

Abstract
Thiazolidinediones (TZDs), pharmacological activators of peroxisome-proliferator-activated receptors γ (PPARγ), significantly improve insulin resistance and lower plasma glucose concentrations. However, the use of TZDs is associated with plasma volume expansion, the mechanism of which has been a matter of controversy. Originally, PPARγ-mediated enhanced transcription of the epithelial Na channel (ENaC) γ subunit was thought to play a central role in TZD-induced volume expansion. However, later studies suggested that the activation of ENaC alone could not explain TZD-induced volume expansion. We have recently shown that TZDs rapidly stimulate sodium-coupled bicarbonate absorption from renal proximal tubule (PT) in vitro and in vivo. TZD-induced transport stimulation was dependent on PPARγ/Src/EGFR/ERK, and observed in rat, rabbit and human. However, this stimulation was not observed in mouse PTs where Src/EGFR is constitutively activated. Analysis in mouse embryonic fibroblast cells confirmed the existence of PPARγ/Src-dependent non-genomic signaling, which requires the ligand binding ability but not the transcriptional activity of PPARγ. The TZD-induced enhancement of association between PPARγ and Src supports an obligatory role for Src in this signaling. These results support the view that TZD-induced volume expansion is multifactorial. In addition to the PPARγ-dependent enhanced expression of the sodium transport system(s) in distal nephrons, the PPARγ-dependent non-genomic stimulation of renal proximal transport may be also involved in TZD-induced volume expansion.
Keywords: Thiazolidinediones, Peroxisome-proliferator-activated receptors γ, Volume expansion, Edema, NBCe1, NHE3, Epithelial Na channel
PHYSIOLOGICAL ROLES OF PEROXISOME-PROLIFERATOR-ACTIVATED RECEPTOR γ
The peroxisome-proliferator-activated receptors (PPARs) are a subfamily of the nuclear-receptor superfamily. Upon ligand binding, these receptors undergo conformational changes, and regulate the expression of multiple genes by recruiting specific sets of co-activators or co-repressors. Among them, PPARγ is most abundantly expressed in adipose tissues, but is also expressed in many tissues including vascular endothelium and kidney[1]. PPARγ is both necessary and sufficient to initiate adipogenesis in fibroblast cell lines, and can be thought of as a master gene in fat cell biology and differentiation[2]. In a key study, homozygous PPARγ-deficient embryos died due to placental deficiency. Unexpectedly, heterozygous PPAΡγ-deficient mice were rather protected from developing insulin resistance under a high-fat diet. These heterozygous mice showed overexpression and hypersecretion of leptin despite the smaller size of their adipocytes, which might explain the protection from insulin resistance[3]. Thus, PPARγ may play dual roles in the regulation of insulin resistance at least in the specific experimental conditions in mice.
By contrast, pharmacological activation of PPARγ by thiazolidinediones (TZDs) consistently improves insulin resistance and lowers plasma glucose concentrations in humans[1]. Interestingly, a recent study suggests that obesity-induced phosphorylation of PPARγ at serine 273 may, by reducing the expression adiponectin, represent a molecular basis of insulin resistance[4]. Thus, PPARγ seems to be an ideal target in the treatment of type 2 diabetes mellitus with insulin resistance.
TZDs such as rosiglitazone and pioglitazone (PGZ) are synthetic ligands functioning as strong activators of PPARγ. The insulin-sensitizing effects of TZDs may involve the decreased expression of insulin-resistant adipokines including tumor necrosis factor (TNF)-α, interleukin (IL)-1 and resistin, as well as the increased production of insulin-sensitizing hormone adiponectin[4]. TZDs may also have beneficial effects on the cardiovascular system, including the reduction of blood pressure, the improvement of vascular function, and the reduction of inflammatory biomarkers[5,6]. These beneficial effects of TZDs on glycemic control and cardiovascular risk factors have lead to their widespread use in the treatment of type 2 diabetes mellitus. However, fluid retention, one of the main side effects of TZDs, precludes the use of TZDs in the setting of severe congestive heart failure (CHF). Moreover, a recent meta-analysis suggests that rosiglitazone may even increase the cardiovascular mortality risk[7].
EDEMA FORMATION AND TZDs IN CLINICAL PRACTICE
When used as monotherapy, the incidence of pedal edema ranges from 3%-5% for both rosiglitazone and pioglitazone[5,8]. The incidence of edema formation significantly increases when either of the TZDs is used in combination with insulin. For example, the use of rosiglitazone in combination with insulin was associated with 13%-16% incidence of edema, compared with 4.7% in those receiving insulin alone[9]. The reasons for fluid retention and peripheral edema with the use of TZDs may involve multiple factors. In addition to vasodilatation, a reduction in renal excretion of sodium and water probably plays a substantial role in plasma volume expansion related to the use of TZDs.
Despite the increased rate of edema formation, direct negative effects of TZDs on cardiac functions are less likely. Actually, the incidence of CHF was less than 1% for rosiglitazone monotherapy[10]. When rosiglitazone was added to insulin therapy, the incidence of CHF increased to 2%-3%, compared with 1% in those receiving insulin alone[5]. Thus, the incidence of CHF in TZD-treated patients is very low. However, the incidence is significantly increased by combination therapy with insulin[5,10,11]. Based on these and other considerations, the American Diabetes Association and the American Heart association evaluated the use of TZDs in patients with heart disease and in those who develop edema or unexpected weight gain. Their recommendations were the evaluation of edema pathogenesis, the careful monitoring of CHF-related symptoms in the first 3 mo of TZD treatment, the evaluation of cardiac functions for the possible existence of CHF, and the reconsideration of TZD use if a new diagnosis of CHF is made[5]. At present, TZDs should not be prescribed to patients with symptoms and signs of New York Heart Association class III or IV cardiac function status.
MECHANISM OF TZD-INDUCED FLUID RETENTION
Molecular mechanism underlying the enhancement of sodium and fluid absorption from the kidney by TZDs has been a matter of controversy. Originally, the studies on mice with the selective deletion of PPARγ from renal collecting ducts suggested that TZD-induced volume expansion is dependent on transport stimulation in the distal nephrons[12,13]. In particular, the PPARγ-mediated enhanced transcription of the epithelial Na channel (ENaC) γ subunit was thought to play a key role[12,13]. Consistent with this view, amiloride, an inhibitor of ENaC, was able to suppress TZD-induced volume expansion in mice[12]. Moreover, TZDs were reported to enhance the surface expression of the ENaC α subunit through the glucocorticoid-inducible kinase SGK1[14].
However, other studies have failed to support a critical role for ENaC in TZD-induced volume expansion. For example, TZDs did not alter the ENaC activity in well-established renal principal cell culture models, although insulin was able to clearly enhance ENaC activity in these cells[15]. TZDs also failed to enhance the stimulatory effect of insulin on ENaC, arguing against a role for ENaC in fluid retention caused by the combined use of TZDs and insulin. In addition, the enhanced expression of ENaC subunits by TZDs was not observed consistently[16]. Furthermore, Vallon et al[17] showed that TZD-induced fluid retention was not suppressed in mice selectively lacking the ENaC α subunit in collecting ducts, strongly suggesting that the enhancement of ENaC activity alone cannot explain TZD-induced volume expansion[17]. Using primary cultured mouse inner medullary collecting ducts, Vallon et al[17] also found that TZDs can increase the activity of a nonselective cation channel. However, they failed to find ENaC activity in these cells[17]. The same group recently found that TZDs can actually repress the ENaC γ subunit promoter via an indirect transcriptional mechanism in M1 cortical collecting duct cells[18]. These results seem to indicate that TZDs may activate sodium transport system(s) other than ENaC in renal distal tubules.
EFFECTS OF TZDs ON RENAL PROXIMAL TUBULE TRANSPORT
Both human[19] and animal[20] studies suggested that renal proximal tubule (PT) transport could be stimulated by TZDs. We therefore speculated that TZD-induced volume expansion is multifactorial, and that PTs may be another target nephron segment for TZDs. In the stimulation of distal nephron transport, the process known as escape phenomenon tends to suppress the sodium reabsorption along PTs and other nephron segments. Therefore, the stimulation of sodium transport in distal nephrons alone, as observed in excessive aldosterone actions, does not usually result in massive volume expansion with edema formation[21].
In PTs, the apical Na+/H+ exchanger NHE3 and the basolateral Na+-HCO3- cotransporter NBCe1 are thought to function in a coordinated fashion to absorb sodium, bicarbonate, and fluid. We found that physiological concentrations of TZDs markedly stimulated sodium-coupled bicarbonate absorption from isolated rabbit, rat, and human PTs by activating both NBCe1 and NHE3 through the PPARγ/Src/EGFR/ERK pathway. Acute PGZ administration in rats also induced changes in renal parameters consistent with the stimulation of in vivo PT transport. However, TZDs failed to stimulate in vitro and in vivo PT transport in mice, where Src/EGFR/ERK is uniquely activated in a constitutive manner[22].
TZDs are known to trigger diverse rapid cellular signaling events including the activation of kinase signaling pathways such as phosphatidylinositol 3-kinase (PI3K)/Akt, ERK, EGFR transactivation, the production of reactive oxygen species, and Ca2+ influx[23]. However, the mechanisms underlying such dose-, time-, and cell type-dependent responses had been largely unknown. By performing transfection experiments on mouse embryonic fibroblast (EF) cells derived from PPARγ-/- mice with the full-length PPARγ or the ligand-binding domain of PPARγ[3], we confirmed the presence of nongenomic signaling, resulting in the activation of ERK and requiring the ligand binding ability but not the transcriptional activity of PPARγ. We also showed that TZDs rapidly facilitated association between PPARγ and Src, which was also dependent on the ligand-binding ability of PPARγ. Together with the rapid kinetics (within minutes) of responses and the non-requirement of transcriptional activity, these results indicated that PPARγ, like another nuclear receptor for estrogen[24], can activate the ERK pathway through a non-genomic mechanism. The dependence on Src, the association between PPARγ and Src, and the negative effect of constitutive Src activation all support the central role of Src in PPARγ-dependent non-genomic signaling. Because the magnitude of enhancement in PT transport by TZDs was actually comparable to or even exceeded that by angiotensin II[25], a hormone with the greatest stimulatory effect in this segment, we concluded that the stimulation of renal PT transport through PPARγ-dependent non-genomic signaling may play a significant role in TZD-induced volume expansion[26].
In human subjects with type 2 diabetes, diuretics targeting the distal nephron were, at least partially, effective in preventing TZD-induced mild volume expansion[27]. On the other hand, massive volume expansion, the clinically more important side effect sometimes observed in human subjects taking TZDs, is known to be resistant to conventional diuretic monotherapy[8]. Although massive volume expansion in human subjects usually occurs after weeks of TZD use, it can also occur as rapidly as 4 d after TZD use[28]. These observations suggest the involvement of multiple mechanisms in TZD-induced edema formation. Consistent with the stimulation of PT transport, a reduction in lithium clearance was indeed reported in human subjected treated with TZDs[19]. Thus, the simultaneous stimulation of sodium transport in PTs and distal nephrons through distinct mechanisms may explain TZD-induced edema formation as shown in Figure 1.
Figure 1.
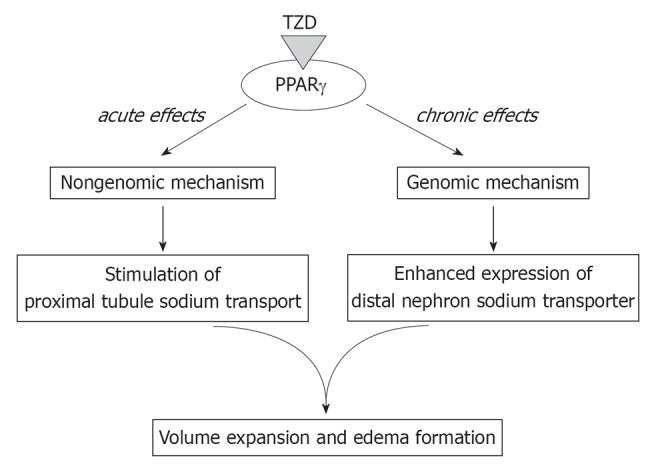
Mechanisms of thiazolidinedione-induced volume expansion. In addition to the genomic stimulation of the sodium transport system(s) in distal nephrons, thiazolidinediones (TZDs) may rapidly stimulate proximal tubule transport through a non-genomic mechanism. PPARγ: Peroxisome-proliferator-activated receptors γ.
On the other hand, the chronic administration of TZDs in rats was reported to enhance the renal abundance of α1 subunit of Na+/K+ ATPase and the apical Na+/H+ exchanger NHE3 in PTs[16]. TZDs were also found to enhance the expression of NHE3 through SGK1-mediated transcriptional activity of PPARγ in cultured human PT cells[29]. These changes in transporter abundance may further amplify the TZD-induced non-genomic stimulation of PT transport.
POTENTIAL SYNERGY BETWEEN TZD-INDUCED VOLUME EXPANSION AND RENAL INSULIN ACTIONS
Insulin action is initiated by activation of tyrosine kinase in cell-surface receptors. The insulin receptor transmits a series of trans-phosphorylation reactions to several docking proteins. Among these insulin receptor substrates (IRSs), IRS1 and IRS2 are the two major substrates. These tyrosine phosphorylated substrates bind other Src homology 2 proteins, resulting in the activation of PI3K and other kinases such as mitogen-activated protein kinase. These kinases mediate multiple actions of insulin, including the translocation of the glucose transporter GLUT4 into the plasma membrane. Defects somewhere in this insulin signaling are thought to be responsible for the occurrence of insulin resistance[30]. Insulin has been known to stimulate sodium absorption along various nephron segments[31]. For example, insulin is known to activate ENaC through the PI3K pathway[32]. In addition, physiological concentrations of insulin can stimulate PT transport through the PI3K pathway[33,34].
We have previously shown that the stimulatory effect of insulin on PT transport requires IRS2 but not IRS1[34]. Interestingly, insulin resistance is often associated with defects in the IRS1-dependent signaling, while IRS2-dependent signaling seems to be sometimes preserved in adipocytes and skeletal muscles[35-38]. These results suggest that the renal actions of insulin may be preserved at least in certain forms of insulin resistance. A recent study in rat models of diabetes and obesity also supports this view[39]. If the renal stimulatory effect of insulin is preserved in human insulin resistance, the renal actions of insulin may work synergistically with the renal actions of TZDs, resulting in the enhanced volume expansion. This scenario may explain the more frequent occurrence of edema and CHF with the combined used of TZDs and insulin. These considerations support the view that the careful observation is required, especially in the combined use of TZDs and insulin[5].
CONCLUSION
TZDs improve insulin resistance by activating PPARγ. However, the use of TZDs is associated with plasma volume expansion, which precludes their use in the setting of CHF. As summarized in Table 1, the mechanism of TZD-induced volume expansion may be multifactorial. Initially, ENaC in collecting ducts was thought to play a key role[12-14]. However, other studies did not support the central role of ENaC[15-18], suggesting that activation of collecting duct Na transporter(s) other than ENaC may play a role. On the other hand, we found that TZDs can activate PT transport through a PPARγ-dependent non-genomic mechanism[26]. Finally, this non-genomic stimulation of PT transport may be amplified by TZD-mediated transcriptional activation of PT transporters such as NHE3[16,29].
Table 1.
Summary of results about mechanism of thiazolidinedione-induced volume expansion
| Ref. | Cells/animals | Results |
| Guan et al[12] | Collecting duct-specific PPARγ KO mouse | TZD-induced volume expansion is dependent on PPARγ in collecting ducts. Increase in collecting duct Na transport and ENaCγ mRNA by TZDs |
| Zhang et al[13] | Collecting duct-specific PPARγ KO mouse | TZD-induced volume expansion is dependent on PPARγ in collecting ducts. Increase in collecting duct Na transport by TZDs |
| Hong et al[14] | Collecting duct cells | Increase in cell surface ENaC expression by TZDs |
| Nofziger et al[15] | Collecting duct cells | No effects of TZDs on ENaC activity |
| Song et al[16] | Rat | Increase in expression of Na/K ATPase and NHE3 but not of ENaC by TZDs |
| Vallon et al[17] | Collecting duct-specific ENaC KO mouse | ENaC-independent volume expansion by TZDs |
| Borsting et al[18] | M1 collecting duct cells | Suppression of ENaCγ promoter by TZDs |
| Endo et al[26] | Isolated PT from rabbit, rat, and human | Non-genomic stimulation of PT transport by TZDs |
| Saad et al[29] | Human PT cells | Sgk-1-dependent stimulation of NHE3 expression by TZDs |
TZDs: Thiazolidinediones; PPARγ: Peroxisome-proliferator-activated receptors γ; PT: Proximal tubule.
Footnotes
Peer reviewer: Pornanong Aramwit, PhD, Department of Pharmacy Practice, Faculty of Pharmaceutical Sciences, Chulalongkorn University, Payathai Road, Bangkok 10330, Thailand
S- Editor Wang JL L- Editor Hughes D E- Editor Zheng XM
References
- 1.Yki-Järvinen H. Thiazolidinediones. N Engl J Med. 2004;351:1106–1118. doi: 10.1056/NEJMra041001. [DOI] [PubMed] [Google Scholar]
- 2.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 3.Kubota N, Terauchi Y, Miki H, Tamemoto H, Yamauchi T, Komeda K, Satoh S, Nakano R, Ishii C, Sugiyama T, et al. PPAR gamma mediates high-fat diet-induced adipocyte hypertrophy and insulin resistance. Mol Cell. 1999;4:597–609. doi: 10.1016/s1097-2765(00)80210-5. [DOI] [PubMed] [Google Scholar]
- 4.Choi JH, Banks AS, Estall JL, Kajimura S, Boström P, Laznik D, Ruas JL, Chalmers MJ, Kamenecka TM, Blüher M, et al. Anti-diabetic drugs inhibit obesity-linked phosphorylation of PPARgamma by Cdk5. Nature. 2010;466:451–456. doi: 10.1038/nature09291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nesto RW, Bell D, Bonow RO, Fonseca V, Grundy SM, Horton ES, Le Winter M, Porte D, Semenkovich CF, Smith S, et al. Thiazolidinedione use, fluid retention, and congestive heart failure: a consensus statement from the American Heart Association and American Diabetes Association. October 7, 2003. Circulation. 2003;108:2941–2948. doi: 10.1161/01.CIR.0000103683.99399.7E. [DOI] [PubMed] [Google Scholar]
- 6.Ryan MJ, Didion SP, Mathur S, Faraci FM, Sigmund CD. PPAR(gamma) agonist rosiglitazone improves vascular function and lowers blood pressure in hypertensive transgenic mice. Hypertension. 2004;43:661–666. doi: 10.1161/01.HYP.0000116303.71408.c2. [DOI] [PubMed] [Google Scholar]
- 7.Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- 8.Mudaliar S, Chang AR, Henry RR. Thiazolidinediones, peripheral edema, and type 2 diabetes: incidence, pathophysiology, and clinical implications. Endocr Pract. 2003;9:406–416. doi: 10.4158/EP.9.5.406. [DOI] [PubMed] [Google Scholar]
- 9.Raskin P, Rendell M, Riddle MC, Dole JF, Freed MI, Rosenstock J. A randomized trial of rosiglitazone therapy in patients with inadequately controlled insulin-treated type 2 diabetes. Diabetes Care. 2001;24:1226–1232. doi: 10.2337/diacare.24.7.1226. [DOI] [PubMed] [Google Scholar]
- 10.Avandia (rosiglitazone maleate) [package insert] Philadelphia: GlaxoSmithKline Pharmaceuticals; 2000. [Google Scholar]
- 11.Actos (pioglitazone hydrochloride) [package insert] Lincolnshire: Takeda Pharmaceuticals America Inc.; 2000. [Google Scholar]
- 12.Guan Y, Hao C, Cha DR, Rao R, Lu W, Kohan DE, Magnuson MA, Redha R, Zhang Y, Breyer MD. Thiazolidinediones expand body fluid volume through PPARgamma stimulation of ENaC-mediated renal salt absorption. Nat Med. 2005;11:861–866. doi: 10.1038/nm1278. [DOI] [PubMed] [Google Scholar]
- 13.Zhang H, Zhang A, Kohan DE, Nelson RD, Gonzalez FJ, Yang T. Collecting duct-specific deletion of peroxisome proliferator-activated receptor gamma blocks thiazolidinedione-induced fluid retention. Proc Natl Acad Sci USA. 2005;102:9406–9411. doi: 10.1073/pnas.0501744102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong G, Lockhart A, Davis B, Rahmoune H, Baker S, Ye L, Thompson P, Shou Y, O'Shaughnessy K, Ronco P, et al. PPARgamma activation enhances cell surface ENaCalpha via up-regulation of SGK1 in human collecting duct cells. FASEB J. 2003;17:1966–1968. doi: 10.1096/fj.03-0181fje. [DOI] [PubMed] [Google Scholar]
- 15.Nofziger C, Chen L, Shane MA, Smith CD, Brown KK, Blazer-Yost BL. PPARgamma agonists do not directly enhance basal or insulin-stimulated Na(+) transport via the epithelial Na(+) channel. Pflugers Arch. 2005;451:445–453. doi: 10.1007/s00424-005-1477-4. [DOI] [PubMed] [Google Scholar]
- 16.Song J, Knepper MA, Hu X, Verbalis JG, Ecelbarger CA. Rosiglitazone activates renal sodium- and water-reabsorptive pathways and lowers blood pressure in normal rats. J Pharmacol Exp Ther. 2004;308:426–433. doi: 10.1124/jpet.103.058008. [DOI] [PubMed] [Google Scholar]
- 17.Vallon V, Hummler E, Rieg T, Pochynyuk O, Bugaj V, Schroth J, Dechenes G, Rossier B, Cunard R, Stockand J. Thiazolidinedione-induced fluid retention is independent of collecting duct alphaENaC activity. J Am Soc Nephrol. 2009;20:721–729. doi: 10.1681/ASN.2008040415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Borsting E, Cheng VP, Glass CK, Vallon V, Cunard R. Peroxisome proliferator-activated receptor-γ agonists repress epithelial sodium channel expression in the kidney. Am J Physiol Renal Physiol. 2012;302:F540–F551. doi: 10.1152/ajprenal.00306.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zanchi A, Chiolero A, Maillard M, Nussberger J, Brunner HR, Burnier M. Effects of the peroxisomal proliferator-activated receptor-gamma agonist pioglitazone on renal and hormonal responses to salt in healthy men. J Clin Endocrinol Metab. 2004;89:1140–1145. doi: 10.1210/jc.2003-031526. [DOI] [PubMed] [Google Scholar]
- 20.Muto S, Miyata Y, Imai M, Asano Y. Troglitazone stimulates basolateral rheogenic Na+/HCO3- cotransport activity in rabbit proximal straight tubules. Exp Nephrol. 2001;9:191–197. doi: 10.1159/000052611. [DOI] [PubMed] [Google Scholar]
- 21.Gonzalez-Campoy JM, Romero JC, Knox FG. Escape from the sodium-retaining effects of mineralocorticoids: role of ANF and intrarenal hormone systems. Kidney Int. 1989;35:767–777. doi: 10.1038/ki.1989.51. [DOI] [PubMed] [Google Scholar]
- 22.Kiley SC, Chevalier RL. Species differences in renal Src activity direct EGF receptor regulation in life or death response to EGF. Am J Physiol Renal Physiol. 2007;293:F895–F903. doi: 10.1152/ajprenal.00227.2007. [DOI] [PubMed] [Google Scholar]
- 23.Burgermeister E, Seger R. PPARgamma and MEK Interactions in Cancer. PPAR Res. 2008;2008:309469. doi: 10.1155/2008/309469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kousteni S, Bellido T, Plotkin LI, O'Brien CA, Bodenner DL, Han L, Han K, DiGregorio GB, Katzenellenbogen JA, Katzenellenbogen BS, et al. Nongenotropic, sex-nonspecific signaling through the estrogen or androgen receptors: dissociation from transcriptional activity. Cell. 2001;104:719–730. [PubMed] [Google Scholar]
- 25.Li Y, Yamada H, Kita Y, Kunimi M, Horita S, Suzuki M, Endo Y, Shimizu T, Seki G, Fujita T. Roles of ERK and cPLA2 in the angiotensin II-mediated biphasic regulation of Na+-HCO3(-) transport. J Am Soc Nephrol. 2008;19:252–259. doi: 10.1681/ASN.2007030289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Endo Y, Suzuki M, Yamada H, Horita S, Kunimi M, Yamazaki O, Shirai A, Nakamura M, Iso-O N, Li Y, et al. Thiazolidinediones enhance sodium-coupled bicarbonate absorption from renal proximal tubules via PPARγ-dependent nongenomic signaling. Cell Metab. 2011;13:550–561. doi: 10.1016/j.cmet.2011.02.015. [DOI] [PubMed] [Google Scholar]
- 27.Karalliedde J, Buckingham R, Starkie M, Lorand D, Stewart M, Viberti G. Effect of various diuretic treatments on rosiglitazone-induced fluid retention. J Am Soc Nephrol. 2006;17:3482–3490. doi: 10.1681/ASN.2006060606. [DOI] [PubMed] [Google Scholar]
- 28.Hirsch IB, Kelly J, Cooper S. Pulmonary edema associated with troglitazone therapy. Arch Intern Med. 1999;159:1811. doi: 10.1001/archinte.159.15.1811. [DOI] [PubMed] [Google Scholar]
- 29.Saad S, Agapiou DJ, Chen XM, Stevens V, Pollock CA. The role of Sgk-1 in the upregulation of transport proteins by PPAR-{gamma} agonists in human proximal tubule cells. Nephrol Dial Transplant. 2009;24:1130–1141. doi: 10.1093/ndt/gfn614. [DOI] [PubMed] [Google Scholar]
- 30.Pessin JE, Saltiel AR. Signaling pathways in insulin action: molecular targets of insulin resistance. J Clin Invest. 2000;106:165–169. doi: 10.1172/JCI10582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Horita S, Seki G, Yamada H, Suzuki M, Koike K, Fujita T. Insulin resistance, obesity, hypertension, and renal sodium transport. Int J Hypertens. 2011;2011:391762. doi: 10.4061/2011/391762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blazer-Yost BL, Esterman MA, Vlahos CJ. Insulin-stimulated trafficking of ENaC in renal cells requires PI 3-kinase activity. Am J Physiol Cell Physiol. 2003;284:C1645–C1653. doi: 10.1152/ajpcell.00372.2002. [DOI] [PubMed] [Google Scholar]
- 33.Baum M. Insulin stimulates volume absorption in the rabbit proximal convoluted tubule. J Clin Invest. 1987;79:1104–1109. doi: 10.1172/JCI112925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zheng Y, Yamada H, Sakamoto K, Horita S, Kunimi M, Endo Y, Li Y, Tobe K, Terauchi Y, Kadowaki T, et al. Roles of insulin receptor substrates in insulin-induced stimulation of renal proximal bicarbonate absorption. J Am Soc Nephrol. 2005;16:2288–2295. doi: 10.1681/ASN.2005020193. [DOI] [PubMed] [Google Scholar]
- 35.Goodyear LJ, Giorgino F, Sherman LA, Carey J, Smith RJ, Dohm GL. Insulin receptor phosphorylation, insulin receptor substrate-1 phosphorylation, and phosphatidylinositol 3-kinase activity are decreased in intact skeletal muscle strips from obese subjects. J Clin Invest. 1995;95:2195–2204. doi: 10.1172/JCI117909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Friedman JE, Ishizuka T, Shao J, Huston L, Highman T, Catalano P. Impaired glucose transport and insulin receptor tyrosine phosphorylation in skeletal muscle from obese women with gestational diabetes. Diabetes. 1999;48:1807–1814. doi: 10.2337/diabetes.48.9.1807. [DOI] [PubMed] [Google Scholar]
- 37.Rondinone CM, Wang LM, Lonnroth P, Wesslau C, Pierce JH, Smith U. Insulin receptor substrate (IRS) 1 is reduced and IRS-2 is the main docking protein for phosphatidylinositol 3-kinase in adipocytes from subjects with non-insulin-dependent diabetes mellitus. Proc Natl Acad Sci USA. 1997;94:4171–4175. doi: 10.1073/pnas.94.8.4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carvalho E, Jansson PA, Axelsen M, Eriksson JW, Huang X, Groop L, Rondinone C, Sjöström L, Smith U. Low cellular IRS 1 gene and protein expression predict insulin resistance and NIDDM. FASEB J. 1999;13:2173–2178. doi: 10.1096/fasebj.13.15.2173. [DOI] [PubMed] [Google Scholar]
- 39.Mima A, Ohshiro Y, Kitada M, Matsumoto M, Geraldes P, Li C, Li Q, White GS, Cahill C, Rask-Madsen C, et al. Glomerular-specific protein kinase C-β-induced insulin receptor substrate-1 dysfunction and insulin resistance in rat models of diabetes and obesity. Kidney Int. 2011;79:883–896. doi: 10.1038/ki.2010.526. [DOI] [PMC free article] [PubMed] [Google Scholar]