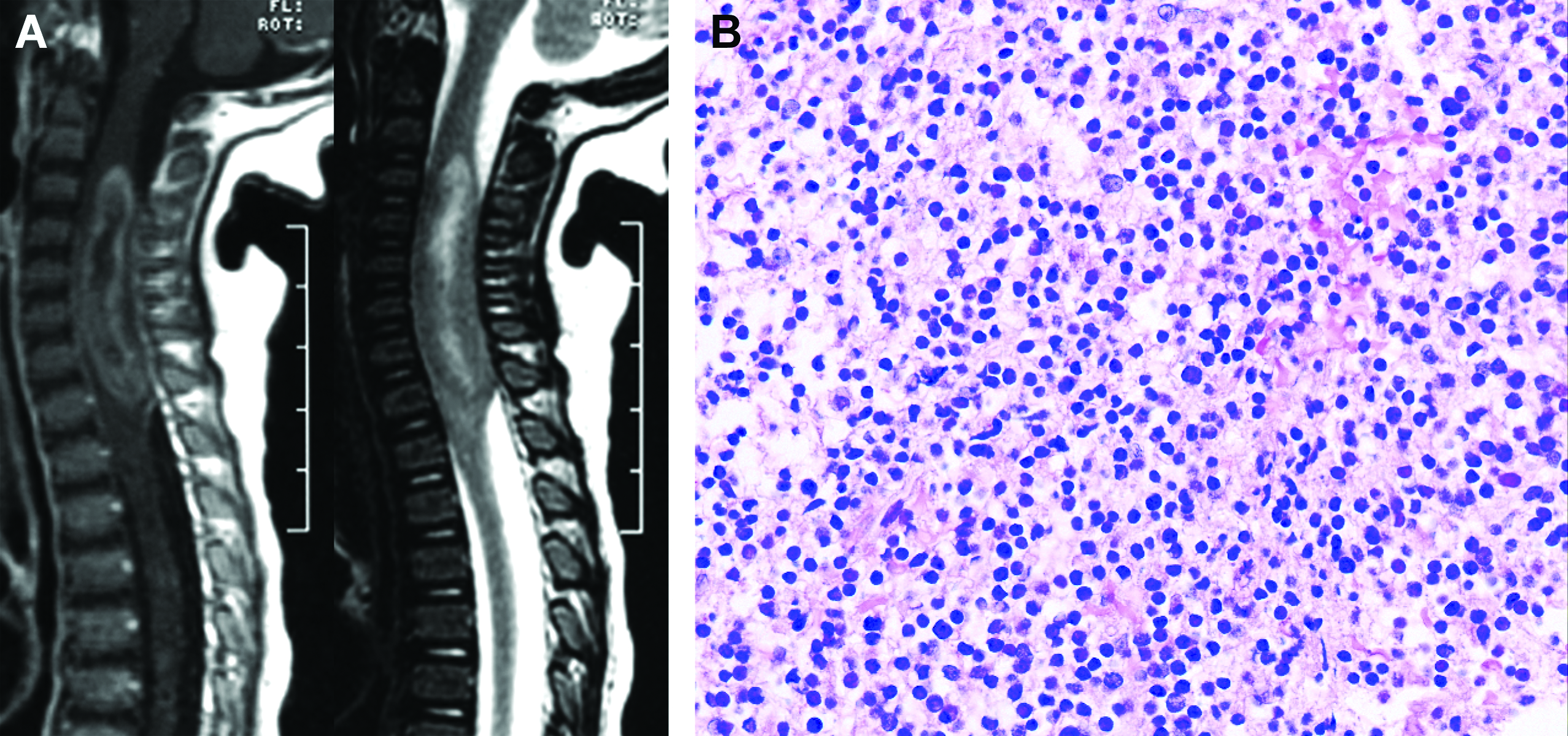
Sibling 2, a 21 month old male, presented with a history of refusal to walk and progressively worsening emesis without focal neurological findings. MRI of the brain at an outside institution revealed a large heterogenous enhancing mass of the right cerebellar hemisphere (Fig. 1A and 1B). Initial surgery occurred at the outside institution. Following referral, residual disease was noted on follow-up MRI; thus, a second surgery occurred at our institution. Pathological evaluation disclosed a diagnosis of desmoplastic nodular medulloblastoma; however, the tumor was an unusual example of this variant (Fig. 1C). A diagnosis of atypical teratoid / rhabdoid tumor was excluded due to the presence of SMARCB1(INI-1) gene product expression. His treatment regimen included high-dose methotrexate (5 g/m2 per dose), vincristine, cisplatin, and cyclophosphamide. Prior to the initiation of chemotherapy, a complete blood cell count revealed a white blood cell count 6,900/mm3, hemoglobin 9.3 g/dL, mean cell volume 77.1 fL (within normal range for age), and platelet count was 349,000/mm3. After the first course of vincristine, cisplatin, and cyclophosphamide, he had prolonged myelosuppression; specifically, an absolute neutrophil count of zero for 2 weeks and thrombocytopenia for 3–4 weeks. His parents reported that his brother (Sibling 1) had similar myelosuppression with his first cycle of chemotherapy and died from toxicity after his third course of chemotherapy. The patient had no other siblings.
Figure 1.

The early age of cancer presentation and unusual chemosensitivity evident in both siblings prompted an investigation for a disorder in DNA damage repair. The diepoxybutane (DEB) test was performed on peripheral blood cells from Sibling 2 and the results were consistent with Fanconi anemia–falling within the range of 1.06–23.9 DEB-induced chromosomal breaks per cell. In contrast, DEB-treated cells of unaffected individuals result in 0–0.10 breaks per cell. Twenty peripheral blood cells from Sibling 2 were treated with DEB, and 18 of the 20 DEB-treated cells (90%) exhibited DEB-induced chromosomal breakage. In addition, spontaneous chromatid breaks in the untreated peripheral blood cells of Sibling 2 (2.4 chromosome breaks per cell) were higher than expected for a patient with Fanconi anemia (0.02–0.80 chromosome breaks per cell) likely due to the clastogenic effect of prior chemotherapeutic exposure in vivo.
Gene-specific BRCA analysis, which included sequencing of all translated exons and adjacent intronic regions of the BRCA2 gene along with a rearrangement test of both BRCA1 and BRCA2 by quantitative polymerase chain reaction analysis, was performed on the peripheral blood of Sibling 2. Results were consistent with germline BRCA2 mutations 6174delT and 886delGT. Further testing confirmed that each parent was a carrier of one of these BRCA2 mutations. The BRCA2 mutation is prevalent in the Ashkenazi Jewish population and of note: the mother was of Jewish decent. 1 As a result of these genetic findings in this family, genetic counseling was offered and highly recommended.
Since conventional medulloblastoma therapy was contraindicated due to the presence of Fanconi anemia, complicated by biallelic BRCA2 mutations, a palliative treatment regimen with limited use of DNA-damaging agents was initiated, which included 4-week cycles of high-dose methotrexate (5 g/m2 per dose), intraventricular methotrexate, and low-dose oral chemotherapy (cyclophosphamide alternating with etoposide). However, Sibling 2 developed symptomatic leukoencephalopathy, significant myelosuppression, and sepsis, which required discontinuation of treatment. Additional radiation therapy was not considered for this patient due to his underlying genetic disorder. Clinical symptoms and diagnostic imaging of leukoencephaolopathy improved after the discontinuation of chemotherapy. Most recent follow-up diagnostic imaging, MRI, 23 months from diagnosis revealed improvement in white matter changes without evidence of disease recurrence.
Discussion
Fanconi anemia is an autosomal recessive cancer susceptibility disorder. Clinical features include short stature, microcephaly, elfin facies, thumb and radial anomalies, renal and ureter abnormalities, endocrinopathies, gastrointestinal abnormalities, and hearing deficits.2,3 Howlett et al. have reported an association between the Fanconi anemia complementation group D1 (FANCD1) and the breast cancer susceptibility gene (BRCA2). 4 Dual mutation in BRCA2 (biallelic inactivation of BRCA2) has been reported to be associated with high spontaneous chromosome breakage rates in cells of patients with Fanconi anemia, along with unusual chemotherapy sensitivity in patients with solid tumors and brain tumors.4 In comparison to other Fanconi anemia subtypes, bone marrow suppression, dysmorphic facial features, skeletal abnormalities, and abnormal mean cell volume (MCV) are less frequent and preclude an early diagnosis in these patients. In addition, a different spectrum of childhood cancers develop in patients with simultaneous Fanconi anemia and biallelic BRCA2 mutations.4,5,6
Reviews of the literature document biallelic BRCA2 mutations not only in a familial Wilms tumor pedigree, but also in a number of other malignancies.7 In particular, of the 17 families studied by Reid et al. the pre-existing diagnosis of Fanconi anemia was not present. Cases of Wilms tumors, brain tumors (including medulloblastoma, glioblastoma multiforme, and astrocytoma), acute myeloid leukemia, and acute lymphoblastic leukemia were identified in 23 biallelic BRCA2 mutation carriers. Twenty-one of the 23 carriers developed childhood cancer, including 9 of the 23 carriers who developed brain tumors. Interestingly, a systematic review of genomic DNA revealed 5 of the 9 brain tumors possessed BRCA2 886delGT, as previously noted in Sibling 2. In addition, Alter and colleagues analyzed 27 published cases with FANCD1 and biallelic mutations of BRCA2 and reported an association of BRCA2 6174delGT with brain tumors.3
All patients in the literature with biallelic BRCA2 mutations and medulloblastoma were diagnosed at a young age (Table I). Most cases did not have the typical clinical features of Fanconi anemia, and as a result, the diagnosis was made in the setting of extreme chemosensitivity. Clinicians must be aware of this association in order to appropriately treat such patients and avoid potentially fatal treatment toxicity. Clinical features such as early age of presentation (< 3 years) in association with markedly delayed peripheral blood count recovery or other evidence of atypical chemosensitivity should warrant further investigation, including chromosomal fragility testing and gene-specific analysis in addition to genetic counseling for family members.
Table I.
BRCA2 mutations and outcomes in children with posterior fossa tumors
| ID | Cancer (age, years) | Nucleotide change I | Nucleotide change II | Outcome | Reference |
|---|---|---|---|---|---|
| 1 | Brain Tumor (3) | 7691/insAT | 9900/insA | Not documented | 4 |
| 2 | Posterior fossa tumor- radiologic findings consistent with medulloblastoma or astrocytoma (4.9) |
6174delT | 9435T→A | Dead | 6 |
| 3 | Astrocytoma (2) | 6174delT | 9435T→A | Dead | 6 |
| 4 | Medulloblastoma (4.5) | 6174delT | 886delGT | Not documented | 6 |
| 5 | Medulloblastoma (2.5) | 5301insA | 7690T→C | Not documented | 6 |
| 6 | Medulloblastoma (3.5) | 4150G→T | 9424C→T | Not documented | 6 |
| 7 | Medulloblastoma (2.3) | 886delGT | 8447T→A | Not documented | 5 |
| 8 | Medulloblastoma (4.3) | 886delGT | 8447T→A | Dead | 5 |
| 9 | Medulloblastom (6) | 886delTG | 5873C→A | Dead * relapse of medulloblastoma at 12 y |
7 |
| 10 | Posterior fossa tumor (1) -VATER associated anamolies |
1548del4 | 1548del4 | Dead | 10 |
| 11 | Medulloblastoma (3.1) - VATER associated anomalies |
6174delT | 9424C→T | Not documented | 3 |
| 12 | Medulloblastoma (1.7) | 6174delT | 886delGT | Alive without evidence of disease |
Current report |
| 13 | Medulloblastoma (2) | 3492insT | IVS19+3A→G | Dead | Personal communication |
Supplementary Material
{kind=link}
Acknowledgment
This research was supported in part by grant CA – 21765 from the National Institutes of Health; the Noyes Brain Tumor Foundation; Musicians Against Childhood Cancer (MACC); and the American Lebanese Syrian Associated Charities (ALSAC).
Footnotes
AUTHORS’ DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST The authors have no potential conflicts of interest.
REFERENCES
- 1.Krainer M, Silva-Arrieta S, FitzGerald MG, et al. Differential contributions of BRCA1 and BRCA2 to early-onset breast cancer. N Engl J Med. 1997;336(20):1416–1421. doi: 10.1056/NEJM199705153362003. [DOI] [PubMed] [Google Scholar]
- 2.Kliegman RM, Behrman RE, Jenson HB, Stanton BF. Fanconi Anemia. 18th edition Saunders; New York, NY: 2007. Nelson Textbook of Pediatrics; pp. 2047–2051. [Google Scholar]
- 3.Alter BP, Rosenberg PS, Brody LC. Clinical and molecular features associated with biallelic mutations in FANCD1/BRCA2. J Med Genet. 2007;44(1):1–9. doi: 10.1136/jmg.2006.043257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howlett NG, Taniguchi T, Olson S, et al. Biallelic inactivation of BRCA2 in Fanconi anemia. Science. 2002;297(5581):606–609. doi: 10.1126/science.1073834. [DOI] [PubMed] [Google Scholar]
- 5.Hirsch B, Shimamura A, Moreau L, et al. Association of biallelic BRCA2/FANCD1 mutations with spontaneous chromosomal instability and solid tumors of childhood. Blood. 2004;103(7):2554–2559. doi: 10.1182/blood-2003-06-1970. [DOI] [PubMed] [Google Scholar]
- 6.Offit K, Levran O, Mullaney B, et al. Shared genetic susceptibility to breast cancer, brain tumors, and Fanconi anemia. J Natl Cancer Inst. 2003;95(20):1548–1551. doi: 10.1093/jnci/djg072. [DOI] [PubMed] [Google Scholar]
- 7.Reid S, Renwick A, Seal S, et al. Biallelic BRCA2 mutations are associated with multiple malignancies in childhood including familial Wilms tumour. J Med Genet. 2005;42(2):147–151. doi: 10.1136/jmg.2004.022673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruud E, Wesenberg F. Microcephalus, medulloblastoma and excessive toxicity from chemotherapy: an unusual presentation of Fanconi anaemia. Acta Paediatr. 2001;90:581–583. [PubMed] [Google Scholar]
- 9.Tischkowitz MD, Chisholm J, Gaze M, et al. Medulloblastoma as the first presentation of Fanconi anemia. J Pediatr Hematol Oncol. 2004;26:52–55. doi: 10.1097/00043426-200401000-00016. [DOI] [PubMed] [Google Scholar]
- 10.Faivre L, Portnoi MF, Pals G, et al. Should chromosome breakage studies be performed in patients with VACTERL association? Am J Med Genet A. 2005;137:55–58. doi: 10.1002/ajmg.a.30853. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.