

Abstract
The anemia of chronic kidney disease and hemodialysis is characterized by chronic inflammation and release of cytokines, resulting in the upregulation of the iron hormone hepcidin, also increased by iron therapy and reduced glomerular filtration, with consequent reduction in iron absorption, recycling, and availability to the erythron. This response proves advantageous in the short-term to restrain iron availability to pathogens, but ultimately leads to severe anemia, and impairs the response to erythropoietin (Epo) and iron. Homozygosity for the common C282Y and H63D HFE polymorphisms influence iron metabolism by hampering hepcidin release by hepatocytes in response to increased iron stores, thereby resulting in inadequate inhibition of the activity of Ferroportin-1, inappropriately high iron absorption and recycling, and iron overload. However, in hemodialysis patients, carriage of HFE mutations may confer an adaptive benefit by decreasing hepcidin release in response to iron infusion and inflammation, thereby improving iron availability to erythropoiesis, anemia control, the response to Epo, and possibly survival. Therefore, anti-hepcidin therapies may improve anemia management in hemodialysis. However, HFE mutations directly favor hemoglobinization independently of hepcidin, and reduce macrophages activation in response to inflammation, whereas hepcidin might also play a beneficial anti-inflammatory and anti-microbic action during sepsis, so that direct inhibition of HFE-mediated regulation of iron metabolism may represent a valuable alternative therapeutic target. Genetic studies may offer a valuable tool to test these hypotheses and guide the research of new therapies.
Keywords: Chronic kidney disease, Hemodialysis, Iron, HFE protein, Iron overload
INTRODUCTION
Anemia represents a common clinical problem in subjects with chronic kidney disease (CKD), and is associated with increased morbidity and mortality related to cardiovascular events, especially in patients undergoing chronic hemodialysis (CHD). The cause is multifactorial[1], but the deficit of the hypoxia-sensing hormone erythropoietin (Epo) that promotes the differentiation of erythroid progenitors into reticulocytes and red blood cells (RBCs) plays a major role. However, the persistence of anemia despite Epo supplementation indicates a concomitant hyporesponsiveness of bone marrow, partly related to inadequate supply of substrates for hemoglobin (Hb) synthesis, including folates, cyanocobalamin, and iron. Furthermore, RBCs life-span is shortened in CKD, and there is an increase tendency of bleeding related to uremia itself. Other causes of anemia include chronic blood loss from gastroenteric system and blood trapping in dialyzers, dietary restrictions, loss of taste for iron-rich foods, secondary hyperparathyroidism determining bone marrow fibrosis, and accumulation of inhibitors of erythropoiesis related to uremia.
Another major contributor of the anemia of CKD is the chronic inflammatory state. Chronic inflammation and cytokines can worsen anemia by shortening erythrocyte life span, induce apoptosis of erythroid precursors, and directly inhibit erythrocyte progenitor proliferation. Inflammation and cytokines are independent predictors of Epo hyporesponsiveness[2], and reduce the availability of iron for hematopoiesis.
Routine monitoring of body iron stores and iron supplementation are therefore essential components of the management of patients with CKD, in particular of those receiving CHD. Iron is an essential nutrient, and besides its role in Hb synthesis, it plays a crucial role in vital biochemical activities, as component of enzymes and other molecular complexes. It nevertheless has to be compartmentalized and maintained at a fixed level to avoid any toxic effects, largely based on its ability to catalyze the generation of reactive oxygen intermediates[3,4].
IRON METABOLISM AND HFE MUTATIONS
The human body contains approximately 3-5 g of iron, the main part of which is employed within Hb in circulating RBCs. Approximately 20%-30% of body iron is stored in hepatocytes and in macrophages, to a large extent within polymers of ferritin. A healthy individual absorbs daily 1-2 mg of iron from the diet, which is utilized to compensate non-specific iron losses by desquamation of enterocytes and epidermis and, in childbearing age women, by period. Erythropoiesis requires approximately 30 mg of iron per day, mainly provided by the recycling of iron via macrophages, which ingest senescent RBCs and release iron, which binds to circulating transferrin (TF).
Iron uptake in the duodenum/jejunum is mediated by specific set of transport proteins and accessory enzymes that change the oxidation state of iron to facilitate the transport process; the most important for systemic regulation is named ferroportin-1 (Fp-1), which allows the transport across basolateral membrane of enterocytes, macrophages and hepatocytes. It works as iron exporter in association with the plasma ferroxidase ceruloplasmin (Cp), even if enterocytes depend heavily on the expression of an analogous transmembrane protein called hephaestin. Ferric iron binds to plasmatic apo-TF to form ferric iron-TF complex, which is the major type of iron present in blood. The TF complex facilitates the transport of iron to cells that express TF receptors (TFR), including erythroid progenitors, and limits the ability of iron to generate toxic radicals. Iron uptake in the cells occurs primarily by the endocytic pathway, which involves the interaction between TF and TFR. Not all absorbed iron is utilized in metabolic processes, but it is partly stored as reserve, both for use when iron levels are low, and to prevent toxic effects of free iron in the cell, and the major part of it is bound to ferritin. Under iron overload conditions, ferritin levels increase dramatically, particularly in the liver.
Systemic iron homeostasis is achieved by modulation of the amount of iron absorbed. Intestinal iron absorption is regulated in response to iron need and availability and erythropoiesis activity, (“storage” and “erythroid” regulators), whereas “inflammatory” regulators communicate signals in response to infection or inflammation. The amount of body storage modulates iron uptake: it is well established that in iron-deficient conditions, iron absorption is stimulated by two- to three-fold compared to basal conditions, which are restored when iron storage are reconstituted. The erythropoietic regulation participates when iron demand for Hb synthesis increases, independently of body iron stores.
Hepcidin, a small antimicrobial peptide synthesized by the liver[5], is the principal effector of the modulation of iron metabolism, via its ability to bind Fp-1 on cellular surface blocking its iron transport activity, and to increase Fp-1 degradation[6]. In enterocytes, Fp-1 internalization on the basolateral surface causes the retention of absorbed iron with subsequent loss by desquamation, while the same process in macrophages causes the failure to release iron[7]. The final effect is the reduction of plasma iron availability. Importantly, hepcidin is upregulated by both increased iron stores and inflammation. On the other hand, hepcidin secretion is reduced in response to signals that cause an increase in iron release from cells, such as iron deprivation, and stimulus to erythropoiesis. Thus, hepcidin represents a common effector of the homeostatic regulation of intercellular iron fluxes in response to the iron stores, erythroid, and inflammatory regulators.
The transcription and secretion of hepcidin is regulated by a mechanism of body iron sensing depending on a complex of interacting proteins, including the hereditary hemochromatosis (HH) protein called HFE, TFR-2, hemojuvelin (HJV), bone morphogenetic protein 6 (BMP6), matriptase-2 (TMPRSS6) and TF (Figure 1). Mutations in HFE, TFR-2, HJV and the hepcidin gene (HAMP) are responsible for HH, a common iron overload disorder characterized by a deficit of hepcidin release or activity[7,8]. HFE mutations represent the most frequent cause of HH in Caucasian adults[9], and the most common is a single nucleotide substitution that causes the substitution of a cysteine with a tyrosine at position 282 (C282Y), which hampers HFE expression on the cell surface. Homozygosity for the C282Y mutation is observed very frequently in Northern Europe (frequency 1/300-400), whereas the prevalence decreases towards Southern Europe. A second and most frequent mutation is a substitution at position 63 of a histidine with an aspartate (H63D). This is a very common polymorphism in the general population, as 25%-30% of the population carries the H63D variant, but it has only a minor effect on HFE function. The penetrance of iron overload depends on age, gender, environmental factors, and on the role of the so-called modifier genes[10], e.g., the β-thalassemic trait that influences eryhropoiesis[11,12]. Variants of one among the various regulatory molecules involved in iron homeostasis can bring to iron overload (BMP6, HJV, SMAD4) or severe iron deficiency (matriptase-2 or TMPRSS6) by interfering with hepcidin secretion. In particular, germline mutations in TMPRSS6, which encodes the type II transmembrane serine protease matriptase-2, are associated to a rare form of iron deficiency anemia refractory to oral iron therapy (iron refractory iron deficiency anemia, IRIDA). The key features of this phenotype are congenital hypochromic, microcytic anemia, low mean corpuscular erythrocyte volume, low TF saturation (TS), abnormal iron absorption characterized by no hematological improvement following treatment with oral iron, abnormal iron utilization consisting of an incomplete response to parenteral iron and inappropriately high hepcidin levels[13]. Moreover, recent genome-wide association studies show that very common polymorphisms in the TMPRSS6 gene represent, together with HFE mutations, a major determinant of the variation in iron status in the general population. The strongest TMPRSS6 association was observed with rs855791, a nonsynonymous coding SNP (resulting in an A736V substitution) in exon 17. In particular, the 736V allele was associated with lower serum iron concentration, lower TS, decreased Hb and reduced erythrocyte mean cell volume[14].
Figure 1.

Role of the proteins and mediators involved in iron homeostasis and impact of hereditary hemochromatosis protein mutations in chronic kidney disease. A: Hereditary hemochromatosis protein (HFE) wild-type, healthy controls; B: HFE wild-type, end-stage renal disease (ESRD); C: HFE mutated, ESRD. Bone morphogenetic protein 6 (BMP-6) binds to the co-receptor membrane hemojuvelin (m-HJV) and to the bone morphogenetic protein receptor (BMP-R) on the membrane of hepatocytes. This initiates phosphorylation of small mother against decapentaplegic (SMAD) proteins and the assembly of heteromeric complexes. After nuclear translocation, the heteromeric SMAD complexes stimulate transcription of the HAMP gene for hepcidin. Another pathway responsible for hepcidin expression involves interleukine (IL)-6/gp130 signaling via signal transducer and activator of transcription 3 (STAT3) nuclear translocation. Negative regulation of the BMP-HJV-hepcidin pathway is achieved through the proteolytic processing of membrane-HJV by matriptase-2 (TMPRSS6). Additional negative regulation of hepcidin transcription is provided by soluble-HJV, which acts as an antagonist of the BMP pathway by competing with m-HJV for BMP-6 binding. The hepcidin regulatory signaling pathway is also regulated by HFE, which interacts either with transferrin receptor 1 (TFR1) or transferrin receptor 1 (TFR2) according to serum transferrin saturation: in the presence of a high transferrin saturation HFE dissociates from TFR1 and binds TFR2 forming an iron sensing complex influencing hepcidin expression via SMAD/ERK signaling. Hepcidin secreted by the liver binds to the extracellular region of ferroportin 1 (Fp-1) on the basolateral membrane of duodenal enterocytes causing Fp-1 internalization, ubiquitination and degradation and consequently impaired intestinal iron absorption. EPO: Erythropoietin; Epo-R: Erythropoietin receptor; s-HJV: Serum hemojuvelin; TSAT: Transferrin saturation.
IRON METABOLISM AND ANEMIA IN CKD
CKD is associated with typical alterations in iron metabolism parameters, i.e., reduced TS and hyperferritinemia (Figure 1). Decreased TS reduces iron availability for erythropoiesis, and is supposed to be related to chronic inflammation, which decreases serum iron by inducing hepcidin, and/or blood losses[15,16]. Hyperferritinemia is also frequently observed and, although in the most severe cases it may reflect excessive iron administration, in the majority of cases has also been ascribed to the state of malnutrition and chronic inflammation, which is associated with a greater risk of cardiovascular disease and with a worse outcome[17]. Both inflammation and oxidative stress induce iron retention in monocytes/macrophages, and the transcription of ferritin[18,19]. However, recent data highlight that ferritin levels reflect also body iron stores in CKD patients[20,21]. Indeed, in this clinical setting, three types of situation related to iron-metabolism are observed: (1) Absolute iron deficiency, due to decreased total body iron stores. It is associated with serum ferritin levels < 100 ng ⁄mL, TS < 20%, and relatively low serum hepcidin. Due to very low iron stores, serum hepcidin is relatively low. This situation is common in CHD, due to low-grade but frequent blood losses (typically 1-3 g of iron/year); (2) Functional iron deficiency, associated with serum ferritin levels higher than 100 ng⁄mL. Iron stores are normal or even increased, but the Epo-stimulated bone marrow needs more iron from TF than the iron output from tissue stores, resulting in Epo resistance in 10%-20% of cases. Hepcidin can aggravate functional iron deficiency by decreasing the release of stored and macrophage iron and intestinal iron absorption, through Fp-1 downregulation; and (3) the most severe form, historically termed “reticuloendothelial blockage”, which usually occurs in the setting of acute or chronic inflammation/infection. It can be considered as an extreme form of functional iron deficiency and is associated with increased C reactive protein levels, TS < 20%, and normal to very high levels of ferritin. Iron stores are locked, partly because of very high hepcidin levels, and there is no release of iron to TF. Resistance to Epo therapy easily develops, especially if iron administration is limited by adherence to the “official” opinion-based upper cutoff. Thus, the pattern of anemia, hyposideremia and decreased TS, Epo resistance, and high ferritin is frequently observed in CHD patients.
Noteworthy, Epo administration reduces hepcidin levels and increases intestinal iron absorption in CHD patients, in order to facilitate iron delivery to the bone marrow when is needed for accelerated RBCs production, suggesting also that the beneficial effect of Epo may be partly mediated by normalization of hepcidin levels and of iron delivery. Indeed, Epo increases the need for iron[22], so that bone marrow requests strip iron off the circulating TF faster than TF can replenish it, resulting in a relative deficit of iron[23]. Evidence now proves that adequate iron availability increases erythropoiesis and reduces Epo requirements[24]. Thus, interpretation of iron status tests should incorporate consideration of the Hb level and Epo dose.
Accordingly, KDOQI guidelines suggest that iron supplementation should be administered during Epo treatment to maintain serum ferritin > 200 ng/mL and TS > 20%, or CHr > 29 pg/cell in CHD and serum ferritin > 100 ng/mL and TS > 20% in CKD or in patients in peritoneal dialysis. The upper limit of serum ferritin besides which there is no recommendation to routinely administer iron was set as 500 ng/mL. When ferritin level is greater than 500 ng/mL, decisions regarding iron administration should weigh Epo responsiveness, Hb and TS level, and the clinical status. The preferred route of iron administration in CHD patients is by intravenous (iv) infusion, since iron absorption from the gastrointestinal tract may be impaired[25-27], likely because of high hepcidin levels. On the other hand, in advanced CKD or peritoneal dialysis patients, iron can generally be administered orally.
IRON TOXICITY IN CKD: THE CARDIOVASCULAR AND INFECTIVE RISKS
Large doses of iron may exceed storage capacity leading to the release of unbound iron into the plasma, which can produce reactive oxygen species[28]. Hyperferritinemia exceeding 800 ng/mL has indeed been associated with imminent death risk, and in particular cardiovascular death in CHD patients[17,29], even after adjustment for the confounding variables[17,30]. In addition to traditional and population specific risk factors such as anemia, hyperhomocysteinemia, hyperphosphatemia and inflammation, iron has been implicated in the pathogenesis of the accelerated atherosclerosis in CHD[31], possibly affecting LDL oxidation and endothelial dysfunction[32-34] via increased oxidative stress[28]. Recent data confirm that in CHD patients hyperferritinemia reflects a relative increase in iron availability and a decrease in iron-specific anti-oxidant activity, is favored by HFE mutations, and represents a risk factor for advanced cardiovascular damage, as evaluated by the presence of plaques, both at carotid and femoral arteries and by the intima media thickness[20]. Furthermore, hepcidin and tumor necrosis factor (TNF) α levels have been correlated with vascular stiffness, another reliable predictor of cardiovascular events in CHD[35]. The underlying mechanism may involve iron trapping into endothelial cells, plaque macrophages, and vascular smooth muscle cells, with activation of the atherogenic process and progression of the plaque lesion. Indeed, a significantly higher iron content has been detected in atherosclerotic plaques than in healthy vascular tissue[36]. Iron mediated vascular damage in CHD seems to be associated with hepcidin upregulation, caused by chronic flogistic state, diminished hemofiltration by damaged kidneys, and also by chronic iron supplementation. This causes an accumulation of iron in macrophages, favoring oxidative stress and the induction of the release of pro-atherogenic cytokines, in particular macrophage chemoattractant protein-1 (MCP-1)[37].
Moreover, iron overload has been associated with an increased incidence and severity of infections, related to the growth-promoting effect of iron on microbial pathogens and also to inhibition of phagocytosis[38], and of the anti-microbial molecule lactoferrin[39]. Infection risk in CKD patients depends not only on iron, but also on the severity of anemia[40], transfusions, and secondary splenic dysfunction. In patients with end stage renal disease, there are additional risk factors, such as the presence of catheters for dialysis (temporary or permanent), malnutrition related to dialytic losses of nutrients, and exposure to extracorporeal circulation. Although human data are not conclusive, it is still recommended that, as a precaution, i.v. iron is stopped in patients with ongoing bacteraemia, as in vitro and in vivo studies demonstrated that administering iron during active infection may contribute to bacterial growth[41,42]. Nevertheless, correction of anemia is effective in reducing oxidative stress and, consequently, cardiovascular risk, decreasing morbidity and mortality and also producing regression of left ventricular hypertrophy in patients with CKD[43], so that a carefully balanced management of iron supplementation is needed in CKD/CHD patients.
HFE MUTATIONS IN CKD
Consistently with previous data[44], our group has shown that the two common C282Y and H63D mutations, present in about one third of subjects, are associated with increased baseline iron stores, a lower requirement of Epo and a trend to a lower requirement of iron supplementation in Italian CHD patients[45], suggesting that the alterations in iron metabolism associated with the presence of these genetic variants include not only increased iron stores, but also iron handling by macrophages after infusion, and iron availability to the erythron. Importantly, iron stores were not lower, and the requirement of iron and Epo were not higher in patients carrying only the minor HFE H63D mutation compared to those positive for the more severe variant C282Y or homozygous for the H63D mutation. It is possible that chronic inflammation and blood losses typical of CHD provide enough environmental pressure to magnify subtle alterations in cellular iron handling also in carriers of the milder and more common H63D mutation, which thus reach clinical significance, unlike to what it is observed in general population[46]. Therefore, HFE mutations possibly attenuate the effects of inflammation on iron metabolism, including reduced iron absorption, iron sequestration in macrophages, and erythroblast resistance to Epo, and protect against iron-related damage by favoring the delivery of intravenously administered iron to the erythron (Figures 1, 2 and 3).
Figure 2.
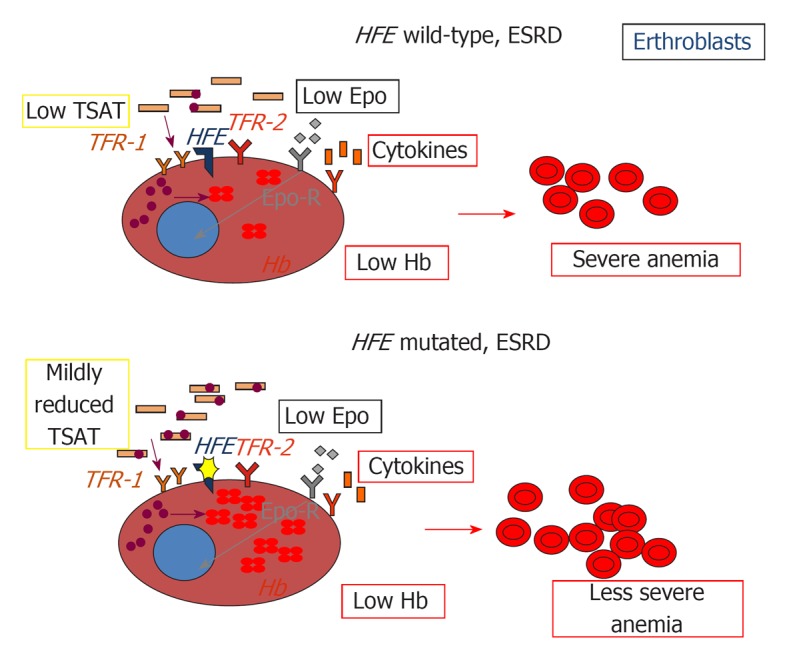
Impact of HFE mutations on erythropoiesis in chronic kidney disease. EPO: Erythropoietin, Epo-R: Erythropoietin receptor; TFR1: Transferrin receptor 1; TFR2: Transferrin receptor 2; HFE: Hereditary hemochromatosis protein; TSAT: Transferrin saturation; Hb: Hemoglobin.
Figure 3.
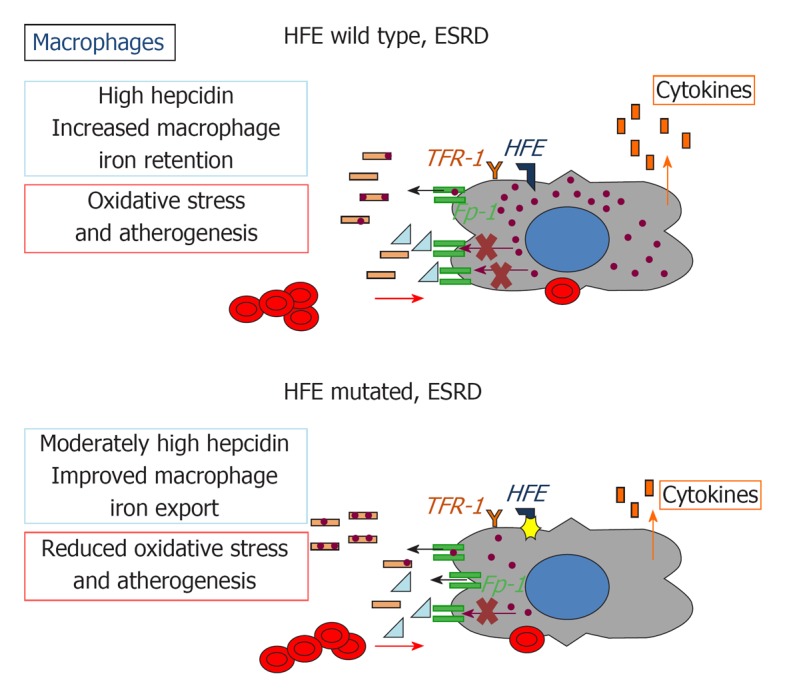
Impact of HFE mutations on macrophage iron recycling, oxidative stress, and atherogenesis in chronic kidney disease. Fp-1: Ferroportin 1; TFR1: Transferrin receptor 1; HFE: Hereditary hemochromatosis protein.
Our data even highlighted that the presence of HFE mutations was possibly associated with a reduced mortality. Indeed, in patients negative for HFE mutations, we observed a higher mortality due to sepsis, previously associated with a higher iron dosage[47,48], and due to cardiovascular disease, possibly linked to hypertension and thromboembolic events related to Epo[49,50] and oxidative stress related to iron[20]. Obviously, these data need confirmation in larger prospective studies and independent cohorts.
In the hypothesis that altered regulation of hepcidin release by iron stores might explain the apparent protective role of the HFE mutations on cardiovascular complications and on the response to Epo[51], we investigated whether the effect of HFE gene mutations on hepcidin-25, the active form of the hormone, could be involved in the pathophysiology of the alterations of iron metabolism and anemia. We confirmed increased levels of hepcidin-25 in CHD patients compared to healthy controls[52,53], and a preserved regulation of hepcidin-25 by iron stores and inflammation in this setting, as demonstrated by the very strong correlation with serum ferritin and CRP levels[52,54]. Interestingly, we also showed a negative correlation between hepcidin-25 and serum iron, and we found that in a subgroup of patients with stable disease, selected to avoid the confounding effect of the frequent presence of acute inflammation, blood losses, cancer, and recent variation in the dosage of therapy, hepcidin-25 negatively correlated with Hb levels. Since anemia and hyposideremia should rather decrease hepcidin levels, these findings suggest that hepcidin-25 plays a causal role in determining anemia by reducing iron availability to the erythron, thus implying that in CHD excessive iron administration may paradoxically hamper iron utilization for erythropoiesis by increasing hepcidin, and that the effect of inflammation on altered iron metabolism and erythropoiesis may be partly mediated by increased hepcidin levels. As a consequence, pharmacological downregulation of hepcidin may be beneficial for anemia control in CHD[55].
However, the protective effect of HFE mutations might not be limited to enhanced erythropoiesis due to decreased hepcidin levels. Very recently, it has indeed been demonstrated that HFE is expressed in erythroblasts and plays a role in the regulation of erythroid differentiation, and that HFE deficiency is associated with increased erythropoiesis partly due to enhanced iron absorption and delivery to the erythron due to decreased hepcidin levels and increased TS, and partly due to a direct effect of HFE on the modulation of iron uptake in erythroid cells[56]. Moreover, contemporary work has shown that TFR-2 is associated to the erythropoietin receptor (EpoR) in the EpoR complex in erythroblasts, and is required for efficient erythroid differentiation and erythropoiesis[57]. These new exciting findings indicate that the TFR-2/HFE complex is directly involved in the regulation of erythropoiesis independently of hepcidin levels, and thus that genetic variations, or pharmacological modulation, of HFE and TFR-2 may influence RBCs production and the development of anemia in conditions characterized by reduced iron availability, such as CHD.
By decreasing hepcidin levels and cytokine release, HFE mutations may also play a protective role in atherogenesis. In patients with metabolic alterations[58], ferritin levels were independently associated with common carotid arteries intima-media thickness, reflecting early vascular damage and a strong predictor of cardiovascular events, and with carotid plaques, but only in patients negative for HFE mutations. This evidence supports the controversial hypothesis that increased hepcidin favors atherosclerosis by inducing iron accumulation in arterial wall macrophages[59], thus promoting transformation into foam cells in the presence of an atherogenic environment[59-61]. Accordingly, HFE mutations would be protective by decreasing hepcidin release and favoring iron egress from macrophages. Thus, it seems that iron mediated vascular damage involves hepcidin upregulation, a mechanism that could be implicated also in the enhanced atherosclerotic process in CHD patients, in whom hepcidin levels are much higher than in general population. In vitro studies on patients with metabolic syndrome confirmed the enhanced release of MCP-1 in monocytes treated with iron[37], but only in the absence of HFE mutations that preclude intracellular iron accumulation[62]. Consistently, patients homozygous for the C282Y HFE mutation had lower MCP-1 levels than those affected by secondary iron overload and healthy controls[63], whereas serum MCP-1 levels were significantly correlated with hepcidin-25, and were an independent predictor of the presence of carotid plaques, indicating an advanced atherosclerotic process, in patients with metabolic alterations[37].
ANTI-HEPCIDIN DRUGS: THE FUTURE OF ANEMIA MANAGEMENT IN CHD
These results may have important clinical implications, as targeting the HFE/hepcidin/Fp-1 axis by pharmacological treatment may further improve the long-term outcomes of patients with CKD and in particular undergoing CHD, by reducing the amount of iron and Epo supplementation needed, improving iron utilization for erythropoiesis[64], and possibly ameliorating iron-induced inflammation. Several drugs are under development for the modulation of the release and activity of hepcidin (Table 1). The approaches under study include neutralizing anti-hepcidin antibodies[65], inhibitors of BMP type I receptors[66], soluble HJV, which acts as decoy receptor inhibiting BMP signaling[55], and Epo itself, which can directly or indirectly inhibit hepcidin release[67].
Table 1.
A list of anti-hepcidin therapies evaluated in experimental models or under pre-clinical development
| Drug | Ref. | Mechanism of action | Experimental model | Effects |
| Anti-hepcidin Abs | [65] | Direct inhibition of hepcidin | Mouse model of ACD | Increased sensitivity to Epo |
| Anti BMP6 Abs | [68] | Inhibition of BMP-6 signaling | Mice | Decrease hepcidin Increase serum iron and transferrin saturation |
| Dorsomorphin | [66] | Inhibition of BMP type 1 receptor kinase function | Cultured mouse pulmonary artery smooth muscle cells Hepatocytes Zebrafish Mice | Decrease hepcidin Increase serum iron |
| Soluble HJV | [55] | Decoy receptor for BMP-6 (competition of membrane HJV and soluble HJV for BMP binding) Inhibition of BMP signaling | Hepatoma-derived HepG2 cells Mice | Decrease hepcidin Increased ferroportin expression Increase serum iron and transferrin saturation Mobilize reticuloendothelial cell iron stores |
| Heparins | [69] | BMP-6 sequestration and consequent inhibition of BMP-6 signaling | Hepatoma-derived HepG2 cells Mice | Decrease hepcidin Increase serum iron Reduce spleen iron concentration |
| Epo | [67] | Increase HIF signaling, direct effect? | Mouse hepatocytes Hepatoma-derived HepG2 cells | Decrease hepcidin |
| Spiegelmers | [71] | Bind and neutralise hepcidin biological activity | Mouse macrophages Mice Cynomolgus monkeys | Inhibition of hepcidin-induced ferroportin degradation Inhibition of hepcidin-induced hypoferremia |
| Anticalins | [72] | Encapsulate and neutralise hepcidin biological activity | Cell system Mice Monkeys | Inhibition of hepcidin-induced ferroportin degradation Inhibition of hepcidin-induced hypoferremia |
Abs: Antibodies; ACD: Anemia of chronic diseases; HJV: Hemojuvelin.
In a mouse model of anemia of chronic disease (ACD), neutralizing antibodies (Abs) directed against hepcidin restored the reticulocyte response and normal Hb levels in combination with Epo, whereas Epo alone, and Epo plus iv iron, were ineffective. Furthermore, over-expression of hepcidin in mice was sufficient to hamper the erythropoietic response to Epo. These preliminary preclinical data suggest that anti-hepcidin therapies could restore susceptibility in patients with Epo resistance or reduce the dose of Epo and iron required to achieve anemia control, thereby minimizing side effects.
A different approach to control hepcidin activity is represented by the modulation of hepcidin expression by targeting bone morphogenetic proteins (BMPs) activity. Administration of soluble forms of HJV (sHJV) inhibits BMP signaling and hepcidin expression, likely by binding to BMP6 and preventing the interaction with BMP receptors[68], and could be exploited as a treatment for ACD[55]. It was also shown that anti-BMP6 Abs were similarly effective[68]. A promising development was provided by the recognition of the inhibitory activity of heparins on hepcidin release. Heparan sulfate proteoglycans (HSPGs) are expressed on the surface of various cell types and in the extracellular matrix, and modulate BMP osteogenic activity by binding BMPs, BMP antagonist, and BMP receptors. Heparin, a proteoglycan analog to HSPGs, strongly down-modulated hepcidin release in vitro and in vivo, resulting in increased serum iron and decreased splenic iron levels[69]. The effect seemed to depend on the sequestration of BMP6, thereby preventing the interaction with HJV and the induction of hepcidin transcription. Importantly, heparin not only hampered BMP6 dependent induction of SMAD phosphorylation and hepcidin transcription, but could also prevent the IL-6 and STAT3 dependent activation of hepcidin promoter, suggesting that it may overcome the effect of inflammation on hepcidin release also in patients with ACD or CKD. Due to the dose-dependent anticoagulant effects and the need for therapeutic monitoring, it is unlikely that unfractioned heparin could ever be implemented as a therapy for the anemia of chronic disease or CHD anemia in patients without thrombophilic conditions. However, it is conceivable that heparin could be modified to improve the anti-hepcidin while decreasing anticoagulant activity[70]. Another possibility consists in the downstream inhibition of BMP receptors. Dorsomorphin has been identified through a large scale in vivo screening approach as a selective inhibitor of BMP signaling through the antagonism of type I receptors activity[66]. Interestingly, this small molecule compound was able to profoundly reduce basal hepcidin levels and increase serum iron in mice. However, due to the many roles of BMP signaling in the regulation of cell differentiation and homeostasis, it is likely that dorsomorphin, that for example effectively inhibits osteogenesis in vitro, would have unacceptable side effects in humans.
It should not also be forgotten that large doses of Epo could reduce hepcidin levels, even if the effect seems to require the effective induction of erythropoietic activity in vivo, and is therefore subjected to clinical resistance, and to side effects. In conclusion, the association of anti-hepcidin Abs, or possibly anti-BMP6 Abs or anti inflammatory/anti-hepcidin heparins, to Epo represents the most promising approach for the treatment of the anemia of CHD and chronic diseases.
Novel approaches include also the exploitation of small-engineered molecules that directly inhibit hepcidin. These include the spiegelmer NOX-H94, that is a RNA molecule with secondary structures complementary to that of hepcidin, demonstrating inhibitory activity in vitro and in vivo in mouse models with a consequent increase in iron levels[71], and PRS-080, a pegylated form of an anticalin, which binds and inactivates hepcidin[72].
In recent years, it became possible to act also on the efficiency of dialysis. The use of new dialysis techniques and more efficient filters, allows the removal of larger quantities and types of toxins. Among all, the haemodiafiltration tecnique (HDF) is used to combine the advantages of the removal of low molecular weight solutes by diffusive transport (determined by the concentration gradient between dialysate and plasma) with the advantages of removing substances to medium-high molecular weight by convective transport. Many studies have shown that, compared to bicarbonate dialysis, HDF allows greater removal of solutes with low-medium molecular weight, including some fraction of complement (C3a, C5a) and proinflammatory cytokines (e.g., TNF-α), substances related with an inflammatory basic status, and possibly also of hepcidin[73,74], even if available data are not conclusive[75].
A NOTE OF CAUTION
As HFE has been demonstrated to be directly involved in the maturation of erythroid progenitors[56], inflammation and atherogenesis, it should be noted that the advantageous effect of the presence of HFE mutations on erythropoiesis in CHD patients may not be completely dependent on the only relatively decreased hepcidin levels in patients carrying HFE mutations[51]. Therefore, even if the degree of hepcidin activity suppression achievable in vivo would likely play a major role in determining iron availability to erythropoiesis, it is possible that anti-hepcidin therapies would result in a lesser improvement in anemia control than that conferred by the presence of protective HFE genotypes. These remarks are reinforced by the recent demonstration that hepcidin, by modulating iron metabolism and STAT3 signaling in leukocytes, may also exert an anti-inflammatory activity in models of acute shock, possibly resulting in an improved survival[76,77]. Therefore, it is possible that while it negatively affects anemia control, at the same time hepcidin improves mortality during severe infections, which are commonly observed in CHD patients. The picture is further complicated by the possibility that increased cytokine release related to low basal hepcidin levels may offer protection against the development of infections[76].
Anyway, the finding that the serine protease TMPRSS6 regulates hepcidin levels has highlighted novel potential applications of hepcidin-targeted therapy for the treatment of iron disorders. For example, inhibition of the protease function of TMPRSS6 might be beneficial for disorders in which hepcidin is inappropriately low such as HH and iron loading anemias. Similarly, treatment with agonists or with the endogenous substrate of TMPRSS6 might be employed in the anemia of chronic disease, such as CKD, in which hepcidin levels are high[13]. However, because of the anti-inflammatory action of hepcidin[76,77], a careful approach is needed when considering to intervene on the hepcidin pathway. Meanwhile, the evaluation of the effect of the common TMPRSS6 polymorphisms, which influence hepcidin release without affecting HFE function, on the outcome of CHD patients may provide some answers to these questions and a possible model for the clinical effect of anti-hepcidin therapies.
A better comprehension of the mechanisms linking HFE mutations with altered protein function both related to iron sensing/hepcidin induction and to the direct control of cellular iron intake would also be needed to design new therapeutic approaches aimed specifically at directly inhibiting this molecule. Furthermore, the possible development of side effects, such as the promotion of carcinogenesis related to chronic exposure to high TS levels, which would result from the inhibition of hepcidin, should be weighted against the potential benefits[78-80].
CONCLUSION
The anemia of CKD and CHD is characterized by chronic inflammation and release of cytokines, resulting in increased hepcidin with consequent reduction in iron absorption, recycling, and availability to the erythron. This response proves advantageous in the short-term to restrain iron availability to pathogens during infections, but in the case of chronicization, it leads to severe anemia, and may impair the response to Epo and iron therapy. Furthermore, as hepcidin is freely filtered by the glomerulus, CKD itself is a contributing factor to increased hepcidin levels.
The common polymorphisms C282Y e H63D in the HFE gene of hemochromatosis represent a major determinant of iron metabolism: these genetic factors act by hampering hepcidin induction in hepatocytes in response to increased iron stores, thereby resulting in reduced serum hepcidin-25 and inadequate inhibition of the activity of the iron exporter Fp-1, inappropriately high duodenal iron absorption, iron recycling from erythrophagocytosis, and increased serum iron. However, in CHD patients HFE mutations may confer an adaptive benefit by decreasing hepcidin release in response to iron and inflammation, thereby improving iron availability to erythropoiesis, anemia control, and the response to Epo and iron. The subtle effect of the C282Y and H63D mutations is likely magnified in CHD patients by the environmental pressure determined by chronic inflammation, and exposure to high amounts of iron and Epo. Although data must be confirmed in large prospective studies, this favorable shift in iron metabolism balance possibly results in reduced mortality, in particular because of cardiovascular and infective diseases. Evidence is also accumulating that HFE mutations directly favor erythroblast maturation and hemoglobinization independently of hepcidin, and reduce activation of macrophages in response to inflammation.
These data suggest that anti-hepcidin therapies such as anti-hepcidin or anti-BMP6 Abs may improve anemia management in CHD patients concomitantly sparing the doses and side-effects of Epo and iron and resulting in better quality-of-life and survival. However, as the beneficial effect of HFE mutations on iron metabolism in CHD does not seem to be fully explained by lower hepcidin levels, and hepcidin might also play a beneficial anti-inflammatory and anti-microbic action during sepsis, direct inhibition of HFE-mediated regulation of iron metabolism may represent a valuable alternative therapeutic target. Genetic studies may offer a valuable tool to test these hypotheses and guide the research of new therapies.
Footnotes
Peer reviewer: Daniel J Canter, MD, Assistant Professor, Emory University, Department of Urology, 1365 Clifton Road, NE, Atlanta, GA 30322, United States
S- Editor Wang JL L- Editor A E- Editor Zheng XM
References
- 1.Lankhorst CE, Wish JB. Anemia in renal disease: diagnosis and management. Blood Rev. 2010;24:39–47. doi: 10.1016/j.blre.2009.09.001. [DOI] [PubMed] [Google Scholar]
- 2.Gunnell J, Yeun JY, Depner TA, Kaysen GA. Acute-phase response predicts erythropoietin resistance in hemodialysis and peritoneal dialysis patients. Am J Kidney Dis. 1999;33:63–72. doi: 10.1016/s0272-6386(99)70259-3. [DOI] [PubMed] [Google Scholar]
- 3.Papanikolaou G, Pantopoulos K. Iron metabolism and toxicity. Toxicol Appl Pharmacol. 2005;202:199–211. doi: 10.1016/j.taap.2004.06.021. [DOI] [PubMed] [Google Scholar]
- 4.Andrews NC. Forging a field: the golden age of iron biology. Blood. 2008;112:219–230. doi: 10.1182/blood-2007-12-077388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pigeon C, Ilyin G, Courselaud B, Leroyer P, Turlin B, Brissot P, Loréal O. A new mouse liver-specific gene, encoding a protein homologous to human antimicrobial peptide hepcidin, is overexpressed during iron overload. J Biol Chem. 2001;276:7811–7819. doi: 10.1074/jbc.M008923200. [DOI] [PubMed] [Google Scholar]
- 6.Nemeth E, Tuttle MS, Powelson J, Vaughn MB, Donovan A, Ward DM, Ganz T, Kaplan J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science. 2004;306:2090–2093. doi: 10.1126/science.1104742. [DOI] [PubMed] [Google Scholar]
- 7.Pietrangelo A. Hemochromatosis: an endocrine liver disease. Hepatology. 2007;46:1291–1301. doi: 10.1002/hep.21886. [DOI] [PubMed] [Google Scholar]
- 8.Camaschella C. Understanding iron homeostasis through genetic analysis of hemochromatosis and related disorders. Blood. 2005;106:3710–3717. doi: 10.1182/blood-2005-05-1857. [DOI] [PubMed] [Google Scholar]
- 9.Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, Dormishian F, Domingo R, Ellis MC, Fullan A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 10.Wood MJ, Powell LW, Ramm GA. Environmental and genetic modifiers of the progression to fibrosis and cirrhosis in hemochromatosis. Blood. 2008;111:4456–4462. doi: 10.1182/blood-2007-11-122374. [DOI] [PubMed] [Google Scholar]
- 11.Piperno A, Mariani R, Arosio C, Vergani A, Bosio S, Fargion S, Sampietro M, Girelli D, Fraquelli M, Conte D, et al. Haemochromatosis in patients with beta-thalassaemia trait. Br J Haematol. 2000;111:908–914. [PubMed] [Google Scholar]
- 12.Valenti L, Canavesi E, Galmozzi E, Dongiovanni P, Rametta R, Maggioni P, Maggioni M, Fracanzani AL, Fargion S. Beta-globin mutations are associated with parenchymal siderosis and fibrosis in patients with non-alcoholic fatty liver disease. J Hepatol. 2010;53:927–933. doi: 10.1016/j.jhep.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 13.Finberg KE. Iron-refractory iron deficiency anemia. Semin Hematol. 2009;46:378–386. doi: 10.1053/j.seminhematol.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 14.Benyamin B, Ferreira MA, Willemsen G, Gordon S, Middelberg RP, McEvoy BP, Hottenga JJ, Henders AK, Campbell MJ, Wallace L, et al. Common variants in TMPRSS6 are associated with iron status and erythrocyte volume. Nat Genet. 2009;41:1173–1175. doi: 10.1038/ng.456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kalantar-Zadeh K, Rodriguez RA, Humphreys MH. Association between serum ferritin and measures of inflammation, nutrition and iron in haemodialysis patients. Nephrol Dial Transplant. 2004;19:141–149. doi: 10.1093/ndt/gfg493. [DOI] [PubMed] [Google Scholar]
- 16.Kalantar-Zadeh K, McAllister CJ, Lehn RS, Liu E, Kopple JD. A low serum iron level is a predictor of poor outcome in hemodialysis patients. Am J Kidney Dis. 2004;43:671–684. doi: 10.1053/j.ajkd.2003.12.029. [DOI] [PubMed] [Google Scholar]
- 17.Kalantar-Zadeh K, Regidor DL, McAllister CJ, Michael B, Warnock DG. Time-dependent associations between iron and mortality in hemodialysis patients. J Am Soc Nephrol. 2005;16:3070–3080. doi: 10.1681/ASN.2005040423. [DOI] [PubMed] [Google Scholar]
- 18.Scaccabarozzi A, Arosio P, Weiss G, Valenti L, Dongiovanni P, Fracanzani AL, Mattioli M, Levi S, Fiorelli G, Fargion S. Relationship between TNF-alpha and iron metabolism in differentiating human monocytic THP-1 cells. Br J Haematol. 2000;110:978–984. doi: 10.1046/j.1365-2141.2000.02280.x. [DOI] [PubMed] [Google Scholar]
- 19.Torti FM, Torti SV. Regulation of ferritin genes and protein. Blood. 2002;99:3505–3516. doi: 10.1182/blood.v99.10.3505. [DOI] [PubMed] [Google Scholar]
- 20.Valenti L, Valenti G, Como G, Burdick L, Santorelli G, Dongiovanni P, Rametta R, Bamonti F, Novembrino C, Fracanzani AL, et al. HFE gene mutations and oxidative stress influence serum ferritin, associated with vascular damage, in hemodialysis patients. Am J Nephrol. 2007;27:101–107. doi: 10.1159/000099635. [DOI] [PubMed] [Google Scholar]
- 21.Rocha LA, Barreto DV, Barreto FC, Dias CB, Moysés R, Silva MR, Moura LA, Draibe SA, Jorgetti V, Carvalho AB, et al. Serum ferritin level remains a reliable marker of bone marrow iron stores evaluated by histomorphometry in hemodialysis patients. Clin J Am Soc Nephrol. 2009;4:105–109. doi: 10.2215/CJN.01630408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cavill I. Iron status as measured by serum ferritin: the marker and its limitations. Am J Kidney Dis. 1999;34:S12–S17. doi: 10.1053/AJKD034s00012. [DOI] [PubMed] [Google Scholar]
- 23.Brugnara C. Reticulocyte cellular indices: a new approach in the diagnosis of anemias and monitoring of erythropoietic function. Crit Rev Clin Lab Sci. 2000;37:93–130. doi: 10.1080/10408360091174196. [DOI] [PubMed] [Google Scholar]
- 24.Besarab A, Amin N, Ahsan M, Vogel SE, Zazuwa G, Frinak S, Zazra JJ, Anandan JV, Gupta A. Optimization of epoetin therapy with intravenous iron therapy in hemodialysis patients. J Am Soc Nephrol. 2000;11:530–538. doi: 10.1681/ASN.V113530. [DOI] [PubMed] [Google Scholar]
- 25.Eschbach JW, Cook JD, Finch CA. Iron absorption in chronic renal disease. Clin Sci. 1970;38:191–196. doi: 10.1042/cs0380191. [DOI] [PubMed] [Google Scholar]
- 26.Donnelly SM, Posen GA, Ali MA. Oral iron absorption in hemodialysis patients treated with erythropoietin. Clin Invest Med. 1991;14:271–276. [PubMed] [Google Scholar]
- 27.Kooistra MP, Marx JJ. The absorption of iron is disturbed in recombinant human erythropoietin-treated peritoneal dialysis patients. Nephrol Dial Transplant. 1998;13:2578–2582. doi: 10.1093/ndt/13.10.2578. [DOI] [PubMed] [Google Scholar]
- 28.Michelis R, Gery R, Sela S, Shurtz-Swirski R, Grinberg N, Snitkovski T, Shasha SM, Kristal B. Carbonyl stress induced by intravenous iron during haemodialysis. Nephrol Dial Transplant. 2003;18:924–930. doi: 10.1093/ndt/gfg031. [DOI] [PubMed] [Google Scholar]
- 29.Kalantar-Zadeh K, Don BR, Rodriguez RA, Humphreys MH. Serum ferritin is a marker of morbidity and mortality in hemodialysis patients. Am J Kidney Dis. 2001;37:564–572. [PubMed] [Google Scholar]
- 30.Feldman HI, Santanna J, Guo W, Furst H, Franklin E, Joffe M, Marcus S, Faich G. Iron administration and clinical outcomes in hemodialysis patients. J Am Soc Nephrol. 2002;13:734–744. doi: 10.1681/ASN.V133734. [DOI] [PubMed] [Google Scholar]
- 31.Griendling KK, FitzGerald GA. Oxidative stress and cardiovascular injury: Part I: basic mechanisms and in vivo monitoring of ROS. Circulation. 2003;108:1912–1916. doi: 10.1161/01.CIR.0000093660.86242.BB. [DOI] [PubMed] [Google Scholar]
- 32.Roest M, van der Schouw YT, de Valk B, Marx JJ, Tempelman MJ, de Groot PG, Sixma JJ, Banga JD. Heterozygosity for a hereditary hemochromatosis gene is associated with cardiovascular death in women. Circulation. 1999;100:1268–1273. doi: 10.1161/01.cir.100.12.1268. [DOI] [PubMed] [Google Scholar]
- 33.Zacharski LR, Chow B, Lavori PW, Howes PS, Bell MR, DiTommaso MA, Carnegie NM, Bech F, Amidi M, Muluk S. The iron (Fe) and atherosclerosis study (FeAST): a pilot study of reduction of body iron stores in atherosclerotic peripheral vascular disease. Am Heart J. 2000;139:337–345. doi: 10.1067/mhj.2000.102909. [DOI] [PubMed] [Google Scholar]
- 34.Wolff B, Völzke H, Lüdemann J, Robinson D, Vogelgesang D, Staudt A, Kessler C, Dahm JB, John U, Felix SB. Association between high serum ferritin levels and carotid atherosclerosis in the study of health in Pomerania (SHIP) Stroke. 2004;35:453–457. doi: 10.1161/01.STR.0000114875.31599.1C. [DOI] [PubMed] [Google Scholar]
- 35.Kuragano T, Itoh K, Shimonaka Y, Kida A, Furuta M, Kitamura R, Yahiro M, Nanami M, Otaki Y, Hasuike Y, et al. Hepcidin as well as TNF-α are significant predictors of arterial stiffness in patients on maintenance hemodialysis. Nephrol Dial Transplant. 2011;26:2663–2667. doi: 10.1093/ndt/gfq760. [DOI] [PubMed] [Google Scholar]
- 36.Stadler N, Lindner RA, Davies MJ. Direct detection and quantification of transition metal ions in human atherosclerotic plaques: evidence for the presence of elevated levels of iron and copper. Arterioscler Thromb Vasc Biol. 2004;24:949–954. doi: 10.1161/01.ATV.0000124892.90999.cb. [DOI] [PubMed] [Google Scholar]
- 37.Valenti L, Dongiovanni P, Motta BM, Swinkels DW, Bonara P, Rametta R, Burdick L, Frugoni C, Fracanzani AL, Fargion S. Serum hepcidin and macrophage iron correlate with MCP-1 release and vascular damage in patients with metabolic syndrome alterations. Arterioscler Thromb Vasc Biol. 2011;31:683–690. doi: 10.1161/ATVBAHA.110.214858. [DOI] [PubMed] [Google Scholar]
- 38.Cieri E. Does iron cause bacterial infections in patients with end stage renal disease. ANNA J. 1999;26:591–596. [PubMed] [Google Scholar]
- 39.Ellison RT, Giehl TJ. Killing of gram-negative bacteria by lactoferrin and lysozyme. J Clin Invest. 1991;88:1080–1091. doi: 10.1172/JCI115407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Hershko C, Peto TE, Weatherall DJ. Iron and infection. Br Med J (Clin Res Ed) 1988;296:660–664. [PMC free article] [PubMed] [Google Scholar]
- 41.Zager RA, Johnson AC, Hanson SY, Lund S. Parenteral iron compounds sensitize mice to injury-initiated TNF-alpha mRNA production and TNF-alpha release. Am J Physiol Renal Physiol. 2005;288:F290–F297. doi: 10.1152/ajprenal.00342.2004. [DOI] [PubMed] [Google Scholar]
- 42.Zager RA. Parenteral iron treatment induces MCP-1 accumulation in plasma, normal kidneys, and in experimental nephropathy. Kidney Int. 2005;68:1533–1542. doi: 10.1111/j.1523-1755.2005.00565.x. [DOI] [PubMed] [Google Scholar]
- 43.Ayus JC, Go AS, Valderrabano F, Verde E, de Vinuesa SG, Achinger SG, Lorenzo V, Arieff AI, Luño J. Effects of erythropoietin on left ventricular hypertrophy in adults with severe chronic renal failure and hemoglobin & lt; 10 g/dL. Kidney Int. 2005;68:788–795. doi: 10.1111/j.1523-1755.2005.00458.x. [DOI] [PubMed] [Google Scholar]
- 44.Canavese C, Bergamo D, Barbieri S, Timbaldi M, Thea A, Martina G, Damiani D, Fenoglio R, Donati-Marella B, Priolo G. Clinical relevance of hemochromatosis-related HFE C282Y/H63D gene mutations in patients on chronic dialysis. Clin Nephrol. 2002;58:438–444. doi: 10.5414/cnp58438. [DOI] [PubMed] [Google Scholar]
- 45.Valenti L, Valenti G, Como G, Santorelli G, Dongiovanni P, Rametta R, Fracanzani AL, Tavazzi D, Messa PG, Fargion S. HFE genotype influences erythropoiesis support requirement in hemodialysis patients: a prospective study. Am J Nephrol. 2008;28:311–316. doi: 10.1159/000111387. [DOI] [PubMed] [Google Scholar]
- 46.Adams PC, Reboussin DM, Barton JC, McLaren CE, Eckfeldt JH, McLaren GD, Dawkins FW, Acton RT, Harris EL, Gordeuk VR, et al. Hemochromatosis and iron-overload screening in a racially diverse population. N Engl J Med. 2005;352:1769–1778. doi: 10.1056/NEJMoa041534. [DOI] [PubMed] [Google Scholar]
- 47.Jean G, Charra B, Chazot C, Vanel T, Terrat JC, Hurot JM, Laurent G. Risk factor analysis for long-term tunneled dialysis catheter-related bacteremias. Nephron. 2002;91:399–405. doi: 10.1159/000064279. [DOI] [PubMed] [Google Scholar]
- 48.Teehan GS, Bahdouch D, Ruthazer R, Balakrishnan VS, Snydman DR, Jaber BL. Iron storage indices: novel predictors of bacteremia in hemodialysis patients initiating intravenous iron therapy. Clin Infect Dis. 2004;38:1090–1094. doi: 10.1086/382878. [DOI] [PubMed] [Google Scholar]
- 49.Miyashita K, Tojo A, Kimura K, Goto A, Omata M, Nishiyama K, Fujita T. Blood pressure response to erythropoietin injection in hemodialysis and predialysis patients. Hypertens Res. 2004;27:79–84. doi: 10.1291/hypres.27.79. [DOI] [PubMed] [Google Scholar]
- 50.Phrommintikul A, Haas SJ, Elsik M, Krum H. Mortality and target haemoglobin concentrations in anaemic patients with chronic kidney disease treated with erythropoietin: a meta-analysis. Lancet. 2007;369:381–388. doi: 10.1016/S0140-6736(07)60194-9. [DOI] [PubMed] [Google Scholar]
- 51.Valenti L, Girelli D, Valenti GF, Castagna A, Como G, Campostrini N, Rametta R, Dongiovanni P, Messa P, Fargion S. HFE mutations modulate the effect of iron on serum hepcidin-25 in chronic hemodialysis patients. Clin J Am Soc Nephrol. 2009;4:1331–1337. doi: 10.2215/CJN.01370209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tomosugi N, Kawabata H, Wakatabe R, Higuchi M, Yamaya H, Umehara H, Ishikawa I. Detection of serum hepcidin in renal failure and inflammation by using ProteinChip System. Blood. 2006;108:1381–1387. doi: 10.1182/blood-2005-10-4043. [DOI] [PubMed] [Google Scholar]
- 53.Kemna EH, Tjalsma H, Willems HL, Swinkels DW. Hepcidin: from discovery to differential diagnosis. Haematologica. 2008;93:90–97. doi: 10.3324/haematol.11705. [DOI] [PubMed] [Google Scholar]
- 54.Kato A, Tsuji T, Luo J, Sakao Y, Yasuda H, Hishida A. Association of prohepcidin and hepcidin-25 with erythropoietin response and ferritin in hemodialysis patients. Am J Nephrol. 2008;28:115–121. doi: 10.1159/000109968. [DOI] [PubMed] [Google Scholar]
- 55.Babitt JL, Huang FW, Xia Y, Sidis Y, Andrews NC, Lin HY. Modulation of bone morphogenetic protein signaling in vivo regulates systemic iron balance. J Clin Invest. 2007;117:1933–1939. doi: 10.1172/JCI31342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramos P, Guy E, Chen N, Proenca CC, Gardenghi S, Casu C, Follenzi A, Van Rooijen N, Grady RW, de Sousa M, et al. Enhanced erythropoiesis in Hfe-KO mice indicates a role for Hfe in the modulation of erythroid iron homeostasis. Blood. 2011;117:1379–1389. doi: 10.1182/blood-2010-09-307462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Forejtnikovà H, Vieillevoye M, Zermati Y, Lambert M, Pellegrino RM, Guihard S, Gaudry M, Camaschella C, Lacombe C, Roetto A, et al. Transferrin receptor 2 is a component of the erythropoietin receptor complex and is required for efficient erythropoiesis. Blood. 2010;116:5357–5367. doi: 10.1182/blood-2010-04-281360. [DOI] [PubMed] [Google Scholar]
- 58.Valenti L, Swinkels DW, Burdick L, Dongiovanni P, Tjalsma H, Motta BM, Bertelli C, Fatta E, Bignamini D, Rametta R, et al. Serum ferritin levels are associated with vascular damage in patients with nonalcoholic fatty liver disease. Nutr Metab Cardiovasc Dis. 2011;21:568–575. doi: 10.1016/j.numecd.2010.01.003. [DOI] [PubMed] [Google Scholar]
- 59.Sullivan JL. Macrophage iron, hepcidin, and atherosclerotic plaque stability. Exp Biol Med (Maywood) 2007;232:1014–1020. doi: 10.3181/0703-MR-54. [DOI] [PubMed] [Google Scholar]
- 60.Yuan XM, Li W, Baird SK, Carlsson M, Melefors O. Secretion of ferritin by iron-laden macrophages and influence of lipoproteins. Free Radic Res. 2004;38:1133–1142. doi: 10.1080/10715760400011692. [DOI] [PubMed] [Google Scholar]
- 61.Kraml PJ, Klein RL, Huang Y, Nareika A, Lopes-Virella MF. Iron loading increases cholesterol accumulation and macrophage scavenger receptor I expression in THP-1 mononuclear phagocytes. Metabolism. 2005;54:453–459. doi: 10.1016/j.metabol.2004.10.012. [DOI] [PubMed] [Google Scholar]
- 62.Garuti C, Tian Y, Montosi G, Sabelli M, Corradini E, Graf R, Ventura P, Vegetti A, Clavien PA, Pietrangelo A. Hepcidin expression does not rescue the iron-poor phenotype of Kupffer cells in Hfe-null mice after liver transplantation. Gastroenterology. 2010;139:315–22.e1. doi: 10.1053/j.gastro.2010.03.043. [DOI] [PubMed] [Google Scholar]
- 63.Lawless MW, White M, Mankan AK, O'Dwyer MJ, Norris S. Elevated MCP-1 serum levels are associated with the H63D mutation and not the C282Y mutation in hereditary hemochromatosis. Tissue Antigens. 2007;70:294–300. doi: 10.1111/j.1399-0039.2007.00895.x. [DOI] [PubMed] [Google Scholar]
- 64.Babitt JL, Lin HY. Molecular mechanisms of hepcidin regulation: implications for the anemia of CKD. Am J Kidney Dis. 2010;55:726–741. doi: 10.1053/j.ajkd.2009.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sasu BJ, Cooke KS, Arvedson TL, Plewa C, Ellison AR, Sheng J, Winters A, Juan T, Li H, Begley CG, et al. Antihepcidin antibody treatment modulates iron metabolism and is effective in a mouse model of inflammation-induced anemia. Blood. 2010;115:3616–3624. doi: 10.1182/blood-2009-09-245977. [DOI] [PubMed] [Google Scholar]
- 66.Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, Lin HY, Bloch KD, Peterson RT. Dorsomorphin inhibits BMP signals required for embryogenesis and iron metabolism. Nat Chem Biol. 2008;4:33–41. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Pinto JP, Ribeiro S, Pontes H, Thowfeequ S, Tosh D, Carvalho F, Porto G. Erythropoietin mediates hepcidin expression in hepatocytes through EPOR signaling and regulation of C/EBPalpha. Blood. 2008;111:5727–5733. doi: 10.1182/blood-2007-08-106195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Andriopoulos B, Corradini E, Xia Y, Faasse SA, Chen S, Grgurevic L, Knutson MD, Pietrangelo A, Vukicevic S, Lin HY, et al. BMP6 is a key endogenous regulator of hepcidin expression and iron metabolism. Nat Genet. 2009;41:482–487. doi: 10.1038/ng.335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Poli M, Girelli D, Campostrini N, Maccarinelli F, Finazzi D, Luscieti S, Nai A, Arosio P. Heparin: a potent inhibitor of hepcidin expression in vitro and in vivo. Blood. 2011;117:997–1004. doi: 10.1182/blood-2010-06-289082. [DOI] [PubMed] [Google Scholar]
- 70.Ceccarelli M, Bani D, Cinci L, Nistri S, Uliva C, Ragazzo E, Vannacci A, Manoni M, Gori AM, Abbate R, et al. Anti-inflammatory effects of low molecular weight heparin derivative in a rat model of carrageenan-induced pleurisy. J Cell Mol Med. 2009;13:2704–2712. doi: 10.1111/j.1582-4934.2008.00658.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Eijk L, Swinkels DW, Aaron J, Schwoebel F, Fliegert F, Summo L, Stéphanie V, Laarakkers C, Riecke K, Pikkers P. Randomized double blind placebo controlled PK/PD study on the effects of a single intravenous dose of the anti-hepcidin spiegelmer Nox-H94 on serum iron during experimental human endotoxemia. 54th meeting American Society of Hematology. 2012. Available from: https://ash.confex.com/ash/2012/webprogram/Paper50672.html. [Google Scholar]
- 72.Hohlbaum AM, Trentman S, Gille H, Allersdorfer A, Siham Belaiba R, Huelsmeyer M, Christian J, Sandal T, Matschiner G, Jensen K, et al. Discovery and preclinical characterization of a novel hepcidin antagonist with tunable PK/PD properties for the treatment of anemia in different patient populations. 53rd meeting American Society of Hematology. 2011. Available from: https://ash.confex.com/ash/2011/webprogram/Paper40699.html. [Google Scholar]
- 73.Ding F, Ahrenholz P, Winkler RE, Ramlow W, Tiess M, Michelsen A, Pätow W. Online hemodiafiltration versus acetate-free biofiltration: a prospective crossover study. Artif Organs. 2002;26:169–180. doi: 10.1046/j.1525-1594.2002.06877.x. [DOI] [PubMed] [Google Scholar]
- 74.Bonforte G, Grillo P, Zerbi S, Surian M. Improvement of anemia in hemodialysis patients treated by hemodiafiltration with high-volume on-line-prepared substitution fluid. Blood Purif. 2002;20:357–363. doi: 10.1159/000063104. [DOI] [PubMed] [Google Scholar]
- 75.Campostrini N, Castagna A, Zaninotto F, Bedogna V, Tessitore N, Poli A, Martinelli N, Lupo A, Olivieri O, Girelli D. Evaluation of hepcidin isoforms in hemodialysis patients by a proteomic approach based on SELDI-TOF MS. J Biomed Biotechnol. 2010;2010:329646. doi: 10.1155/2010/329646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Pagani A, Nai A, Corna G, Bosurgi L, Rovere-Querini P, Camaschella C, Silvestri L. Low hepcidin accounts for the proinflammatory status associated with iron deficiency. Blood. 2011;118:736–746. doi: 10.1182/blood-2011-02-337212. [DOI] [PubMed] [Google Scholar]
- 77.De Domenico I, Zhang TY, Koening CL, Branch RW, London N, Lo E, Daynes RA, Kushner JP, Li D, Ward DM, et al. Hepcidin mediates transcriptional changes that modulate acute cytokine-induced inflammatory responses in mice. J Clin Invest. 2010;120:2395–2405. doi: 10.1172/JCI42011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Zacharski LR, Chow BK, Howes PS, Shamayeva G, Baron JA, Dalman RL, Malenka DJ, Ozaki CK, Lavori PW. Decreased cancer risk after iron reduction in patients with peripheral arterial disease: results from a randomized trial. J Natl Cancer Inst. 2008;100:996–1002. doi: 10.1093/jnci/djn209. [DOI] [PubMed] [Google Scholar]
- 79.Dongiovanni P, Fracanzani AL, Cairo G, Megazzini CP, Gatti S, Rametta R, Fargion S, Valenti L. Iron-dependent regulation of MDM2 influences p53 activity and hepatic carcinogenesis. Am J Pathol. 2010;176:1006–1017. doi: 10.2353/ajpath.2010.090249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fargion S, Valenti L, Fracanzani AL. Hemochromatosis gene (HFE) mutations and cancer risk: expanding the clinical manifestations of hereditary iron overload. Hepatology. 2010;51:1119–1121. doi: 10.1002/hep.23541. [DOI] [PubMed] [Google Scholar]