

Abstract
Renal insults are considered a public health problem and are linked to increased rates of morbidity and mortality worldwide. The heme oxygenase (HO) system consists of evolutionary specialized machinery that degrades free heme and produces carbon monoxide, biliverdin and free iron. In this sense, the inducible isoform HO-1 seems to develop an important role and is widely studied. The reaction involved with the HO-1 molecule provides protection to injured tissue, directly by reducing the toxic heme molecule and indirectly by the release of its byproducts. The up regulation of HO-1 enzyme has largely been described as providing antioxidant, antiapoptotic, anti-inflammatory and immunomodulatory properties. Several works have explored the importance of HO-1 in renal diseases and they have provided consistent evidence that its overexpression has beneficial effects in such injuries. So, in this review we will focus on the role of HO-1 in kidney insults, exploring the protective effects of its up regulation and the enhanced deleterious effects of its inhibition or gene deletion.
Keywords: Heme oxigenase-1, Renal cytoprotection, Antioxidants, Anti-inflammatory, Renal diseases
INTRODUCTION
Renal injuries can occur as a consequence of a number of factors, like hypoxia, nephrotoxicity, diabetes, renin-angiotensin system activation, among others. Most of these lesions are characterized by an increased amount of oxidative stress, inflammatory milieu and pro fibrotic stimuli. These factors lead to a breakdown of renal homeostasis and promote cell damage, with increased cell death and/or transdifferentiation. The ability of reducing this cellular damage could be crucial to a better outcome of the disease and, in this manner, the enzyme heme oxygenase (HO) could provide an important protective effect against renal insult[1].
Heme molecule (iron protoporphyrin IX) represents the prosthetic group of various proteins and enzymes, including hemoglobin, nitric oxide synthase, cytochrome P-450, cyclooxygenase, and catalase, among others. It is involved in critical functions, such as oxygen supply, mitochondrial respiratory burst and signal transduction[2,3]. In this sense, HO is the rate limiting enzyme responsible for heme degradation. HO cleaves to the heme ring in a reaction requiring oxygen and nicotinamide adenine dinucleotide phosphate and, as a result, biliverdin is produced, releasing iron and carbon monoxide (CO) in equimolar quantities. Later, biliverdin is converted to bilirubin by the enzyme bilirubin reductase[4,5].
HO was previously described by Tenhunen et al[6] in 1968 and interest in it has increased every year. This information is based on the fact that, since its discovery, more than 10 000 publications have been reported, and in 2010 alone, almost 1000 papers were published with this theme.
The HO system consists of two distinct isoforms, HO-1 (inducible) and HO-2 (constitutive), which are products of different genes. HO-1 is localized in microsomes and is ubiquitously present in mammalian tissue. Moreover, in physiological conditions, its expression is relatively low. The only exception comes from the spleen, where HO-1 is important for recycling iron from senescent erythrocytes. Recent studies showed that HO-1 deficiency affects stress erythropoiesis and leads to reduced function and viability of erythrophagocytosing macrophages, resulting in tissue damage and iron redistribution[7,8].
On the other hand, HO-2 seems to work as a physiological regulator of cell function. It is present in mitochondria and generally expressed in brain, testis, endothelium, nephron distal segments, liver and the gastrointestinal tract[9]. It seems to share 40% of amino acid homology with HO-1. Finally, formerly known as an isoform, HO-3 now is recognized as a pseudogene[10].
PROTECTIVE EFFECTS OF HO-1
Of these two isoforms, HO-1 is the most studied and seems to provide higher cytoprotection; so, hereafter, we will mostly discuss this isoform. The protection beyond HO-1 is observed in a variety of processes and these will be discussed in more detail in the next paragraphs.
HO-1 acts as an antioxidant in a direct and indirect manner. Directly, the enzyme contributes withdrawal of excessive heme molecule, which is a pro oxidant agent[11]. Indirectly, the free iron released from the reaction stimulates the expression of ferritin, an intracellular iron reservoir, diminishing the generation of hydroxyl radicals[12]. Furthermore, the biliverdin and, consequently, bilirubin formation displays an important antioxidant effect, as both molecules are peroxyl radicals scavengers[13].
The antiproliferative effects are based mainly on vascular smooth muscle cells experiments. A recent study showed that rapamycin could induce HO-1 expression and this up regulation led to protection in a model of pulmonary disease. The same work showed that smooth muscle cells derived from deficient animals for HO-1 were not responsive to the antiproliferative or cell cycle inhibition actions of rapamycin[14]. Moreover, studies have shown that a possible mechanism related to inhibition of cell growth by HO-1 could be up regulation of inhibitory protein p21cip. Interestingly, this pathway also contributes to an anti-apoptotic property of HO-1[15,16].
HO-1 can also act as an immunomodulatory enzyme, especially in T lymphocytes mediated diseases[17]. Burt et al[18] proposed that HO-1 contributes to T cells homeostasis, maintaining these lymphocytes in a nonactivated state, and the pharmacological inhibition of HO-1 leads to T cell activation and proliferation. The importance of HO-1 in Treg cells were described by a couple of works which stated that CD4+CD25+ Treg cells constitutively expressed HO-1 and that this enzyme could be induced after FoxP3 expression in CD4+CD25- cells, conferring a regulatory phenotype to these[19,20]. Another study showed that, in a murine model of colitis, treatment with hemin, a HO-1 inducer, resulted in expansion of Treg cells and decreased the levels of Th17 related molecules. On the other hand, inhibition of HO-1 led to opposite effects and aggravated the disease[21]. Still, the immunomodulatory effect of HO-1 also influences the priming of T cells. Cheng et al[22] showed that deletion of HO-1 gene or use of small interfering RNA silence in dendritic cells promoted up regulation of major histocompatibility complex class II, enhancing the alloantigen presentation to CD4+ T lymphocytes.
Finally, the anti-inflammatory property of HO-1 could be due to the enzymatic degradation of the pro-inflammatory heme molecule, as well as the production of its byproducts, which have the capacity to suppress the inflammatory process. In the first case, free heme is a highly toxic compound and may cause oxidative stress. Furthermore, its presence led to increased influx of leukocytes into organs during an experimental inflammation[23]. In addition, heme is part of many pro-inflammatory enzymes, like cytochrome p450 mono-oxygenases, inducible nitric oxide synthase and cyclooxygenase[24]. So, once HO-1 removes excessive free heme, it will impair the optimal activity of those enzymes, attenuating the inflammation[25]. On the other hand, some studies have shown that the up regulation of HO-1 can directly inhibit the inflammatory process. A recent work indicated that when HO-1 is induced, there is a negative modulation of inflammation, with decreased gene expression and protein production of tumor necrosis factor α, interleukin (IL)-6 and IL-1β, with concomitant increased protein levels of the immunomodulatory cytokine IL-10[26]. One of the pathways involved in this suppression may be related to p38 mitogen activated protein kinase. Lee et al[27] have shown that when there is an inhibition of this kinase, HO-1 induction is diminished and, consequently, the protection on human proximal tubular epithelial cells is abrogated.
HO EXPRESSION IN THE KIDNEY
The first paper describing the role of HO in kidney was published by Pimstone et al[28] in 1971 and it provided evidence that HO induction could contribute to renal cytoprotection. As stated earlier, HO-2 is a constitutive enzyme, so its expression is important for maintenance of kidney functions. In this sense, a study published by Da Silva and colleagues documented that this isoform is present in vascular and tubular compartments[29]. More specifically, HO-2 was observed in the medullar thick ascending limb, distal convoluted tubule, connecting tubule segments, principal cells of the collecting duct, renal interlobar arteries and preglomerular arterioles in the kidney[29]. On the other hand, HO-1 is weakly expressed in the kidney under normal conditions. But, confirming its role as a stress induced enzyme, after an acute or chronic renal insult, it is rapidly expressed[30,31]. Immunolocalization of HO-1 in rat kidneys showed that this enzyme was identified on proximal and distal tubules, as well as in medullar collecting tubules and loops of Henle[29]. Still, in a model of streptozotocin-induced diabetic nephropathy, HO-1 was also expressed in glomeruli[9]. Moreover, according to Jarmi and Agarwal’s work, HO-1 expression is also observed in human renal diseases, especially in proximal tubules[32]. Localization of HO-1 expression sites in different diseases could be important for development of specific therapeutic drugs.
Also, one of the main chemoattractant proteins is MCP-1, which can recruit leucocytes to the site of injury[5]. A recent study has shown that renal epithelial cells that constitutively overexpressed HO-1 presented decreased production of MCP-1 after stimulation with albumin[33]. Moreover, in HO-1 deficient mice, the basal levels of MCP-1 are significantly increased when compared to wild type animals and it becomes even higher after a stress condition[34]. Finally, as HO-1 is classified as a stress responsiveness enzyme, a recent work showed that urinary HO-1 could be useful and a sensitive biomarker for tubule interstitial inflammatory damage in renal diseases[35].
HO-1 AND RENAL INSULTS
In the following section, we will be discussing more about the protective effects of HO-1 in some renal diseases. For a better didactic comprehension, we divided it into four topics: acute kidney injury (AKI), diabetic nephropathy, renal transplantation and chronic kidney disease. Also, Figure 1 shows a summary of the role of HO-1 in these renal diseases.
Figure 1.
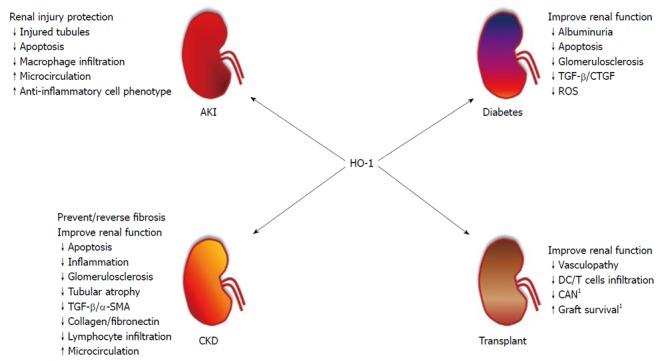
Overview of heme oxygenase-1 in renal diseases. Heme oxygenase (HO)-1 induction prevents renal damage in diverse renal diseases, such as acute kidney injury (AKI), chronic kidney disease (CKD), diabetic nephropathy (Diabetes) and renal Transplant. Arrows followed by text indicate increase (↑) or decrease (↓). 1Requires further study. TGF: Transforming growth factor; SMA: Smooth muscle actin; CTGF: Connective tissue growth factor; CAN: Chronic allograft nephropathy.
AKI
AKI, previously called acute renal failure, is defined by a rapid decrease of renal function, resulting in retention of urea and creatinine levels in serum as well as changes in cellular volumes and electrolyte imbalance. AKI is associated with high mortality rates and ischemia and reperfusion injury (IRI)[36] is one of its causes.
IRI starts with the break of ionic homeostasis due to ATP depletion in proximal tubular cells, which lose polarity and undergo the cell death process. All this is accompanied by intense vasoconstriction, increase of adhesion molecules, reactive oxygen species, pro-inflammatory cytokines and chemokines[37]. This inflammatory process also comprises of cells from innate (macrophages, dendritic cells and neutrophils) and adaptive immune response (B cells, CD4+ and CD8+ T cells)[36]. As HO-1 was seen to modulate immune cells to a regulatory profile[38] and to favor anti-inflammatory response, it obviously questioned the role of HO-1 in AKI.
Shimizu et al[39] used an HO-1 inhibitor, tin mesoporphyrin, in the unilateral renal ischemia and reperfusion model and observed an increase in microsomal heme, which is toxic to the cell. Different from non-treated animals, levels of heme were sustained in tin treated rats and renal injury was exacerbated in this group. In the opposite way, tin chloride treatment, a HO-1 inducer, protected animals from IRI[39,40]. These results demonstrated that HO-1 is associated with IRI protection. In the same direction, other HO-1 inducers led to protection in this model: hemin treatment improved microcirculation in an isogenic kidney transplantation model and also reduced IRI[41], and the use of Cobalt protoporphyrin (CoPPIX) in rapamycin-induced renal dysfunction after IRI increased HO-1 levels and eased renal injury[42]. Administration of cobalt chloride also protected rats from IRI, with an increase of hypoxia inducible factor (HIF)-1α, HO-1, erythropoietin, glucose transporter 1 and vascular endothelial growth factor, and diminished macrophage infiltration into the kidney[43]. More recently, Wu et al[44] showed that the induction of bardoxolone methyl increased HO-1 and nuclear factor erythroid 2-related factor 2 (Nrf2) expression, and protected mice from unilateral IRI. Ferenbach et al[45] produced macrophages that overexpressed HO-1, which presented an anti-inflammatory phenotype. Intravenous injection of these cells 20 min after IRI improved renal function outcome, suggesting a new tool to control injury in this model. The byproduct of HO-1, CO, was able by itself to protect animals from IRI with less injured tubules with the pre-administration of Tricarbonylchloro(glycinato) ruthenium(II) ([Ru(CO)3Cl-(glycinate)], the CORM-3[46], indicating that HO-1 and its byproducts were able to protect kidney from ischemic AKI.
Other AKI models were also protected after HO-1 induction. Rhabdomyolysis is characterized by heme and myoglobin release from damaged muscle, which allows the excessive exposure of the kidney to these proteins and leads to AKI. Glycerol-induced rhabdomyolysis presented renal injury protection with hemoglobin injection, while exacerbated renal damage was observed when tin protoporphyrin, a HO-1 inhibitor, was administered[47]. HO-1 induction by granulocyte colony-stimulating factor also protected animals from rhabdomyolysis kidney injury accompanied by increased survival and diminished apoptosis[48]. The same model of AKI presented worse renal function and higher mortality when using HO-1 knockout mice[49], which reinforces the importance of HO-1 to diminish the toxic heme accumulation.
Nephrotoxic-induced AKI occurs mostly as a side-effect of chemotherapy and other treatment drugs. In the cyclosporine-renal injury model, the treatment with CoPPIX led to a kidney morphological pattern similar to control[50]. Similar results were observed in animals that received cisplatin, a chemotherapy drug, with the concomitant treatment of CORM-3, presenting renal injury protection[51]. These results demonstrated that HO-1 has a role in nephrotoxic-induced AKI and highlight the importance of CO, HO-1 byproduct, in kidney injury protection.
All these works show that HO-1 induction can improve the outcome in AKI and, although this subject needs further investigation, suggests that the HO-1 manipulation could be an interesting tool to diminish AKI related mortality.
Diabetic nephropathy
Diabetic nephropathy is the most common cause of end-stage renal failure in developed countries[52]. Although the beginning of the disease in type I and type II diabetes is distinct, the changes observed in renal physiology caused by excessive glucose are quite similar and usually lead to alterations in kidney architecture accompanied by renal failure[53]. The role of HO-1 system in diabetes has been widely reported and its upregulation has been proved to mediate insulin release by pancreatic cells, conferring a protective effect[54-57].
Specifically talking about renal involvement of high glucose levels, some studies have shown that treatment with HO-1 inducers provides a better renal function outcome. Ohtomo et al[58] used a model of obese, hypertensive and diabetes type II rats (SHR/NDmcr-cp). These animals presented with severe proteinuria and renal histological changes. The up regulation of HO-1 with CoPPIX in such rats improved proteinuria levels and significantly decreased histological abnormalities. Moreover, the treatment also reduced the gene expression of pro-fibrotic molecules transforming growth factor (TGF)-β and CTGF.
Podocytes are important cells present in the glomerular compartment, being specialized in the good maintenance of the renal filtration[59]. A recent study showed that HO-1 inhibition promoted increased albuminuria and reduced podocyte numbers in diabetic rats. In vitro experiments showed that podocytes exposed to high glucose and to the HO-1 inhibitor ZnPP presented increased apoptosis. Induction of HO-1 protected these cells from pro apoptotic stimuli under diabetic conditions[60].
Finally, the most important antioxidant cellular regulator is Nrf2, which promotes expression of detoxifying and antioxidant enzymes that are a downstream Nrf2 gene, including HO-1[61]. Some recent studies have studied this pathway and its strict relationship with diabetic nephropathy. Li et al[62] showed that when streptozotocin-induced diabetic mice were treated with a Nrf2 activator, HO-1 gene and protein expressions were up regulated, with concomitant decreased levels of albuminuria, proteinuria and glomerulosclerosis index. Another study showed that mesangial cells submitted to high glucose levels presented increased reactive oxygen species production, proliferation and TGF-β production. When these cells were transfected with Nrf2-plasmid, HO-1 was induced and such parameters were ameliorated. On the other hand, when mesangial cells were exposed to Nrf2 specific siRNA, this protection was abrogated[63]. All these works provide support that one of the most important mechanisms of renal disease of diabetes, the pro-oxidant axis, can be attenuated by HO-1 induction.
Renal transplantation
Organ transplants are required in end state organ failure; however, in renal diseases, it might be the best treatment considering cost/benefit and quality of life, which encourages the increase of numbers in renal transplants. According to the World Health Organization, renal transplant comprises the majority of organ transplants performed in 98 countries. In 2005, 66 000 kidneys were transplanted, while 21 000 livers and 6000 hearts were transplanted in the same year[64]. Although it seems easy and advisable to do, renal transplant comes with a series of complications. Delayed graft function (DGF) is one and frequently occurs after a kidney transplant in almost 50% of the cases[65]. Ischemia and reperfusion is associated with DGF once cadaveric organs undergo an ischemic period before transplant and the incidence of DGF is increased in this case. DGF is also involved in acute rejection and can leads to chronic allograft nephropathy (CAN) development and chronic rejection[65,66], which is why the search for new therapies is needed.
As HO-1 was seen to have a protective role in IRI, to induce an anti-inflammatory environment, it was thought to have the same role in renal transplants. Formerly known as a stress induced enzyme, HO-1 was observed when protein expression was analyzed in biopsies. An increase of HO-1 expression in kidney biopsies prior[67] and post-reperfusion[68] was associated with DGF. Moreover, Avihingsanon et al[69] demonstrated that nonrejecting grafts as well as chronic rejected grafts presented no expression of HO-1, while acute rejected grafts presented higher expression of the enzyme, suggesting that the increase of HO-1 in acute rejection is an attempt to maintain graft viability and function and limit tissue injury. On the other hand, Lemos et al[70] showed that cadaveric donor kidneys presented decreased expression of HO-1 and worse renal function compared to living donor kidneys, reinforcing the protective role of this enzyme. These inconsistencies might be explained by the different population analyzed in each study and the environmental conditions of the transplantation that may influence the outcome of the graft.
The consequence of genetic polymorphism has also been described in the literature. A polymorphism based on dinucleotide repeat (GT)n was identified in HO-1 gene promoter. The short (S)-allele (< 27) has increased transcription of HO-1 in comparison to the long (L)-allele (> 27)[71]. Studies about the influence of this polymorphism in renal transplant are controversial: improvement on graft survival was seen in S-allele, as well as less CAN[71] and better renal function[71-73], while different groups observed no influence of donors and/or recipient S-allele kidneys in graft survival and CAN[73,74].
Animal models at least minimize the conflict of genetic background. A HO-1 inducer, CoPPIX, was administered in recipient rats and prevented allograft rejection, presumably due to less vasculopathy[75]. Administration of CoPPIX in donor rats also reduced DC, T CD4+ and T CD8+ in the graft, indicating less immunogenicity[76]. Not only the induction of HO-1, but also the use of methylene chloride, a CO donor, was able to improve graft function[77], corroborating the idea of HO-1 as an immune modulator in favor of graft acceptance. Human studies require further investigation, but they suggest so far that HO-1 works as a biomarker for acute rejection and that it also has a role in kidney homeostasis, which might help the acceptance and the better outcome of the graft. Animal models contribute to the better understanding of the HO-1 role in renal transplant, confirming the protective and immune regulatory function that culminates in graft acceptance.
Chronic kidney disease
Renal fibrosis results from a complex process of extracellular matrix production that ultimately leads to end stage renal disease. In this sense, the fibrotic process in the tubule-interstitial compartment has been estimated as a value predictor of irreversible loss of renal function[78]. Some studies have addressed the role of HO-1 in this progressive renal disease and will be further discussed.
An important study, published by Kie and colleagues, highlighted the role of basal HO-1 expression in renal fibrosis. The authors performed the experimental model of unilateral ureter obstruction (UUO) in wild-type and HO-1 deficient mice. The latter exhibited increased fibrosis deposition, macrophage infiltration, extracellular matrix synthesis and TGF-β production. In vitro experiments using proximal tubular cells isolated from kidneys of both animals showed that, after TGF-β treatment, the HO-1 deficient mice derived cells presented increased epithelial-to-mesenchymal transition, a process that occurs in fibrotic states[79]. Such work confirmed the information that HO-1, acting as a stress induced enzyme, is up regulated after a renal insult[80] in an attempt by the injured tissue to attenuate the incipient damage.
If the basal levels of HO-1 are important for kidney homeostasis, its overexpression seems to have important cytoprotective effects on progressive renal disease. Iwai et al[81] described that rats pre-treated with CoPPIX and submitted to obstructive nephropathy had, when compared to untreated animals, less expression of fibrosis markers α smooth muscle actin, fibronectin and collagen. Furthermore, they presented decreased mRNA for TGF-β and lymphocyte infiltration. Interestingly, the treated group showed increased macrophage infiltration, but co-localization techniques evidenced that these cells were also positive for HO-1, which was not seen on untreated rats. Probably, such macrophages display an immune suppressor profile.
Another work addressed the role of HO-1 as an anti-apoptotic molecule. In this case, animals were treated with hemin 48 h prior to chronic renal disease surgical induction. The group that received HO-1 inducer presented less pro-apoptotic proteins Bad and cleaved caspase-3. In contrast, the protein expression of anti-apoptotic molecule Bcl-2 was enhanced in this group[82]. A previous work from our group described that modulation of the inflammatory process by HO-1 up regulation also provides protection on the UUO model. Our experiments support the idea that inflammation is an important mediator of the fibrotic process development. After the treatment of rats with hemin, the inflammatory pattern was reduced and, consequently, it promoted a better renal function outcome and decreased fibrosis deposition and TGF-β production. A great finding in this work was that if the treatment with hemin was given after the effective establishment of the fibrosis, it could also reverse the progressive renal disease development[26].
Most of the studies that explored the role HO-1 in chronic kidney disease were performed using the UUO model. But, some work was also done in different experimental models. Desbuards et al[83] showed that induction of HO-1 in the remnant kidney model was able to improve systemic blood pressure and proteinuria, when compared to untreated animals. They also showed that the hemin-treated group had less glomerulosclerosis and tubular atrophy, which was accompanied by decreased TGF-β expression and increased BMP-7 levels (an anti fibrotic protein). Moreover, another work using this experimental model addressed the fact that induction of HO-1 with CoPPIX promotes a beneficial angiogenesis in renal tissue, with concomitant decrease of vimentin expression and tubular apoptosis[84]. Finally, Tanaka et al[85] demonstrated that HO-1 overexpression activates HIF-regulated genes and protects the hypoxic tubule interstitium compartment from injury in the nephrectomized Thy1 nephritis model.
Taken together, all these works support the idea that HO-1 up regulation can prevent and even reverse chronic kidney disease. The mechanism underlying this protection seems to be mainly by down regulating the inflammatory and apoptotic processes.
FINAL CONSIDERATIONS
In this review, we have explored the role of HO-1 in a variety of renal diseases. The cytoprotection behind it seems to be extremely relevant and provides an interesting feedback for future clinical trials. Nowadays, many studies have been performed to improve our knowledge upon HO-1 byproducts. The information that such studies are providing can be relevant for the development of specific drugs. We propose here that the HO-1 system plays an important role in the maintenance of renal homeostasis after an insult.
Footnotes
Supported by FAPESP (07/07139-3 and 09/54474-8), CAPES/PNPD, INCT Complex Fluids and CNPq
Peer reviewer: Dominique Guerrot, MD, PhD, Nephrology Department - Rouen University Hospital, 1 Avenue de Germont, 76031 Rouen, France
S- Editor Wang JL L- Editor Roemmele A E- Editor Zheng XM
References
- 1.Sikorski EM, Hock T, Hill-Kapturczak N, Agarwal A. The story so far: Molecular regulation of the heme oxygenase-1 gene in renal injury. Am J Physiol Renal Physiol. 2004;286:F425–F441. doi: 10.1152/ajprenal.00297.2003. [DOI] [PubMed] [Google Scholar]
- 2.Ibraham NG, Friedland ML, Levere RD. Heme metabolism in erythroid and hepatic cells. Prog Hematol. 1983;13:75–130. [PubMed] [Google Scholar]
- 3.Hill-Kapturczak N, Chang SH, Agarwal A. Heme oxygenase and the kidney. DNA Cell Biol. 2002;21:307–321. doi: 10.1089/104454902753759726. [DOI] [PubMed] [Google Scholar]
- 4.Abraham NG, Cao J, Sacerdoti D, Li X, Drummond G. Heme oxygenase: the key to renal function regulation. Am J Physiol Renal Physiol. 2009;297:F1137–F1152. doi: 10.1152/ajprenal.90449.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Courtney AE, Maxwell AP. Heme oxygenase 1: does it have a role in renal cytoprotection? Am J Kidney Dis. 2008;51:678–690. doi: 10.1053/j.ajkd.2007.11.033. [DOI] [PubMed] [Google Scholar]
- 6.Tenhunen R, Marver HS, Schmid R. The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc Natl Acad Sci U S A. 1968;61:748–755. doi: 10.1073/pnas.61.2.748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cao YA, Kusy S, Luong R, Wong RJ, Stevenson DK, Contag CH. Heme oxygenase-1 deletion affects stress erythropoiesis. PLoS One. 2011;6:e20634. doi: 10.1371/journal.pone.0020634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kovtunovych G, Eckhaus MA, Ghosh MC, Ollivierre-Wilson H, Rouault TA. Dysfunction of the heme recycling system in heme oxygenase 1-deficient mice: effects on macrophage viability and tissue iron distribution. Blood. 2010;116:6054–6062. doi: 10.1182/blood-2010-03-272138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Agarwal A, Nick HS. Renal response to tissue injury: lessons from heme oxygenase-1 GeneAblation and expression. J Am Soc Nephrol. 2000;11:965–973. doi: 10.1681/ASN.V115965. [DOI] [PubMed] [Google Scholar]
- 10.Hayashi S, Omata Y, Sakamoto H, Higashimoto Y, Hara T, Sagara Y, Noguchi M. Characterization of rat heme oxygenase-3 gene. Implication of processed pseudogenes derived from heme oxygenase-2 gene. Gene. 2004;336:241–250. doi: 10.1016/j.gene.2004.04.002. [DOI] [PubMed] [Google Scholar]
- 11.Ryter SW, Tyrrell RM. The heme synthesis and degradation pathways: role in oxidant sensitivity. Heme oxygenase has both pro- and antioxidant properties. Free Radic Biol Med. 2000;28:289–309. doi: 10.1016/s0891-5849(99)00223-3. [DOI] [PubMed] [Google Scholar]
- 12.Juckett MB, Balla J, Balla G, Jessurun J, Jacob HS, Vercellotti GM. Ferritin protects endothelial cells from oxidized low density lipoprotein in vitro. Am J Pathol. 1995;147:782–789. [PMC free article] [PubMed] [Google Scholar]
- 13.Stocker R. Antioxidant activities of bile pigments. Antioxid Redox Signal. 2004;6:841–849. doi: 10.1089/ars.2004.6.841. [DOI] [PubMed] [Google Scholar]
- 14.Zhou H, Liu H, Porvasnik SL, Terada N, Agarwal A, Cheng Y, Visner GA. Heme oxygenase-1 mediates the protective effects of rapamycin in monocrotaline-induced pulmonary hypertension. Lab Invest. 2006;86:62–71. doi: 10.1038/labinvest.3700361. [DOI] [PubMed] [Google Scholar]
- 15.Abraham NG, Scapagnini G, Kappas A. Human heme oxygenase: cell cycle-dependent expression and DNA microarray identification of multiple gene responses after transduction of endothelial cells. J Cell Biochem. 2003;90:1098–1111. doi: 10.1002/jcb.10736. [DOI] [PubMed] [Google Scholar]
- 16.Inguaggiato P, Gonzalez-Michaca L, Croatt AJ, Haggard JJ, Alam J, Nath KA. Cellular overexpression of heme oxygenase-1 up-regulates p21 and confers resistance to apoptosis. Kidney Int. 2001;60:2181–2191. doi: 10.1046/j.1523-1755.2001.00046.x. [DOI] [PubMed] [Google Scholar]
- 17.Xia ZW, Zhong WW, Meyrowitz JS, Zhang ZL. The role of heme oxygenase-1 in T cell-mediated immunity: the all encompassing enzyme. Curr Pharm Des. 2008;14:454–464. doi: 10.2174/138161208783597326. [DOI] [PubMed] [Google Scholar]
- 18.Burt TD, Seu L, Mold JE, Kappas A, McCune JM. Naive human T cells are activated and proliferate in response to the heme oxygenase-1 inhibitor tin mesoporphyrin. J Immunol. 2010;185:5279–5288. doi: 10.4049/jimmunol.0903127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi BM, Pae HO, Jeong YR, Kim YM, Chung HT. Critical role of heme oxygenase-1 in Foxp3-mediated immune suppression. Biochem Biophys Res Commun. 2005;327:1066–1071. doi: 10.1016/j.bbrc.2004.12.106. [DOI] [PubMed] [Google Scholar]
- 20.Pae HO, Oh GS, Choi BM, Chae SC, Chung HT. Differential expressions of heme oxygenase-1 gene in CD25- and CD25+ subsets of human CD4+ T cells. Biochem Biophys Res Commun. 2003;306:701–705. doi: 10.1016/s0006-291x(03)01037-4. [DOI] [PubMed] [Google Scholar]
- 21.Zhong W, Xia Z, Hinrichs D, Rosenbaum JT, Wegmann KW, Meyrowitz J, Zhang Z. Hemin exerts multiple protective mechanisms and attenuates dextran sulfate sodium-induced colitis. J Pediatr Gastroenterol Nutr. 2010;50:132–139. doi: 10.1097/MPG.0b013e3181c61591. [DOI] [PubMed] [Google Scholar]
- 22.Cheng C, Noorderloos M, van Deel ED, Tempel D, den Dekker W, Wagtmans K, Duncker DJ, Soares MP, Laman JD, Duckers HJ. Dendritic cell function in transplantation arteriosclerosis is regulated by heme oxygenase 1. Circ Res. 2010;106:1656–1666. doi: 10.1161/CIRCRESAHA.110.216945. [DOI] [PubMed] [Google Scholar]
- 23.Wagener FA, Abraham NG, van Kooyk Y, de Witte T, Figdor CG. Heme-induced cell adhesion in the pathogenesis of sickle-cell disease and inflammation. Trends Pharmacol Sci. 2001;22:52–54. doi: 10.1016/s0165-6147(00)01609-6. [DOI] [PubMed] [Google Scholar]
- 24.White KA, Marletta MA. Nitric oxide synthase is a cytochrome P-450 type hemoprotein. Biochemistry. 1992;31:6627–6631. doi: 10.1021/bi00144a001. [DOI] [PubMed] [Google Scholar]
- 25.Gozzelino R, Jeney V, Soares MP. Mechanisms of cell protection by heme oxygenase-1. Annu Rev Pharmacol Toxicol. 2010;50:323–354. doi: 10.1146/annurev.pharmtox.010909.105600. [DOI] [PubMed] [Google Scholar]
- 26.Correa-Costa M, Semedo P, Monteiro AP, Silva RC, Pereira RL, Gonçalves GM, Marques GD, Cenedeze MA, Faleiros AC, Keller AC, et al. Induction of heme oxygenase-1 can halt and even reverse renal tubule-interstitial fibrosis. PLoS One. 2010;5:e14298. doi: 10.1371/journal.pone.0014298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee HT, Xu H, Ota-Setlik A, Emala CW. Oxidant preconditioning protects human proximal tubular cells against lethal oxidant injury via p38 MAPK and heme oxygenase-1. Am J Nephrol. 2003;23:324–333. doi: 10.1159/000072914. [DOI] [PubMed] [Google Scholar]
- 28.Pimstone NR, Engel P, Tenhunen R, Seitz PT, Marver HS, Schmid R. Inducible heme oxygenase in the kidney: a model for the homeostatic control of hemoglobin catabolism. J Clin Invest. 1971;50:2042–2050. doi: 10.1172/JCI106697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.da Silva JL, Zand BA, Yang LM, Sabaawy HE, Lianos E, Abraham NG. Heme oxygenase isoform-specific expression and distribution in the rat kidney. Kidney Int. 2001;59:1448–1457. doi: 10.1046/j.1523-1755.2001.0590041448.x. [DOI] [PubMed] [Google Scholar]
- 30.Semedo P, Correa-Costa M, Antonio Cenedeze M, Maria Avancini Costa Malheiros D, Antonia dos Reis M, Shimizu MH, Seguro AC, Pacheco-Silva A, Saraiva Camara NO. Mesenchymal stem cells attenuate renal fibrosis through immune modulation and remodeling properties in a rat remnant kidney model. Stem Cells. 2009;27:3063–3073. doi: 10.1002/stem.214. [DOI] [PubMed] [Google Scholar]
- 31.Toda N, Takahashi T, Mizobuchi S, Fujii H, Nakahira K, Takahashi S, Yamashita M, Morita K, Hirakawa M, Akagi R. Tin chloride pretreatment prevents renal injury in rats with ischemic acute renal failure. Crit Care Med. 2002;30:1512–1522. doi: 10.1097/00003246-200207000-00020. [DOI] [PubMed] [Google Scholar]
- 32.Jarmi T, Agarwal A. Heme oxygenase and renal disease. Curr Hypertens Rep. 2009;11:56–62. doi: 10.1007/s11906-009-0011-z. [DOI] [PubMed] [Google Scholar]
- 33.Murali NS, Ackerman AW, Croatt AJ, Cheng J, Grande JP, Sutor SL, Bram RJ, Bren GD, Badley AD, Alam J, et al. Renal upregulation of HO-1 reduces albumin-driven MCP-1 production: implications for chronic kidney disease. Am J Physiol Renal Physiol. 2007;292:F837–F844. doi: 10.1152/ajprenal.00254.2006. [DOI] [PubMed] [Google Scholar]
- 34.Pittock ST, Norby SM, Grande JP, Croatt AJ, Bren GD, Badley AD, Caplice NM, Griffin MD, Nath KA. MCP-1 is up-regulated in unstressed and stressed HO-1 knockout mice: Pathophysiologic correlates. Kidney Int. 2005;68:611–622. doi: 10.1111/j.1523-1755.2005.00439.x. [DOI] [PubMed] [Google Scholar]
- 35.Yokoyama T, Shimizu M, Ohta K, Yuno T, Okajima M, Wada T, Toma T, Koizumi S, Yachie A. Urinary heme oxygenase-1 as a sensitive indicator of tubulointerstitial inflammatory damage in various renal diseases. Am J Nephrol. 2011;33:414–420. doi: 10.1159/000327020. [DOI] [PubMed] [Google Scholar]
- 36.Kinsey GR, Li L, Okusa MD. Inflammation in acute kidney injury. Nephron Exp Nephrol. 2008;109:e102–e107. doi: 10.1159/000142934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Devarajan P. Update on mechanisms of ischemic acute kidney injury. J Am Soc Nephrol. 2006;17:1503–1520. doi: 10.1681/ASN.2006010017. [DOI] [PubMed] [Google Scholar]
- 38.Soares MP, Marguti I, Cunha A, Larsen R. Immunoregulatory effects of HO-1: how does it work? Curr Opin Pharmacol. 2009;9:482–489. doi: 10.1016/j.coph.2009.05.008. [DOI] [PubMed] [Google Scholar]
- 39.Shimizu H, Takahashi T, Suzuki T, Yamasaki A, Fujiwara T, Odaka Y, Hirakawa M, Fujita H, Akagi R. Protective effect of heme oxygenase induction in ischemic acute renal failure. Crit Care Med. 2000;28:809–817. doi: 10.1097/00003246-200003000-00033. [DOI] [PubMed] [Google Scholar]
- 40.Akagi R, Takahashi T, Sassa S. Cytoprotective effects of heme oxygenase in acute renal failure. Contrib Nephrol. 2005;148:70–85. doi: 10.1159/000086044. [DOI] [PubMed] [Google Scholar]
- 41.Hölzen JP, August C, Bahde R, Minin E, Lang D, Heidenreich S, Dietl KH, Spiegel HU. Influence of heme oxygenase-1 on microcirculation after kidney transplantation. J Surg Res. 2008;148:126–135. doi: 10.1016/j.jss.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 42.Gonçalves GM, Cenedeze MA, Feitoza CQ, Wang PM, Bertocchi AP, Damião MJ, Pinheiro HS, Antunes Teixeira VP, dos Reis MA, Pacheco-Silva A, et al. The role of heme oxygenase 1 in rapamycin-induced renal dysfunction after ischemia and reperfusion injury. Kidney Int. 2006;70:1742–1749. doi: 10.1038/sj.ki.5001893. [DOI] [PubMed] [Google Scholar]
- 43.Matsumoto M, Makino Y, Tanaka T, Tanaka H, Ishizaka N, Noiri E, Fujita T, Nangaku M. Induction of renoprotective gene expression by cobalt ameliorates ischemic injury of the kidney in rats. J Am Soc Nephrol. 2003;14:1825–1832. doi: 10.1097/01.asn.0000074239.22357.06. [DOI] [PubMed] [Google Scholar]
- 44.Wu QQ, Wang Y, Senitko M, Meyer C, Wigley WC, Ferguson DA, Grossman E, Chen J, Zhou XJ, Hartono J, et al. Bardoxolone methyl (BARD) ameliorates ischemic AKI and increases expression of protective genes Nrf2, PPARγ, and HO-1. Am J Physiol Renal Physiol. 2011;300:F1180–F1192. doi: 10.1152/ajprenal.00353.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferenbach DA, Ramdas V, Spencer N, Marson L, Anegon I, Hughes J, Kluth DC. Macrophages expressing heme oxygenase-1 improve renal function in ischemia/reperfusion injury. Mol Ther. 2010;18:1706–1713. doi: 10.1038/mt.2010.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vera T, Henegar JR, Drummond HA, Rimoldi JM, Stec DE. Protective effect of carbon monoxide-releasing compounds in ischemia-induced acute renal failure. J Am Soc Nephrol. 2005;16:950–958. doi: 10.1681/ASN.2004090736. [DOI] [PubMed] [Google Scholar]
- 47.Nath KA, Balla G, Vercellotti GM, Balla J, Jacob HS, Levitt MD, Rosenberg ME. Induction of heme oxygenase is a rapid, protective response in rhabdomyolysis in the rat. J Clin Invest. 1992;90:267–270. doi: 10.1172/JCI115847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei Q, Hill WD, Su Y, Huang S, Dong Z. Heme oxygenase-1 induction contributes to renoprotection by G-CSF during rhabdomyolysis-associated acute kidney injury. Am J Physiol Renal Physiol. 2011;301:F162–F170. doi: 10.1152/ajprenal.00438.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nath KA, Haggard JJ, Croatt AJ, Grande JP, Poss KD, Alam J. The indispensability of heme oxygenase-1 in protecting against acute heme protein-induced toxicity in vivo. Am J Pathol. 2000;156:1527–1535. doi: 10.1016/S0002-9440(10)65024-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rezzani R, Rodella L, Buffoli B, Goodman AA, Abraham NG, Lianos EA, Bianchi R. Change in renal heme oxygenase expression in cyclosporine A-induced injury. J Histochem Cytochem. 2005;53:105–112. doi: 10.1177/002215540505300112. [DOI] [PubMed] [Google Scholar]
- 51.Tayem Y, Johnson TR, Mann BE, Green CJ, Motterlini R. Protection against cisplatin-induced nephrotoxicity by a carbon monoxide-releasing molecule. Am J Physiol Renal Physiol. 2006;290:F789–F794. doi: 10.1152/ajprenal.00363.2005. [DOI] [PubMed] [Google Scholar]
- 52.United States Renal Data System. USRDS 2003 annual data report: atlas of end-stage renal disease in the United States. Bethesda, MD: National Institute of Health, National Institute of Diabetes and Kidney Disease; 2003. [Google Scholar]
- 53.Wolfe RA, Port FK, Webb RL, Bloembergen WE, Hirth R, Young EW, Ojo AO, Strawderman RL, Parekh R, Stack A, et al. Introduction to the excerpts from the United States Renal Data System 1999 Annual Data Report. Am J Kidney Dis. 1999;34:S1–S3. doi: 10.1053/AJKD034s00001. [DOI] [PubMed] [Google Scholar]
- 54.Fu SH, Hsu BR, Juang JH, Chen ST, Yang TY, Hsu S. Cobalt-protoporphyrin treatment enhances murine isoislets engraftment. Transplant Proc. 2004;36:2205–2206. doi: 10.1016/j.transproceed.2004.06.050. [DOI] [PubMed] [Google Scholar]
- 55.Hsu BR, Chen ST, Fu SH. A single-dose of cobalt-protoporphyrin protects islet beta cells from glucocorticoid suppression. Transplant Proc. 2005;37:1826–1827. doi: 10.1016/j.transproceed.2005.02.085. [DOI] [PubMed] [Google Scholar]
- 56.Ndisang JF, Jadhav A. The heme oxygenase system attenuates pancreatic lesions and improves insulin sensitivity and glucose metabolism in deoxycorticosterone acetate hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R211–R223. doi: 10.1152/ajpregu.91000.2008. [DOI] [PubMed] [Google Scholar]
- 57.Ndisang JF, Lane N, Syed N, Jadhav A. Up-regulating the heme oxygenase system with hemin improves insulin sensitivity and glucose metabolism in adult spontaneously hypertensive rats. Endocrinology. 2010;151:549–560. doi: 10.1210/en.2009-0471. [DOI] [PubMed] [Google Scholar]
- 58.Ohtomo S, Nangaku M, Izuhara Y, Takizawa S, Strihou CY, Miyata T. Cobalt ameliorates renal injury in an obese, hypertensive type 2 diabetes rat model. Nephrol Dial Transplant. 2008;23:1166–1172. doi: 10.1093/ndt/gfm715. [DOI] [PubMed] [Google Scholar]
- 59.Fligny C, Barton M, Tharaux PL. Endothelin and podocyte injury in chronic kidney disease. Contrib Nephrol. 2011;172:120–138. doi: 10.1159/000328692. [DOI] [PubMed] [Google Scholar]
- 60.Lee SC, Han SH, Li JJ, Lee SH, Jung DS, Kwak SJ, Kim SH, Kim DK, Yoo TH, Kim JH, et al. Induction of heme oxygenase-1 protects against podocyte apoptosis under diabetic conditions. Kidney Int. 2009;76:838–848. doi: 10.1038/ki.2009.286. [DOI] [PubMed] [Google Scholar]
- 61.Cheng X, Siow RC, Mann GE. Impaired redox signaling and antioxidant gene expression in endothelial cells in diabetes: a role for mitochondria and the nuclear factor-E2-related factor 2-Kelch-like ECH-associated protein 1 defense pathway. Antioxid Redox Signal. 2011;14:469–487. doi: 10.1089/ars.2010.3283. [DOI] [PubMed] [Google Scholar]
- 62.Li H, Zhang L, Wang F, Shi Y, Ren Y, Liu Q, Cao Y, Duan H. Attenuation of glomerular injury in diabetic mice with tert-butylhydroquinone through nuclear factor erythroid 2-related factor 2-dependent antioxidant gene activation. Am J Nephrol. 2011;33:289–297. doi: 10.1159/000324694. [DOI] [PubMed] [Google Scholar]
- 63.Li H, Wang F, Zhang L, Cao Y, Liu W, Hao J, Liu Q, Duan H. Modulation of Nrf2 expression alters high glucose-induced oxidative stress and antioxidant gene expression in mouse mesangial cells. Cell Signal. 2011;23:1625–1632. doi: 10.1016/j.cellsig.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 64.Bagozzi D. WHO proposes global agenda on transplantation. Geneva: World Health Organization; 2007. Available from: http://www.who.int/mediacentre/news/releases/2007/pr12/en/index.html. [Google Scholar]
- 65.Perico N, Cattaneo D, Sayegh MH, Remuzzi G. Delayed graft function in kidney transplantation. Lancet. 2004;364:1814–1827. doi: 10.1016/S0140-6736(04)17406-0. [DOI] [PubMed] [Google Scholar]
- 66.McLaren AJ, Jassem W, Gray DW, Fuggle SV, Welsh KI, Morris PJ. Delayed graft function: risk factors and the relative effects of early function and acute rejection on long-term survival in cadaveric renal transplantation. Clin Transplant. 1999;13:266–272. doi: 10.1034/j.1399-0012.1999.130308.x. [DOI] [PubMed] [Google Scholar]
- 67.August C, Brockmann J, Vowinkel T, Wolters H, Dietl KH, Levkau B, Heidenreich S, Lang D, Baba HA. Stress associated proteins metallothionein, HO-1 and HSP 70 in human zero-hour biopsies of transplanted kidneys. Virchows Arch. 2006;449:192–199. doi: 10.1007/s00428-006-0216-3. [DOI] [PubMed] [Google Scholar]
- 68.Ollinger R, Kogler P, Biebl M, Sieb M, Sucher R, Bösmüller C, Troppmair J, Mark W, Weiss H, Margreiter R. Protein levels of heme oxygenase-1 during reperfusion in human kidney transplants with delayed graft function. Clin Transplant. 2008;22:418–423. doi: 10.1111/j.1399-0012.2008.00800.x. [DOI] [PubMed] [Google Scholar]
- 69.Avihingsanon Y, Ma N, Csizmadia E, Wang C, Pavlakis M, Giraldo M, Strom TB, Soares MP, Ferran C. Expression of protective genes in human renal allografts: a regulatory response to injury associated with graft rejection. Transplantation. 2002;73:1079–1085. doi: 10.1097/00007890-200204150-00011. [DOI] [PubMed] [Google Scholar]
- 70.Lemos FB, Ijzermans JN, Zondervan PE, Peeters AM, van den Engel S, Mol WM, Weimar W, Baan CC. Differential expression of heme oxygenase-1 and vascular endothelial growth factor in cadaveric and living donor kidneys after ischemia-reperfusion. J Am Soc Nephrol. 2003;14:3278–3287. doi: 10.1097/01.asn.0000098683.92538.66. [DOI] [PubMed] [Google Scholar]
- 71.Baan C, Peeters A, Lemos F, Uitterlinden A, Doxiadis I, Claas F, Ijzermans J, Roodnat J, Weimar W. Fundamental role for HO-1 in the self-protection of renal allografts. Am J Transplant. 2004;4:811–818. doi: 10.1111/j.1600-6143.2004.00420.x. [DOI] [PubMed] [Google Scholar]
- 72.Ozaki KS, Marques GM, Nogueira E, Feitoza RQ, Cenedeze MA, Franco MF, Mazzali M, Soares MP, Pacheco-Silva A, Câmara NO. Improved renal function after kidney transplantation is associated with heme oxygenase-1 polymorphism. Clin Transplant. 2008;22:609–616. doi: 10.1111/j.1399-0012.2008.00832.x. [DOI] [PubMed] [Google Scholar]
- 73.Katana E, Skoura L, Giakoustidis D, Takoudas D, Malisiovas N, Daniilidis M. Association between the heme oxygenase-1 promoter polymorphism and renal transplantation outcome in Greece. Transplant Proc. 2010;42:2479–2485. doi: 10.1016/j.transproceed.2010.05.161. [DOI] [PubMed] [Google Scholar]
- 74.Courtney AE, McNamee PT, Nelson WE, Maxwell AP. Does angiotensin blockade influence graft outcome in renal transplant recipients with IgA nephropathy? Nephrol Dial Transplant. 2006;21:3550–3554. doi: 10.1093/ndt/gfl506. [DOI] [PubMed] [Google Scholar]
- 75.Bédard EL, Jiang J, Parry N, Wang H, Liu W, Garcia B, Kim P, Chakrabarti S, Buelow R, Zhong R. Peritransplant treatment with cobalt protoporphyrin attenuates chronic renal allograft rejection. Transpl Int. 2005;18:341–349. doi: 10.1111/j.1432-2277.2004.00062.x. [DOI] [PubMed] [Google Scholar]
- 76.Martins PN, Kessler H, Jurisch A, Reutzel-Selke A, Kramer J, Pascher A, Pratschke J, Neuhaus P, Volk HD, Tullius SG. Induction of heme oxygenase-1 in the donor reduces graft immunogenicity. Transplant Proc. 2005;37:384–386. doi: 10.1016/j.transproceed.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 77.Martins PN, Reuzel-Selke A, Jurisch A, Atrott K, Pascher A, Pratschke J, Buelow R, Neuhaus P, Volk HD, Tullius SG. Induction of carbon monoxide in the donor reduces graft immunogenicity and chronic graft deterioration. Transplant Proc. 2005;37:379–381. doi: 10.1016/j.transproceed.2004.11.079. [DOI] [PubMed] [Google Scholar]
- 78.Liu Y. Cellular and molecular mechanisms of renal fibrosis. Nat Rev Nephrol. 2011;7:684–696. doi: 10.1038/nrneph.2011.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kie JH, Kapturczak MH, Traylor A, Agarwal A, Hill-Kapturczak N. Heme oxygenase-1 deficiency promotes epithelial-mesenchymal transition and renal fibrosis. J Am Soc Nephrol. 2008;19:1681–1691. doi: 10.1681/ASN.2007101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pat B, Yang T, Kong C, Watters D, Johnson DW, Gobe G. Activation of ERK in renal fibrosis after unilateral ureteral obstruction: modulation by antioxidants. Kidney Int. 2005;67:931–943. doi: 10.1111/j.1523-1755.2005.00157.x. [DOI] [PubMed] [Google Scholar]
- 81.Iwai T, Kitamoto K, Teramoto K, Machida Y, Tamada S, Yukimura T, Iwao H, Nakatani T, Miura K. Cobalt protoporphyrin attenuates rat obstructive nephropathy: role of cellular infiltration. Urology. 2008;72:432–438. doi: 10.1016/j.urology.2007.11.123. [DOI] [PubMed] [Google Scholar]
- 82.Kim JH, Yang JI, Jung MH, Hwa JS, Kang KR, Park DJ, Roh GS, Cho GJ, Choi WS, Chang SH. Heme oxygenase-1 protects rat kidney from ureteral obstruction via an antiapoptotic pathway. J Am Soc Nephrol. 2006;17:1373–1381. doi: 10.1681/ASN.2005091001. [DOI] [PubMed] [Google Scholar]
- 83.Desbuards N, Hyvelin JM, Machet MC, Eder V, Garrigue MA, Halimi JM, Antier D. Heme oxygenase-1 inducer hemin attenuates the progression of remnant kidney model. Nephron Exp Nephrol. 2009;113:e35–e44. doi: 10.1159/000228081. [DOI] [PubMed] [Google Scholar]
- 84.Tanaka T, Kojima I, Ohse T, Ingelfinger JR, Adler S, Fujita T, Nangaku M. Cobalt promotes angiogenesis via hypoxia-inducible factor and protects tubulointerstitium in the remnant kidney model. Lab Invest. 2005;85:1292–1307. doi: 10.1038/labinvest.3700328. [DOI] [PubMed] [Google Scholar]
- 85.Tanaka T, Matsumoto M, Inagi R, Miyata T, Kojima I, Ohse T, Fujita T, Nangaku M. Induction of protective genes by cobalt ameliorates tubulointerstitial injury in the progressive Thy1 nephritis. Kidney Int. 2005;68:2714–2725. doi: 10.1111/j.1523-1755.2005.00742.x. [DOI] [PubMed] [Google Scholar]