

Abstract
Structural changes in neuroanatomical subregions can be measured using serial magnetic resonance imaging scans, and provide powerful biomarkers for detecting and monitoring Alzheimer's disease. The Alzheimer's Disease Neuroimaging Initiative (ADNI) has made a large database of longitudinal scans available, with one of its primary goals being to explore the utility of structural change measures for assessing treatment effects in clinical trials of putative disease‐modifying therapies. Several ADNI‐funded research laboratories have calculated such measures from the ADNI database and made their results publicly available. Here, using sample size estimates, we present a comparative analysis of the overall results that come from the application of each laboratory's extensive processing stream to the ADNI database. Obtaining accurate measures of change requires correcting for potential bias due to the measurement methods themselves; and obtaining realistic sample size estimates for treatment response, based on longitudinal imaging measures from natural history studies such as ADNI, requires calibrating measured change in patient cohorts with respect to longitudinal anatomical changes inherent to normal aging. We present results showing that significant longitudinal change is present in healthy control subjects who test negative for amyloid‐β pathology. Therefore, sample size estimates as commonly reported from power calculations based on total structural change in patients, rather than change in patients relative to change in healthy controls, are likely to be unrealistically low for treatments targeting amyloid‐related pathology. Of all the measures publicly available in ADNI, thinning of the entorhinal cortex quantified with the Quarc methodology provides the most powerful change biomarker. Hum Brain Mapp, 2012. © 2011 Wiley Periodicals, Inc.
Keywords: MCI, bias, biomarker, clinical trial, disease‐specific effect, amyloid, aging, Alzheimer's disease, entorhinal cortex, hippocampus
INTRODUCTION
Structural magnetic resonance imaging (MRI) is highly sensitive to the neurodegeneration that occurs in Alzheimer's disease (AD), even in prodromal stages [McEvoy et al., 2009; Vemuri et al., 2009]. Atrophy measures in neuroanatomical subregions correlate well with disease stage determined from histopathology [Vemuri et al., 2008], and with clinical measures of disease severity [Jack et al., 2004; McDonald et al., 2009]. They are predictive of clinical decline and conversion to AD in individuals with mild cognitive impairment (MCI) [Fan et al., 2008; Jack et al., 1999, 2004; Kovacevic et al., 2009; McEvoy et al., 2011; Vemuri et al., 2009]. Structural changes over time in neuroanatomical subregions can be quantified from serial MRI scans [Holland et al., 2009], and provide powerful biomarkers for tracking disease progression or slowing of progression with treatment. As a reflection of the progressive neurodegeneration that underlies the cognitive and functional decline in AD, anatomical change measures have high face validity as outcome measures for evaluating putative disease‐modifying effects of new therapeutic interventions, and are being evaluated in clinical trial settings as potential surrogates for standard clinical or cognitive outcomes.
To be useful as primary or secondary outcome measures in clinical trials, longitudinal MRI analysis methods must be able to detect with high fidelity subtle structural changes over time. Multiple methodologies have been developed to address this challenge. The Alzheimer's Disease Neuroimaging Initiative (ADNI), a large‐scale, multisite, longitudinal study of the natural history of AD [Mueller et al., 2005] was launched in 2003 with an overarching goal of determining the best set of in vivo biomarkers for early detection and tracking of AD (http://www.adni-info.org) [Mueller et al., 2005]. A related goal is to determine which methods provide maximum power for detecting treatment effects in clinical trials of potential disease‐modifying therapies [Cummings, 2010]. A unique aspect of ADNI is that all raw data are being made available publicly as they are collected (http://adni.loni.ucla.edu). Research groups funded by ADNI [Jack et al., 2010] have made their derived data publicly available as well, enabling a direct comparison of the relative sensitivity of different methods for detecting and tracking neuropathological changes related to AD. Here, we use ADNI's publicly available derived data to present a comparative analysis of measures of whole‐brain and subregional change, obtained from several widely used analysis methods. These methods amount to extensive processing streams and involve many factors, such as image exclusion decisions, quality control, the choice of using the pair of scans or choosing just the best single scan available for subject‐timepoints, and the quality of gradient‐field nonlinearity unwarping employed, and thus are composed of far more than change‐measurement algorithms. Given all these differences, it is only practical to evaluate each methodology based on the overall results individually available from applying them to the same large ADNI database. To compare methods, we use estimated sample size requirements, as these have become a standard metric for evaluating biomarkers, and are directly relevant to clinical trial design. We further provide statistical significance results (P values) for differences in sample size estimates obtained in strict head‐to‐head pairwise comparisons among all measures.
To obtain realistic sample size estimates from longitudinal neuroimaging measures, it is essential to control for potential bias that can arise in image analysis [Thompson and Holland, 2011]. The problem of bias in image registration has been known since the early days of nonrigid morphometric methods [Ashburner and Friston, 2000; Christensen, 1999; Christensen and Johnson, 2001] and has received a great deal of attention recently, including the development of some general and implementation‐specific solutions [Leow et al., 2007; Reuter et al., 2010; Yanovsky et al., 2009; Yushkevich et al., 2010]. Sources of bias include asymmetries in image smoothing and/or interpolation, and asymmetry in the image matching or regularization term in the cost function used in image registration. Such bias can be accentuated to varying degrees depending on the minimization scheme used [Yushkevich et al., 2010].
Another critical consideration when estimating sample sizes for treatment response is whether to include effects seen in normal aging as treatable effects. When performing power calculations based on a natural history (nonintervention) trial such as ADNI, sample size estimates are typically calculated for a hypothesized treatment effect expressed in terms of a percentage of the total disease‐related effect, for example, a 25% slowing in rate of decline on the outcome measure.
Compared to clinical or cognitive measures, neuroimaging measures are very sensitive to changes that occur over time in cognitively healthy older adults [Fjell et al., 2009; Fotenos et al., 2005; Fox et al., 2000; Jack et al., 2008]. When effect sizes are estimated based on absolute change measures, for example, 25% reduction in the total atrophy rate of a given anatomical structure, the usually implicit and probably false [Herrup, 2010] assumption is that all change over time is due to AD. Several ADNI presentations, the ADNI‐2 grant proposal (available at http://www.adni-info.org/Scientists/ADNIScientistsHome.aspx), and most ADNI studies evaluating or comparing sample size estimates for neuroimaging outcome measures make this assumption implicitly [Beckett et al., 2010; Cummings, 2010; Ho et al., 2010; Hua et al., 2009, 2010; Kohannim et al., 2010; Lorenzi et al., 2010; Nestor et al., 2008; Risacher et al., 2010; Schuff et al., 2009; Vemuri et al., 2010], with a few notable exceptions [Fox et al., 2000; Holland et al., 2009; Leung et al., 2010; McEvoy et al., 2010; Schott et al., 2010]. This is a critically important issue since sample size estimates will be substantially smaller if effect sizes are calculated based on absolute change measures rather than on the difference in change measures between patients and controls.
The use of absolute change measures is valid if all atrophy over time in cognitively healthy older individuals is due to AD, that is, if all cognitively healthy older individuals are in a preclinical state of AD. Current research suggests, however, that only 18% of cognitively healthy older individuals aged 60–69 show signs of amyloid‐β (Aβ) pathology, one of the key necessary features for AD, rising to 65% in those over 80 years [Rowe et al., 2010]. Pathological levels of cortical Aβ can be assessed directly through positron emission tomography (PET) imaging of amyloid‐sensitive ligands [Rabinovici and Jagust, 2009] or indirectly through cerebrospinal fluid levels of Aβ42 [Blennow et al., 2010]. CSF and PET measures of Aβ pathology correlate highly with each other [Fagan et al., 2009] and with measures of Aβ at autopsy [Ikonomovic et al., 2008]. Proposed models of the trajectories of different AD biomarkers [Aisen et al., 2010; Frisoni et al., 2010; Jack et al., 2010] postulate that Aβ pathology is the earliest detectable sign of AD pathology, and may be apparent a decade or more before other signs of AD occur, such as neurodegeneration and cognitive impairment. These models further postulate that neurodegenerative changes, reflected in atrophy on structural MRIs, are downstream events that occur closer in time to, and underlie, the functional and cognitive impairment that characterize AD—and indeed for familial AD, gradual atrophy acceleration has been found in the prodromal stages [Ridha et al., 2006].
According to these models, atrophy observed in individuals who do not show signs of Aβ pathology would not be due to AD, as Aβ pathology appears prior to, and presumably triggers [Hardy and Selkoe, 2002], the AD‐related neurodegeneration. There would thus be no reason to expect that a treatment, such as an anti‐amyloid therapy, aimed at slowing progression of AD pathology, would affect atrophy that stems from causes other than AD pathology. Therefore, determination of disease‐specific effect sizes for neuroimaging outcome measures, based on a natural history trial of AD, would be best estimated as the difference in atrophy rates experienced by MCI or AD patients relative to atrophy rates observed in cognitively healthy individuals without Aβ pathology. It should be noted, however, that there is little evidence to date for differences in atrophy rates in AD‐vulnerable regions between Aβ‐positive and Aβ‐negative healthy older adults (or healthy older controls, HCs) [Chetelat et al., 2010; Fjell et al., 2010], though using variants of Boundary Shift Integral (BSI) for whole brain, ventricles, and hippocampus, a significant difference was found between 65 Aβ‐negative HCs (including two converters) and 40 Aβ‐positive HCs (including four converters) [Schott et al., 2010]. Here we compare atrophy rates in ADNI's full HC group, and in HCs separated into two subgroups, those who test negative for Aβ pathology and those who test positive, based on CSF Aβ42 levels, and examine the implications of these findings for sample size estimation.
In this study, we analyze publicly available ADNI data from the application of five methodologies to serial brain scans to determine which method provides the most sensitive detection of anatomical change over time. These methodologies are: (1) Quarc (quantitative anatomical regional change, developed in our laboratory) [Holland and Dale, 2011; Holland et al., 2009], (2) FreeSurfer Longitudinal v.4.4, (3) FreeSurfer Cross‐sectional v.4.3 [Dale and Sereno, 1993; Dale et al., 1999; Fischl et al., 1999, 2002; Jack et al., 2010], (4) BSI [Freeborough and Fox, 1997; Leung et al., 2010], and (5) Tensor‐Based Morphometry (TBM) [Hua et al., 2008a, b, 2009, 2010]. (We note that “Tensor‐Based Morphometry” is sometimes used to refer to any nonlinear registration method, even if the only tensors involved are rank 1, that is, vectors. “Tensor‐Based Morphometry” is used in the title of several of the Hua et al. articles, referring to the full processing stream developed and implemented by those authors at LONI, UCLA; the tensors in question comprise the set of 3 × 3 Jacobian matrices, one at each voxel, which result from morphometric registration—the registration itself is not based on these tensors, but the analysis of structural change is based on statistical properties of the Jacobian field. In agreement with Hua et al., here we use “Tensor‐Based Morphometry” and “TBM” as identifiers referring exclusively to the complete UCLA‐LONI methodology.) Official ADNI data for Voxel‐Based Morphometry [Alexander et al., 2010; Ashburner and Friston, 2000; Chetelat et al., 2005; Tzourio‐Mazoyer et al., 2002] did not yield meaningful results and so was not included in our analysis. We assess the impact of measurement bias on sample size estimates derived from these neuroimage analysis methodologies and provide sample size estimates, with confidence intervals, for the bias‐corrected data, along with P values for pairwise head‐to‐head comparisons. We also evaluate the impact of failing to control for changes observed in healthy aging. Finally, to determine the sensitivity of neuroimaging variables as outcome measures, we compare sample size estimates derived using change in the neuroimaging measures to those derived using change on a standard clinical outcome measure, the Clinical Dementia Rating–Sum of Boxes (CDR‐SB) score.
The five methodologies under discussion have some aspects in common, and some unique features. FreeSurfer‐cross‐sectional performs independent tissue segmentations at each timepoint for each subject, and is the only method considered here that does not use any form of registration between longitudinal images; FreeSurfer‐longitudinal is conceptually an extension of the cross‐sectional variant, where input from all available time points for a subject is used to update the segmentations at each timepoint. Quarc and TBM are nonlinear registration methods, and estimate change by integrating a volume‐change field over anatomically predefined tissue ROIs or statistically defined ROIs; they differ significantly in their details, for example, image matching and regularization terms, minimization schemes, use or not of Jacobians, use or not of an atlas, intensity normalization, and so forth. BSI has evolved through several versions, but essentially estimates tissue (contrast) boundary displacements between pairs of affine registered images. Altogether, this is a rich set of methodologies that result in some remarkable similarities and differences, which we discuss below.
METHODS
ADNI
All data used in the preparation of this article were obtained from the ADNI database (http://www.loni.ucla.edu/ADNI). ADNI was launched in 2003 by the National Institute on Aging (NIA), the National Institute of Biomedical Imaging and Bioengineering (NIBIB), the Food and Drug Administration (FDA), private pharmaceutical companies and nonprofit organizations, as a $60 million, 5‐year public–private partnership. ADNI's goal is to test whether serial MRI, PET, other biological markers, and clinical and neuropsychological assessment can be combined to measure the progression of MCI and early AD. Determination of sensitive and specific markers of very early AD progression is intended to aid researchers and clinicians to develop new treatments and monitor their effectiveness, as well as lessen the time and cost of clinical trials.
ADNI is the result of efforts of many coinvestigators from a broad range of academic institutions and private corporations. ADNI has recruited 227 cognitively normal individuals to be followed for 3 years, 396 people with MCI to be followed for 3 years, and 193 with mild AD to be followed for 2 years (see http://www.adni-info.org). The research protocol was approved by each local institutional review board and written informed consent is obtained from each participant.
Participants
The ADNI general eligibility criteria have been described elsewhere [Petersen et al., 2010]. Briefly, subjects are not depressed, have a modified Hachinski score of 4 or less, and have a study partner able to provide an independent evaluation of functioning. HC subjects have a Clinical Dementia Rating (CDR) of 0. Subjects with MCI have a subjective memory complaint, objective memory loss measured by education‐adjusted scores on Wechsler Memory Scale Logical Memory II, a CDR of 0.5, preserved activities of daily living, and absence of dementia. Subjects with AD have a CDR of 0.5 or 1.0 and meet National Institute of Neurological Disorders and Stroke and Alzheimer's Disease and Related Disorders Association criteria for probable AD.
Data Processing
Analyses were performed on data sets available from http://www.loni.ucla.edu/ADNI/Data through April 16, 2011. These data sets comprise measures derived from longitudinal structural MRI processed with: Quarc; FreeSurfer‐longitudinal (FS); FreeSurfer‐cross‐sectional (FSx); BSI; and TBM. The measures in these data sets are for various ROIs, both predefined tissue regions and data‐driven regions, at baseline and follow‐up (generally 6‐months apart) through 36‐months. Images for Quarc were preprocessed locally, similarly to the preprocessing at Mayo Clinic performed by ADNI for the other methodologies, but using both images per time point where available, and using image correction procedures for site‐specific distortion effects updated for recent scanner changes. The other methodologies used only a single scan per timepoint—the best scan in the event of artifactual degradation in the other. Since one of the goals of ADNI is to identify biomarkers that are more powerful than current standard outcomes for tracking early disease progression, sample sizes were also determined using CDR‐SB as an outcome variable. Change with respect to baseline was the measure used in all cases (follow‐up images were directly registered with baseline). All data that passed quality control (as defined by the several methodologies) for all available time points were used. This provides for a global overview when comparing the full methodological processing streams, and implicitly takes into account differences in the methods' failure rates. We also carried out pairwise comparisons on identical subject‐timepoint data sets for a more narrowly focused assessment of relative performance.
For the FreeSurfer‐related methods, we focused primarily on the entorhinal, hippocampus, and whole brain, these being the ROIs traditionally of interest in AD studies, but we also provide results for other ROIs in Tables II and III; for BSI we used the whole‐brain measure “KN‐BSI,” described in [Leung et al., 2010]; and for TBM we used the “Stat‐ROI,” the statistically defined ROI in the temporal lobe that was designed to undergo a high degree of change from AD, as described in [Hua et al., 2009]. An earlier analysis of the TBM Stat‐ROI showed that the differences between 0 to 6 month and subsequent interval atrophy rates were highly significant for this measure for all diagnostic groups [Thompson and Holland, 2011], and an attempt to redress the issues raised [Hua et al., 2011] resulted in alternative sample size estimates, which we discuss below. Individual subject data from the modified method are not available on the ADNI website, but since a large number of publications have reported on the problematic TBM Stat ROI results [Beckett et al., 2010; Cummings, 2010; Ho et al., 2010; Hua et al., 2008a, b, 2009, 2010; Jack et al., 2010; Kohannim et al., 2010], and results from the modified method are similar, we provide a more detailed analysis here of this method, and compare results from the two approaches.
Table II.
Bias‐corrected sample size estimates for MCI
| Measure | N MCI | N MCI‐HC(Aβ−) | N MCI‐HC |
|---|---|---|---|
| Quarc entorhinal | 134 [110 171] | 286 [201 446] | 293 [214 432] |
| Quarc amygdala | 165 [133 214] | 297 [215 444] | 366 [264 549] |
| Quarc hippocampus | 164 [133 213] | 388 [273 605] | 444 [314 687] |
| Quarc fusiform | 150 [121 194] | 440 [302 707] | 479 [338 742] |
| Quarc inf temporal | 186 [149 246] | 490 [334 800] | 480 [340 742] |
| Quarc mid temporal | 200 [159 266] | 515 [347 856] | 522 [364 825] |
| Quarc whole brain | 149 [121 193] | 555 [341 1057] | 657 [433 1137] |
| Quarc ventricles | 183 [146 241] | 813 [465 1785] | 1008 [623 1934] |
| FS hippocampus | 175 [142 223] | 327 [209 585] | 576 [386 963] |
| FS entorhinal | 413 [315 578] | 694 [400 1505] | 745 [490 1286] |
| FS inf temporal | 449 [337 636] | 988 [555 2240] | 986 [617 1846] |
| FS mid temporal | 488 [364 701] | 825 [491 1679] | 1013 [626 1936] |
| FS fusiform | 518 [385 750] | 799 [437 1932] | 1121 [663 2326] |
| FS ventricles | 164 [133 211] | 1095 [542 3328] | 1179 [695 2461] |
| FS amygdala | 1113 [755 1842] | 716 [423 1480] | 1799 [974 4444] |
| FS whole brain | 384 [294 531] | 1769 [743 8642] | 2179 [1053 7010] |
| FSx hippocampus | 233 [190 298] | 620 [377 1206] | 771 [513 1310] |
| FSx ventricles | 153 [126 194] | 721 [434 1443] | 991 [632 1803] |
| FSx mid temporal | 549 [417 774] | 1162 [701 2327] | 1082 [703 1917] |
| FSx entorhinal | 656 [494 935] | 1592 [777 4928] | 1121 [706 2080] |
| FSx inf temporal | 661 [492 955] | 2188 [992 8424] | 1452 [863 2993] |
| FSx fusiform | 763 [559 1129] | 1870 [915 5740] | 1661 [961 3608] |
| FSx whole brain | 381 [299 512] | 1445 [717 4353] | 1961 [1052 4944] |
| FSx amygdala | 1863 [1194 3371] | 1856 [805 8164] | 5169 [2114 27691] |
| KN‐BSI | 142 [115 182] | 500 [330 853] | 715 [472 1221] |
| VBSI | 181 [145 235] | 778 [449 1675] | 1173 [703 2368] |
| TBM Stat. ROI | 287 [223 395] | 1184 [523 4825] | 1358 [712 3624] |
| CDR‐SB | 542 [404 775] | 608 [443 898] | 638 [464 946] |
See Methods section for methodology nomenclature; references are given in the Introduction. Values for selected measures are plotted in Figure 3. The 95% confidence intervals of the estimated sample sizes are shown in brackets. N MCI refers to sample size estimates required to detect 25% slowing in the total or absolute change seen in MCI, with 80% power and 5% significance. N MCI‐HC are sample size estimates for 25% slowing in the rate of change in MCI that is in excess of that seen in all HCs; ditto for N MCI‐HC(Aβ−), but with respect to the Aβ‐negative subgroup of HCs. P‐values for head‐to‐head pairwise comparisons of relative change sample sizes are in Table IV.
Table III.
Bias‐corrected sample size estimates for AD
| Measure | N AD | N AD‐HC(Aβ−) | N AD‐HC |
|---|---|---|---|
| Quarc entorhinal | 44 [33 63] | 73 [53 113] | 74 [54 113] |
| Quarc fusiform | 57 [44 81] | 98 [72 147] | 102 [75 151] |
| Quarc inf temporal | 73 [55 105] | 118 [85 181] | 117 [85 177] |
| Quarc mid temporal | 76 [57 109] | 123 [88 189] | 124 [90 188] |
| Quarc hippocampus | 71 [54 103] | 118 [85 181] | 126 [91 195] |
| Quarc amygdala | 78 [59 113] | 116 [83 179] | 132 [94 205] |
| Quarc whole brain | 84 [63 123] | 179 [121 302] | 196 [134 321] |
| Quarc ventricles | 92 [69 135] | 219 [144 382] | 243 [164 409] |
| FS hippocampus | 105 [79 152] | 157 [105 265] | 217 [150 355] |
| FS ventricles | 90 [68 128] | 247 [159 450] | 255 [174 427] |
| FS entorhinal | 240 [169 383] | 326 [206 617] | 340 [225 597] |
| FS inf temporal | 226 [160 356] | 352 [223 658] | 352 [232 623] |
| FS fusiform | 236 [167 375] | 299 [188 564] | 354 [231 637] |
| FS mid temporal | 261 [182 423] | 346 [223 631] | 383 [248 692] |
| FS whole brain | 252 [175 408] | 629 [332 1658] | 696 [393 1617] |
| FS amygdala | 1410 [729 4074] | 879 [441 2635] | 2380 [971 13548] |
| FSx hippocampus | 119 [91 169] | 230 [153 396] | 264 [183 428] |
| FSx ventricles | 95 [73 131] | 240 [163 396] | 280 [195 451] |
| FSx mid temporal | 347 [241 564] | 516 [328 959] | 498 [323 897] |
| FSx fusiform | 360 [249 593] | 547 [334 1092] | 522 [333 969] |
| FSx whole brain | 228 [162 359] | 504 [295 1077] | 586 [354 1193] |
| FSx entorhinal | 429 [291 730] | 713 [404 1645] | 591 [370 1139] |
| FSx inf temporal | 456 [305 790] | 903 [489 2287] | 729 [437 1515] |
| FSx amygdala | 1399 [742 3747] | 1395 [611 5996] | 2604 [1095 13180] |
| KN‐BSI | 75 [58 104] | 152 [109 234] | 180 [129 276] |
| VBSI | 92 [70 128] | 213 [144 353] | 257 [177 417] |
| TBM Stat. ROI | 110 [82 165] | 243 [143 516] | 260 [168 472] |
| CDR‐SB | 279 [198 437] | 292 [206 464] | 298 [210 474] |
Quality control information, using acronymic nomenclature, was explicitly provided by the individual research groups in the publicly available ADNI spreadsheets for FS, FSx, BSI, and Quarc data, and used here for filtering out subject visits that did not have values as follows: FS and FSx QVERALLQC = “Pass” or “Partial”; BSI VENTACCEPT = 1, REGRATING ≤ 3, KMNREGRATING ≤ 3; and Quarc QCPASS = 1. The total numbers of remaining subjects, categorized by diagnosis (and CSF‐Aβ status for HCs), for all methodologies and CDR‐SB are shown in Table I.
Table I.
Number of subjects
| Methodology | AD | MCI | HCa | HC(Aβ+) | HC(Aβ−) |
|---|---|---|---|---|---|
| Quarc | 131 | 311 | 182 | 35 | 58 |
| FS | 135 | 320 | 182 | 36 | 59 |
| FSx | 169 | 365 | 199 | 37 | 64 |
| BSI | 156 | 346 | 192 | 35 | 65 |
| TBM | 133 | 291 | 148 | 31 | 48 |
| CDR‐SB | 177 | 371 | 200 | 38 | 66 |
HC does not include 13 converters, based on clinical records up to and including the visit 36 months from baseline. Only 113 of 227 subjects diagnosed as HC at baseline had CSF Aβ‐status determined; six (2 Aβ−, 4 Aβ+) of these converted.
Bias Estimation
All measures were evaluated for potential bias by estimating the intercept based on a linear fit to the 6‐ and 12‐month timepoints [Yushkevich et al., 2010]. This linear fit was performed simultaneously across groups (AD, MCI, and HC), allowing different slopes for each group but requiring constant intercept, based on the assumption that additive bias, if arising from methodology, should equally affect measures from all groups.
More formally, we used the following linear model to fit for additive bias (intercept) b, and slopes (rates of change) s H for HCs, s M for MCI subjects, and s A for AD subjects: Y = s H × T H + s M × T M + s A × T A + b. Here, Y, T H, T M, and T A are vectors of length equal to the total number of all subject‐visits at 6‐ and 12‐months: Y is the vector of response measurements (percent volume change from baseline) for all subjects‐visits; T H is the vector of times from baseline for all HC subject‐visits, with zeros at positions corresponding to non‐HCs; T M, and T A are similar vectors but for MCI and AD subjects, respectively. The general linear model was fit using Matlab, and the null hypothesis that the y‐intercept is zero, indicating no bias, was tested.
Measures were corrected for bias by subtracting the estimated b at all follow‐up timepoints. Bias‐corrected measures were then used for subsequent power calculations.
Power Calculations
Power calculations, modeling linear change over time, were performed for each methodology with standard methods briefly described in [Holland et al., 2009], using all available timepoints through 36‐months for each subject. Since we are measuring change from baseline, in plots of measured change versus time of measurement all intercepts are zero at baseline. Each subject, however, is assumed to change at an independent rate. Thus we have a linear mixed‐effects model (fixed intercepts, fixed group slopes, random individual subject slopes, random within‐subject additive or observational error) where, for a specific diagnostic group, the measurement Y ij at time t ij for subject i at follow‐up timepoint j is Y ij = m i t ij+εij. Here, εij is the within‐subject error, assumed to be independent and identically normally distributed with zero mean and variance σ; m i = m + γi, where m is the fixed effect slope (mean rate of change for the group) and γi is the between‐subject random effect slope with variance σ. We use the Matlab (version R2009b) nlmefit function in the Statistics Toolbox (http://www.mathworks.com) to obtain maximum likelihood point estimates of σ, σ, and m. These fixed and random effects parameter estimates can be used in power calculations to obtain point estimates of the sample sizes N, per arm, required for a hypothetical placebo‐controlled longitudinal study, as described in [Fitzmaurice et al., 2004]. This approach was used to calculate sample sizes required to detect a 25% slowing in mean rate of decline for a hypothetical disease‐modifying treatment versus placebo for a 24‐month, two‐arm, equal allocation trial, with 6‐month assessment intervals. Power calculations were performed with the requirement that the trial have 80% power to detect the treatment effect using a two‐sided significance level of 5%. When correcting for normal aging, the sample size estimates were calculated using the variance parameters (σ, σ) from the patient cohort, while the treatment effect size of interest was assumed to be 25% of the difference between the mean rates of change in the patient and healthy populations.
To determine 95% confidence intervals on the sample size estimates, the joint a posteriori probability density function for the mixed effects model parameters (σ, σ, and m) was computed based on the multivariate Gaussian likelihood function of the observed data, given the model parameters, evaluated at a regular mesh of points in the space spanned by the model parameters. The sample size values at the mesh points were then sorted, and the cumulative distribution calculated from the correspondingly sorted a posteriori probability values. The 95% confidence intervals were then computed from the cumulative distribution function (cdf) values of 0.025 and 0.975.
To calculate P values for pairwise comparisons of sample sizes required for different measures, we carried out a two‐tailed test for the null hypothesis of equal sample sizes (N 1 − N 2 = 0). We used the a posteriori probability distributions described above to compute the probability distribution for the difference between the sample sizes for the two measures; the latter is given by the convolution of the two sample size distributions, since N 1 and N 2 are independent random variables. Thus, p = 2 × min[P (N 1 ≤ N 2), P (N 2 ≤ N 1)], where P (N 1 ≤ N 2) = cdfN1‐N2(0), and P (N 2 ≤ N 1) = 1‐cdfN1‐N2(0) [Casella and Berger, 2002].
RESULTS
Bias
The TBM Stat‐ROI measure showed statistically significant bias, as illustrated in Figure 1. This figure shows the average cumulative atrophy detected by this method, as a percentage of baseline volume, for HC, MCI, and AD subjects up through 36‐months, along with the additive bias estimate. Without accounting for bias, the cumulative atrophy plots show that all three groups undergo an initial high rate of change (an average of 1.64% for HC, 1.93% for MCI, and 2.28% for AD over the first 6‐month interval of the study), with substantially lower rates of change after that (e.g., average change in the second 6‐month period of the study is 0.34% for HC, 0.55% for MCI, and 0.76% for AD subjects; for the final 12‐months of the study, the average change was 0.09% for MCI and −0.01% for HC—a pronounced deceleration that is indicative of higher‐order contributions to bias in TBM). Based on the simultaneous linear fit to the 6‐ and 12‐month timepoints of all three diagnostic groups, estimated additive bias was 1.31%, which is equal to 68% of the observed change in the MCI cohort and 57% of the observed change in the AD cohort at 6‐months. The P value for the null hypothesis of no bias was, to the precision of the Matlab numerical libraries, 0. Although subject data from the modified TBM Stat‐ROI method are not available, Hua et al. [ 2011] report a bias of 0.29% in the modified method, which is substantially reduced from 1.31% reported above. However, the new measures of change for the Stat‐ROI are also substantially reduced (average change measurement of 0.5% for HC, 0.6% for MCI, and 0.9% for AD over the first 6‐month interval of the study), so that the bias as a percentage of 6‐month change in MCI remains large: 48%. (We note that the bias reported in [Hua et al., 2011] would be slightly reduced if actual visit times were used, that is, not assuming that all visits happened exactly at 6‐month intervals.)
Figure 1.
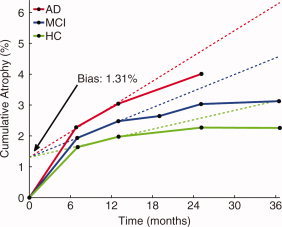
Average cumulative atrophy for TBM Stat‐ROI, as a percentage of baseline volume, for HC, MCI, and AD subjects, along with linear fits for the additive bias estimate. The additive bias of 1.31% is large, equal to 68% of the change in the MCI cohort, and 57% of the change in the AD cohort, at 6‐months, and highly statistically significant (P = 0 to precision of Matlab numerical libraries). The linear fits are conservatively restricted to the 6‐ and 12‐month visits. Note the slight shift to the right for follow‐up visits. On average, actual visit dates occurred later than the nominal interval dates.
The KN‐BSI measure also showed significant bias, accounting for 18% of the change observed in MCI subjects and 12% of the change observed in AD subjects at 6‐months (P = 0.042). None of the other methods showed significant bias.
Estimated sample sizes for absolute‐change were smallest for the bias‐uncorrected TBM Stat‐ROI measure. For example, using 291 MCI subjects, the sample size estimate to detect 25% slowing in the MCI population was N = 84, and the 95% confidence interval was CI = [71 103]. After bias correction, however, this estimate more than tripled, to N = 287, CI = [223 395], rendering this method significantly less powerful for detecting change than bias‐corrected KN‐BSI (head‐to‐head comparison of 266 MCI subjects: TBM N = 319, CI = [239 457]; KN‐BSI N = 147, CI = [117 197]; P value for difference in sample‐size estimates = 0.0002), Quarc entorhinal (236 MCI subjects: TBM N = 233, CI = [179 327]; ERC N = 150, CI = [118 202]; P = 0.029), and Quarc hippocampus (Hipp. N = 156, CI = [122 210]; P = 0.047). Sample size estimates for all methods, after bias‐correction, are shown in Table II for MCI and Table III for AD.
For the modified TBM Stat‐ROI method, the sample size estimate reported for absolute change in MCI is N = 129; correcting this for the 0.29% bias (and using the new estimated MCI annual rate of change of 1%, along with the standard sample size formula, ibid.), gives N = 129/(1 − 0.29)2 = 256, close to the value N = 287, CI = [223 395], reported above and in Table II for the original TBM Stat‐ROI method.
Relative vs. Absolute Change: Defining the Potentially Treatable Effect
To determine whether atrophy rates in HCs are due to preclinical AD (the implicit assumption in numerous published studies that use absolute rather than relative change measures in sample size calculations), we examined atrophy rates in the two ROIs affected earliest in AD–entorhinal cortex and hippocampus—as well as in the whole brain in HCs who tested negative for Aβ pathology, based on the cut‐off value of CSF Aβ42 levels >192 pg/mL as determined by Shaw et al. [ 2009], for all methodologies for which these ROIs are defined; we also similarly examined the TBM temporal lobe Stat‐ROI (note that CSF measures were obtained only on a subset of ADNI subjects—see Table I). Figure 2 shows bias‐corrected annual atrophy rates with 95% confidence intervals for the full HC group, the two HC subgroups who were, respectively, Aβ‐negative and Aβ‐positive, and the MCI group; these atrophy rates were calculated using a mixed‐effects regression model on all baseline data and follow‐up data available up to 3‐years, as described in the Methods section.
Figure 2.
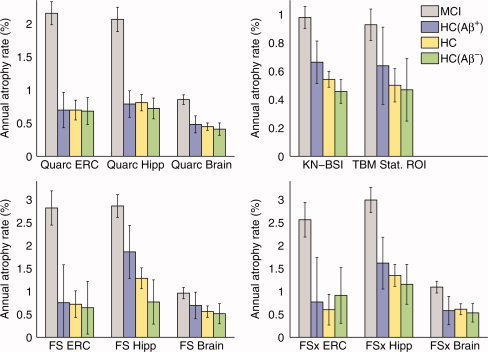
Annual rate of volume change in entorhinal cortex (ERC), hippocampus (Hipp), and whole brain (Brain and KN‐BSI), with 95% confidence intervals, calculated for all methodologies, along with the TBM Stat‐ROI. Aβ‐negative HCs are almost the same as all HCs, but have approximately a third to a half the change seen in MCI; Aβ‐positive HCs atrophy at a slightly higher rate than Aβ‐negative HCs. Thus, most HC change is not AD‐related; assuming it is leads to seriously underpowered clinical trials. All rates are corrected for bias, if any. Note that only a subset of all HCs had Aβ status determined.
As shown in Figure 2, longitudinal volumetric changes in entorhinal cortex, hippocampus, and whole brain are clearly present in Aβ‐negative HC subjects, and the annual percentage changes in these subjects do not substantially differ from those observed in the full HC group. Atrophy rates in Aβ‐negative HC subjects for all ROIs, regardless of methodology, are a substantial fraction (one‐third to one‐half) of the atrophy rates seen in MCI subjects. As the anatomical changes seen in Aβ‐negative HC subjects are not likely to be due to Aβ pathology, there is no reason to expect that they would be affected by therapeutic agents designed specifically to target amyloid pathology. The potentially treatable effect therefore would be most realistically defined as the amyloid‐negative aging‐corrected rate of change in the patient cohort, that is, the difference in rates of change between the patient group and the Aβ‐negative HC subjects. However, since there is little difference between the atrophic rates of change in the Aβ‐negative HC group and the full HC group (compared with the differences between either of these groups and the MCI or AD groups), the potentially treatable effect could conservatively be defined as the aging‐corrected rate of change in the patient cohort, that is, the difference in rates of change between the patient group and the full HC group–to take advantage of the larger N of the full control group.
The annual rates of change for the TBM Stat‐ROI shown in Figure 2 are very similar to those for the whole brain measure KN‐BSI, which in turn are fairly consistent with the whole brain measures for Quarc, FS, and FSx. (Note that the statistics for the HC(Aβ+) group are poorest, reflecting the relatively small number of subjects in that group—see Table I.) This is, on the face of it, an unexpected result because the TBM Stat‐ROI was specifically designed to identify the brain subregion undergoing the highest rate of change, yet the resulting rates are essentially the same as those for whole brain change obtained by the other methods. The estimates reported in [Hua et al., 2011] imply even smaller rates of change; for example, the TBM Stat‐ROI annual rate of change for MCI, when correcting for the remaining bias, is 1 − 0.29 = 0.71%, compared with 0.98%, CI = [0.90% 1.06%], for KN‐BSI.
Sample Size Estimates
Sample size estimates using disease‐specific (aging‐corrected) and absolute (aging‐uncorrected) rates of change for bias‐corrected data are shown in Figure 3 for MCI subjects and Figure 4 for AD subjects, for representative measures for each methodology (numerical values for these and other neuroimaging measures are shown in Tables II and III). For reference, the sample size estimate using CDR‐SB, the most sensitive standard clinical outcome measure, is also shown. P‐values for the significance of the difference in sample size estimates from all head‐to‐head pairwise comparisons of relative‐change (aging‐corrected) measures are in Tables IV and V, for MCI and AD, respectively. In the figures, data are arranged in ascending order for the conservative aging‐corrected sample size estimate, “MCI‐HC” or “AD‐HC.” CDR‐SB represents a demarcation for those neuroimaging measures that are competitive with current clinical outcome measure in tracking longitudinal change over time in MCI and mild AD. In the case of MCI, only the entorhinal cortex (ERC) as measured by Quarc has a conservative disease‐specific 95% confidence interval that is wholly below the CDR‐SB confidence intervals (head‐to‐head comparison involving 311 MCI subjects and 181 HC subjects gives: Quarc ERC N = 297, CI = [216 439]; CDR‐SB N = 582, CI = [417 884]; P value for difference in N's = 0.01). Head‐to‐head comparisons of the Quarc measures with measures from the other methodologies are shown in Figure 5 for MCI and Figure 6 for AD; P values presented in the first row in Table IV show that for MCI the Quarc entorhinal is significantly more powerful than all other measures of change, except the Quarc Hippocampus. From either the last column (MCI‐HC) or last row (MCI‐HC(Aβ−)) in Table IV, Quarc ERC is the only measure that is statistically significantly superior to CDR‐SB. From Figure 2, the measurement of rates of change for FS Hippocampus appears to be anomalous for the HC(Aβ−) and HC(Aβ+) groups, which show substantial and significant difference. This is in contrast with the much higher degree of similarity among these subject groups for all other measures. Furthermore, one would expect the point estimates for FS and FSx to be similar, as indeed they are for the hippocampus measured for all MCIs and all HCs, and for all subject groups for the whole brain. It should be noted that there are much fewer subjects in the HC(Aβ−) and, particularly, the HC(Aβ+) groups compared to all HCs (see Table I), so the estimates for these subgroups are not as robust.
Figure 3.
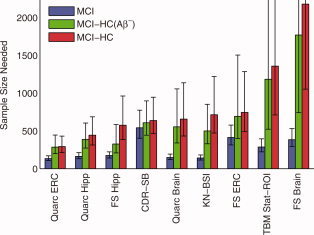
Estimated sample sizes, per arm, to detect a 25% reduction in rate of change in MCI subjects, at the P < 0.05 level with 80% power assuming a 24‐month trial with scans every 6 months. Sample sizes are estimated using a linear mixed effects model with fixed intercepts (no relative change at baseline) and random slopes applied to all data available up through 36 months. Results for the conservative aging‐corrected rates of change are shown in red; results for the more realistic amyloid‐negative aging‐corrected rates of change are shown in green; and results for absolute (aging uncorrected) rates of change are shown in blue. Error bars show the 95% confidence intervals. All numerical values are shown Table II. P‐values for all head‐to‐head pairwise comparisons of measures are in Table IV. FS is FreeSurfer‐longitudinal. FreeSurfer‐cross‐sectional (FSx) generally performs poorer than FS; it is not shown here, but values for it are in Tables II and IV.
Figure 4.
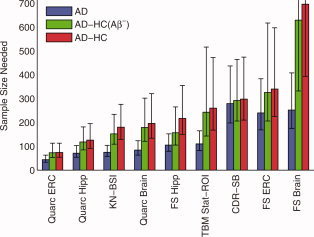
Estimated sample sizes, per arm, to detect a 25% reduction in rate of change in mild AD subjects, at the P < 0.05 level with 80% power assuming a 24‐month trial with scans every 6 months. See caption of Figure 3 for further details. Numerical values are shown in Table III. P‐values for all head‐to‐head pairwise comparisons of measures are in Table V.
Table IV.
P‐values for significance of difference in head‐to‐head comparisons of sample size estimates: MCI‐HC above diagonal, MCI‐HC(Aβ−) below diagonal
| Measure | Quarc‐ Entorhinal | Quarc‐ Hippocampus | Quarc‐Whole‐ Brain | FS‐ Entorhinal | FS‐ Hippocampus | FS‐Whole‐ Brain | FSx‐ Entorhinal | FSx‐ Hippocampus | FSx‐Whole‐ Brain | KN‐ BSI | TBM‐ Stat‐ROI | CDR‐SB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Quarc‐ERC | 0.12 | 0.005 | 0.005 | 0.014 | 4e‐6 | 1e‐5 | 0.013 | 7e‐6 | 0.008 | 2e‐4 | 0.01 | |
| Quarc‐Hipp | 0.29 | 0.2 | 0.2 | 0.36 | 0.002 | 0.004 | 0.33 | 0.003 | 0.31 | 0.037 | 0.37 | |
| Quarc‐Brain | 0.049 | 0.3 | 0.91 | 0.83 | 0.056 | 0.19 | 0.57 | 0.14 | 0.8 | 0.24 | 0.67 | |
| FS‐ERC | 0.005 | 0.1 | 0.51 | 0.44 | 0.02 | 0.11 | 0.86 | 0.019 | 0.88 | 0.4 | 0.77 | |
| FS‐Hipp | 0.24 | 0.95 | 0.41 | 0.065 | 0.003 | 0.02 | 0.39 | 0.003 | 0.77 | 0.074 | 0.88 | |
| FS‐Brain | 3e‐4 | 0.015 | 0.16 | 0.12 | 3e‐4 | 0.43 | 0.031 | 0.95 | 0.026 | 0.49 | 0.004 | |
| FSx‐ERC | 4e‐6 | 3e‐4 | 0.051 | 0.047 | 6e‐6 | 0.83 | 0.29 | 0.21 | 0.063 | 0.72 | 0.05 | |
| FSx‐Hipp | 0.076 | 0.39 | 0.61 | 0.86 | 0.1 | 0.065 | 0.057 | 0.025 | 0.8 | 0.076 | 0.54 | |
| FSx‐Brain | 0.004 | 0.04 | 0.48 | 0.13 | 4e‐4 | 0.84 | 0.88 | 0.085 | 0.04 | 0.51 | 0.003 | |
| KN‐BSI | 0.026 | 0.53 | 0.56 | 0.3 | 0.26 | 0.16 | 0.006 | 0.93 | 0.23 | 0.23 | 0.96 | |
| TBM‐St.ROI | 0.013 | 0.15 | 0.37 | 0.76 | 0.003 | 0.81 | 0.48 | 0.17 | 0.6 | 0.15 | 0.51 | |
| CDR‐SB | 0.015 | 0.16 | 0.92 | 0.94 | 0.06 | 0.051 | 0.02 | 0.91 | 0.04 | 0.42 | 0.63 |
Table V.
P‐values for significance of difference in head‐to‐head comparisons of sample size estimates: AD‐HC above diagonal, AD‐HC(Aβ−) below diagonal
| Measure | Quarc‐ Entorhinal | Quarc‐ Hippocampus | Quarc‐Whole‐ Brain | FS‐ Entorhinal | FS‐ Hippocampus | FS‐Whole‐ Brain | FSx‐ Entorhinal | FSx‐ Hippocampus | FSx‐Whole‐ Brain | KN‐ BSI | TBM‐ Stat‐ROI | CDR‐ SB |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Quarc‐ERC | 0.048 | 7e‐4 | <e‐6 | 7e‐5 | 4e‐6 | <e‐6 | 6e‐5 | 3e‐6 | 0.009 | 1e‐4 | 4e‐6 | |
| Quarc‐Hipp | 0.085 | 0.13 | 9e‐4 | 0.034 | 0.005 | 9e‐6 | 0.039 | 0.005 | 0.46 | 0.32 | 0.01 | |
| Quarc‐Brain | 0.003 | 0.16 | 0.044 | 0.39 | 0.12 | 0.005 | 0.64 | 0.23 | 0.37 | 0.19 | 0.29 | |
| FS‐ERC | <e‐6 | 5e‐4 | 0.028 | 0.17 | 0.08 | 0.1 | 0.49 | 0.62 | 0.072 | 0.46 | 0.48 | |
| FS‐Hipp | 0.001 | 0.13 | 0.74 | 0.039 | 0.002 | 0.003 | 0.51 | 0.072 | 0.39 | 0.45 | 0.43 | |
| FS‐Brain | 5e‐5 | 0.013 | 0.18 | 0.16 | 8e‐4 | 0.92 | 0.03 | 0.33 | 0.047 | 0.032 | 0.012 | |
| FSx‐ERC | <e‐6 | 1e‐6 | 0.002 | 0.047 | 7e‐5 | 0.51 | 0.019 | 0.99 | 7e‐4 | 0.073 | 0.03 | |
| FSx‐Hipp | 4e‐4 | 0.053 | 0.71 | 0.4 | 0.25 | 0.043 | 0.005 | 0.025 | 0.1 | 0.45 | 0.7 | |
| FSx‐Brain | 1e‐4 | 0.022 | 0.44 | 0.79 | 0.027 | 0.34 | 0.46 | 0.044 | 0.006 | 0.042 | 0.04 | |
| KN‐BSI | 0.012 | 0.57 | 0.53 | 0.026 | 0.89 | 0.13 | 1e‐4 | 0.21 | 0.027 | 0.094 | 0.062 | |
| TBM‐St.ROI | 0.002 | 0.48 | 0.26 | 0.45 | 0.1 | 0.11 | 0.032 | 0.49 | 0.082 | 0.086 | 0.4 | |
| CDR‐SB | 6e‐6 | 0.005 | 0.21 | 0.57 | 0.071 | 0.038 | 0.015 | 0.49 | 0.1 | 0.023 | 0.41 |
Figure 5.
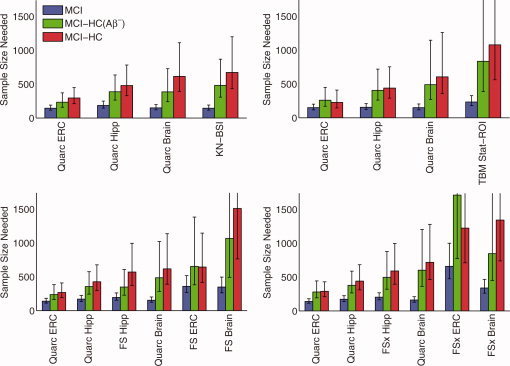
Estimated sample sizes for MCI, as in Figure 3, but from pairwise head‐to‐head comparison of Quarc with: BSI (subjects in common: 287 MCI, 170 HC, 55 HC(Aβ−)); TBM (236 MCI, 132 HC, 41 HC(Aβ−)); FreeSurfer‐longitudinal (267 MCI, 161 HC, 49 HC(Aβ−)); and FreeSurfer‐cross‐sectional (305 MCI, 178 HC, 56 HC(Aβ−)). P‐values for all head‐to‐head pairwise comparisons of measures are in Table IV.
Figure 6.
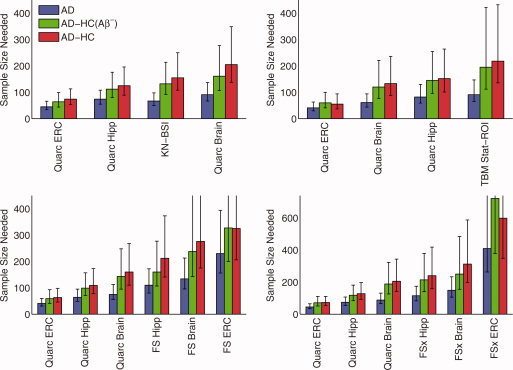
Estimated sample sizes for AD, as in Figure 4, but from pairwise head‐to‐head comparison of Quarc with BSI (123 AD subjects in common—see also Figure 5); TBM (97 AD); FreeSurfer‐longitudinal (FS; 107 AD); and FreeSurfer‐cross‐sectional (FSx; 129 AD; note different scale due to relatively poor performance for entorhinal cortex). P‐values for all head‐to‐head pairwise comparisons of measures are in Table V.
Sample size estimates for the more realistic amyloid‐negative aging‐corrected rates of change, that is, for “MCI‐HC(Aβ−)”, are generally lower compared with those for “MCI‐HC”, reflecting the slightly smaller atrophic rates for HC(Aβ−) compared with those for HC in Figure 2. Note that the point estimates for both the Quarc and FS entorhinal remain unchanged.
Using the estimated annual rates of change reported in [Hua et al., 2011] for MCI, and noting that additive bias is essentially eliminated when calculating relative change between groups, the sample size estimate from the modified TBM Stat‐ROI method for the change in MCI relative to that in all HCs is N = 129/(1 − 0.7)2 = 1,433, close to the value N = 1,358, CI = [712 3624], reported in Table II for the original TBM Stat‐ROI method.
Sample size estimates for a trial involving mild AD patients, shown in Figure 4 and Table III, are substantially smaller, with most neuroimaging outcome measures yielding smaller estimated sample sizes than the CDR‐SB (significance of differences are in Table V). Sample size estimates based on absolute change are also shown for comparison. As expected, for any given neuroimaging measure, they are substantially smaller with much tighter confidence intervals than their disease‐specific counterparts.
Figures 5 and 6 show direct head‐to‐head comparisons of all methodologies with Quarc. In all cases, the entorhinal and hippocampal measures from Quarc were the strongest measures of change.
DISCUSSION
This study compared five widely used methodologies for quantifying longitudinal change measures from structural MRIs and examined two critical issues that significantly impact sample size estimation when neuroimaging measures are used as outcome variables: bias in image analysis and the definition of the potentially treatable effect. Failure to control for either of these factors can lead to dramatic underestimation of sample sizes needed to detect a potentially beneficial effect of a disease‐modifying therapy.
Potential bias in image registration is a well‐known problem in the analysis of serial MRIs that has received much attention in recent literature [Thompson and Holland, 2011]. Although most methodologies employ procedures to minimize bias, our results show that some commonly used methods, particularly TBM Stat‐ROI, are significantly affected. For TBM Stat‐ROI, correction for bias tripled (or doubled, using the alternative results from the modified method) the sample size estimates for detection of change in MCI subjects. Acquiring scans on the same day, where no deformation is expected, would provide ideal images for testing the presence of additive bias—though multiplicative bias would remain undetectable using such images. A simple way to eliminate bias due to registration methodology is to make the entire procedure symmetric by construction: register image A to image B, independently register image B to image A, and then combine the changes measured in both directions by algebraic or geometric averaging.
The definition of the potentially treatable effect is another critical factor that profoundly affects estimated sample sizes. The majority of publications in the ADNI literature, notably [Beckett et al., 2010; Cummings, 2010; Hua et al., 2010; Vemuri et al., 2010], implicitly define the potentially treatable effect as the absolute change from baseline, although as mentioned earlier there are exceptions [Fox et al., 2000; Holland et al., 2009; McEvoy et al., 2010; Schott et al., 2010]. The rationale for use of absolute change measures has not clearly been articulated in published reports, but presumably it arises from the assumption that atrophy in HCs is dominated by a subset of HCs who are in a preclinical stage of AD and experiencing disease‐related elevated rates of decline. We directly tested this assumption by examining atrophy rates in HCs who tested negative for Aβ pathology on the basis of CSF Aβ42 levels and comparing these rates with those seen in all HCs. Since amyloid lesions neuropathologically partly characterize AD [Mirra et al., 1991], and CSF and PET measures of their prevalence are believed to be the earliest detectable signs of possible AD [Morris et al., 2010] (in which case clinical decline might not occur until a decade or so after the lesions become manifest [Price and Morris, 1999]), elderly individuals without Aβ pathology are highly unlikely to be in a preclinical stage of AD—and indeed since many elderly HCs have elevated plaque burden while remaining cognitively normal [Price et al., 2009], it might be that “Alzheimer's is not a part of normal aging any more than breaking your hip is a part of normal aging” [Herrup, 2010]. Our results show, however, that the Aβ‐negative HCs experienced an approximately similar rate of whole brain, entorhinal, and hippocampal atrophy as the full, undifferentiated HC group. Furthermore, as can be seen in Figure 2, apart from the anomalous “FS Hipp” result discussed above, hippocampal and entorhinal atrophy rates are similar for HC(Aβ+) and HC(Aβ−). This result is in agreement with neuropathological studies that show no significant difference in total entorhinal [Price et al., 2001] and hippocampal [West et al., 2004] neuron counts—even in CA1, the hippocampal sector most affected by neuron loss in the early stages of AD—between cognitively normal subjects essentially free of amyloid pathology and those exhibiting significant amounts of amyloid deposition, to a degree consistent with a neuropathological diagnosis of possible AD. Thus, the preponderance of atrophy in HCs must arise from causes other than Aβ pathology, and it would not be reasonable to expect an AD therapy, in particular one targeting Aβ pathology, to reduce atrophy rates for atrophy that occurs in the absence of such pathology. Neuropathological analysis of HCs who do not fulfill criteria for the neuropathological diagnosis of AD or other neurodegenerative disease [Freeman et al., 2008] supports this conclusion. Atrophy in such individuals can persist over an age range of five decades, likely reflects loss of dendritic complexity in neuropil and/or changes in neuronal size, but in contrast with AD‐related atrophy, preserves neuronal number. The neuropathological study shows further that in these subjects the presence of diffuse plaques did not correlate with cortical atrophy; that cortical atrophy correlates with age; and though neuritic plaque burden also correlates with age, the small number of plaques and tangles had no direct influence on cortical atrophy. Therefore, “cortical changes seen in aging are not simply the result of early AD changes but are related to aging itself” (ibid.).
It should be noted that in a clinical trial employing biomarkers for the natural history of the disease, care must be taken in assessing the disease modifying ability of the therapy [Citron, 2010; Salloway et al., 2008]. Correlation is not sufficient [Baker and Kramer, 2003]: the biomarker must also be in the causal pathway of the disease, and directly relate to clinically meaningful endpoints [Mani, 2004]. By the same token, therapy might affect atrophy in unexpected ways, as shown by the AN1792 Aβ immunotherapy trial [Gilman et al., 2005] where whole brain atrophy was greater in the approximately one‐fifth of subjects who were antibody responders than in the placebo group, a result possibly due to brain hydration state related to therapy, or to negative effects of the vaccination on fiber or white matter volume, and that was not reflected in worsening cognitive performance [Fox et al., 2005]. Though clinical improvement in this trial largely may have been precluded because the patient cohort was at a relatively advanced stage of the disease [Holmes et al., 2008; Hyman, 2011], this outcome nevertheless argues in favor of analyzing subregional, in particular cortical, change rather than global change when monitoring disease‐modifying effects of therapy.
Defining the potentially treatable effect based on absolute change from baseline is attractive in that it leads to small sample size requirements—often much smaller than those based on standard clinical outcome measures. This approach represents the most optimistic assessment of the potentially treatable effect. The more conservative approach of defining the potentially treatable effect relative to change experienced by all HCs, or the slightly more realistic approach of defining it relative to change experienced by Aβ‐negative HCs, may represent a more achievable goal, particularly since most current therapies target amyloid pathology. Requiring the treatment to slow all change, even that unrelated to the targeted mechanism of the drug, is likely to result in a trial that is substantially underpowered to detect slowing of disease‐specific atrophy. An additional advantage of using relative rather than absolute change measures is that any purely additive systematic bias in the results arising from errors in image acquisition or analysis methods will, by definition, be removed upon subtraction.
Consideration of these two factors together–bias correction and defining the potentially treatable effect as a measure of relative rather than absolute change–substantially alters the conclusions regarding the relative sensitivity of different neuroimaging biomarkers from what has been published in the literature [Beckett et al., 2010; Cummings, 2010; Jack et al., 2010]. From data available on the ADNI website through April 16, 2011, estimates of change in subregional cortical areas, as determined by Quarc, produce the smallest estimated sample sizes when change relative to HCs is taken into account. In particular, for the entorhinal cortex, the area known to be first affected by AD pathology, N = 293 CI = [214 432] for MCI‐HC, N = 74 CI = [54 113] for AD‐HC, and as can be seen in Table IV, sample size estimates based on the Quarc entorhinal are the only ones that are significantly smaller than those achieved using CDR‐SB as the outcome measure for either MCI‐HC or MCI‐HC(Aβ−). Several other temporal lobe structures quantified with Quarc (Tables II and III) also provided powerful relative‐change biomarkers, including the amygdala (N = 366 CI = [264 549] for MCI‐HC, N = 132 CI = [94 205] for AD‐HC) which suffers from a significant increase in the numbers of both neuritic plaques and NFTs when transitioning from HC to amnestic MCI, and again when transitioning from amnestic MCI to early AD [Markesbery et al., 2006]. Quarc was designed not only to capture large‐scale structural change, but also to measure change in small regions with high precision [Holland and Dale, 2011], and generally provides significantly improved measures of change compared with those of the standard (cross‐sectional) FreeSurfer and FreeSurfer‐longitudinal, the only other methods that attempt to quantify change in cortical regions. The results presented here for Quarc were derived using both the back‐to‐back scans per timepoint (which should improve signal to noise); the other methods used a single scan per timepoint (choosing the best scan from each pair should reduce the degrading impact of image artifacts). Though these effects have not been assessed here, when considering future trials the requirement for acquiring two scans to achieve these sample sizes should be borne in mind. Also, sample size estimates have not been modeled to account for attrition due to QC. (The methods used in Quarc are fully documented in [Holland and Dale, 2011], and are available to other researchers on a not‐for‐profit recharge basis through the UCSD Multimodal Imaging Laboratory, mmil.ucsd.edu.)
Although the current study is primarily concerned with issues that affect the use of neuroimaging biomarkers as outcome measures in clinical trials, it is important to point out that longitudinal MRI measures as provided by ADNI are also being used in the comparative investigation of disease‐related trajectories of various AD biomarkers [Caroli and Frisoni, 2010; Frisoni et al., 2010; Jack et al., 2010; Perrin et al., 2009; Trojanowski et al., 2010]. To ensure fidelity of the serial measurements, it has been proposed in a recent consensus article [Klein et al., 2009] that registration procedures be validated with respect to linearity (the inverse consistency of forward and reverse transformations between image pairs), and transitivity (e.g., the total change calculated when registering visit 3 to visit 1 should equal the sum calculated when registering visit 3 to visit 2, and visit 2 to visit 1). The methodologies used in ADNI, including Quarc, have not been validated in this respect. Therefore, caution is advised when considering the validity of the nonlinear trajectories that have been published based on ADNI data.
CONCLUSION
ADNI has been revolutionary in its pioneering of the open source model of data sharing, making raw data and derived measures freely accessible to the scientific community and industry as soon as they become available. It has been highly successful in advancing research on biomarkers in AD. ADNI results are being used by the pharmaceutical industry to aid in decision‐making on the choice of biomarkers for use as outcome measures, and for powering clinical trials. Ultimately, ADNI data and analyses may form the basis for regulatory qualification for imaging biomarkers.
It is thus essential that these biomarkers are validated and that the models used for power calculations be consistent with the biological mechanisms targeted by the therapy under investigation. It should be noted, however, that establishing imaging biomarkers as surrogates for clinical‐cognitive outcomes cannot be achieved with natural history studies, but will require successful clinical trials, that is, ones where cognitive outcomes are improved and where there is a clear and cogent biological connection with the imaging measures [Carrillo et al., 2009; Katz, 2004]. Establishing the surrogacy of biomarkers will be all the more difficult if those biomarkers are not correctly calibrated for non‐disease‐related effects. For therapies targeting Aβ pathology, it is not reasonable to expect that they will affect atrophy rates observed in healthy individuals without evidence of Aβ pathology. Since such individuals show atrophy rates in AD‐vulnerable structures equivalent to those of the larger HC group, the potentially treatable effect is best conservatively defined as change relative to that experience by HCs. It is also essential to provide confidence intervals for any sample size estimates to enable selection of biomarkers that estimate change with the highest certainty [Holland et al., 2009; McEvoy et al., 2010; Schott et al., 2010].
As part of the stated goals of the study, ADNI has been charged with statistically evaluating and comparing the different biomarkers and analysis methods to inform clinical trial design. To date, results presented and published by ADNI have not taken these critical issues into account. They report sample size estimates based only on absolute change measures; they have not considered issues of potential bias in image registration; and they have not provided a clear index of uncertainty of the results. If left uncorrected, these findings could lead to the adoption of suboptimal biomarkers for outcome measures, and to trials that are substantially underpowered for detecting potential disease‐modifying effects.
Acknowledgements
The authors thank Yoon Chung, Trevor Cooper, Rahul Desikan, Matt Erhart, Donald Hagler, Robin Jennings, Alan Koyama, and Chris Pung. This study is coordinated by the Alzheimer's Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of California, Los Angeles. Anders M. Dale is a founder and holds equity in CorTechs Labs, Inc, and also serves on its Scientific Advisory Board. The terms of this arrangement have been reviewed and approved by the University of California, San Diego in accordance with its conflict of interest policies. Linda K. McEvoy's spouse is President of CorTechs Labs, Inc. A patent application for Quarc has been filed through the UCSD Technology Transfer Office.
Data used in the preparation of this article were obtained from the Alzheimer's Disease Neuroimaging Initiative (ADNI) database (http://www.loni.ucla.edu/ADNI). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. Complete listing of ADNI investigators available at, http://www.loni.ucla.edu/ADNI/Data/ADNI_Authorship_List.pdf
REFERENCES
- Aisen PS, Petersen RC, Donohue MC, Gamst A, Raman R, Thomas RG, Walter S, Trojanowski JQ, Shaw LM, Beckett LA, Jack CR, Jr. , Jagust W, Toga AW, Saykin AJ, Morris JC, Green RC, Weiner MW ( 2010): Clinical Core of the Alzheimer's Disease Neuroimaging Initiative: progress and plans. Alzheimers Dement 6: 239–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander GE, Chen K, Reiman EM ( 2010): adni_uaspmvbm_ dict_2010–05‐23.csv. www.loni.ucla.edu/ADNI.
- Ashburner J, Friston KJ ( 2000): Voxel‐based morphometry–the methods. Neuroimage 11: 805–821. [DOI] [PubMed] [Google Scholar]
- Baker SG, Kramer BS ( 2003): A perfect correlate does not a surrogate make. BMC Med Res Methodol 3: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckett LA, Harvey DJ, Gamst A, Donohue M, Kornak J, Zhang H, Kuo JH ( 2010): The Alzheimer's Disease Neuroimaging Initiative: Annual change in biomarkers and clinical outcomes. Alzheimers Dement 6: 257–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K, Hampel H, Weiner M, Zetterberg H ( 2010): Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol 6: 131–144. [DOI] [PubMed] [Google Scholar]
- Caroli A, Frisoni GB ( 2010): The dynamics of Alzheimer's disease biomarkers in the Alzheimer's Disease Neuroimaging Initiative cohort. Neurobiol Aging 31: 1263–1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrillo MC, Blackwell A, Hampel H, Lindborg J, Sperling R, Schenk D, Sevigny JJ, Ferris S, Bennett DA, Craft S, Hsu T, Klunk W ( 2009): Early risk assessment for Alzheimer's disease. Alzheimers Dement 5: 182–196. [DOI] [PubMed] [Google Scholar]
- Casella G, Berger RL ( 2002): Statistical inference. Australia; Pacific Grove, CA: Thomson Learning; xxviii, 660 p. p. [Google Scholar]
- Chetelat G, Landeau B, Eustache F, Mezenge F, Viader F, de la Sayette V, Desgranges B, Baron JC ( 2005): Using voxel‐based morphometry to map the structural changes associated with rapid conversion in MCI: a longitudinal MRI study. Neuroimage 27: 934–946. [DOI] [PubMed] [Google Scholar]
- Chetelat G, Villemagne VL, Pike KE, Baron JC, Bourgeat P, Jones G, Faux NG, Ellis KA, Salvado O, Szoeke C, Martins RN, Ames D, Masters CL, Rowe CC ( 2010): Larger temporal volume in elderly with high versus low beta‐amyloid deposition. Brain 133: 3349–3358. [DOI] [PubMed] [Google Scholar]
- Christensen GE ( 1999): Consistent linear‐elastic transformations for image matching. Information Processing in Medical Imaging, Proceedings 1613: 224–237. [Google Scholar]
- Christensen GE, Johnson HJ ( 2001): Consistent image registration. IEEE Trans Med Imaging 20: 568–582. [DOI] [PubMed] [Google Scholar]
- Citron M ( 2010): Alzheimer's disease: strategies for disease modification. Nat Rev Drug Discov 9: 387–398. [DOI] [PubMed] [Google Scholar]
- Cummings JL ( 2010): Integrating ADNI results into Alzheimer's disease drug development programs. Neurobiol Aging 31: 1481–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dale AM, Fischl B, Sereno MI ( 1999): Cortical surface‐based analysis. I. Segmentation and surface reconstruction. Neuroimage 9: 179–194. [DOI] [PubMed] [Google Scholar]
- Dale AM, Sereno MI ( 1993): Improved localization of cortical activity by combining EEG and MEG with MRI cortical surface reconstruction. Journal of Cognitive Neuroscience 5: 162–176. [DOI] [PubMed] [Google Scholar]
- Fagan AM, Mintun MA, Shah AR, Aldea P, Roe CM, Mach RH, Marcus D, Morris JC, Holtzman DM ( 2009): Cerebrospinal fluid tau and ptau(181) increase with cortical amyloid deposition in cognitively normal individuals: implications for future clinical trials of Alzheimer's disease. EMBO Mol Med 1: 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Y, Batmanghelich N, Clark CM, Davatzikos C ( 2008): Spatial patterns of brain atrophy in MCI patients, identified via high‐dimensional pattern classification, predict subsequent cognitive decline. Neuroimage 39: 1731–1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl B, Salat DH, Busa E, Albert M, Dieterich M, Haselgrove C, van der Kouwe A, Killiany R, Kennedy D, Klaveness S, Montillo A, Makris N, Rosen B, Dale AM ( 2002): Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 33: 341–355. [DOI] [PubMed] [Google Scholar]
- Fischl B, Sereno MI, Dale AM ( 1999): Cortical surface‐based analysis. II: Inflation, flattening, and a surface‐based coordinate system. Neuroimage 9: 195–207. [DOI] [PubMed] [Google Scholar]
- Fitzmaurice GM, Laird NM, Ware JH ( 2004): Applied longitudinal analysis. Hoboken, N.J.: Wiley‐Interscience; xix, 506 p. p. [Google Scholar]
- Fjell AM, Walhovd KB, Fennema‐Notestine C, McEvoy LK, Hagler DJ, Holland D, Blennow K, Brewer JB, Dale AM ( 2010): Brain atrophy in healthy aging is related to CSF levels of Abeta1–42. Cereb Cortex 20: 2069–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fjell AM, Walhovd KB, Fennema‐Notestine C, McEvoy LK, Hagler DJ, Holland D, Brewer JB, Dale AM ( 2009): One‐year brain atrophy evident in healthy aging. J Neurosci 29: 15223–15231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fotenos AF, Snyder AZ, Girton LE, Morris JC, Buckner RL ( 2005): Normative estimates of cross‐sectional and longitudinal brain volume decline in aging and AD. Neurology 64: 1032–1039. [DOI] [PubMed] [Google Scholar]
- Fox NC, Black RS, Gilman S, Rossor MN, Griffith SG, Jenkins L, Koller M ( 2005): Effects of Abeta immunization (AN1792) on MRI measures of cerebral volume in Alzheimer disease. Neurology 64: 1563–1572. [DOI] [PubMed] [Google Scholar]
- Fox NC, Cousens S, Scahill R, Harvey RJ, Rossor MN ( 2000): Using serial registered brain magnetic resonance imaging to measure disease progression in Alzheimer disease: power calculations and estimates of sample size to detect treatment effects. Arch Neurol 57: 339–344. [DOI] [PubMed] [Google Scholar]
- Freeborough PA, Fox NC ( 1997): The boundary shift integral: an accurate and robust measure of cerebral volume changes from registered repeat MRI. IEEE Trans Med Imaging 16: 623–629. [DOI] [PubMed] [Google Scholar]
- Freeman SH, Kandel R, Cruz L, Rozkalne A, Newell K, Frosch MP, Hedley‐Whyte ET, Locascio JJ, Lipsitz LA, Hyman BT ( 2008): Preservation of neuronal number despite age‐related cortical brain atrophy in elderly subjects without Alzheimer disease. J Neuropathol Exp Neurol 67: 1205–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisoni GB, Fox NC, Jack CR, Jr. , Scheltens P, Thompson PM ( 2010): The clinical use of structural MRI in Alzheimer disease. Nat Rev Neurol 6: 67–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM ( 2005): Clinical effects of Abeta immunization (AN1792) in patients with AD in an interrupted trial. Neurology 64: 1553–1562. [DOI] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ ( 2002): The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science 297: 353–356. [DOI] [PubMed] [Google Scholar]
- Herrup K ( 2010): Reimagining Alzheimer's disease–an age‐based hypothesis. J Neurosci 30: 16755–16762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho AJ, Hua X, Lee S, Leow AD, Yanovsky I, Gutman B, Dinov ID, Lepore N, Stein JL, Toga AW, Jack CR, Jr. , Bernstein MA, Reiman EM, Harvey DJ, Kornak J, Schuff N, Alexander GE, Weiner MW, Thompson PM ( 2010): Comparing 3 T and 1.5 T MRI for tracking Alzheimer's disease progression with tensor‐based morphometry. Hum Brain Mapp 31: 499–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland D, Brewer JB, Hagler DJ, Fenema‐Notestine C, Dale AM ( 2009): Subregional neuroanatomical change as a biomarker for Alzheimer's disease. Proc Natl Acad Sci U S A 106: 20954–20959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland D, Dale AM ( 2011): Nonlinear registration of longitudinal images and measurement of change in regions of interest. Med Image Anal 15: 489–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Boche D, Wilkinson D, Yadegarfar G, Hopkins V, Bayer A, Jones RW, Bullock R, Love S, Neal JW, Zotova E, Nicoll JA ( 2008): Long‐term effects of Abeta42 immunisation in Alzheimer's disease: follow‐up of a randomised, placebo‐controlled phase I trial. Lancet 372: 216–223. [DOI] [PubMed] [Google Scholar]
- Hua X, Gutman B, Boyle CP, Rajagopalan P, Leow AD, Yanovsky I, Kumar AR, Toga AW, Jack CR, Jr. , Schuff N, Alexander GE, Chen K, Reiman EM, Weiner MW, Thompson PM ( 2011): Accurate measurement of brain changes in longitudinal MRI scans using tensor‐based morphometry. Neuroimage 57: 5–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Hibar DP, Lee S, Toga AW, Jack CR, Jr. , Weiner MW, Thompson PM ( 2010): Sex and age differences in atrophic rates: an ADNI study with n=1368 MRI scans. Neurobiol Aging 31: 1463–1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Lee S, Yanovsky I, Leow AD, Chou YY, Ho AJ, Gutman B, Toga AW, Jack CR, Jr. , Bernstein MA, Reiman EM, Harvey DJ, Kornak J, Schuff N, Alexander GE, Weiner MW, Thompson PM ( 2009): Optimizing power to track brain degeneration in Alzheimer's disease and mild cognitive impairment with tensor‐based morphometry: an ADNI study of 515 subjects. Neuroimage 48: 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Leow AD, Lee S, Klunder AD, Toga AW, Lepore N, Chou YY, Brun C, Chiang MC, Barysheva M, Jack CR, Jr. , Bernstein MA, Britson PJ, Ward CP, Whitwell JL, Borowski B, Fleisher AS, Fox NC, Boyes RG, Barnes J, Harvey D, Kornak J, Schuff N, Boreta L, Alexander GE, Weiner MW, Thompson PM, Alzheimer's Disease Neuroimaging I ( 2008a): 3D characterization of brain atrophy in Alzheimer's disease and mild cognitive impairment using tensor‐based morphometry. Neuroimage 41: 19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua X, Leow AD, Parikshak N, Lee S, Chiang MC, Toga AW, Jack CR,Jr. , Weiner MW, Thompson PM ( 2008b): Tensor‐based morphometry as a neuroimaging biomarker for Alzheimer's disease: an MRI study of 676 AD, MCI, and normal subjects. Neuroimage 43: 458–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman BT ( 2011): Amyloid‐Dependent and Amyloid‐Independent Stages of Alzheimer Disease. Arch Neurol. [DOI] [PubMed] [Google Scholar]
- Ikonomovic MD, Klunk WE, Abrahamson EE, Mathis CA, Price JC, Tsopelas ND, Lopresti BJ, Ziolko S, Bi W, Paljug WR, Debnath ML, Hope CE, Isanski BA, Hamilton RL, DeKosky ST ( 2008): Post‐mortem correlates of in vivo PiB‐PET amyloid imaging in a typical case of Alzheimer's disease. Brain 131: 1630–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr. , Bernstein MA, Borowski BJ, Gunter JL, Fox NC, Thompson PM, Schuff N, Krueger G, Killiany RJ, Decarli CS, Dale AM, Carmichael OW, Tosun D, Weiner MW ( 2010a): Update on the magnetic resonance imaging core of the Alzheimer's disease neuroimaging initiative. Alzheimers Dement 6: 212–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR,Jr. , Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, Petersen RC, Trojanowski JQ ( 2010b): Hypothetical model of dynamic biomarkers of the Alzheimer's pathological cascade. Lancet Neurol 9: 119–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr. , Petersen RC, Xu YC, O'Brien PC, Smith GE, Ivnik RJ, Boeve BF, Waring SC, Tangalos EG, Kokmen E ( 1999): Prediction of AD with MRI‐based hippocampal volume in mild cognitive impairment. Neurology 52: 1397–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr. , Shiung MM, Gunter JL, O'Brien PC, Weigand SD, Knopman DS, Boeve BF, Ivnik RJ, Smith GE, Cha RH, Tangalos EG, Petersen RC ( 2004): Comparison of different MRI brain atrophy rate measures with clinical disease progression in AD. Neurology 62: 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, Jr. , Weigand SD, Shiung MM, Przybelski SA, O'Brien PC, Gunter JL, Knopman DS, Boeve BF, Smith GE, Petersen RC ( 2008): Atrophy rates accelerate in amnestic mild cognitive impairment. Neurology 70: 1740–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz R ( 2004): Biomarkers and surrogate markers: an FDA perspective. NeuroRx 1: 189–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein A, Andersson J, Ardekani BA, Ashburner J, Avants B, Chiang MC, Christensen GE, Collins DL, Gee J, Hellier P, Song JH, Jenkinson M, Lepage C, Rueckert D, Thompson P, Vercauteren T, Woods RP, Mann JJ, Parsey RV ( 2009): Evaluation of 14 nonlinear deformation algorithms applied to human brain MRI registration. Neuroimage 46: 786–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohannim O, Hua X, Hibar DP, Lee S, Chou YY, Toga AW, Jack CR, Jr. , Weiner MW, Thompson PM ( 2010): Boosting power for clinical trials using classifiers based on multiple biomarkers. Neurobiol Aging 31: 1429–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacevic S, Rafii MS, Brewer JB ( 2009): High‐throughput, fully automated volumetry for prediction of MMSE and CDR decline in mild cognitive impairment. Alzheimer Dis Assoc Disord 23: 139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leow AD, Yanovsky I, Chiang MC, Lee AD, Klunder AD, Lu A, Becker JT, Davis SW, Toga AW, Thompson PM ( 2007): Statistical properties of Jacobian maps and the realization of unbiased large‐deformation nonlinear image registration. IEEE Trans Med Imaging 26: 822–832. [DOI] [PubMed] [Google Scholar]
- Leung KK, Barnes J, Ridgway GR, Bartlett JW, Clarkson MJ, Macdonald K, Schuff N, Fox NC, Ourselin S ( 2010a): Automated cross‐sectional and longitudinal hippocampal volume measurement in mild cognitive impairment and Alzheimer's disease. Neuroimage 51: 1345–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung KK, Clarkson MJ, Bartlett JW, Clegg S, Jack CR, Jr. , Weiner MW, Fox NC, Ourselin S ( 2010b): Robust atrophy rate measurement in Alzheimer's disease using multi‐site serial MRI: tissue‐specific intensity normalization and parameter selection. Neuroimage 50: 516–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzi M, Donohue M, Paternico D, Scarpazza C, Ostrowitzki S, Blin O, Irving E, Frisoni GB ( 2010): Enrichment through biomarkers in clinical trials of Alzheimer's drugs in patients with mild cognitive impairment. Neurobiol Aging 31: 1443–1451. [DOI] [PubMed] [Google Scholar]
- Mani RB ( 2004): The evaluation of disease modifying therapies in Alzheimer's disease: a regulatory viewpoint. Stat Med 23: 305–314. [DOI] [PubMed] [Google Scholar]
- Markesbery WR, Schmitt FA, Kryscio RJ, Davis DG, Smith CD, Wekstein DR ( 2006): Neuropathologic substrate of mild cognitive impairment. Arch Neurol 63: 38–46. [DOI] [PubMed] [Google Scholar]
- McDonald CR, McEvoy LK, Gharapetian L, Fennema‐Notestine C, Hagler DJ, Jr. , Holland D, Koyama A, Brewer JB, Dale AM ( 2009): Regional rates of neocortical atrophy from normal aging to early Alzheimer disease. Neurology 73: 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy LK, Edland SD, Holland D, Hagler DJ, Jr. , Roddey JC, Fennema‐Notestine C, Salmon DP, Koyama AK, Aisen PS, Brewer JB, Dale AM ( 2010): Neuroimaging enrichment strategy for secondary prevention trials in Alzheimer disease. Alzheimer Dis Assoc Disord 24: 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy LK, Fennema‐Notestine C, Roddey JC, Hagler DJ, Jr. , Holland D, Karow DS, Pung CJ, Brewer JB, Dale AM ( 2009): Alzheimer disease: quantitative structural neuroimaging for detection and prediction of clinical and structural changes in mild cognitive impairment. Radiology 251: 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McEvoy LK, Holland D, Hagler DJ,Jr. , Fennema‐Notestine C, Brewer JB, Dale AM ( 2011): Mild Cognitive Impairment: Baseline and Longitudinal Structural MR Imaging Measures Improve Predictive Prognosis. Radiology. 259: 834–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L ( 1991): The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41: 479–486. [DOI] [PubMed] [Google Scholar]
- Morris JC, Roe CM, Xiong C, Fagan AM, Goate AM, Holtzman DM, Mintun MA ( 2010): APOE predicts amyloid‐beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol 67: 122–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller SG, Weiner MW, Thal LJ, Petersen RC, Jack CR, Jagust W, Trojanowski JQ, Toga AW, Beckett L ( 2005): Ways toward an early diagnosis in Alzheimer's disease: the Alzheimer's Disease Neuroimaging Initiative (ADNI). Alzheimers Dement 1: 55–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestor SM, Rupsingh R, Borrie M, Smith M, Accomazzi V, Wells JL, Fogarty J, Bartha R ( 2008): Ventricular enlargement as a possible measure of Alzheimer's disease progression validated using the Alzheimer's disease neuroimaging initiative database. Brain 131: 2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrin RJ, Fagan AM, Holtzman DM ( 2009): Multimodal techniques for diagnosis and prognosis of Alzheimer's disease. Nature 461: 916–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen RC, Aisen PS, Beckett LA, Donohue MC, Gamst AC, Harvey DJ, Jack CR, Jr. , Jagust WJ, Shaw LM, Toga AW, Trojanowski JQ, Weiner MW ( 2010): Alzheimer's Disease Neuroimaging Initiative (ADNI): clinical characterization. Neurology 74: 201–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC ( 2001): Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol 58: 1395–1402. [DOI] [PubMed] [Google Scholar]
- Price JL, McKeel DW, Jr. , Buckles VD, Roe CM, Xiong C, Grundman M, Hansen LA, Petersen RC, Parisi JE, Dickson DW, Smith CD, Davis DG, Schmitt FA, Markesbery WR, Kaye J, Kurlan R, Hulette C, Kurland BF, Higdon R, Kukull W, Morris JC ( 2009): Neuropathology of nondemented aging: presumptive evidence for preclinical Alzheimer disease. Neurobiol Aging 30: 1026–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Morris JC ( 1999): Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol 45: 358–368. [DOI] [PubMed] [Google Scholar]
- Rabinovici GD, Jagust WJ ( 2009): Amyloid imaging in aging and dementia: testing the amyloid hypothesis in vivo. Behav Neurol 21: 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reuter M, Rosas HD, Fischl B ( 2010): Highly accurate inverse consistent registration: a robust approach. Neuroimage 53: 1181–1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridha BH, Barnes J, Bartlett JW, Godbolt A, Pepple T, Rossor MN, Fox NC ( 2006): Tracking atrophy progression in familial Alzheimer's disease: a serial MRI study. Lancet neurology 5: 828–834. [DOI] [PubMed] [Google Scholar]
- Risacher SL, Shen L, West JD, Kim S, McDonald BC, Beckett LA, Harvey DJ, Jack CR, Jr. , Weiner MW, Saykin AJ ( 2010): Longitudinal MRI atrophy biomarkers: relationship to conversion in the ADNI cohort. Neurobiol Aging 31: 1401–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe CC, Ellis KA, Rimajova M, Bourgeat P, Pike KE, Jones G, Fripp J, Tochon‐Danguy H, Morandeau L, O'Keefe G, Price R, Raniga P, Robins P, Acosta O, Lenzo N, Szoeke C, Salvado O, Head R, Martins R, Masters CL, Ames D, Villemagne VL ( 2010): Amyloid imaging results from the Australian Imaging, Biomarkers and Lifestyle (AIBL) study of aging. Neurobiology of aging 31: 1275–1283. [DOI] [PubMed] [Google Scholar]
- Salloway S, Mintzer J, Weiner MF, Cummings JL ( 2008): Disease‐modifying therapies in Alzheimer's disease. Alzheimers Dement 4: 65–79. [DOI] [PubMed] [Google Scholar]
- Schott JM, Bartlett JW, Barnes J, Leung KK, Ourselin S, Fox NC ( 2010a): Reduced sample sizes for atrophy outcomes in Alzheimer's disease trials: baseline adjustment. Neurobiol Aging 31: 1452–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schott JM, Bartlett JW, Fox NC, Barnes J ( 2010b): Increased brain atrophy rates in cognitively normal older adults with low cerebrospinal fluid Abeta1–42. Ann Neurol 68: 825–834. [DOI] [PubMed] [Google Scholar]
- Schuff N, Woerner N, Boreta L, Kornfield T, Shaw LM, Trojanowski JQ, Thompson PM, Jack CR, Jr. , Weiner MW ( 2009): MRI of hippocampal volume loss in early Alzheimer's disease in relation to ApoE genotype and biomarkers. Brain 132: 1067–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik‐Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM, Trojanowski JQ ( 2009): Cerebrospinal fluid biomarker signature in Alzheimer's disease neuroimaging initiative subjects. Ann Neurol 65: 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson WK, Holland D ( 2011): Bias in tensor based morphometry Stat‐ROI measures may result in unrealistic power estimates. Neuroimage 57: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trojanowski JQ, Vandeerstichele H, Korecka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter WZ, Weiner MW, Jack CR, Jr. , Jagust W, Toga AW, Lee VM, Shaw LM ( 2010): Update on the biomarker core of the Alzheimer's Disease Neuroimaging Initiative subjects. Alzheimers Dement 6: 230–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzourio‐Mazoyer N, Landeau B, Papathanassiou D, Crivello F, Etard O, Delcroix N, Mazoyer B, Joliot M ( 2002): Automated anatomical labeling of activations in SPM using a macroscopic anatomical parcellation of the MNI MRI single‐subject brain. Neuroimage 15: 273–289. [DOI] [PubMed] [Google Scholar]
- Vemuri P, Whitwell JL, Kantarci K, Josephs KA, Parisi JE, Shiung MS, Knopman DS, Boeve BF, Petersen RC, Dickson DW, Jack CR, Jr. ( 2008): Antemortem MRI based STructural Abnormality iNDex (STAND)‐scores correlate with postmortem Braak neurofibrillary tangle stage. Neuroimage 42: 559–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Knopman DS, Trojanowski JQ, Shaw LM, Bernstein MA, Aisen PS, Weiner M, Petersen RC, Jack CR, Jr. , ( 2010): Serial MRI and CSF biomarkers in normal aging, MCI, and AD. Neurology 75: 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, Knopman DS, Petersen RC, Jack CR, Jr. , ( 2009a): MRI and CSF biomarkers in normal, MCI, and AD subjects: diagnostic discrimination and cognitive correlations. Neurology 73: 287–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vemuri P, Wiste HJ, Weigand SD, Shaw LM, Trojanowski JQ, Weiner MW, Knopman DS, Petersen RC, Jack CR, Jr. , ( 2009b): MRI and CSF biomarkers in normal, MCI, and AD subjects: predicting future clinical change. Neurology 73: 294–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West MJ, Kawas CH, Stewart WF, Rudow GL, Troncoso JC ( 2004): Hippocampal neurons in pre‐clinical Alzheimer's disease. Neurobiol Aging 25: 1205–1212. [DOI] [PubMed] [Google Scholar]
- Yanovsky I, Leow AD, Lee S, Osher SJ, Thompson PM ( 2009): Comparing registration methods for mapping brain change using tensor‐based morphometry. Med Image Anal 13: 679–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yushkevich PA, Avants BB, Das SR, Pluta J, Altinay M, Craige C ( 2010): Bias in estimation of hippocampal atrophy using deformation‐based morphometry arises from asymmetric global normalization: an illustration in ADNI 3 T MRI data. Neuroimage 50: 434–445. [DOI] [PMC free article] [PubMed] [Google Scholar]