

Abstract
Platelets are the first peripheral source of amyloid precursor protein (APP). They possess the proteolytic machinery to produce Aβ and fragments similar to those produced in neurons, and thus offer an ex-vivo model to study APP processing and changes associated with Alzheimer’s disease (AD). Platelet process APP mostly through the α-secretase pathway to release soluble APP (sAPP). They produce small amounts of Aβ, predominantly Aβ40 over Aβ42. sAPP and Aβ are stored in α-granules and are released upon platelet activation by thrombin and collagen, and agents inducing platelet degranulation. A small proportion of full-length APP is present at the platelet surface and this increases by 3-fold upon platelet activation. Immunoblotting of platelet lysates detects APP as isoforms of 130 kDa and 106-110 kDa. The ratio of these of APP isoforms is significantly lower in patients with AD and mild cognitive impairment (MCI) than in healthy controls. This ratio follows a decrease that parallels cognitive decline and can predict conversion from MCI to AD. Alterations in the levels of α-secretase ADAM10 and in the enzymatic activities of α- and β-secretase observed in platelets of patients with AD are consistent with increased processing through the amyloidogenic pathway. β-APP cleaving enzyme activity is increased by 24% in platelet membranes of patients with MCI and by 17% in those with AD. Reports of changes in platelet APP expression with MCI and AD have been promising so far and merit further investigation as the search for blood biomarkers in AD, in particular at the prodromal stage, remains a priority and a challenge.
Keywords: Alzheimer’s disease, Platelet, Biomarker, Amyloid precursor protein, Aβ amyloid, β-amyloid precursor protein cleaving enzyme, Secretase, Protease-nexin 2
ALZHEIMER’S DISEASE PATHOLOGY AND BIOMARKERS
Alzheimer’s disease (AD) is the most common cause of dementia in the aging population and a major socio-economic burden for the developed countries[1]. Disease-modifying therapies are being developed and these are most likely to be successful if administered at a prodromal or pre-clinical stage. Therefore, there is a crucial need for early biomarkers for AD.
AD pathological diagnosis is established post-mortem by the presence of characteristic lesions consisting of extracellular amyloid plaques and intracellular neurofibrillary tangles in cortical and hippocampal regions, and a major loss of grey matter[2]. The amyloid plaques, as well as congophilic deposits found around blood vessels, are mostly constituted of self-aggregating Aβ peptides[3,4]. The neurofibrillary tangles result from the self-association of hyperphosphorylated, microtubule-associated tau protein[5]. Thus, methods and assays to detect changes in Aβ and tau proteins have been investigated as AD biomarkers.
Aβ amyloid is a 40-43 amino acids heterogeneous fragment that derives from the amyloid precursor protein (APP)[6]. It is produced by the sequential action of two proteolytic enzymes termed β-APP cleaving enzyme (BACE) and γ-secretase[7]. Although the molecular mechanisms leading to AD are not fully understood, genetic and molecular evidence support the amyloid cascade theory and point to the important role of the proteolytic processing of APP in the disease pathogenesis[8]. The major genes implicated in familial AD are APP itself, with a cluster of disease-causing missense mutations located near the proteolytic cleavage sites that concern Aβ, and the presenilin genes, Psen1 and Psen2 that encode γ-secretase proteolytic subunits which, in their mutated forms, promote the production of aggregating forms of Aβ[9,10]. Extensive experimental studies have documented the cellular toxicity of Aβ or soluble oligomers, which correspond to an early stage of Aβ aggregation that precedes amyloid plaque formation[11,12]. Recently, positron-emission tomography (PET) imaging has been applied to the detection of Aβ amyloid in the brain and has revealed that Aβ peptide accumulates in the frontal cortex of patients with mild cognitive impairment (MCI), the prodromal stage of AD[13]. Thus, PET imaging of Aβ represents a promising tool for the early diagnosis of AD, but it is a sophisticated technique that requires special equipment, and cannot be widely used.
Measurements of Aβ and tau isoforms in the cerebrospinal fluid (CSF) provide the most reliable biomarker to facilitate AD diagnosis in live patients[14]. Low CSF Aβ42 levels reflect the decreased clearance of Aβ42 and its deposition in the brain, but this is not absolutely specific for AD and is also observed in patients with dementia with Lewy bodies. Elevated phosphorylated tau (p-tau) is a more specific marker, and measurements of either p181-tau, or p231-tau give similar diagnosis accuracy[15]. The combination of Aβ42, total tau and p-tau provides a diagnosis for AD with a sensitivity of 80% and a specificity of 90%, and can help predict the conversion from MCI to AD[16]. However, these markers remain insufficiently used due to the delicate procedure of CSF collection by lumbar puncture. The measurements of Aβ in plasma have so far provided inconsistent results[14]. Other methods to diagnose AD are based on brain structural imaging and involve complex techniques such as magnetic resonance spectroscopy and computed tomography that require access to expensive instrumentation (reviewed by Hampel et al[17]). Therefore, there is a critical need for blood biomarkers for AD.
Human platelets offer an ex-vivo model to study and understand APP metabolism and function[18]. In human, platelets are the second source of APP protein after the brain, and they possess their own proteolytic machinery to produce Aβ and other APP secretase products similar to those observed in neuronal cells[19,20]. This review will discuss the potential of platelet APP and secretase-derived fragments as biomarkers for the early diagnosis of AD.
STRUCTURAL FEATURES OF THE AMYLOID PRECURSOR PROTEIN
Cloning of the gene coding for the Aβ APP revealed a new family of type I membrane integral glycoproteins[6], consisting of at least ten alternative gene products.
The major APP isoforms result from alternative splicing of exon 7 that encodes a Kunitz serine protease inhibitor domain (KPI), exon 8 that codes for a domain with homology to the MRC OX-2 antigen (OX-2), and exon 15 (Figure 1). The APP695 isoform, which lacks the KPI (APP-KPI-) and OX-2 domains, is expressed predominantly in neuronal cells. Peripheral cells and platelets, preferably express APP isoforms that contain the KPI domain (APP-KPI+), including APP751 (lacking the OX-2 domain) and APP770 (expressing all exons)[21-24]. The ratio of APP-KPI+/APP-KPI- was reported to be increased in AD brain[25].
Figure 1.
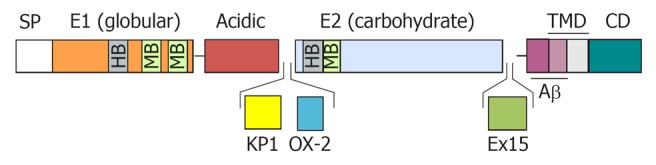
Schematic diagram of the amyloid precursor protein. The diagram represents the structural domain arrangement of the amyloid precursor protein (APP) gene products. All domains are expressed in the APP770 isoform. The OX-2 domain is missing in APP751, whereas both the Kunitz protease inhibitor domain (KPI) and the OX-2 domain are missing in APP695. SP: Signal peptide; E1: Ectodomain 1; E2: Ectodomain 2; TMD: Transmembrane domain; CD: Cytosolic domain; OX-2: Domain with homology to OX-2 leukocyte antigen; Ex 15: Exon 15 product; HB: Heparin-binding domain; MB: Metal ion-binding domain.
Splicing out of exon 15 produces L-APP isoforms, which are expressed in the lymphocyte/monocyte lineage and in non-neuronal cells within the central nervous system (CNS)[26,27].The L-APP isoforms contain an attachment site for chondroitin sulfate proteoglycans that is created by the deletion of exon 15 and this binding of a glycosaminoglycan chain close to Aβ N-terminus causes an hindrance likely to impair the amyloidogenic processing of these APP variants[28].
The APP family also consists of the two homologues, APLP1 and APLP2, which are produced by alternative genes[29,30], and undergo proteolytic processing similar to APP[31,32]. However, these are unlikely to play a major role in AD pathogenesis as the Aβ sequence is not conserved among the APP homologues[33,34].
The schematic structure of APP proteins is depicted in Figure 1 and reviewed in ref[7]. APP contains an N-terminal globular domain, or E1 domain[35], which includes binding sites for heparin, zinc, and copper. This is connected to an unstructured acidic domain followed by the KPI domain (except in APP695) and the OX-2 domain (spliced out in most isoforms) and a large E2 glycosylated domain[36] (also termed carbohydrate domain), comprising binding sites for heparin and collagen, and which is involved in homo- and hetero-dimerization[37,38]. APP is anchored in the membrane through a single transmembrane domain, followed by a cytosolic domain. The Aβ region comprises the C-terminal moiety of the extracellular domain and part of the membrane-spanning domain.
APP maturation involves post-translational modifications such as glycosylation, sulfation and phosphorylation that take place during transit through the Golgi[39], as it traffics through the secretory pathway. APP is endocytosed from the cell surface, and processed by the endosomal-lysosomal pathway. The cytoplasmic domain of APP contains two consensus motifs, YENPTY and YTS, which mediate interactions with adaptor proteins that control endocytosis[40]. YENPTY is a recognition sequence for proteins with a Src-homology 2 (SH2) or with a phosphotyrosine binding domain (PTB). A detailed review of APP interacting proteins can be found in Perreau et al[41]. APP interactors with a PTB domain include the Fe65 (or APBB1) and X11/Mint families of brain proteins which modulate its trafficking and processing, as X11 stabilizes APP and decreases its amyloidogenic processing, whereas Fe65 promotes Aβ secretion[42]. The cytoplasmic domain of APP also interacts with disrupted-in-schizophrenia 1 (DISC1) to help regulate neuronal migration during brain development[43].
SECRETASE PROCESSING OF APP AND Aβ PRODUCTION
APP can be proteolytically processed by an α-secretase-dependent, non-amyloidogenic pathway, which involves cleavage within the Aβ sequence (Figure 2) (reviewed in ref[40]). This takes place in the secretory pathway, at the plasma membrane and in secretory vesicles, and can be stimulated by protein kinase C activation. It involves α-secretase enzymes that belong to the family of “a disintegrin and metalloprotease” (ADAM proteinases) and include ADAM 10 and ADAM 17[44,45]. Cleavage of APP by α-secretase generates soluble N-terminal fragments of 100 - 130 kDa (sAPPα) and an 83 amino acid, membrane-associated C-terminal fragment (C83), which consists of the C-terminal portion of Aβ, and APP cytosolic domain.
Figure 2.
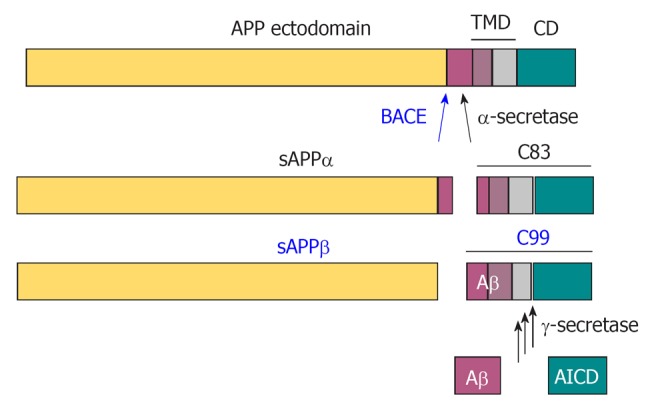
Proteolytic processing of amyloid precursor protein by the secretases. In a non-amyloidogenic pathway, α-secretase cleaves amyloid precursor protein (APP) within the Aβ domain to release the α-cleaved soluble N-terminal fragment, sAPPα. β-Amyloid precursor cleaving enzyme (BACE) cleaves APP at the N-terminus of Aβ to release the β-cleaved soluble N-terminal fragment sAPPβ, and thereby initiates the amyloidogenic pathway. The remaining 99 amino acid-long C-terminal fragment (C99) is further processed at multiple sites by γ-secretase to release the APP intracellular domain (AICD) in the cytosol and Aβ peptides in the extracellular space.
The alternative, amyloidogenic pathway involves cleavage of APP by β-secretase at the amino terminus of Aβ to release the soluble N-terminal fragment, sAPPβ, followed by processing of the remaining 99 amino acid membrane-tethered C-terminal fragment (C99) by γ-secretase to release Aβ[7]. β-Secretase has been identified as the membrane-anchored aspartyl protease β-APP-cleaving enzyme (BACE, or BACE1)[46]. The sAPPβ fragment released by BACE can be further processed by an unknown mechanism to produce a ligand that can activate the death-receptor, DR6 to regulate axon pruning during brain development[47].
γ-Secretase activity is contained within a molecular complex formed by the association of presenilins (either PS1 or PS2) with three other subunits, termed nicastrin, presenilin-enhancer-2 (PEN-2) and anterior-pharynx-defective 1 (APH-1). The presenilins are ubiquitously expressed, including in platelets[48], and are the catalytic subunits of γ-secretase, whereas the other partners help stabilize the complex and contribute to recruiting the substrates to be cleaved[49]. γ-Secretase cleaves APP transmembrane domain at multiple sites, in a sequential order beginning on the cytosolic side, to produce alternative Aβ peptides, mostly the fairly soluble Aβ40, and minor amounts of aggregating and toxic Aβ42/43[50]. Autosomal dominant single point mutations in the presenilin genes, which are the most frequent cause of inherited AD, increase the production of the longer isoforms, Aβ42/43[50].
γ-Secretase also releases the APP intracellular domain (AICD) in the cytosol and allows its diffusion to the nucleus together with transcription-factor binding partners[51,52]. Crossing APLP2 knockout mice with mice that express APP lacking the cytosolic domain, but capable of being processed into sAPP and Aβ, results in a neuromuscular phenotype similar to double APP/APLP2 knockout, supporting that the AICD, which is highly conserved in the APP homologues, fulfils an important individual function during development[53].
Both sAPPβ and Aβ are secreted from normal cells and can be detected in the plasma and CSF[14,39,54-60]. The use of alternative processing pathways is regulated by signal transduction (reviewed in ref[61]), and may dependent on cell types and APP isoforms. Non-neuronal cells and platelets process APP mostly through the α-secretase pathway[62], whereas neurons and neuronal cell lines preferentially process APP by the β-secretase pathway and produce higher amounts of Aβ[55,63].
FUNCTIONS OF APP AND ITS DERIVED FRAGMENTS
Many biological functions have been attributed to APP and its proteolytic fragments, which may depend on APP isoforms, tissue/cellular expression, and interacting partners.
The identification of APP suggested it was a cell surface receptor[6], and this hypothesis was strengthened when its was established that APP proteolytic processing resembles that of Notch and could potentially transduce transmembrane signalling. Many proteins have been shown to interact with APP[41]. however the search for a specific ligand has remained elusive, although the brain protein F-spondin represents a possible candidate[64]. APP has been proposed to be a cell-adhesion molecule. Indeed, it can establish cell-cell interactions through homodimerization or through heterodimerization with its APP homologues[38]. Moreover, cell surface APP can bind to cell surface sulfated proteoglycans (glypican), laminin, collagen, or integrin-like receptors through its specific binding domains[65-69]. A contribution of APP to neurite outgrowth was identified nearly two decades ago[70,71], and it has now been shown that this is mediated through interactions with reelin and integrin β[72]. Binding to the trophic factor netrin-1 has also been demonstrated[73]. Furthermore, APP was found to form part of a macromolecular complex that includes Fe65 and is involved in axon branching during development[74]. There is mounting evidence that APP plays a role in synaptogenesis and synaptic plasticity (reviewed in ref[75]).
The soluble, sAPPα has neurotrophic properties and can also protect neurons against excitotoxic or ischemic insults by stabilizing intracellular calcium[70,76-79]. As noted earlier, sAPPβ is the precursor of a ligand for DR6 and involved in axon pruning, and may therefore have an antagonistic function to sAPPα in the developing brain[47]. Soluble APP that contains the KPI domain was identified as protease nexin-2, a chymotrypsin inhibitor and a regulator of blood coagulation[80]. This will be further developed in the platelet section.
APP has also been involved in the homeostasis of the zinc, copper and iron metal ions[81-83], and a recent study has demonstrated APP’s ferroxidase activity and its involvement in cellular iron export[84].
The physiological function of APP has been further addressed by gene knockout experiments in animal models. Mice with a single gene knockout (APP-/-, APLP1-/-, APLP2-/-) as well as the APLP1-/-/APP-/- double knockout mice are viable and fertile. In contrast, the double knockout of APP-/-/APLP2-/- causes perinatal lethality and profound defects in neuromuscular junction, suggesting that APP and APLP2 have overlapping and non-redundant functions[85-88]. The identification of a role for APP and APLP2 in the modulation of glucose and insulin plasma levels may explain the low survival of the APP-/-/APLP2-/- double knockout mice[89]. These display 66% decrease in plasma glucose levels combined with hyperinsulinemia, and do not live more than 24 h after birth.
Apart from the pathogenic role of its aggregated forms, Aβ itself has been attributed physiological functions in cholesterol metabolism, and as an antimicrobial protective agent[90,91].Therefore, alterations in APP function and proteolytic processing, as observed in AD, would contribute to cognitive impairment, and defects in synaptic plasticity, brain glucose levels, and metal homeostasis, as well as a dysregulation of blood coagulation factors.
APP EXPRESSION AND PHYSIOLOGICAL FUNCTION IN PLATELETS
Platelets are a readily accessible source of human tissue and have been used as an ex vivo model to study APP processing and function. They contain APP levels comparable to those expressed in the brain and the second highest among all peripheral tissues[92].
Although platelets contain mRNA for the three major APP variants, APP695, APP751, and APP770, they express mostly the sAPPα-KPI+ isoforms (APP751 and APP770) and contain no detectable amounts of L-APP isoforms or APLP proteins[93]. Ninety percent of total platelet APP corresponds to the soluble form and only 10% consists of full-length APP (APPFL-KPI+) and C-terminally truncated membrane-associated APP (APPMem-KPI+)[24]. This is in contrast with the brain where -50% of APP is present as a soluble form[94]. Activation of platelets by thrombin increases the surface expression of APP by up to 3-fold, suggesting that APP-KPI+ may be involved in the regulation of haemostatic protease activity on the platelet surface[24].
Platelets are the primary source of APP in the blood circulation. They produce more than 90% of circulating APP[95] and up to 90% of circulating Aβ[24,57,96]. Both APP (including-90% as the soluble forms, sAPPα, sAPPβ, and 10% of full-length APP) and the Aβ fragment are stored in α-granules[24,97], and become released upon platelet activation by agents that induce platelet degranulation, like the physiological agonists thrombin and collagen, or non-physiological agonists, such as the ionophores A23187 and ionomycin[56,92,96,98,99].
The released sAPP isoforms are potent inhibitors of the coagulation factors XIa and IXa[100-102], and, to a lesser extent, of factor VIIa-tissue factor complex, supporting that they contribute to haemostasis[103]. Indeed, APP inhibits factor XIa with a Ki value of 450 ± 50 pmol/L, and this inhibitory effect is enhanced by heparin and zinc[104]. Structural information derived from co-crystallization of the catalytic domain of factor XIa with the APP KPI domain has identified the critical amino acids of the KPI domain that are involved in the inhibition of factor XIa[105]. APP also inhibits factor IXa with a high affinity, with a Ki of 79 pmol/L in the absence of heparin, and of 39 pmol/L in the presence of heparin. It inhibits to a lesser extent factor VIIa (FVIIa) and the factor VIIa-tissue factor complex (FVIIa-TF), with apparent Ki of 11 ± 2 μmol/L and 15 ± 1 μmol/L, respectively, and this inhibition can be increased by more than two orders of magnitude in the presence of soluble tissue factor[104].
Our group has reported that exogenous sAPP can inhibit platelet aggregation and secretion induced by ADP or adrenaline via the arachidonic acid pathway, suggesting that platelet degranulation and APP secretion may provide a negative regulation to platelet activation[106]. Aβ appears to exert an antagonistic effect as it can counteract sAPP inhibition of platelet aggregation. Indeed, Aβ was shown to augment ADP-dependent platelet aggregation and to support platelet adhesion[107,108]. Therefore, a balance between the levels of sAPP and Aβ may be required in haemostasis. Similarly, a balance between APP and Aβ may also be important in the brain, as Aβ is thought to be toxic to neurons by disrupting Ca2+ homeostasis, whereas sAPP has been shown to protect neurons against excitotoxic insults by stabilizing intracellular Ca2+ concentration[77].
PLATELET APP ISOFORMS AND AD
As APP plays a key role in the aetiology of Alzheimer’s disease and is abundantly expressed in platelets, it was important to examine changes in its expression with AD to determine its potential as a peripheral biomarker. Davies et al[109] examined APP secretion from isolated platelets after activation by α-thrombin and found increased retention of APP in the platelets of AD patients compared to controls, including age-matched controls and patients with other brain neurodegenerative conditions. They showed by western blotting that AD platelets accumulated unprocessed, 120-130 kDa APP at their surface and released less sAPP, suggesting a defect in proteolytic processing. This finding was observed in patients at advanced stages of AD, but not in those with moderate AD[110].
Rosenberg et al[111] analysed APP expression in intact platelets by western blotting and detected a predominant 120-130 kDa signal plus a 110 kDa minor band. When calculating the ratio of the 120-130 kDa and 110 kDa isoforms, they showed that this was significantly lower in patients with AD (n = 15) compared to controls (n = 19), and that this decreased ratio was more pronounced in patients who carry the ApoE epsilon4 allele. The same group further demonstrated that the decrease in platelet APP isoform ratio was specific for AD, and in a follow-up study of ten patients with moderate AD, they showed a correlation between declines in mini-mental score examination (MMSE) performance and declines in platelet APP ratios at a 3-year interval[112].
Di Luca et al[113] and colleagues described western blot detection of platelet APP as 130 kDa and 106-110 kDa signals, of comparable intensity in control subjects (0.84 ± 0.2; n = 25) and showed that the ratio of APP 130/106-110 isoforms (APPr) was significantly lower in AD patients (0.31 ± 0.15; n = 32) compared to age-matched controls or to patients with other neurological disorders (0.97 ± 0.4; n = 16). They found no difference in APP mRNA transcripts levels between experimental groups, a fact that may suggest the abnormal proteolytic processing of platelet APP in AD. The diagnostic value of platelet APPr was further supported by analysis of a cohort of 85 probable AD and 95 age-matched healthy controls which indicated a marked decrease in APPr in the AD group (0.35 ± 0.18) compared to the controls (0.92 ± 0.38)[114]. Setting a cut-off value at 0.57, they showed that APPr score was able to detect AD with 88% sensitivity and 89% specificity. They also established that APPr value correlated with the disease severity and could be used as an early biomarker of AD. In a study involving 35 patients with mild AD (MMSE score = 20.0 ± 1.8), 21 with very mild AD (MMSE score = 24.9 ± 0.9), 30 with MCI (MMSE score = 27.9 ± 1.2) and 25 age-matched controls (MMSE score = 29.4 ± 1.0), Padovani et al[115] reported that APPr was decreased by 59% in the group with mild AD, by 47% in the group with very mild AD, and by 33% in the group with MCI, compared to the controls. Using a threshold value of 0.60, they showed that APPr score had a sensitivity of 88.6% at detecting mild AD and 85.7% at detecting very mild AD, with a specificity of 88% for the control group. Therefore, AD-specific alteration of platelet APP isoforms occurs as an early event in AD and correlates with progression of clinical symptoms and cognitive decline, suggesting that this assay could be used as a peripheral biochemical marker for the early detection of AD.
Platelet APPr was next examined for its predictive value in the conversion from MCI to AD. When thirty patients with MCI were tested by MMSE over a period of 2-year and the data compared to APPr measurement at the beginning of the study, Borroni et al[116] found that the twelve patients who converted to Alzheimer-type dementia during the study were those with lower platelet APPr score (0.36 ± 0.28), whereas patients who remained stable or converted to other types of dementia had platelet APPr values closer to normal (0.73 for MCI and 0.86 for other dementia). By setting a cut-off score at 0.6, APPr could detect preclinical AD with a sensitivity of 83% and a specificity of 71%. In a further study, they showed that the preclinical diagnostic value of APPr could be enhanced when combined with measurement of regional cerebral blood flow by SPECT scan, reaching a sensitivity of 95% and a specificity of 75% at predicting MCI conversion to AD[117].
In an independent study, Zainaghi et al[118] have also reported a decreased ratio of APP 130/110 kDa isoforms in patients with AD (n = 23) compared to control subjects (n = 29), but no difference between patients with MCI (n = 25) and controls. The smaller cohort in this study, but also differences in methodology may account for these apparently conflicting results. In Zainaghi’s study, platelet APP in control subjects was detected as a predominant band of 130 kDa and a minor form of 110 kDa, whereas Borroni et al[119], detected APP in platelets of healthy individuals as 130 kDa and 106-110 kDa signals of similar intensity. Nevertheless, in a 4-year follow-up study that involved 34 patients with MCI and 21 patients with AD, Zainaghi and colleagues have recently reported that the baseline levels of APP 130/110 isoform ratio were significantly lower in platelets from patients with MCI who converted to AD than in subjects who remained stable MCI[120], further supporting the value of APP isoform ratio for predicting the conversion from MCI to AD.
Measurement of APPr may also prove valuable for monitoring the effect of therapies in patients with AD. After 30 d treatment with the acetylcholinesterase inhibitor donepezil (5 mg daily), patients with mild to moderate AD showed an increase in platelet APPr, which was influenced by ApoE genotype as the non-epsilon4 carriers showed a higher APPr recovery[121]. As hypercholesterolemia was shown to increase Aβ secretion from platelets[122], it was interesting to examine the effect of high cholesterol levels on platelet APP. Borroni et al[123] found that AD patients with high cholesterol levels had lower APPr scores. Interestingly, a group of AD patients who received the anticholesterol drug statin for 6 wk showed their APPr increase as their cholesterol levels decreased[124].
Since resting platelets contain mostly the secreted forms of APP, the difference in molecular mass between the 130 kDa and the 106-110 kDa isoforms has been attributed to the presence or absence of KPI (APP-KPI+/APP-KPI-), although the full characterization of these protein fragments is yet to be performed. The declining ratio of APP isoforms in platelets may result from increased release of the 130 kDa species in plasma during platelet activation. This would be consistent with our finding that the 130 kDa sAPP species are elevated in the plasma of AD patients with moderate to severe dementia compared to age-matched controls[125]. Abnormal release of APP-KPI+ from platelets would contribute to the inhibition of coagulation factors in the blood circulation, reduce platelet aggregation, and thus could contribute to microvasculature deficiencies in the AD brain[126].
SECRETASE PROCESSING AND APP FRAGMENTS IN PLATELETS
Our studies have demonstrated that platelets possess α-, β-, and γ-secretase activities and are able to produce APP fragments similar to those produced by neurons: sAPPα and sAPPβ soluble isoforms, the C99 amyloidogenic fragment, and Aβ peptides[56,62]. Furthermore, α-secretase ADAM-10 and BACE, as well as the γ-secretase catalytic subunit presenilin are expressed in platelets[19,48,127]. As APP processing plays an important role in AD aetiology, investigations have been conducted to compare secretase levels and APP proteolytic fragments in the platelets of patients with AD and control subjects in order to determine potential changes associated with the disease.
α-Secretase cleavage is the predominant pathway for APP processing in platelets, as the levels of sAPPα levels detected are much higher than those of Aβ[62] and, in particular, high amounts of sAPPα are stored in the platelet α-granules[128]. The calmodulin antagonist W7 stimulates the proteolysis of platelet APP by the non-amyloidogenic pathway, supporting that calmodulin is a regulator of APP processing by α-secretase[128]. Several studies support a decrease of α-secretase candidate, ADAM 10 in platelets from AD patients[127,129,130]. Western blot analysis of platelets homogenates from 33 patients with sporadic AD and 26 age-matched controls showed a significant decrease in ADAM10 in the patients group[129]. Release of platelets α-granule contents by thrombin activation showed that platelets from AD patients (n = 15) contained -50% less sAPPα than platelets from controls (n = 12), which is consistent with reduced α-secretase activity. A similar reduction in sAPPα levels was observed in the CSF of the AD group. In a subsequent study, Colciaghi and colleagues investigated the expression of ADAM10 and BACE in platelets of patients with mild and very mild AD[127]. They observed decreased levels of ADAM10 in both patients groups[127,129]. Platelet BACE was detected as two bands, at 57 kDa and 36 kDa, suggesting that platelet BACE undergoes endoproteolysis, a fact previously observed in peripheral tissues but not in human brain, and which may represent a regulatory mechanism[131]. The 57 kDa signal, which is consistent with the apparent molecular weight of full-length BACE, was not significantly different between the controls and two patient groups. The 36 kDa endoproteolytic fragment was markedly decreased in the AD groups, and the ratio of the 36/57 kDa BACE signals was decreased by about 50% in the platelets of patients with mild and very mild AD. This result was corroborated by Tang et al[130] who reported a significant -75% decrease in BACE 37/57 kDa ratio in patients with mild (n = 18) and severe (n = 13) AD compared to controls (n = 10). Therefore, measurement of secretases and their APP proteolytic fragments can provide further tools for early detection of AD. They may also help monitoring the effect of therapeutic treatments. Indeed, the levels of platelet ADAM10 and sAPPα, were rescued and a simultaneous decrease in β-secretase cleavage product, C99 was observed when patients with AD patients were treated with a cholinesterase inhibitor for 30 d[132]. Di Luca et al[133] showed that integration of APPr with ADAM10 and BACE values for 37 patients with mild AD and 25 control subjects using a complex software analysis could provide a very specific diagnostic method that discriminate patients with early AD in 94% cases and controls in 92%.
Although we have shown by western blotting the presence of sAPPβ in platelets[62], its levels are low and near detection limits, and thus have not been compared between AD and controls. β-Secretase enzymatic activity is easier to quantify, and since it has been reported that BACE is elevated in AD brain cortex[134-136] and AD CSF[137,138], several studies have compared β-secretase activity in platelets from patients with AD to healthy controls. Measurement of β-secretase activity in CHAPS-solubilised platelet membranes using a fluorogenic substrate cleavage assay has revealed a highly significant 17% increase in the platelets of AD patients (n = 86) compared to age-matched controls (n = 115)[139]. The activity measured in this assay was established to derive from BACE by its sensitivity to a specific BACE inhibitor. Johnston’s group also reported that platelet membrane BACE activity was increased by 24% in subjects with MCI (n = 52) compared to controls (n = 75)[140].This finding suggests that BACE activity increases in the early stage of AD and further supports the lack of correlation between β-secretase activity and MMSE in AD, as an increase in platelet BACE activity would precede the loss of cognitive function[139].
In contrast, Gorham et al[141], showed no difference in either α- or β-secretase activities in platelets of AD sufferers (n = 20), patients with MCI (n = 6) and controls (n = 30). This latest study involved a smaller number of cases, used total platelet lysates, and measured secretase activities with commercial kits, which may lack specificity. Liu et al[142], identified a biphasic relationship between platelet membrane cholesterol and β-secretase activity, with a positive correlation at high membrane cholesterol levels and a negative correlation at lower membrane cholesterol levels. Studying platelets from 172 patients with AD or MCI and 171 controls they found that this relationship was disrupted in AD patients, as these were more likely to lie within the negative correlation zone than the controls.
Unlike neurons that produce significant amounts of Aβ42 peptide, platelets produce mainly the less aggregating Aβ40, and they release part of their Aβ40 content upon activation with thrombin and collagen, thereby contributing to most Aβ present in the blood circulation[56]. Specific ELISA measurements using platelets from healthy control individuals have determined the levels of Aβ40 to be 83.8 ng/mL in lysates of quiescent platelets and 56.8 ng/mL in lysates from activated platelets, whereas Aβ42 levels are 1.7 mg/mL in resting platelets, and 1.6 mg/mL in activated platelets[143]. Therefore, it appears that thrombin and collagen-activated platelets release approximately 30% of their Aβ40 content, whereas they release little Aβ42. Interestingly, high platelet density induces activation and the release of Aβ40 stores[144]. Conversely, treatment with the proapoptotic calcium ionophore ionomycin increased the intracellular levels of platelet Aβ40[99].
There is no report so far of a direct comparison of Aβ40 levels in platelets from patients with AD and controls, as most Aβ quantitative analyses have been carried out on plasma, and these have been so far inconclusive[14,145]. Recent studies have shown that platelet activation parallels cognitive decline, thus it may be postulated that the consequent increased release of Aβ40 upon platelet activation would contribute to cerebral amyloid angiopathy[146]. Indeed, the levels of thrombin and collagen activated platelets (or “coated platelets”) are higher in amnestic than non-amnestic MCI patients and may represent an early AD biomarker[147,148]. A longitudinal study showed that elevated coated-platelets levels in patients with amnestic MCI could help predict their conversion to AD[149]. Platelet activation is also a marker for the rate of cognitive decline in AD patients[150].
Changes in another uncharacterized fragment of APP have also been reported. A study comparing APP expression in platelets of 31 patients with and 10 healthy controls has revealed in the patients group a two-fold increase in a 22 kDa fragment that encompasses Aβ[130]. This may represent the proteolytic fragment of APP that we, and others have described and is the product of APP processing by the calcium-dependent cysteine protease, calpain during platelet activation[20,151].
CONCLUSION
Platelets express high levels of APP and possess the secretase proteolytic machinery to produce Aβ and fragments similar to those identified in neurons, therefore they represent a useful model to study changes in APP expression and processing associated with AD. A significant decrease in the ratio of APP isoforms (130 kDa/106-110 kDa kDa) has been observed in platelets of patients with AD and MCI and found to parallel cognitive decline[119]. This offers promise that the measurement of APP isoforms could provide a tool for diagnosing AD at its early stages, for monitoring the disease progression, and for evaluating the response of patients to a therapeutic intervention. Recently, a correlation was identified between platelet activation and cognitive decline[149], and since platelets release sAPP and Aβ upon degranulation, it is likely that simultaneous changes in sAPP and Aβ levels due to platelet activation would be observed if analysed. Decreased levels of sAPPα, ADAM10 protein, and α-secretase activity in platelets from AD patients and increased β-secretase activity support that the balance between non-amyloidogenic and amyloidogenic processing of APP is disrupted, and this mirrors changes occurring in the brain.
The investigation of platelet APP also helps improve our understanding of APP function, and of its complex processing in general, therefore of its role in the brain, in health and AD. Although the amyloid cascade theory has been highly debated, it remains widely accepted in a reappraised form, as it is supported by a wealth of experimental evidence that demonstrates Aβ toxicity, and by genetic studies that converge to a same cellular pathway involving Aβ formation, aggregation and clearance[152]. The initial postulate that Aβ deposition represents the critical event that prompts neurodegeneration[8] had to be reconsidered when it was found that Aβ brain load did not correlate with neuronal loss and cognitive impairment, whereas tau pathology could reflect more closely the disease progression, with neurofibrillary tangles correlating to neuronal loss and cognitive deficits[153]. The reassessed amyloid hypothesis states that soluble Aβ oligomers represent the toxic species that trigger neuronal death or/and synaptic dysfunction, and possibly, that a threshold level of Aβ has to be reached to induce toxicity[152]. This means that Aβ production is an early event in the disease process that precedes the appearance of AD pathological hallmarks that are the plaques and tangles, and also precedes clinical symptoms and cognitive deficits caused by neuronal loss. Therefore, identifying early changes in APP and secretase activities in platelets may alert to similar changes occurring in the brain that will lead to a patient’s progression from pre-clinical stage to MCI and AD.
The full characterization and sequence analysis of the different APP isoforms and fragments expressed in platelets, which should be enable by the latest progress in proteomics technologies, would provide the basis for the development of standardized quantitative assays with improved accuracy and simplicity of use over the current western blot. Changes observed in APP expression in the platelets of patients with MCI and pre-clinical AD are encouraging and merit further investigation, as developing biomarkers is crucial for the accurate diagnosis of patients at risk of developing AD and who would benefit from the early administration of novel disease-modifying therapies, such as anti-amyloid drugs and secretase inhibitors that are under clinical trials. Used in combination with other biomarkers and cognitive tests, platelet APP measurements may provide a non-invasive, easy to access, and cost-effective mean to accrue the reliability, sensitivity and specificity for the early diagnosis of AD.
Footnotes
Supported by (in part) The Judith Jane Mason and Harold Stannett Williams Memorial Foundation (ANZ Mason Foundation); and the National Health and Medical Research Council of Australia (NHMRC project 566520)
Peer reviewer: Subho Chakrabarti, MD, MAMS, FRCPsych, Professor, Department of Psychiatry, Postgraduate Institute of Medical Education and Research, Chandigarh 160012, India
S- Editor Lu YJ L- Editor A E- Editor Zheng XM
References
- 1.Querfurth HW, LaFerla FM. Alzheimer's disease. N Engl J Med. 2010;362:329–344. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 2.Reitz C, Brayne C, Mayeux R. Epidemiology of Alzheimer disease. Nat Rev Neurol. 2011;7:137–152. doi: 10.1038/nrneurol.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA. 1985;82:4245–4249. doi: 10.1073/pnas.82.12.4245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Glenner GG, Wong CW. Alzheimer's disease: initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem Biophys Res Commun. 1984;120:885–890. doi: 10.1016/s0006-291x(84)80190-4. [DOI] [PubMed] [Google Scholar]
- 5.Goedert M. Tau protein and the neurofibrillary pathology of Alzheimer's disease. Trends Neurosci. 1993;16:460–465. doi: 10.1016/0166-2236(93)90078-z. [DOI] [PubMed] [Google Scholar]
- 6.Kang J, Lemaire HG, Unterbeck A, Salbaum JM, Masters CL, Grzeschik KH, Multhaup G, Beyreuther K, Müller-Hill B. The precursor of Alzheimer's disease amyloid A4 protein resembles a cell-surface receptor. Nature. 1987;325:733–736. doi: 10.1038/325733a0. [DOI] [PubMed] [Google Scholar]
- 7.Evin G, Weidemann A. Biogenesis and metabolism of Alzheimer's disease Abeta amyloid peptides. Peptides. 2002;23:1285–1297. doi: 10.1016/s0196-9781(02)00063-3. [DOI] [PubMed] [Google Scholar]
- 8.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer's disease. Trends Pharmacol Sci. 1991;12:383–388. doi: 10.1016/0165-6147(91)90609-v. [DOI] [PubMed] [Google Scholar]
- 9.Tanzi RE, Bertram L. New frontiers in Alzheimer's disease genetics. Neuron. 2001;32:181–184. doi: 10.1016/s0896-6273(01)00476-7. [DOI] [PubMed] [Google Scholar]
- 10.Selkoe DJ. Alzheimer's disease: genes, proteins, and therapy. Physiol Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 11.McLean CA, Cherny RA, Fraser FW, Fuller SJ, Smith MJ, Beyreuther K, Bush AI, Masters CL. Soluble pool of Abeta amyloid as a determinant of severity of neurodegeneration in Alzheimer's disease. Ann Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 12.Xia W, Yang T, Shankar G, Smith IM, Shen Y, Walsh DM, Selkoe DJ. A specific enzyme-linked immunosorbent assay for measuring beta-amyloid protein oligomers in human plasma and brain tissue of patients with Alzheimer disease. Arch Neurol. 2009;66:190–199. doi: 10.1001/archneurol.2008.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villemagne VL, Rowe CC. Amyloid imaging. Int Psychogeriatr. 2011;23 Suppl 2:S41–S49. doi: 10.1017/S1041610211000895. [DOI] [PubMed] [Google Scholar]
- 14.Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131–144. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- 15.Hampel H, Buerger K, Zinkowski R, Teipel SJ, Goernitz A, Andreasen N, Sjoegren M, DeBernardis J, Kerkman D, Ishiguro K, et al. Measurement of phosphorylated tau epitopes in the differential diagnosis of Alzheimer disease: a comparative cerebrospinal fluid study. Arch Gen Psychiatry. 2004;61:95–102. doi: 10.1001/archpsyc.61.1.95. [DOI] [PubMed] [Google Scholar]
- 16.Mattsson N, Zetterberg H, Hansson O, Andreasen N, Parnetti L, Jonsson M, Herukka SK, van der Flier WM, Blankenstein MA, Ewers M, et al. CSF biomarkers and incipient Alzheimer disease in patients with mild cognitive impairment. JAMA. 2009;302:385–393. doi: 10.1001/jama.2009.1064. [DOI] [PubMed] [Google Scholar]
- 17.Hampel H, Frank R, Broich K, Teipel SJ, Katz RG, Hardy J, Herholz K, Bokde AL, Jessen F, Hoessler YC, et al. Biomarkers for Alzheimer's disease: academic, industry and regulatory perspectives. Nat Rev Drug Discov. 2010;9:560–574. doi: 10.1038/nrd3115. [DOI] [PubMed] [Google Scholar]
- 18.Di Luca M, Colciaghi F, Pastorino L, Borroni B, Padovani A, Cattabeni F. Platelets as a peripheral district where to study pathogenetic mechanisms of alzheimer disease: the case of amyloid precursor protein. Eur J Pharmacol. 2000;405:277–283. doi: 10.1016/s0014-2999(00)00559-8. [DOI] [PubMed] [Google Scholar]
- 19.Evin G, Zhu A, Holsinger RM, Masters CL, Li QX. Proteolytic processing of the Alzheimer's disease amyloid precursor protein in brain and platelets. J Neurosci Res. 2003;74:386–392. doi: 10.1002/jnr.10745. [DOI] [PubMed] [Google Scholar]
- 20.Li QX, Evin G, Small DH, Multhaup G, Beyreuther K, Masters CL. Proteolytic processing of Alzheimer's disease beta A4 amyloid precursor protein in human platelets. J Biol Chem. 1995;270:14140–14147. doi: 10.1074/jbc.270.23.14140. [DOI] [PubMed] [Google Scholar]
- 21.Tanzi RE, McClatchey AI, Lamperti ED, Villa-Komaroff L, Gusella JF, Neve RL. Protease inhibitor domain encoded by an amyloid protein precursor mRNA associated with Alzheimer's disease. Nature. 1988;331:528–530. doi: 10.1038/331528a0. [DOI] [PubMed] [Google Scholar]
- 22.Golde TE, Estus S, Usiak M, Younkin LH, Younkin SG. Expression of beta amyloid protein precursor mRNAs: recognition of a novel alternatively spliced form and quantitation in Alzheimer's disease using PCR. Neuron. 1990;4:253–267. doi: 10.1016/0896-6273(90)90100-t. [DOI] [PubMed] [Google Scholar]
- 23.Mönning U, König G, Banati RB, Mechler H, Czech C, Gehrmann J, Schreiter-Gasser U, Masters CL, Beyreuther K. Alzheimer beta A4-amyloid protein precursor in immunocompetent cells. J Biol Chem. 1992;267:23950–23956. [PubMed] [Google Scholar]
- 24.Li QX, Berndt MC, Bush AI, Rumble B, Mackenzie I, Friedhuber A, Beyreuther K, Masters CL. Membrane-associated forms of the beta A4 amyloid protein precursor of Alzheimer's disease in human platelet and brain: surface expression on the activated human platelet. Blood. 1994;84:133–142. [PubMed] [Google Scholar]
- 25.Moir RD, Lynch T, Bush AI, Whyte S, Henry A, Portbury S, Multhaup G, Small DH, Tanzi RE, Beyreuther K, et al. Relative increase in Alzheimer's disease of soluble forms of cerebral Abeta amyloid protein precursor containing the Kunitz protease inhibitory domain. J Biol Chem. 1998;273:5013–5019. doi: 10.1074/jbc.273.9.5013. [DOI] [PubMed] [Google Scholar]
- 26.König G, Mönning U, Czech C, Prior R, Banati R, Schreiter-Gasser U, Bauer J, Masters CL, Beyreuther K. Identification and differential expression of a novel alternative splice isoform of the beta A4 amyloid precursor protein (APP) mRNA in leukocytes and brain microglial cells. J Biol Chem. 1992;267:10804–10809. [PubMed] [Google Scholar]
- 27.Sandbrink R, Masters CL, Beyreuther K. Beta A4-amyloid protein precursor mRNA isoforms without exon 15 are ubiquitously expressed in rat tissues including brain, but not in neurons. J Biol Chem. 1994;269:1510–1517. [PubMed] [Google Scholar]
- 28.Pangalos MN, Efthimiopoulos S, Shioi J, Robakis NK. The chondroitin sulfate attachment site of appican is formed by splicing out exon 15 of the amyloid precursor gene. J Biol Chem. 1995;270:10388–10391. doi: 10.1074/jbc.270.18.10388. [DOI] [PubMed] [Google Scholar]
- 29.Wasco W, Bupp K, Magendantz M, Gusella JF, Tanzi RE, Solomon F. Identification of a mouse brain cDNA that encodes a protein related to the Alzheimer disease-associated amyloid beta protein precursor. Proc Natl Acad Sci USA. 1992;89:10758–10762. doi: 10.1073/pnas.89.22.10758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wasco W, Gurubhagavatula S, Paradis MD, Romano DM, Sisodia SS, Hyman BT, Neve RL, Tanzi RE. Isolation and characterization of APLP2 encoding a homologue of the Alzheimer's associated amyloid beta protein precursor. Nat Genet. 1993;5:95–100. doi: 10.1038/ng0993-95. [DOI] [PubMed] [Google Scholar]
- 31.Eggert S, Paliga K, Soba P, Evin G, Masters CL, Weidemann A, Beyreuther K. The proteolytic processing of the amyloid precursor protein gene family members APLP-1 and APLP-2 involves alpha-, beta-, gamma-, and epsilon-like cleavages: modulation of APLP-1 processing by n-glycosylation. J Biol Chem. 2004;279:18146–18156. doi: 10.1074/jbc.M311601200. [DOI] [PubMed] [Google Scholar]
- 32.Li Q, Südhof TC. Cleavage of amyloid-beta precursor protein and amyloid-beta precursor-like protein by BACE 1. J Biol Chem. 2004;279:10542–10550. doi: 10.1074/jbc.M310001200. [DOI] [PubMed] [Google Scholar]
- 33.Sprecher CA, Grant FJ, Grimm G, O'Hara PJ, Norris F, Norris K, Foster DC. Molecular cloning of the cDNA for a human amyloid precursor protein homolog: evidence for a multigene family. Biochemistry. 1993;32:4481–4486. doi: 10.1021/bi00068a002. [DOI] [PubMed] [Google Scholar]
- 34.Wasco W, Brook JD, Tanzi RE. The amyloid precursor-like protein (APLP) gene maps to the long arm of human chromosome 19. Genomics. 1993;15:237–239. doi: 10.1006/geno.1993.1047. [DOI] [PubMed] [Google Scholar]
- 35.Dahms SO, Hoefgen S, Roeser D, Schlott B, Gührs KH, Than ME. Structure and biochemical analysis of the heparin-induced E1 dimer of the amyloid precursor protein. Proc Natl Acad Sci USA. 2010;107:5381–5386. doi: 10.1073/pnas.0911326107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lee S, Xue Y, Hu J, Wang Y, Liu X, Demeler B, Ha Y. The E2 domains of APP and APLP1 share a conserved mode of dimerization. Biochemistry. 2011;50:5453–5464. doi: 10.1021/bi101846x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Scheuermann S, Hambsch B, Hesse L, Stumm J, Schmidt C, Beher D, Bayer TA, Beyreuther K, Multhaup G. Homodimerization of amyloid precursor protein and its implication in the amyloidogenic pathway of Alzheimer's disease. J Biol Chem. 2001;276:33923–33929. doi: 10.1074/jbc.M105410200. [DOI] [PubMed] [Google Scholar]
- 38.Soba P, Eggert S, Wagner K, Zentgraf H, Siehl K, Kreger S, Löwer A, Langer A, Merdes G, Paro R, et al. Homo- and heterodimerization of APP family members promotes intercellular adhesion. EMBO J. 2005;24:3624–3634. doi: 10.1038/sj.emboj.7600824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weidemann A, König G, Bunke D, Fischer P, Salbaum JM, Masters CL, Beyreuther K. Identification, biogenesis, and localization of precursors of Alzheimer's disease A4 amyloid protein. Cell. 1989;57:115–126. doi: 10.1016/0092-8674(89)90177-3. [DOI] [PubMed] [Google Scholar]
- 40.Thinakaran G, Koo EH. Amyloid precursor protein trafficking, processing, and function. J Biol Chem. 2008;283:29615–29619. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Perreau VM, Orchard S, Adlard PA, Bellingham SA, Cappai R, Ciccotosto GD, Cowie TF, Crouch PJ, Duce JA, Evin G, et al. A domain level interaction network of amyloid precursor protein and Abeta of Alzheimer's disease. Proteomics. 2010;10:2377–2395. doi: 10.1002/pmic.200900773. [DOI] [PubMed] [Google Scholar]
- 42.King GD, Scott Turner R. Adaptor protein interactions: modulators of amyloid precursor protein metabolism and Alzheimer's disease risk. Exp Neurol. 2004;185:208–219. doi: 10.1016/j.expneurol.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 43.Young-Pearse TL, Suth S, Luth ES, Sawa A, Selkoe DJ. Biochemical and functional interaction of disrupted-in-schizophrenia 1 and amyloid precursor protein regulates neuronal migration during mammalian cortical development. J Neurosci. 2010;30:10431–10440. doi: 10.1523/JNEUROSCI.1445-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin metalloprotease. Proc Natl Acad Sci USA. 1999;96:3922–3927. doi: 10.1073/pnas.96.7.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumor necrosis factor alpha converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem. 1998;273:27765–27767. doi: 10.1074/jbc.273.43.27765. [DOI] [PubMed] [Google Scholar]
- 46.Vassar R, Citron M. Abeta-generating enzymes: recent advances in beta- and gamma-secretase research. Neuron. 2000;27:419–422. doi: 10.1016/s0896-6273(00)00051-9. [DOI] [PubMed] [Google Scholar]
- 47.Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Vidal R, Ghiso J, Wisniewski T, Frangione B. Alzheimer's presenilin 1 gene expression in platelets and megakaryocytes. Identification of a novel splice variant. FEBS Lett. 1996;393:19–23. doi: 10.1016/0014-5793(96)00845-9. [DOI] [PubMed] [Google Scholar]
- 49.Iwatsubo T. Assembly and activation of the gamma-secretase complex: roles of presenilin cofactors. Mol Psychiatry. 2004;9:8–10. doi: 10.1038/sj.mp.4001438. [DOI] [PubMed] [Google Scholar]
- 50.Qi-Takahara Y, Morishima-Kawashima M, Tanimura Y, Dolios G, Hirotani N, Horikoshi Y, Kametani F, Maeda M, Saido TC, Wang R, et al. Longer forms of amyloid beta protein: implications for the mechanism of intramembrane cleavage by gamma-secretase. J Neurosci. 2005;25:436–445. doi: 10.1523/JNEUROSCI.1575-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weidemann A, Eggert S, Reinhard FB, Vogel M, Paliga K, Baier G, Masters CL, Beyreuther K, Evin G. A novel epsilon-cleavage within the transmembrane domain of the Alzheimer amyloid precursor protein demonstrates homology with Notch processing. Biochemistry. 2002;41:2825–2835. doi: 10.1021/bi015794o. [DOI] [PubMed] [Google Scholar]
- 52.Cao X, Südhof TC. A transcriptionally [correction of transcriptively] active complex of APP with Fe65 and histone acetyltransferase Tip60. Science. 2001;293:115–120. doi: 10.1126/science.1058783. [DOI] [PubMed] [Google Scholar]
- 53.Li H, Wang Z, Wang B, Guo Q, Dolios G, Tabuchi K, Hammer RE, Südhof TC, Wang R, Zheng H. Genetic dissection of the amyloid precursor protein in developmental function and amyloid pathogenesis. J Biol Chem. 2010;285:30598–30605. doi: 10.1074/jbc.M110.137729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Esch FS, Keim PS, Beattie EC, Blacher RW, Culwell AR, Oltersdorf T, McClure D, Ward PJ. Cleavage of amyloid beta peptide during constitutive processing of its precursor. Science. 1990;248:1122–1124. doi: 10.1126/science.2111583. [DOI] [PubMed] [Google Scholar]
- 55.Fuller SJ, Storey E, Li QX, Smith AI, Beyreuther K, Masters CL. Intracellular production of beta A4 amyloid of Alzheimer's disease: modulation by phosphoramidon and lack of coupling to the secretion of the amyloid precursor protein. Biochemistry. 1995;34:8091–8098. doi: 10.1021/bi00025a015. [DOI] [PubMed] [Google Scholar]
- 56.Li QX, Whyte S, Tanner JE, Evin G, Beyreuther K, Masters CL. Secretion of Alzheimer's disease Abeta amyloid peptide by activated human platelets. Lab Invest. 1998;78:461–469. [PubMed] [Google Scholar]
- 57.Seubert P, Vigo-Pelfrey C, Esch F, Lee M, Dovey H, Davis D, Sinha S, Schlossmacher M, Whaley J, Swindlehurst C. Isolation and quantification of soluble Alzheimer's beta-peptide from biological fluids. Nature. 1992;359:325–327. doi: 10.1038/359325a0. [DOI] [PubMed] [Google Scholar]
- 58.Suzuki N, Cheung TT, Cai XD, Odaka A, Otvos L, Eckman C, Golde TE, Younkin SG. An increased percentage of long amyloid beta protein secreted by familial amyloid beta protein precursor (beta APP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 59.Blennow K. Biomarkers in Alzheimer's disease drug development. Nat Med. 2010;16:1218–1222. doi: 10.1038/nm.2221. [DOI] [PubMed] [Google Scholar]
- 60.Haass C, Schlossmacher MG, Hung AY, Vigo-Pelfrey C, Mellon A, Ostaszewski BL, Lieberburg I, Koo EH, Schenk D, Teplow DB. Amyloid beta-peptide is produced by cultured cells during normal metabolism. Nature. 1992;359:322–325. doi: 10.1038/359322a0. [DOI] [PubMed] [Google Scholar]
- 61.Gandy S, Petanceska S. Regulation of Alzheimer beta-amyloid precursor trafficking and metabolism. Biochim Biophys Acta. 2000;1502:44–52. doi: 10.1016/s0925-4439(00)00031-4. [DOI] [PubMed] [Google Scholar]
- 62.Li QX, Cappai R, Evin G, Tanner JE, Gray CW, Beyreuther K, Masters CL. Products of the Alzheimer's disease amyloid precursor protein generated by b-secretase are present in human platelets, and secreted upon degranulation. Am J Alzheimers Dis. 1998;13:236–244. [Google Scholar]
- 63.Simons M, de Strooper B, Multhaup G, Tienari PJ, Dotti CG, Beyreuther K. Amyloidogenic processing of the human amyloid precursor protein in primary cultures of rat hippocampal neurons. J Neurosci. 1996;16:899–908. doi: 10.1523/JNEUROSCI.16-03-00899.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ho A, Südhof TC. Binding of F-spondin to amyloid-beta precursor protein: a candidate amyloid-beta precursor protein ligand that modulates amyloid-beta precursor protein cleavage. Proc Natl Acad Sci USA. 2004;101:2548–2553. doi: 10.1073/pnas.0308655100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Beher D, Hesse L, Masters CL, Multhaup G. Regulation of amyloid protein precursor (APP) binding to collagen and mapping of the binding sites on APP and collagen type I. J Biol Chem. 1996;271:1613–1620. doi: 10.1074/jbc.271.3.1613. [DOI] [PubMed] [Google Scholar]
- 66.Ghiso J, Rostagno A, Gardella JE, Liem L, Gorevic PD, Frangione B. A 109-amino-acid C-terminal fragment of Alzheimer's-disease amyloid precursor protein contains a sequence, -RHDS-, that promotes cell adhesion. Biochem J. 1992;288(Pt 3):1053–1059. doi: 10.1042/bj2881053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Small DH, Nurcombe V, Moir R, Michaelson S, Monard D, Beyreuther K, Masters CL. Association and release of the amyloid protein precursor of Alzheimer's disease from chick brain extracellular matrix. J Neurosci. 1992;12:4143–4150. doi: 10.1523/JNEUROSCI.12-11-04143.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Williamson TG, Mok SS, Henry A, Cappai R, Lander AD, Nurcombe V, Beyreuther K, Masters CL, Small DH. Secreted glypican binds to the amyloid precursor protein of Alzheimer's disease (APP) and inhibits APP-induced neurite outgrowth. J Biol Chem. 1996;271:31215–31221. doi: 10.1074/jbc.271.49.31215. [DOI] [PubMed] [Google Scholar]
- 69.Young-Pearse TL, Chen AC, Chang R, Marquez C, Selkoe DJ. Secreted APP regulates the function of full-length APP in neurite outgrowth through interaction with integrin beta1. Neural Dev. 2008;3:15. doi: 10.1186/1749-8104-3-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Milward EA, Papadopoulos R, Fuller SJ, Moir RD, Small D, Beyreuther K, Masters CL. The amyloid protein precursor of Alzheimer's disease is a mediator of the effects of nerve growth factor on neurite outgrowth. Neuron. 1992;9:129–137. doi: 10.1016/0896-6273(92)90228-6. [DOI] [PubMed] [Google Scholar]
- 71.Mattson MP. Secreted forms of beta-amyloid precursor protein modulate dendrite outgrowth and calcium responses to glutamate in cultured embryonic hippocampal neurons. J Neurobiol. 1994;25:439–450. doi: 10.1002/neu.480250409. [DOI] [PubMed] [Google Scholar]
- 72.Hoe HS, Lee KJ, Carney RS, Lee J, Markova A, Lee JY, Howell BW, Hyman BT, Pak DT, Bu G, et al. Interaction of reelin with amyloid precursor protein promotes neurite outgrowth. J Neurosci. 2009;29:7459–7473. doi: 10.1523/JNEUROSCI.4872-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lourenço FC, Galvan V, Fombonne J, Corset V, Llambi F, Müller U, Bredesen DE, Mehlen P. Netrin-1 interacts with amyloid precursor protein and regulates amyloid-beta production. Cell Death Differ. 2009;16:655–663. doi: 10.1038/cdd.2008.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ikin AF, Sabo SL, Lanier LM, Buxbaum JD. A macromolecular complex involving the amyloid precursor protein (APP) and the cytosolic adapter FE65 is a negative regulator of axon branching. Mol Cell Neurosci. 2007;35:57–63. doi: 10.1016/j.mcn.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aydin D, Weyer SW, Müller UC. Functions of the APP gene family in the nervous system: insights from mouse models. Exp Brain Res. 2012;217:423–434. doi: 10.1007/s00221-011-2861-2. [DOI] [PubMed] [Google Scholar]
- 76.Clarris HJ, Cappai R, Heffernan D, Beyreuther K, Masters CL, Small DH. Identification of heparin-binding domains in the amyloid precursor protein of Alzheimer's disease by deletion mutagenesis and peptide mapping. J Neurochem. 1997;68:1164–1172. doi: 10.1046/j.1471-4159.1997.68031164.x. [DOI] [PubMed] [Google Scholar]
- 77.Mattson MP, Barger SW, Cheng B, Lieberburg I, Smith-Swintosky VL, Rydel RE. beta-Amyloid precursor protein metabolites and loss of neuronal Ca2+ homeostasis in Alzheimer's disease. Trends Neurosci. 1993;16:409–414. doi: 10.1016/0166-2236(93)90009-b. [DOI] [PubMed] [Google Scholar]
- 78.Saitoh T, Sundsmo M, Roch JM, Kimura N, Cole G, Schubert D, Oltersdorf T, Schenk DB. Secreted form of amyloid beta protein precursor is involved in the growth regulation of fibroblasts. Cell. 1989;58:615–622. doi: 10.1016/0092-8674(89)90096-2. [DOI] [PubMed] [Google Scholar]
- 79.Small DH, Nurcombe V, Reed G, Clarris H, Moir R, Beyreuther K, Masters CL. A heparin-binding domain in the amyloid protein precursor of Alzheimer's disease is involved in the regulation of neurite outgrowth. J Neurosci. 1994;14:2117–2127. doi: 10.1523/JNEUROSCI.14-04-02117.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Van Nostrand WE, Wagner SL, Suzuki M, Choi BH, Farrow JS, Geddes JW, Cotman CW, Cunningham DD. Protease nexin-II, a potent antichymotrypsin, shows identity to amyloid beta-protein precursor. Nature. 1989;341:546–549. doi: 10.1038/341546a0. [DOI] [PubMed] [Google Scholar]
- 81.Bush AI. The metallobiology of Alzheimer's disease. Trends Neurosci. 2003;26:207–214. doi: 10.1016/S0166-2236(03)00067-5. [DOI] [PubMed] [Google Scholar]
- 82.Maynard CJ, Cappai R, Volitakis I, Cherny RA, White AR, Beyreuther K, Masters CL, Bush AI, Li QX. Overexpression of Alzheimer's disease amyloid-beta opposes the age-dependent elevations of brain copper and iron. J Biol Chem. 2002;277:44670–44676. doi: 10.1074/jbc.M204379200. [DOI] [PubMed] [Google Scholar]
- 83.White AR, Reyes R, Mercer JF, Camakaris J, Zheng H, Bush AI, Multhaup G, Beyreuther K, Masters CL, Cappai R. Copper levels are increased in the cerebral cortex and liver of APP and APLP2 knockout mice. Brain Res. 1999;842:439–444. doi: 10.1016/s0006-8993(99)01861-2. [DOI] [PubMed] [Google Scholar]
- 84.Duce JA, Tsatsanis A, Cater MA, James SA, Robb E, Wikhe K, Leong SL, Perez K, Johanssen T, Greenough MA, et al. Iron-export ferroxidase activity of β-amyloid precursor protein is inhibited by zinc in Alzheimer's disease. Cell. 2010;142:857–867. doi: 10.1016/j.cell.2010.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heber S, Herms J, Gajic V, Hainfellner J, Aguzzi A, Rülicke T, von Kretzschmar H, von Koch C, Sisodia S, Tremml P, et al. Mice with combined gene knock-outs reveal essential and partially redundant functions of amyloid precursor protein family members. J Neurosci. 2000;20:7951–7963. doi: 10.1523/JNEUROSCI.20-21-07951.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.von Koch CS, Zheng H, Chen H, Trumbauer M, Thinakaran G, van der Ploeg LH, Price DL, Sisodia SS. Generation of APLP2 KO mice and early postnatal lethality in APLP2/APP double KO mice. Neurobiol Aging. 1997;18:661–669. doi: 10.1016/s0197-4580(97)00151-6. [DOI] [PubMed] [Google Scholar]
- 87.Zheng H, Jiang M, Trumbauer ME, Sirinathsinghji DJ, Hopkins R, Smith DW, Heavens RP, Dawson GR, Boyce S, Conner MW, et al. beta-Amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell. 1995;81:525–531. doi: 10.1016/0092-8674(95)90073-x. [DOI] [PubMed] [Google Scholar]
- 88.Wang P, Yang G, Mosier DR, Chang P, Zaidi T, Gong YD, Zhao NM, Dominguez B, Lee KF, Gan WB, et al. Defective neuromuscular synapses in mice lacking amyloid precursor protein (APP) and APP-Like protein 2. J Neurosci. 2005;25:1219–1225. doi: 10.1523/JNEUROSCI.4660-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Needham BE, Wlodek ME, Ciccotosto GD, Fam BC, Masters CL, Proietto J, Andrikopoulos S, Cappai R. Identification of the Alzheimer's disease amyloid precursor protein (APP) and its homologue APLP2 as essential modulators of glucose and insulin homeostasis and growth. J Pathol. 2008;215:155–163. doi: 10.1002/path.2343. [DOI] [PubMed] [Google Scholar]
- 90.Grösgen S, Grimm MO, Friess P, Hartmann T. Role of amyloid beta in lipid homeostasis. Biochim Biophys Acta. 2010;1801:966–974. doi: 10.1016/j.bbalip.2010.05.002. [DOI] [PubMed] [Google Scholar]
- 91.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, et al. The Alzheimer's disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5:e9505. doi: 10.1371/journal.pone.0009505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Bush AI, Martins RN, Rumble B, Moir R, Fuller S, Milward E, Currie J, Ames D, Weidemann A, Fischer P. The amyloid precursor protein of Alzheimer's disease is released by human platelets. J Biol Chem. 1990;265:15977–15983. [PubMed] [Google Scholar]
- 93.Li QX, Whyte S, Birchall I, Beyreuther K, Masters CL. The amyloid protein precursor of Alzheimer's disease in human platelets and kidney. In: Zatta P, Nicolini M, editors. Non-neuronal cells in Alzheimer's disease. Singapore: World Scientific; 1995. pp. 62–70. [Google Scholar]
- 94.Moir RD, Martins RN, Bush AI, Small DH, Milward EA, Rumble BA, Multhaup G, Beyreuther K, Masters CL. Human brain beta A4 amyloid protein precursor of Alzheimer's disease: purification and partial characterization. J Neurochem. 1992;59:1490–1498. doi: 10.1111/j.1471-4159.1992.tb08465.x. [DOI] [PubMed] [Google Scholar]
- 95.Van Nostrand WE, Schmaier AH, Farrow JS, Cines DB, Cunningham DD. Protease nexin-2/amyloid beta-protein precursor in blood is a platelet-specific protein. Biochem Biophys Res Commun. 1991;175:15–21. doi: 10.1016/s0006-291x(05)81193-3. [DOI] [PubMed] [Google Scholar]
- 96.Chen M, Inestrosa NC, Ross GS, Fernandez HL. Platelets are the primary source of amyloid beta-peptide in human blood. Biochem Biophys Res Commun. 1995;213:96–103. doi: 10.1006/bbrc.1995.2103. [DOI] [PubMed] [Google Scholar]
- 97.Van Nostrand WE, Schmaier AH, Farrow JS, Cunningham DD. Protease nexin-II (amyloid beta-protein precursor): a platelet alpha-granule protein. Science. 1990;248:745–748. doi: 10.1126/science.2110384. [DOI] [PubMed] [Google Scholar]
- 98.Smith RP, Broze GJ. Characterization of platelet-releasable forms of beta-amyloid precursor proteins: the effect of thrombin. Blood. 1992;80:2252–2260. [PubMed] [Google Scholar]
- 99.Casoli T, Di Stefano G, Giorgetti B, Balietti M, Recchioni R, Moroni F, Marcheselli F, Bernardini G, Fattoretti P, Bertoni-Freddari C. Platelet as a physiological model to investigate apoptotic mechanisms in Alzheimer beta-amyloid peptide production. Mech Ageing Dev. 2008;129:154–162. doi: 10.1016/j.mad.2007.11.004. [DOI] [PubMed] [Google Scholar]
- 100.Schmaier AH, Dahl LD, Hasan AA, Cines DB, Bauer KA, Van Nostrand WE. Factor IXa inhibition by protease nexin-2/amyloid beta-protein precursor on phospholipid vesicles and cell membranes. Biochemistry. 1995;34:1171–1178. doi: 10.1021/bi00004a010. [DOI] [PubMed] [Google Scholar]
- 101.Smith RP, Higuchi DA, Broze GJ. Platelet coagulation factor XIa-inhibitor, a form of Alzheimer amyloid precursor protein. Science. 1990;248:1126–1128. doi: 10.1126/science.2111585. [DOI] [PubMed] [Google Scholar]
- 102.Mahdi F, Van Nostrand WE, Schmaier AH. Protease nexin-2/amyloid beta-protein precursor inhibits factor Xa in the prothrombinase complex. J Biol Chem. 1995;270:23468–23474. doi: 10.1074/jbc.270.40.23468. [DOI] [PubMed] [Google Scholar]
- 103.Mahdi F, Rehemtulla A, Van Nostrand WE, Bajaj SP, Schmaier AH. Protease nexin-2/Amyloid beta-protein precursor regulates factor VIIa and the factor VIIa-tissue factor complex. Thromb Res. 2000;99:267–276. doi: 10.1016/s0049-3848(00)00245-0. [DOI] [PubMed] [Google Scholar]
- 104.Komiyama Y, Murakami T, Egawa H, Okubo S, Yasunaga K, Murata K. Purification of factor XIa inhibitor from human platelets. Thromb Res. 1992;66:397–408. doi: 10.1016/0049-3848(92)90289-m. [DOI] [PubMed] [Google Scholar]
- 105.Navaneetham D, Jin L, Pandey P, Strickler JE, Babine RE, Abdel-Meguid SS, Walsh PN. Structural and mutational analyses of the molecular interactions between the catalytic domain of factor XIa and the Kunitz protease inhibitor domain of protease nexin 2. J Biol Chem. 2005;280:36165–36175. doi: 10.1074/jbc.M504990200. [DOI] [PubMed] [Google Scholar]
- 106.Henry A, Li QX, Galatis D, Hesse L, Multhaup G, Beyreuther K, Masters CL, Cappai R. Inhibition of platelet activation by the Alzheimer's disease amyloid precursor protein. Br J Haematol. 1998;103:402–415. doi: 10.1046/j.1365-2141.1998.01005.x. [DOI] [PubMed] [Google Scholar]
- 107.Kowalska MA, Badellino K. beta-Amyloid protein induces platelet aggregation and supports platelet adhesion. Biochem Biophys Res Commun. 1994;205:1829–1835. doi: 10.1006/bbrc.1994.2883. [DOI] [PubMed] [Google Scholar]
- 108.Wolozin B, Maheshwari S, Jones C, Dukoff R, Wallace W, Racchi M, Nagula S, Shulman NR, Sunderland T, Bush A. Beta-amyloid augments platelet aggregation: reduced activity of familial angiopathy-associated mutants. Mol Psychiatry. 1998;3:500–507. doi: 10.1038/sj.mp.4000451. [DOI] [PubMed] [Google Scholar]
- 109.Davies TA, Long HJ, Sgro K, Rathbun WH, McMenamin ME, Seetoo K, Tibbles H, Billingslea AM, Fine RE, Fishman JB, et al. Activated Alzheimer disease platelets retain more beta amyloid precursor protein. Neurobiol Aging. 1997;18:147–153. doi: 10.1016/s0197-4580(97)00013-4. [DOI] [PubMed] [Google Scholar]
- 110.Davies TA, Long HJ, Tibbles HE, Sgro KR, Wells JM, Rathbun WH, Seetoo KF, McMenamin ME, Smith SJ, Feldman RG, et al. Moderate and advanced Alzheimer's patients exhibit platelet activation differences. Neurobiol Aging. 1997;18:155–162. doi: 10.1016/s0197-4580(97)00016-x. [DOI] [PubMed] [Google Scholar]
- 111.Rosenberg RN, Baskin F, Fosmire JA, Risser R, Adams P, Svetlik D, Honig LS, Cullum CM, Weiner MF. Altered amyloid protein processing in platelets of patients with Alzheimer disease. Arch Neurol. 1997;54:139–144. doi: 10.1001/archneur.1997.00550140019007. [DOI] [PubMed] [Google Scholar]
- 112.Baskin F, Rosenberg RN, Iyer L, Hynan L, Cullum CM. Platelet APP isoform ratios correlate with declining cognition in AD. Neurology. 2000;54:1907–1909. doi: 10.1212/wnl.54.10.1907. [DOI] [PubMed] [Google Scholar]
- 113.Di Luca M, Pastorino L, Bianchetti A, Perez J, Vignolo LA, Lenzi GL, Trabucchi M, Cattabeni F, Padovani A. Differential level of platelet amyloid beta precursor protein isoforms: an early marker for Alzheimer disease. Arch Neurol. 1998;55:1195–1200. doi: 10.1001/archneur.55.9.1195. [DOI] [PubMed] [Google Scholar]
- 114.Padovani A, Borroni B, Colciaghi F, Pastorino L, Archetti S, Cottini E, Caimi L, Cattabeni F, Di Luca M. Platelet amyloid precursor protein forms in AD: a peripheral diagnostic tool and a pharmacological target. Mech Ageing Dev. 2001;122:1997–2004. doi: 10.1016/s0047-6374(01)00315-3. [DOI] [PubMed] [Google Scholar]
- 115.Padovani A, Borroni B, Colciaghi F, Pettenati C, Cottini E, Agosti C, Lenzi GL, Caltagirone C, Trabucchi M, Cattabeni F, et al. Abnormalities in the pattern of platelet amyloid precursor protein forms in patients with mild cognitive impairment and Alzheimer disease. Arch Neurol. 2002;59:71–75. doi: 10.1001/archneur.59.1.71. [DOI] [PubMed] [Google Scholar]
- 116.Borroni B, Colciaghi F, Caltagirone C, Rozzini L, Broglio L, Cattabeni F, Di Luca M, Padovani A. Platelet amyloid precursor protein abnormalities in mild cognitive impairment predict conversion to dementia of Alzheimer type: a 2-year follow-up study. Arch Neurol. 2003;60:1740–1744. doi: 10.1001/archneur.60.12.1740. [DOI] [PubMed] [Google Scholar]
- 117.Borroni B, Perani D, Broli M, Colciaghi F, Garibotto V, Paghera B, Agosti C, Giubbini R, Di Luca M, Padovani A. Pre-clinical diagnosis of Alzheimer disease combining platelet amyloid precursor protein ratio and rCBF spect analysis. J Neurol. 2005;252:1359–1362. doi: 10.1007/s00415-005-0867-z. [DOI] [PubMed] [Google Scholar]
- 118.Zainaghi IA, Forlenza OV, Gattaz WF. Abnormal APP processing in platelets of patients with Alzheimer's disease: correlations with membrane fluidity and cognitive decline. Psychopharmacology (Berl) 2007;192:547–553. doi: 10.1007/s00213-007-0748-5. [DOI] [PubMed] [Google Scholar]
- 119.Borroni B, Agosti C, Marcello E, Di Luca M, Padovani A. Blood cell markers in Alzheimer Disease: Amyloid Precursor Protein form ratio in platelets. Exp Gerontol. 2010;45:53–56. doi: 10.1016/j.exger.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 120.Zainaghi IA, Talib LL, Diniz BS, Gattaz WF, Forlenza OV. Reduced platelet amyloid precursor protein ratio (APP ratio) predicts conversion from mild cognitive impairment to Alzheimer's disease. J Neural Transm. 2012;119:815–819. doi: 10.1007/s00702-012-0807-x. [DOI] [PubMed] [Google Scholar]
- 121.Borroni B, Colciaghi F, Pastorino L, Archetti S, Corsini P, Cattabeni F, Di Luca M, Padovani A. ApoE genotype influences the biological effect of donepezil on APP metabolism in Alzheimer disease: evidence from a peripheral model. Eur Neuropsychopharmacol. 2002;12:195–200. doi: 10.1016/s0924-977x(02)00013-5. [DOI] [PubMed] [Google Scholar]
- 122.Smith CC, Hyatt PJ, Stanyer L, Betteridge DJ. Platelet secretion of beta-amyloid is increased in hypercholesterolaemia. Brain Res. 2001;896:161–164. doi: 10.1016/s0006-8993(01)02080-7. [DOI] [PubMed] [Google Scholar]
- 123.Borroni B, Colciaghi F, Lenzi GL, Caimi L, Cattabeni F, Di Luca M, Padovani A. High cholesterol affects platelet APP processing in controls and in AD patients. Neurobiol Aging. 2003;24:631–636. doi: 10.1016/s0197-4580(02)00190-2. [DOI] [PubMed] [Google Scholar]
- 124.Baskin F, Rosenberg RN, Fang X, Hynan LS, Moore CB, Weiner M, Vega GL. Correlation of statin-increased platelet APP ratios and reduced blood lipids in AD patients. Neurology. 2003;60:2006–2007. doi: 10.1212/01.wnl.0000068029.56740.96. [DOI] [PubMed] [Google Scholar]
- 125.Bush AI, Whyte S, Thomas LD, Williamson TG, Van Tiggelen CJ, Currie J, Small DH, Moir RD, Li QX, Rumble B. An abnormality of plasma amyloid protein precursor in Alzheimer's disease. Ann Neurol. 1992;32:57–65. doi: 10.1002/ana.410320110. [DOI] [PubMed] [Google Scholar]
- 126.Borroni B, Akkawi N, Martini G, Colciaghi F, Prometti P, Rozzini L, Di Luca M, Lenzi GL, Romanelli G, Caimi L, et al. Microvascular damage and platelet abnormalities in early Alzheimer's disease. J Neurol Sci. 2002;203-204:189–193. doi: 10.1016/s0022-510x(02)00289-7. [DOI] [PubMed] [Google Scholar]
- 127.Colciaghi F, Marcello E, Borroni B, Zimmermann M, Caltagirone C, Cattabeni F, Padovani A, Di Luca M. Platelet APP, ADAM 10 and BACE alterations in the early stages of Alzheimer disease. Neurology. 2004;62:498–501. doi: 10.1212/01.wnl.0000106953.49802.9c. [DOI] [PubMed] [Google Scholar]
- 128.Canobbio I, Catricalà S, Balduini C, Torti M. Calmodulin regulates the non-amyloidogenic metabolism of amyloid precursor protein in platelets. Biochim Biophys Acta. 2011;1813:500–506. doi: 10.1016/j.bbamcr.2010.12.002. [DOI] [PubMed] [Google Scholar]
- 129.Colciaghi F, Borroni B, Pastorino L, Marcello E, Zimmermann M, Cattabeni F, Padovani A, Di Luca M. [alpha]-Secretase ADAM10 as well as [alpha]APPs is reduced in platelets and CSF of Alzheimer disease patients. Mol Med. 2002;8:67–74. [PMC free article] [PubMed] [Google Scholar]
- 130.Tang K, Hynan LS, Baskin F, Rosenberg RN. Platelet amyloid precursor protein processing: a bio-marker for Alzheimer's disease. J Neurol Sci. 2006;240:53–58. doi: 10.1016/j.jns.2005.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Huse JT, Byant D, Yang Y, Pijak DS, D'Souza I, Lah JJ, Lee VM, Doms RW, Cook DG. Endoproteolysis of beta-secretase (beta-site amyloid precursor protein-cleaving enzyme) within its catalytic domain. A potential mechanism for regulation. J Biol Chem. 2003;278:17141–17149. doi: 10.1074/jbc.M213303200. [DOI] [PubMed] [Google Scholar]
- 132.Zimmermann M, Borroni B, Cattabeni F, Padovani A, Di Luca M. Cholinesterase inhibitors influence APP metabolism in Alzheimer disease patients. Neurobiol Dis. 2005;19:237–242. doi: 10.1016/j.nbd.2005.01.002. [DOI] [PubMed] [Google Scholar]
- 133.Di Luca M, Grossi E, Borroni B, Zimmermann M, Marcello E, Colciaghi F, Gardoni F, Intraligi M, Padovani A, Buscema M. Artificial neural networks allow the use of simultaneous measurements of Alzheimer disease markers for early detection of the disease. J Transl Med. 2005;3:30. doi: 10.1186/1479-5876-3-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Holsinger RM, McLean CA, Beyreuther K, Masters CL, Evin G. Increased expression of the amyloid precursor beta-secretase in Alzheimer's disease. Ann Neurol. 2002;51:783–786. doi: 10.1002/ana.10208. [DOI] [PubMed] [Google Scholar]
- 135.Fukumoto H, Cheung BS, Hyman BT, Irizarry MC. Beta-secretase protein and activity are increased in the neocortex in Alzheimer disease. Arch Neurol. 2002;59:1381–1389. doi: 10.1001/archneur.59.9.1381. [DOI] [PubMed] [Google Scholar]
- 136.Yang LB, Lindholm K, Yan R, Citron M, Xia W, Yang XL, Beach T, Sue L, Wong P, Price D, et al. Elevated beta-secretase expression and enzymatic activity detected in sporadic Alzheimer disease. Nat Med. 2003;9:3–4. doi: 10.1038/nm0103-3. [DOI] [PubMed] [Google Scholar]
- 137.Holsinger RM, McLean CA, Collins SJ, Masters CL, Evin G. Increased beta-Secretase activity in cerebrospinal fluid of Alzheimer's disease subjects. Ann Neurol. 2004;55:898–899. doi: 10.1002/ana.20144. [DOI] [PubMed] [Google Scholar]
- 138.Zhong Z, Ewers M, Teipel S, Bürger K, Wallin A, Blennow K, He P, McAllister C, Hampel H, Shen Y. Levels of beta-secretase (BACE1) in cerebrospinal fluid as a predictor of risk in mild cognitive impairment. Arch Gen Psychiatry. 2007;64:718–726. doi: 10.1001/archpsyc.64.6.718. [DOI] [PubMed] [Google Scholar]
- 139.Johnston JA, Liu WW, Coulson DT, Todd S, Murphy S, Brennan S, Foy CJ, Craig D, Irvine GB, Passmore AP. Platelet beta-secretase activity is increased in Alzheimer's disease. Neurobiol Aging. 2008;29:661–668. doi: 10.1016/j.neurobiolaging.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 140.Liu WW, Todd S, Craig D, Passmore AP, Coulson DT, Murphy S, Irvine GB, Johnston JA. Elevated platelet beta-secretase activity in mild cognitive impairment. Dement Geriatr Cogn Disord. 2007;24:464–468. doi: 10.1159/000110739. [DOI] [PubMed] [Google Scholar]
- 141.Gorham P, Bark N, Björkhem I, Meaney S, Crisby M. Platelet alpha- and beta- secretase activities are not significantly affected by dementia or mild cognitive impairment in Swedish patients. Curr Alzheimer Res. 2010;7:134–139. doi: 10.2174/156720510790691254. [DOI] [PubMed] [Google Scholar]
- 142.Liu WW, Todd S, Coulson DT, Irvine GB, Passmore AP, McGuinness B, McConville M, Craig D, Johnston JA. A novel reciprocal and biphasic relationship between membrane cholesterol and beta-secretase activity in SH-SY5Y cells and in human platelets. J Neurochem. 2009;108:341–349. doi: 10.1111/j.1471-4159.2008.05753.x. [DOI] [PubMed] [Google Scholar]
- 143.Roher AE, Esh CL, Kokjohn TA, Castaño EM, Van Vickle GD, Kalback WM, Patton RL, Luehrs DC, Daugs ID, Kuo YM, et al. Amyloid beta peptides in human plasma and tissues and their significance for Alzheimer's disease. Alzheimers Dement. 2009;5:18–29. doi: 10.1016/j.jalz.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Casoli T, Di Stefano G, Giorgetti B, Grossi Y, Balietti M, Fattoretti P, Bertoni-Freddari C. Release of beta-amyloid from high-density platelets: implications for Alzheimer's disease pathology. Ann N Y Acad Sci. 2007;1096:170–178. doi: 10.1196/annals.1397.082. [DOI] [PubMed] [Google Scholar]
- 145.Hansson O, Zetterberg H, Vanmechelen E, Vanderstichele H, Andreasson U, Londos E, Wallin A, Minthon L, Blennow K. Evaluation of plasma Abeta(40) and Abeta(42) as predictors of conversion to Alzheimer's disease in patients with mild cognitive impairment. Neurobiol Aging. 2010;31:357–367. doi: 10.1016/j.neurobiolaging.2008.03.027. [DOI] [PubMed] [Google Scholar]
- 146.Casoli T, Di Stefano G, Balietti M, Solazzi M, Giorgetti B, Fattoretti P. Peripheral inflammatory biomarkers of Alzheimer's disease: the role of platelets. Biogerontology. 2010;11:627–633. doi: 10.1007/s10522-010-9281-8. [DOI] [PubMed] [Google Scholar]
- 147.Prodan CI, Ross ED, Vincent AS, Dale GL. Rate of progression in Alzheimer's disease correlates with coated-platelet levels--a longitudinal study. Transl Res. 2008;152:99–102. doi: 10.1016/j.trsl.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 148.Prodan CI, Ross ED, Vincent AS, Dale GL. Differences in coated-platelet production between frontotemporal dementia and Alzheimer disease. Alzheimer Dis Assoc Disord. 2009;23:234–237. doi: 10.1097/WAD.0b013e318199dd1a. [DOI] [PubMed] [Google Scholar]
- 149.Prodan CI, Ross ED, Stoner JA, Cowan LD, Vincent AS, Dale GL. Coated-platelet levels and progression from mild cognitive impairment to Alzheimer disease. Neurology. 2011;76:247–252. doi: 10.1212/WNL.0b013e3182074bd2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Stellos K, Panagiota V, Kögel A, Leyhe T, Gawaz M, Laske C. Predictive value of platelet activation for the rate of cognitive decline in Alzheimer's disease patients. J Cereb Blood Flow Metab. 2010;30:1817–1820. doi: 10.1038/jcbfm.2010.140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Chen M, Durr J, Fernandez HL. Possible role of calpain in normal processing of beta-amyloid precursor protein in human platelets. Biochem Biophys Res Commun. 2000;273:170–175. doi: 10.1006/bbrc.2000.2919. [DOI] [PubMed] [Google Scholar]
- 152.Karran E, Mercken M, De Strooper B. The amyloid cascade hypothesis for Alzheimer's disease: an appraisal for the development of therapeutics. Nat Rev Drug Discov. 2011;10:698–712. doi: 10.1038/nrd3505. [DOI] [PubMed] [Google Scholar]
- 153.Hyman BT, Gomez-Isla T. The natural history of Alzheimer neurofibrillary tangles and amyloid deposits. Neurobiol Aging. 1997;18:386–387;. doi: 10.1016/s0197-4580(97)00054-7. [DOI] [PubMed] [Google Scholar]