

Abstract
Tumor protein D52 (D52) is constitutively expressed in healthy tissues and overexpressed in multiple cancers, including (but not limited to) breast, prostate and ovarian carcinomas. Although the normal functions of D52 are unknown, it is clear that increased D52 expression levels not only stimulate cell proliferation and metastasis, but also correlate with poor prognosis in a subset of breast cancer patients. The murine orthologs of D52 (mD52) shares 86% identity with its human counterpart (hD52) and mirrors hD52 expression patterns. The forced overexpression of mD52 induces anchorage-independent growth in vitro and promotes tumor formation as well as spontaneous metastasis in vivo. We have previously reported that the intramuscular administration of recombinant mD52 elicits immune responses capable of rejecting a challenge with tumor cells and preventing spontaneous metastasis only in 50% of mice. We hypothesized that mechanisms of peripheral tolerance dampen immune responses against mD52, thus limiting the protective effects of vaccination. To test this hypothesis, mice were depleted of CD25+ regulatory T cells (Tregs) and subcutaneously immunized with mD52 prior to a tumor challenge. The subcutaneous immunization failed to induce protective antitumor immunity unless accompanied by Treg depletion, which resulted in a rate of protection of 70% as compared with
Keywords: CD25+ Treg depletion, CD8+ Tregs, TPD52, mD52 protein, mouse, vaccine
Introduction
Human and murine tumor protein D52 (TP52, hereafter referred to as D52) are expressed at detectable levels in multiple normal tissues and cells.1-4 The human D52 gene family5 comprises four distinct genes, D52 or PRLZ6,7 D53 or TPD52L1,5,8 D54 or TPD52L29,10 and D55 or TPD52L3.11 Importantly, human D52 (hD52), was found to be overexpressed in a wide range of cancers.6,7,12-17 Studies suggest that hD52 encodes a marker or regulator of cancer cell proliferation,5 supporting the notion that the expression of D52 may be important for initiating or maintaining a tumorigenic and/or metastatic phenotype. In this respect, we have previously demonstrated that the knockdown of hD52 in human cancer cells limits cell proliferation and stimulates apoptosis. In addition, D52 overexpression was found to be associated with a poor overall survival of human breast carcinoma patients, suggesting a strong potential for the therapeutic targeting of D52 functions in cancer.5
The cloning of the murine ortholog of D52 (mD52), which is approximately 86% identical to hD52 at the amino acid level, has proven critical for preclinical studies on D52 functions. We evaluated the effect of increased mD52 expression on non-transformed cells using contact-inhibited murine NIH-3T3 fibroblasts18 transfected with the full-length mD52-coding cDNA. mD52 overexpression was confirmed by RT-PCR and immunoblotting. mD52-transformed NIH-3T3 (3T3.mD52) cells exhibited a 2-fold increase in growth rate, lost contact inhibition, displayed marked morphological alterations and acquired the ability for anchorage-independent cell growth. Importantly, 3T3.mD52 cells formed tumors when injected s.c. into naïve, immunocompetent syngeneic mice. Remarkably, when the lungs from 3T3.mD52 tumor-bearing mice were analyzed, numerous neoplastic nodules were observed, demonstrating the ability of these cells to spontaneously metastasize.
We have previously demonstrated that the administration of recombinant mD52 i.m. together with CpG oligonucleotides (ODNs) confers a partial protection to mice against a subsequent challenge with tumor cells.1 We have also reported that the vaccination of transgenic adenocarcinoma of the mouse prostate (TRAMP) mice with an mD52-coding DNA induces an immune response that promotes tumor rejection. T-cell cytokine secretion patterns indicated that a TH1 cellular immune response was involved in tumor rejection in these models.1,19 Mirshahidi and colleagues reported an mD52-overlapping peptide vaccine to be partially effective in a murine breast cancer model.20 Taken together, these preclinical vaccine studies demonstrate that mD52 is immunogenic in murine tumor models. However, the levels of protection achieved in these settings were never maximal. To further explore the immunogenicity of recombinant mD52 as an anticancer vaccine and attempt to increase its therapeutic potential, we sought to alter adjuvant and injection route.
In the present study, mice were immunized s.c. with recombinant mD52 admixed with CpG ODNs as a water in oil emulsion in incomplete Freund’s adjuvant (IFA). Contrary to our previous studies, simply switching the adjuvant from alum to IFA and the route from i.m. to s.c. failed to protect >10% mice from a challenge with tumor cells. In order to generate immune response that were indeed capable of protecting mice against tumor challenges, this vaccine had to be accompanied by the modulation of regulatory T cells (Tregs). This was accomplished by the antibody-mediated depletion of CD25+ T cells in vivo.
Results
Treg depletion enhances the protective effects of subcutaneous mD52 vaccination
We have previously reported that the intramuscular administration of recombinant mD52 admixed with CpG ODNs in alum protects approximately 50% of mice from a challenge with mKSA tumor cells, whereas CpG ODNs in alum, recombinant mD52 in alum or alum alone all failed to protect mice from a tumor challenge.1 In an attempt to increase the efficacy of this mD52-based vaccines and inspired by the results of several preclinical and clinical studies, we were interested in studying the subcutaneous route of vaccination and oil in water as an adjuvant in this model. Keeping parameters the same with the exception of administration route and adjuvant, we immunized mice s.c. with recombinant mD52 admixed with CpG ODNs in IFA (Fig. 1A), followed by a challenge with tumor cells. To our surprise, this approach was not as effective at protecting mice from the tumor challenge as in our previous study. In fact, immunizing animals s.c. in IFA failed to protect the majority of mice from tumor development. We hypothesized that this lack of protection could be due to a robust induction of Tregs by subcutaneous Langerhans cells (a subset of dendritic cells, DCs), which perhaps are more efficient at doing so than DCs activated by intramuscular injections.

Figure 1. Immunization and regulatory T-cell depletion. (A) BALB/c mice were immunized s.c. four times every 10–14 d with recombinant mD52 (rmD52) admixed with the CpG oligodeoxynucleotide (ODN) 1826 in incomplete Freund’s adjuvant (IFA). Animals were challenged s.c. with 5 × 105 live, syngeneic mKSA tumor cells approximately 14 d after the final immunization. Animals surviving this challenge were re-challenged 103 d later with 5 × 105 mKSA tumor cells, administered s.c. to the opposite flank. To deplete CD25+ cells, a CD25-specific antibody (PC-61) was administered i.p. with the first (200–300 μg per mouse) and fourth immunization (400 μg per mouse). Irrelevant IgGs (mock depletion) were used to provide appropriate control conditions. (B) Peripheral blood lymphocytes (PBLs) were collected from BALB/c mice immunized and challenged as described above 7 d following the 4th immunization. Lymphocytes from animals in the same experimental group were pooled (n = 10 animals per pooled sample) and stained with anti-CD4-FITC and anti-CD25-PE antibodies. Representative dot plots from two repeated experiments are shown.
To assess the role of Tregs in suppressing mD52-induced immunity, mice were immunized and depleted of CD25+ T cells with specific monoclonal antibodies prior to the tumor challenge (Fig. 1A). CD4+CD25+ Tregs were reduced to nearly 50% in mice receiving anti-CD25 antibodies as compared with mock-depleted animals (Fig. 1B). None of immunized and mock-depleted mice were capable of rejecting a challenge with mKSA cells (Fig. 2A). Conversely, 70% (7/10) of immunized mice that were also depleted of CD25+ Treg cells rejected such a tumor challenge (Fig. 2B). In line with our previous results, mice immunized with recombinant mD52 in IFA (in the absence of CpG ODNs) failed to reject a challenge with mKSA cells (data not shown). To study immunological memory and the duration of vaccine-induced immune responses, mice immunized and depleted of CD25+ Treg cells that survived the first tumor challenge were inoculated with mKSA tumor cells s.c., in the opposite flank, approximately 100 d later. All mice (7/7) that were protected from the primary challenge also failed to develop tumors in response to the secondary challenge, with a follow-up time of 91 d (Fig. 2C). These data demonstrate that the subcutaneous administration of recombinant mD52 and CpG ODNs in IFA alone fail to protect mice from a tumor challenge, unless immunization is accompanied by an at least partial depletion (50%) of CD4+CD25+ Tregs.

Figure 2. Tumor growth in BALB/c mice immunized with recombinant mD52 protein and challenged with mKSA tumor cells. (A–C) BALB/c mice were immunized four times, depleted of CD25+ cells and challenged with tumor cells as described in Figure 1A. (A) Tumor growth following immunization, mock depletion with irrelevant IgGs and primary tumor challenge. (B) Tumor growth following immunization, CD25+ cell depletion with anti-CD25 antibodies (PC-61) and primary tumor challenge. (C) Tumor growth in mice surviving the primary tumor challenge and re-challenged 103 d later. Data are representative of two repeated experiments.
Induction of cytotoxic T cells following subcutaneous vaccination with mD52
Given that mD52 is an intracellular protein, it was of interest to determine if MHC class I-restricted cytotoxic T lymphocytes (CTLs) would be induced by vaccination, and thus be involved in the rejection of tumor challenges. To this aim, splenocytes were harvested from immunized mice and analyzed for their tumor-specific cytotoxic activity. Effector cells generated by a 5–7 d mixed lymphocyte tumor culture (MLTC) with mKSA cells were demonstrated to be a relatively even mix of CD4+ and CD8+ T cells by flow cytometry (data not shown). Targets consisted of syngeneic MHC Class I-matched 3T3.mD52 tumor cells, mKSA tumor cells (the same used for tumor challenges) and allogeneic MHC Class I-mismatched TRAMP-C2 tumor cells. The natural killer (NK) cell-specific target cell line Yac-1 was also tested but not lysed, suggesting the absence of NK cells (data not shown). We have previously reported that 3T3.mD52, mKSA and TRAMP-C2 cells all overexpress mD52 as compared with normal cells of the same type.1
CTLs generated from mice immunized s.c. with mD52 and CpG ODNs in IFA and challenged with mKSA cells demonstrated tumor cell-specific cytotoxic activity (Fig. 3). Both mKSA cells (H-2Kd) and MHC Class-I matched (H-2Kd) mD52-expressing 3T3.mD52 cells were lysed by CTLs (approximately by 40–60% and 20–50%, respectively, for an effector-to-target cell ratio of 12:1). Conversely, the specific lytic activity of CTLs against TRAMP-C2 cells, which lack matched MHC Class I molecules, and MHC Class I-matched non-transformed NIH-3T3 cells failed to exceed the lower limit of detection of our assay (Fig. 3). Although tumor-specific CTLs were detected in immunized mice, irrespective of whether they were depleted or not of CD25+ Tregs, the extent of specific lysis by CTLs obtained from Treg-depleted animals was 50–75% higher than that by CTLs isolated from mock-depleted mice (Fig. 3A and B). Taken together, these data suggest that CTLs generated by a subcutaneous vaccine combined with Treg depletion were either more potent and/or more abundant than those elicited by vaccination alone. Most importantly, only immunized mice concomitantly subjected to Treg depletion rejected tumor challenges.
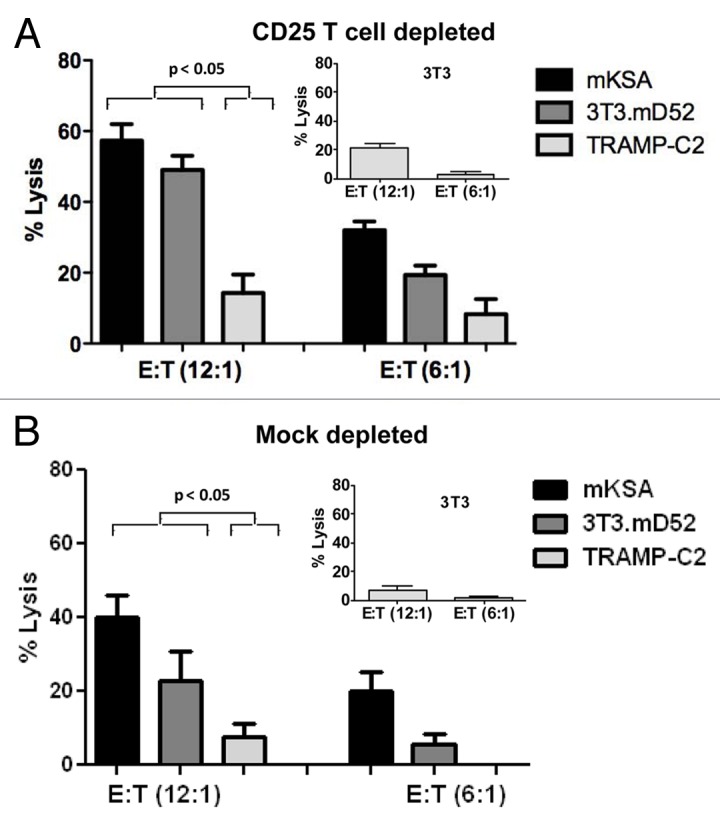
Figure 3. Cytotoxic T-lymphocyte responses following immunization and tumor challenge. (A and B) Bar graph showing specific lysis at two effector-to-target cell (E:T) ratios (12:1) and (6:1), respectively. BALB/c-derived mD52-expressing mKSA cells were used for the in vivo challenge. BALB/c-derived mD52-expressing 3T3.mD52 cells were engineered to overexpress mD52 and served as a positive control target. C57Bl/6-derived mD52-expressing TRAMP-C2 cells served as an MHC Class I-mismatched target. (A) Specific lysis of tumor cells by T cells from CD25+ cell-depleted mice. (B) Specific lysis of tumor cells by T cells from mock-depleted mice. NIH-3T3 cells were used as a normal MHC Class I-matched cell type in both (A and B, insets) Data are reported as means ± SEM of triplicates assessments and are representative of two repeated experiments. Statistical significance was determined using one-way ANOVA and Tukey–Kramer multiple comparisons post-hoc test.
Cytokine profile induced by Treg depletion and subcutaneous vaccination with mD52
As stated above, mD52 is expressed as an intracellular protein by tumor cells. Thus, it was important to determine what type of immune responses were induced in mice that were immunized by a subcutaneous mD52-targeting vaccine combined with Treg depletion and rejected primary and secondary tumor challenges. As we established that the vaccination with mD52 induced tumor-specific CTLs (Fig. 3), it was interesting to determine if the mD52 vaccine combined with Treg depletion would induce a TH1 immune response supporting CTL generation. Splenocytes were harvested from mice that survived both two tumor challenges upon vaccination with mD52 and Treg depletion and cultured with irradiated mKSA cells for 5–7 d in MLTCs. Thereafter, splenocytes were isolated by means of Lympholyte-M® density separation medium and incubated in vitro with irradiated tumor cells for additional 24 h. Finally, supernatants were collected and assayed for the presence of cytokines representing TH1, TH2 and TH17 cellular immune responses using a Multi-Analyte ELISArray (Fig. 4). Of the 12 cytokines tested, only interleukin (IL)-10, IL-13 and interferon (IFN)γ were detected in homotypic T-cell cultures (Fig. 4A), while tumor cells cultured alone secreted undetectable levels of all cytokines tested (Fig. 4B and C). IL-6, IL-10, IL-13 and IFNγ were detected in the supernatants of T cells cultured with irrelevant control targets (Fig. 4D) or mKSA tumor cells (Fig. 4E). However, the amount of cytokines secreted by T cells exposed to irrelevant targets was very low (Fig. 4D), presumably constituting a baseline secretion from T cells. Though neither of the prototypical TH2 cytokines IL-4 nor IL-5 was detected, the small of amount of IL-6 and IL-13 produced suggests that TH2 cells may be activated in this setting. This is not surprising given the nature (soluble whole protein) and administration route of the vaccine. In line with this notion, we have previously described the generation of anti-mD52 antibodies in response to immunization with recombinant mD52.1 TH17 cytokines were not detected, nor were they found previously in this vaccine model.1,19 The dominant cytokine produced by T cells exposed to relevant tumor cells (Fig. 4E) was IFNγ, with IL-6 and IL-10 representing the only other cyokines detected at increased levels as compared with control conditions. These findings support the notion that a TH1 cellular response dominates in immunized and Treg-depleted mice. Since these data were obtained with a cytokine array, it was interesting to quantitatively assess the concentrations of induced cytokines. This was accomplished using specific cytokine capture ELISAs. We assayed supernatants from T cells cultured as described above for IFNγ, IL-4, IL-10, IL-17 and transforming growth factor β1 (TGFβ1) using this method. Only IL-10 and IFNγ were produced by MHC Class-I-restricted T cells, as summarized in Table 1. These data suggest that neither a TH2 nor a TH17 immune response was elicited by vaccination and played a role in tumor protection. This finding was not surprising, given that we detected tumor-specific CTL responses as a result of mD52 vaccination combined Treg depletion (Fig. 3). The amount of IFNγ produced upon 24 h of cell culture was significantly increased when T cells were exposed to relevant, rather than to control, targets (Fig. 5A). The amount of IFNγ produced by T cells cultured with (antigen negative, MHC class I-matched) NIH-3T3 cells (Fig. 5A) and (antigen positive, MHC Class I-mismatched) TRAMP-C2 cells (data not shown) was negligible. These data indicate that tumor cell-specific CD8+ CTL responses develop in immunized mice that are concurrently depleted of CD25+ Tregs. This was confirmed by the ability of H-2d-specific monoclonal antibodies to significantly inhibit the production of IFNγ by T cells co-cultured for 24 h with mKSA target cells (p < 0.05) (Fig. 5A). Conversely, control anti-H-2b monoclonal antibodies failed to inhibit IFNγ production by T cells cultured with mKSA cells (data not shown). Taken together, these data suggest that mD52 vaccination combined with Treg depletion induces antigen-specific, MHC Class I-restricted cellular immune responses that mediate efficient tumor protection.

Figure 4. T-cell cytokine production profile. (A–E) Mice were were immunized four times, depleted of CD25+ cells and challenged with tumor cells as described in Figure 1A. Lymphocytes were isolated from harvested splenocytes and cultured for 24 h with or without tumor cell targets, after which supernatants were harvested and assayed at a 1:5 dilution for cytokines using an ELISA-based array kit specific for murine TH1/TH2/TH17 cells. (A) T cells alone. (B) TRAMP-C2 tumor cells alone. (C) mKSA tumor cells alone. (D) T cells plus TRAMP-C2 tumor cells. (E) T cells plus mKSA tumor cells.
Table 1. Cytokine production of T cells upon immunization, regulatory T cell depletion and challenge with tumor cells*.
| |
Vaccine approach |
|
|
|---|---|---|---|
| Cytokine production | mD52+ODN mock deplete |
mD52+ODN CD25 deplete |
MHC-I mAB blocking |
|
IL-4 |
negative |
negative |
not done |
|
IL-17 |
negative |
negative |
not done |
|
TGF-β1 |
negative |
negative |
not done |
|
IL-10 |
positive |
positive |
yes |
| IFN-γ | positive | positive | yes |
As measured by cytokine-specific standard antigen capture ELISAs in the supernatants of lymphocytes harvested from mice that were immunized as indicated and survived both primary and secondary tumor challenges, cultured with relevant tumor cell targets. Data are representative of two repeated experiments. IFNγ, interferon γ; IL, interleukin; TGFβ1, transforming growth factor β1; negative, below lower detection limit of the assay (applies also to tumor cells alone, T cells alone and T cells exposed to control cell targets; positive, values within the linear range of the standard curve for the indicated cytokine. See also Figure 5.

Figure 5. Production of interferon γ and interleukin-10 by T cells following efficient immunization. (A and B) Results of a standard antigen capture ELISA measuring either interferon γ (IFNγ) (A) or interleukin-10 (IL-10) (B) production in the supernatants of lymphocytes harvested from mice that were immunized with recombinant mD52 plus CpG oligodeoxynucleotides in incomplete Freund’s adjuvant (IFA), and survived both primary and secondary tumor challenges with mKSA tumor cells. Data are presented as pg cytokine per mL supernatant (upon 1:5 dilution) per 24 h, for T cells alone, tumor cells alone or T cells cultured in the presence of (i) MHC Class I-matched (mD52+) mKSA, (mD52+) 3T3.mD52 cells as well as parent NIH-3T3 cells, as an MHC Class I-matched antigen-negative control; or MHC Class I-mismatched TRAMP-C2 tumor cells, as a negative control (data not shown). To confirm MHC Class I restriction, T cells were cultured with mKSA cells in the presence of monoclonal antibodies specific for H-2d (T cells + mKSA + MHC-I mAb). Data were analyzed using one-way ANOVA with Bonferoni multiple comparison post-hoc test. *+p < 0.05 between alike samples, demonstrating reduced IFNγ production in the presence of MHC Class I-blocking antibodies. Numbers in both (A and B) refer to individual mice.
The T cells that were elicited by mD52 immunization plus Treg depletion also produced IL-10 (Fig. 5B). However, the amount of IL-10 was about 3-fold lower than that of IFNγ. This suggests that IFNγ plays a dominant role in tumor protection, as in survivor mice the vaccine combined to Treg depletion was sufficient to induce immune responses capable of rejecting a further tumor challenge. Similar to IFNγ-producing T cells, IL-10-producing T cells were MHC Class-I restricted in all five animals tested in this respect, suggesting that CD8+ Tregs are responsible for the lack of complete protection in mice that were immunized and depleted of CD4+CD25+ Tregs (Fig. 5B). However, the inhibition of IFNγ production by H-2d-specific monoclonal antibodies reached a statistically significant levels only in 2/5 animals tested (Fig. 5A). This may be due to the presence of IFNγ-producing CD4+ T cells. Since the efficiency of CD25+ Treg depletion was assessed on pooled lymphocytes (Fig. 1B), it was not possible to evaluate cytokine production in cells from vaccinated mice that failed to reject the primary tumor challenge. It is possible that immunized mice failing to reject the first tumor challenge were not efficiently depleted of CD25+ Tregs, resulting in a lack of protection. It is also tempting to speculate that production of IL-10 observed in MHC Class I-restricted T cells from protected mice might have been higher in non-protected animals, pointing to the dominance of CD8+ Tregs. Of note, both T cells and tumor cells cultured alone failed to secrete detectable levels of IFNγ and IL-10 (Fig. 5A and B).
Prevention of spontaneous lung metastases by subcutaneous mD52 vaccination and Treg depletion
We have previously reported that 3T3.mD52 tumor cells spontaneously metastasize to the lungs following subcutaneous inoculation, forming lethal tumor burdens.21 Further, we have demonstrated that the intramuscular administration of recombinant mD52 admixed with CpG ODNs in alum efficiently prevents the metastatic spread of 3T3.mD52 cells.1 Since mKSA tumor cells do not spontaneously metastasize, we employed the 3T3.mD52 tumor cell model also in this study. To determine whether subcutaneous mD52 immunization combined with Treg depletion was capable of preventing lethal lung metastases, mice were immunized as described above and challenged s.c. with a dose of 3T3.mD52 tumor cells that results in rapid tumor growth at the injection site. The enumeration of pulmonary tumor nodules was then performed in the lungs of mice bearing subcutaneous tumors with a surface area > 0.25 cm2 (days 42–50 post-inoculation), upon staining with an India ink solution. Similar to previous findings with mKSA tumor cells, the administration of the mD52-targeting vaccine in the absence of Treg depletion failed to prevent the growth of 3T3.mD52 tumor cells in the majority of mice. Conversely, ~40% of mice that were immunized and concomitantly depleted of Tregs failed to develop tumors upon the inoculation of 3T3.mD52 cells (data not shown). We were not interested in employing the 3T3.mD52 model to assess protection from primary tumor growth, but specifically to determine the efficacy of our vaccination protocol against spontaneous lung metastasis. The subcutaneous mD52 immunization combined with Treg depletion completely prevented spontaneous lung metastases, an effect that was not seen in control immunized mice or in Treg-competent mD52-immunized mice (Fig. 6). These data indicate that a vaccine based on recombinant mD52 and CpG ODNs administered s.c. as an oil in water emulsion in IFA may require Treg depletion to efficiently prevent spontaneous lethal lung metastases.

Figure 6. Spontaneous 3T3.mD52 lung metastases following immunization and CD25+ cell cell depletion. (A and B) Representative lungs from 10 BALB/c mice following immunization, CD25+ cell depletion (as described in Fig. 1A) and a subcutaneous challenge with 1 × 106 3T3.mD52 tumor cells. Lungs were harvested when the primary tumors reached a surface area of approximately 1 cm2. (A) From left to right: lungs from a naïve mouse; a control immunized mouse (incomplete Freund’s adjuvant, IFA, only), following a tumor challenge; a control immunized mouse (CpG oligodeoxynucleotides in IFA), following a tumor challenge and a mouse immunized with recombinant mD52 in IFA, following a tumor challenge. (B) From left to right: lungs from mice immunized with recombinant mD52 plus CpG oligodeoxynucleotides in IFA and treated with irrelevant IgGs, following a tumor challenge; lungs from mice immunized with recombinant mD52 plus CpG oligodeoxynucleotides in IFA and depleted of CD25+ cells, following a tumor challenge. Each set of lungs is representative of two repeated experiments.
Discussion
D52 is an intracellular protein expressed by normal cells and tissues with an hitherto undefined function. D52 is naturally overexpressed by numerous human and murine tumors. In murine tumor models, mD52 represents a bona fide overexpressed self-tumor-associated antigen (TAA). In addition, we have previously demonstrated not only that D52 is involved in increased cell proliferation and tumor progression to metastasis, but also that high expression levels of D52 are associated with poor prognosis in breast cancer patients.3,21 We believe that these characteristics of D52 make it an ideal target for prophylactic or therapeutic anticancer vaccination.
Recently, the National Cancer Institute sponsored a pilot project to prioritize cancer vaccine target antigens for translational research. The study involved the development of a set of nine “ideal” criteria for TAAs, with therapeutic function, immunogenicity and a role in oncogenesis/tumor progression deemed as the most important.22 From a representative list of antigens, overexpressed self-TAAs stand out as the group with the largest number of candidate targets for immunotherapy. It is arguable that tumor-specific antigens represent “ideal” targets for active immunotherapy. Nevertheless, most cancer antigens that have been identified to date constitute self-proteins.23 This class of antigens is generated by the re-expression of developmental/embryonic genes that are normally silenced in adult tissues (e.g., CEA),24 by the production of normal proteins with abnormal sugar moieties (e.g., MUC1),25 by the expression of tissue-specific differentiation antigens (e.g., multiple melanoma-associated antigens)26 or by the overexpression of self-proteins (e.g., ERBB2/HER2, hTERT, survivin).27-29 Of these common examples, only ERBB2/HER2 can be classified as an overexpressed self-TAA with a role in oncogenesis and tumor progression. These properties have been proposed to be important characteristics for the next generation of cancer vaccine target antigens.22 D52 is similar to hTERT and survivin in that it is associated with a wide range of cancers including (but not limited to) breast, prostate and ovarian cancers.30 In addition, D52 exhibits oncogenic functions similar to ERBB2/HER2.3,21
In the study described herein, we examined the role of the immunization site in immune responses to murine tumor cells that endogenously overexpress D52 (mKSA cells) as elicited by recombinant mD52 administered s.c. We have previously reported that mD52 administered i.m. together with CpG ODNs induces a cellular immune response capable of protecting 50% of mice from a challenge with mKSA cells.1 We hypothesized that this partial level of protection may be due to the intramuscular route being less efficient than the subcutaneous one at inducing potent antitumor immunity. To address this issue, we immunized mice s.c. with recombinant mD52 admixed with CpG ODNs in IFA. Following four immunizations, animals were challenged s.c. with a tumorigenic dose of syngeneic mKSA tumor cells and tumor growth was monitored (Fig. 1A). Surprisingly, <10% of immunized mice were protected from the tumor challenge (Fig. 2A). This result was rather unexpected as the subcutaneous route is generally accepted as one of the most potent administration routes for vaccines against microbial pathogens, and as the majority of clinical trials currently testing anticancer vaccines are based on some TAA in oil and water emulsion administered s.c. In light of this finding, we postulated that Treg modulation might be needed to achieve a protection from tumor challenge upon the administration of vaccines s.c. This hypothesis was based on the fact that D52 is a self-protein expressed by normal cells and tissues, although overexpressed by tumor cells.1,21 To address this issue, we immunized mice along with the intraperitoneal injection of anti-mouse CD25 monoclonal antibodies (Fig. 1A and B). The combination of the subcutaneous mD52-targeting vaccine and CD25+ Treg depletion resulted in a dramatic increase in the rate of tumor protection to nearly 70% of mice (Fig. 2B). Importantly, the protection generated by subcutaneous vaccination combined with Treg modulation resulted in durable immune responses, as demonstrated by the 100% rate of rejection of a secondary challenge with mKSA cells given more than 3 mo after the rejection of the primary challenge (Fig. 2C). These findings are corroborated by published data demonstrating that the administration of anti-CD25 monoclonal antibodies efficiently depletes Tregs, hence enhancing primary immune responses as well as the development of immunological memory.31-34 The increase in the efficacy and duration of vaccine-elicited immune response, which was largely dependent on Treg depletion, was therefore not surprising. However, the complete lack of efficacy that ensued the shift from the intramuscular (50% protection rate)1 to the subcutaneous route without Treg depletion (<10% protection rate) was surprising. This may reflect differences in the type of antigen-presenting cells residing in (or being attracted to) the deep muscle vs. the epidermis/dermis. It is accepted that epidermal DCs exist in a naïve state as compared with DCs from other anatomical sites. This may impart a qualitative difference in their ability to prime T cells against poorly immunogenic self-proteins like D52. In contrast, subcutaneous DCs may be more effective at eliciting Tregs to dampen immune responses against self-proteins, perhaps explaining the induction of protective antitumor immunity by our s.c. vaccine only upon the concomitant depletion of Tregs. Though the role of DCs was not directly assessed in this study, it may be important to address this point for future studies on anticancer vaccines, especially in light of the results or recent clinical trials based on the subcutaneous administration route.
Importantly, our subcutaneous vaccine was able to induce measurable tumor-specific CTL responses irrespective of Treg depletion, although Treg depletion did result in CTL responses that were as much as 2-fold greater in magnitude (Fig. 3A and B). In addition, Treg depletion was clearly needed for our vaccine to induce protective antitumor immune responses. In addition, our vaccine combined with Treg depletion efficiently prevented metastases was evaluated in a spontaneous lung metastasis model (Fig. 6). In addition to examining tumor-specific killing by CTLs from protected (immunized and Treg-depleted) mice, we studied cytokine production by T cells using a multi-analyte ELISArray for mouse TH1/TH2/TH17 cells. We found that only 4 out of 12 cytokines tested were produced by T cells from protected animals, including IFNγ, IL-10, IL-6 and IL-13 (Fig. 4). We confirmed these observations with cytokine-specific ELISAs (Fig. 5; Table 1). Most importantly, we demonstrated that both IFNγ and IL-10 were only produced by T cells exposed in response to syngeneic, antigen-positive tumor cells. The production of IL-10 was significantly blocked by anti-MHC Class I monoclonal antibodies by T cells from all five animals tested in this respect, while IFNγ secretion was significantly blocked in 2/5 samples (Fig. 5A and B). The suboptimal inhibition of cytokine production by anti-MHC Class I monoclonal antibodies was likely due to the presence of cytokine-producing CD4+ T cells. The production of IFNγ was expected, demonstrating that a TH1 cellular response dominated in this scenario. The fact that anti-MHC Class I monoclonal antibodies could block IL-10 production indicates that CD8+ MHC Class I-restricted antigen-specific T cells are present and are likely involved in the suppression of vaccine-induced antitumor immune responses via IL-10 production. We have previously reported that mD52 expression by tumor cells is associated with an increased secretion of TGF-β1,21 and it is widely accepted that TGF-β1 induces Tregs that may dampen immune responses to self-antigens. mD52 is a self-TAA and mD52-expressing tumors secrete TGF-β1. These facts support the notion that IL-10 is involved in suppressing vaccine-induced immune responses and may (at least part) explain the lack of complete protection (Fig. 2A). Though not tested here, it is tempting to speculate that the amount of IL-10 may have been greater than that of IFNγ in vaccinated mice that eventually developed tumors. Others have reported on an under-studied CD8+ subset of Tregs in autoimmunity,35 infectious disease36 and cancer.37 However, unlike for CD4+ Tregs, a consensus is lacking on the phenotype and mechanism of action of these cells, which have been proposed to be CD25+FOXP3+ cells,38 CD122+IL-10+ cells,39,40 Qa-1-restricted cells41 as well as unspecific cells.42 Consequently, there is a gap in the knowledge about CD8+ Tregs and their negative effects on cancer immunotherapy. Future studies on this immunosuppressive cell type may help to understand how CD8+ Tregs inhibit vaccine-induced immune responses targeting overexpressed self-TAAs like D52. It is possible, however, that this CD8+ T-cell population might constitute a unique subset of IL-10 secreting effector cells.43 This possibility will have to be considered in future studies.
Here, we determined whether the subcutaneous route of immunization would increase antitumor immune responses as induced by vaccination with a recombinant TAA, mD52. Contrary to our previous results in intramuscular vaccination settings, switching the adjuvant from alum to IFA and the route from intramuscular to subcutaneous failed to protect mice from a tumor challenge. To generate immune responses capable of protecting mice against tumor challenges, subcutaneous mD52-targeting vaccination had to be combined with the depletion of CD25+ T cells. Importantly, CD25+ Treg depletion did not interfere with the development of immunological memory in this context. Furthermore, this combination therapy revealed the presence of a population of IL-10-producing CD8+ T cells that may be involved in suppressing vaccine-induced antitumor immunity. This finding is unique to our prophylactic model, as opposed to therapeutic models that clearly demonstrate that the tumor microenvironment plays an active immunosuppressive role through the activation of multiple cell types. The vaccine-mediated immunosuppression that we observed in response to recombinant mD52 (as a model of overexpressed self-TAA) involved CD25+ Tregs and perhaps CD8+ regulatory cells. This occurred prior to tumor exposure, precluding any influence from the tumor microenvironment. In this light, our study represents a model to induce immune responses against a self-TAA. Our results could prove useful for solving problems related to weak immune responses elicited by vaccines against overexpressed self-TAAs and for leading the design of new anticancer vaccines against this large and understudied group of candidate immunotherapeutic targets.
Materials and Methods
Mice and tumor cells
Female, 6- to 8-week-old BALB/c mice were purchased from Charles River Laboratories. All animals were cared for and treated according to Institutional Animal Care and Use Committee guidelines at Texas Tech University Health Sciences Center. The tumorigenic BALB/c 3T3.mD52 cell line21 and the tumorigenic SV40-transformed BALB/c murine kidney mKSA cell line were used for tumor challenge following immunization procedures. C57BL/6 TRAMP-C2 tumor cells were used as MHC Class I-mismatched, mD52-expressing controls in immunological assays. mKSA and 3T3.mD52 cells were cultured in RPMI 1640 medium (Fisher Scientific) supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 250 ng/mL fungizone, 50 IU/mL penicillin, 50 μg/mL streptomycin, 50 μg/mL gentamicin sulfate and 10 mM HEPES buffer. TRAMP-C2 cells were cultured as previously reported.44
Recombinant mD52 purification
The B-PER GST Fusion Protein Purification kit (Thermo Scientific) was used to affinity purify a recombinant mD52-glutathion S transferase (GST) fusion protein, according to manufacturer’s instructions and our previously published methods.1 This method yields a purified protein that is free of bacterial contamination including lipopolysaccharide. Briefly, a 250 mL culture of recombinant Escherichia coli expressing mD52-GST fusion protein was grown to log phase and protein expression induced with 100 mM isopropylthio-β-D-galactoside (IPTG). Cells were harvested and lysed in B-PER 1 X protease inhibitor cocktail (Thermo Scientific) 250 U benzonase (Novagen) and the mD52 fusion protein was purified using supplied pre-packed, disposable columns, according to the manufacturer’s instructions. The eluted protein was dialyzed for two 30-min cycles in PBS at room temperature. Following a final overnight dialysis in PBS, the protein was concentrated using aquacide (Calbiochem, EMD Biosciences). Protein concentration was determined by BCA analysis, according to the manufacturer’s instructions (Sigma Aldrich). Protein purity was assessed by SDS-PAGE and Coomassie Brilliant blue staining or silver staining, as well as by immunoblotting based on an anti-TPD52 polyclonal antibody (generated by immunizing rabbits with the N-terminal, carrier-conjugated peptide GCAYKKTSETLSQAGQKAS; italic letters represents the region of TPD52 that is identical in humans and mice) (BioSynthesis, Inc.).
Immunization, Treg depletion and tumor challenge
Individual mice were immunized via subcutaneous injection every 10–14 d with 50 μg of recombinant mD52 admixed with 50 μg of the CpG ODN 1826 (TCCATGACGTTCCTGACGTT)45 in incomplete IFA as an oil in water emulsion, for a total of four injections. CpG ODNs in IFA or IFA alone served as control immunizations. To deplete CD25+ cells, mice were injected i.p. with approximately 200–300 μg anti-CD25 monoclonal antibodies in 200 μL PBS on day 0, and again on day 35, following the fourth and final immunization, with 400 μg anti-CD25 monoclonal antibodies in 200 μL PBS. As a control condition, mock depletion was performed with a similar amount of isotype-matched IgGs injected i.p. on day 0 and day 35. Mice in all groups were bled from the dorsal tail vein prior to each immunization. Two weeks after the final immunization, mice were challenged with 5 × 105 mKSA tumor cells. For spontaneous lung metastasis studies, mice were challenged with 1 × 106 3T3.mD52 tumor cells. Tumor cells were harvested, counted and re-suspended in PBS and 100 μL of viable cell suspension were injected subcutaneously in the right flank of each mouse for the primary tumor challenge. The same procedure was used for the secondary tumor challenge, which was administered to the left (opposite) flank. Tumor size was determined by taking perpendicular measurements with common calipers every 2 to 3 d, and tumor volume (mm3) was calculated using the following formula: (A × B2)/2, where B is the smaller of the two measurements.
Analysis of CTL-mediated tumor cell lysis
T cells were isolated from the spleens of immunized mice that survived tumor challenge and subjected to standard CTL cytotoxicity assays. CTLs were generated by culturing splenocytes in the presence of irradiated mKSA tumor cells in the presence of 10 ng/mL IL-2, 5 ng/mL IL-7 and 5 ng/mL IL-12 at 37 °C for 5–7 d. Specificity was evaluated by mixing various amounts of CTLs with a constant number (5 × 103 cells) of target cells in 96-well round bottom plates. Specific lysis was determined using a Europium time-resolved, fluorescence-based, 2-h method, and measured using a Victor3™ plate reader (Perkin Elmer). Percent lysis was calculated as % specific lysis = 1 − (E − S)/(M − S) × 100, where E represents Eu release in the presence of effector cells, S is spontaneous Eu release in medium alone and M represents maximum Eu released in lysis buffer.46,47
T-cell culture and ELISAs for cytokine production
T cells were obtained from the spleen of mD52-immunized by gradient separation and stimulated in vitro by culture with irradiated mKSA tumor cells in the presence of 10 ng/mL IL-2, 5 ng/mL IL-7 and 5 ng/mL IL-12 at 37 °C for 5–7 d. After 24 h, supernatants were harvested from T cells (1×106 cells/mL in 200 μL medium in 96-well plates) cultured alone or together with various targets (1:1 ratio): mKSA cells (H-2d, mD52+) used for challenge; TRAMP-C1 and TRAMP-C2 tumor cells (both H-2b, mD52+), which served as control MHC Class I-mismatched targets. To confirm MHC Class I-restricted tumor recognition, blocking assays were performed by incubating mKSA tumor cells with anti-H-2d or anti-H-2b (negative control) monoclonal antibodies prior to incubation with T cells, as previously described.19 The assessment of cytokine secretion by tumor-specific T cells was performed by applying culture supernatants to commercially available sandwich ELISAs specific for IFNγ, IL-4, IL-10 and IL-17 (R&D Systems) or to commercially available Multi-Analyte ELISArray specific for mouse TH1/TH2/TH17 cells (SABiosciences), as per the manufacturer’s instructions. Assays were analyzed using the Victor3™ plate reader (Perkin Elmer). Standard curves to determine the concentration of IFNγ, IL-4, IL-10 and IL-17 were generated based on provided internal controls.
Flow cytometry
For the determination of CD25+ cell depletion, BALB/c mice were immunized and challenged as described above. Seven days after the final immunization, peripheral blood mononuclear cells (PBMCs) were collected by tail vein bleed, and lymphocytes were isolated using the Lympholyte-M® density separation medium (Cedarlane Labs). Lymphocytes from animals in the same experimental group were pooled (n = 10 per pooled sample) and stained with 1 μg of anti-CD4-FITC and anti-CD25-PE monoclonal antibodies (BD Bioscience) per 1 × 106 cells. Antibodies were purchased from). Cells were fixed in 1% paraformaldehyde at 4 °C for 1 h and then analyzed by flow cytometry using an LSRII flow cytometer (BD Bioscience).
Enumeration of 3T3.mD52 spontaneous lung metastases
The analysis of 3T3.mD52 metastatic spread was performed by removing the lungs of animals following euthanasia and injecting the lungs with India ink to visualize individual tumor nodules.21 Briefly, an India ink solution was injected through the trachea and allowed to fill the lungs. The lungs were then placed in Fekete’s solution for de-staining. Tumor nodules do not absorb the India ink, which results in the normal lung tissue staining black with tumor nodules remaining white. Tumor nodules were counted blindly and size was noted by three individuals (J.D.B., H.N.S., R.K.B.).
Statistical analyses
ELISA data were analyzed using one-way ANOVA with Bonferroni multiple comparison post-hoc test. CTL cytotoxicity data were analyzed using one-way ANOVA with Tukey–Kramer post-hoc test. In both cases, p values < 0.05 were considered as statistically significant (GraphPad Prism 5.0). When required, tumor challenge data were analyzed with a Student’s t-test to determine whether significant differences existed between mean tumor volumes in mD52-immunized vs. control immunized mice.
Acknowledgments
This work was supported by funds from Texas Tech University Health Sciences Center.
Glossary
Abbreviations:
- 3T3.mD52
mD52-transformed NIH-3T3
- D52
tumor protein D52
- hD52
human D52
- mD52
murine D52
- ODN
oligodeoxynucleotide
- Treg
T regulatory cell
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/25049
References
- 1.Payton LA, Lewis JD, Byrne JA, Bright RK. Vaccination with metastasis-related tumor associated antigen TPD52 and CpG/ODN induces protective tumor immunity. Cancer Immunol Immunother. 2008;57:799–811. doi: 10.1007/s00262-007-0416-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petrova DT, Asif AR, Armstrong VW, Dimova I, Toshev S, Yaramov N, et al. Expression of chloride intracellular channel protein 1 (CLIC1) and tumor protein D52 (TPD52) as potential biomarkers for colorectal cancer. Clin Biochem. 2008;41:1224–36. doi: 10.1016/j.clinbiochem.2008.07.012. [DOI] [PubMed] [Google Scholar]
- 3.Shehata M, Bieche I, Boutros R, Weidenhofer J, Fanayan S, Spalding L, et al. Non-redundant functions for tumor protein D52-like proteins support specific targeting of TPD52. Clin Ca Res. 2008;14:5050–60. doi: 10.1158/1078-0432.CCR-07-4994. [DOI] [PubMed] [Google Scholar]
- 4.Tiacci E, Orvietani PL, Bigerna B, Pucciarini A, Corthals GL, Pettirossi V, et al. Tumor protein D52 (TPD52): a novel B-cell/plasma-cell molecule with unique expression pattern and Ca(2+)-dependent association with annexin VI. Blood. 2005;105:2812–20. doi: 10.1182/blood-2004-07-2630. [DOI] [PubMed] [Google Scholar]
- 5.Byrne JA, Mattei MG, Basset P. Definition of the D52 (TPD52) gene family through cloning of D52 homologues in human (hD53) and mouse (mD52) Genomics. 1996;35:523–32. doi: 10.1006/geno.1996.0393. [DOI] [PubMed] [Google Scholar]
- 6.Byrne JA, Tomasetto C, Garnier JM, Rouyer N, Mattei MG, Bellocq JP, et al. A screening method to identify genes commonly overexpressed in carcinomas and the identification of a novel complementary DNA sequence. Cancer Res. 1995;55:2896–903. [PubMed] [Google Scholar]
- 7.Wang R, Xu J, Saramäki O, Visakorpi T, Sutherland WM, Zhou J, et al. PrLZ, a novel prostate-specific and androgen-responsive gene of the TPD52 family, amplified in chromosome 8q21.1 and overexpressed in human prostate cancer. Cancer Res. 2004;64:1589–94. doi: 10.1158/0008-5472.CAN-03-3331. [DOI] [PubMed] [Google Scholar]
- 8.Byrne JA, Mattei MG, Basset P, Gunning P. Identification and in situ hybridization mapping of a mouse Tpd52l1 (D53) orthologue to chromosome 10A4-B2. Cytogenet Cell Genet. 1998;81:199–201. doi: 10.1159/000015029. [DOI] [PubMed] [Google Scholar]
- 9.Byrne JA, Nourse CR, Basset P, Gunning P. Identification of homo- and heteromeric interactions between members of the breast carcinoma-associated D52 protein family using the yeast two-hybrid system. Oncogene. 1998;16:873–81. doi: 10.1038/sj.onc.1201604. [DOI] [PubMed] [Google Scholar]
- 10.Nourse CR, Mattei MG, Gunning P, Byrne JA. Cloning of a third member of the D52 gene family indicates alternative coding sequence usage in D52-like transcripts. Biochim Biophys Acta. 1998;1443:155–68. doi: 10.1016/S0167-4781(98)00211-5. [DOI] [PubMed] [Google Scholar]
- 11.Cao Q, Chen J, Zhu L, Liu Y, Zhou Z, Sha J, et al. A testis-specific and testis developmentally regulated tumor protein D52 (TPD52)-like protein TPD52L3/hD55 interacts with TPD52 family proteins. Biochem Biophys Res Commun. 2006;344:798–806. doi: 10.1016/j.bbrc.2006.03.208. [DOI] [PubMed] [Google Scholar]
- 12.Chen S-L, Maroulakou IG, Green JE, Romano-Spica V, Modi W, Lautenberger J, et al. Isolation and characterization of a novel gene expressed in multiple cancers. Oncogene. 1996;12:741–51. [PubMed] [Google Scholar]
- 13.Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, et al. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001;412:822–6. doi: 10.1038/35090585. [DOI] [PubMed] [Google Scholar]
- 14.Rubin MA, Varambally S, Beroukhim R, Tomlins SA, Rhodes DR, Paris PL, et al. Overexpression, amplification, and androgen regulation of TPD52 in prostate cancer. Cancer Res. 2004;64:3814–22. doi: 10.1158/0008-5472.CAN-03-3881. [DOI] [PubMed] [Google Scholar]
- 15.Malek RL, Irby RB, Guo QM, Lee K, Wong S, He M, et al. Identification of Src transformation fingerprint in human colon cancer. Oncogene. 2002;21:7256–65. doi: 10.1038/sj.onc.1205900. [DOI] [PubMed] [Google Scholar]
- 16.Byrne JA, Balleine RL, Schoenberg Fejzo M, Mercieca J, Chiew YE, Livnat Y, et al. Tumor protein D52 (TPD52) is overexpressed and a gene amplification target in ovarian cancer. Int J Cancer. 2005;117:1049–54. doi: 10.1002/ijc.21250. [DOI] [PubMed] [Google Scholar]
- 17.Byrne JA, Chen Y, Martin La Rotta N, Peters GB. Challenges in identifying candidate amplification targets in human cancers: chromosome 8q21 as a case study. Genes Cancer. 2012;3:87–101. doi: 10.1177/1947601912456287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Todaro GJ, Green H. Quantitative studies of the growth of mouse embryo cells in culture and their development into established lines. J Cell Biol. 1963;17:299–313. doi: 10.1083/jcb.17.2.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lewis JD, Sullivan LA, Byrne JA, de Riese W, Bright RK. Memory and cellular immunity induced by a DNA vaccine encoding self antigen TPD52 administered with soluble GM-CSF. Cancer Immunol Immunother. 2009;58:1337–49. doi: 10.1007/s00262-009-0659-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mirshahidi S, Kramer VG, Whitney JB, Essono S, Lee S, Dranoff G, et al. Overlapping synthetic peptides encoding TPD52 as breast cancer vaccine in mice: prolonged survival. Vaccine. 2009;27:1825–33. doi: 10.1016/j.vaccine.2009.01.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lewis JD, Payton LA, Whitford JG, Byrne JA, Smith DI, Yang L, et al. Induction of tumorigenesis and metastasis by the murine orthologue of tumor protein D52. Mol Cancer Res. 2007;5:133–44. doi: 10.1158/1541-7786.MCR-06-0245. [DOI] [PubMed] [Google Scholar]
- 22.Cheever MA, Allison JP, Ferris AS, Finn OJ, Hastings BM, Hecht TT, et al. The prioritization of cancer antigens: a national cancer institute pilot project for the acceleration of translational research. Clin Cancer Res. 2009;15:5323–37. doi: 10.1158/1078-0432.CCR-09-0737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schreiber TH, Raez L, Rosenblatt JD, Podack ER. Tumor immunogenicity and responsiveness to cancer vaccine therapy: the state of the art. Semin Immunol. 2010;22:105–12. doi: 10.1016/j.smim.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Arlen PM, Gulley JL, Madan RA, Hodge JW, Schlom J. Preclinical and clinical studies of recombinant poxvirus vaccines for carcinoma therapy. Crit Rev Immunol. 2007;27:451–62. doi: 10.1615/CritRevImmunol.v27.i5.40. [DOI] [PubMed] [Google Scholar]
- 25.Acres B, Limacher JM. MUC1 as a target antigen for cancer immunotherapy. Expert Rev Vaccines. 2005;4:493–502. doi: 10.1586/14760584.4.4.493. [DOI] [PubMed] [Google Scholar]
- 26.Rosenberg SA. Cancer vaccines based on the identification of genes encoding cancer regression antigens. Immunol Today. 1997;18:175–82. doi: 10.1016/S0167-5699(97)84664-6. [DOI] [PubMed] [Google Scholar]
- 27.Cavallo F, Calogero RA, Forni G. Are oncoantigens suitable targets for anti-tumour therapy? Nat Rev Cancer. 2007;7:707–13. doi: 10.1038/nrc2208. [DOI] [PubMed] [Google Scholar]
- 28.Beatty GL, Vonderheide RH. Telomerase as a universal tumor antigen for cancer vaccines. Expert Rev Vaccines. 2008;7:881–7. doi: 10.1586/14760584.7.7.881. [DOI] [PubMed] [Google Scholar]
- 29.Andersen MH, Svane IM, Becker JC, Straten PT. The universal character of the tumor-associated antigen survivin. Clin Cancer Res. 2007;13:5991–4. doi: 10.1158/1078-0432.CCR-07-0686. [DOI] [PubMed] [Google Scholar]
- 30.Shehata M, Weidenhofer J, Thamotharampillai K, Hardy JR, Byrne JA. Tumor protein D52 overexpression and gene amplification in cancers from a mosaic of microarrays. Crit Rev Oncog. 2008;14:33–55. doi: 10.1615/CritRevOncog.v14.i1.30. [DOI] [PubMed] [Google Scholar]
- 31.Van Meirvenne S, Dullaers M, Heirman C, Straetman L, Michiels A, Thielemans K. In vivo depletion of CD4+CD25+ regulatory T cells enhances the antigen-specific primary and memory CTL response elicited by mature mRNA-electroporated dendritic cells. Mol Ther. 2005;12:922–32. doi: 10.1016/j.ymthe.2005.02.029. [DOI] [PubMed] [Google Scholar]
- 32.Carvalho-Gaspar M, Jones ND, Luo S, Martin L, Brook MO, Wood KJ. Location and time-dependent control of rejection by regulatory T cells culminates in a failure to generate memory T cells. J Immunol. 2008;180:6640–8. doi: 10.4049/jimmunol.180.10.6640. [DOI] [PubMed] [Google Scholar]
- 33.Sharma S, Dominguez AL, Manrique SZ, Cavallo F, Sakaguchi S, Lustgarten J. Systemic targeting of CpG-ODN to the tumor microenvironment with anti-neu-CpG hybrid molecule and T regulatory cell depletion induces memory responses in BALB-neuT tolerant mice. Cancer Res. 2008;68:7530–40. doi: 10.1158/0008-5472.CAN-08-1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bos R, van Duikeren S, Morreau H, Franken K, Schumacher TNM, Haanen JB, et al. Balancing between antitumor efficacy and autoimmune pathology in T-cell-mediated targeting of carcinoembryonic antigen. Cancer Res. 2008;68:8446–55. doi: 10.1158/0008-5472.CAN-08-1864. [DOI] [PubMed] [Google Scholar]
- 35.Pomié C, Ménager-Marcq I, van Meerwijk JPM. Murine CD8+ regulatory T lymphocytes: the new era. Hum Immunol. 2008;69:708–14. doi: 10.1016/j.humimm.2008.08.288. [DOI] [PubMed] [Google Scholar]
- 36.Joosten SA, Ottenhoff THM. Human CD4 and CD8 regulatory T cells in infectious diseases and vaccination. Hum Immunol. 2008;69:760–70. doi: 10.1016/j.humimm.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 37.Wang RF. CD8+ regulatory T cells, their suppressive mechanisms, and regulation in cancer. Hum Immunol. 2008;69:811–4. doi: 10.1016/j.humimm.2008.08.276. [DOI] [PubMed] [Google Scholar]
- 38.Sugita S, Futagami Y, Horie S, Mochizuki M. Transforming growth factor beta-producing Foxp3(+)CD8(+)CD25(+) T cells induced by iris pigment epithelial cells display regulatory phenotype and acquire regulatory functions. Exp Eye Res. 2007;85:626–36. doi: 10.1016/j.exer.2007.07.015. [DOI] [PubMed] [Google Scholar]
- 39.Suzuki H, Shi Z, Okuno Y, Isobe K-I. Are CD8+CD122+ cells regulatory T cells or memory T cells? Hum Immunol. 2008;69:751–4. doi: 10.1016/j.humimm.2008.08.285. [DOI] [PubMed] [Google Scholar]
- 40.Wang L-X, Li Y, Yang G, Pang P-Y, Haley D, Walker EB, et al. CD122+CD8+ Treg suppress vaccine-induced antitumor immune responses in lymphodepleted mice. Eur J Immunol. 2010;40:1375–85. doi: 10.1002/eji.200839210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lu L, Cantor H. Generation and regulation of CD8(+) regulatory T cells. Cell Mol Immunol. 2008;5:401–6. doi: 10.1038/cmi.2008.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fenoglio D, Ferrera F, Fravega M, Balestra P, Battaglia F, Proietti M, et al. Advancements on phenotypic and functional characterization of non-antigen-specific CD8+CD28- regulatory T cells. Hum Immunol. 2008;69:745–50. doi: 10.1016/j.humimm.2008.08.282. [DOI] [PubMed] [Google Scholar]
- 43.Mocellin S, Marincola FM, Young HA. Interleukin-10 and the immune response against cancer: a counterpoint. J Leukoc Biol. 2005;78:1043–51. doi: 10.1189/jlb.0705358. [DOI] [PubMed] [Google Scholar]
- 44.Foster BA, Gingrich JR, Kwon ED, Madias C, Greenberg NM. Characterization of prostatic epithelial cell lines derived from transgenic adenocarcinoma of the mouse prostate (TRAMP) model. Cancer Res. 1997;57:3325–30. [PubMed] [Google Scholar]
- 45.Davis HL, Weeratna R, Waldschmidt TJ, Tygrett L, Schorr J, Krieg AM. CpG DNA is a potent enhancer of specific immunity in mice immunized with recombinant hepatitis B surface antigen. J Immunol. 1998;160:870–6. [PubMed] [Google Scholar]
- 46.Lewis JD, Shearer MH, Kennedy RC, Bright RK. Surrogate tumor antigen vaccination induces tumor-specific immunity and the rejection of spontaneous metastases. Cancer Res. 2005;65:2938–46. doi: 10.1158/0008-5472.CAN-04-2874. [DOI] [PubMed] [Google Scholar]
- 47.Bright RK, Kimchi ET, Shearer MH, Kennedy RC, Pass HI. SV40 Tag-specific cytotoxic T lymphocytes generated from the peripheral blood of malignant pleural mesothelioma patients. Cancer Immunol Immunother. 2002;50:682–90. doi: 10.1007/s00262-001-0240-8. [DOI] [PMC free article] [PubMed] [Google Scholar]