

Abstract
Pulmonary arterial hypertension (PAH) and hepatopulmonary syndrome (HPS) are rare pulmonary vascular complications of type 1 Gaucher disease (GD1). We examined GBA1 genotype, spleen status, Severity Score Index (SSI), and other patient characteristics as determinants of GD/PAH-HPS phenotype. We also examined the long-term outcomes of imiglucerase enzyme replacement therapy (ERT) +/− adjuvant therapies in 14 consecutive patients. We hypothesized a role of BMPR2 and ALK1 as genetic modifiers underlying GD/PAH-HPS phenotype. Median age at diagnosis of GD1 was 5 yrs (2–22); PAH was diagnosed at median 36 yrs (22–63). There was a preponderance of females (ratio 5:2). ERT was commenced at median 36.5 yrs (16–53) and adjuvant therapy at 36 yrs (24–57). GBA1 genotype was N370S homozygous in two patients, N370S heteroallelic in 12. Median SSI was 15 (7–20). All patients had undergone splenectomy at median age 12 yrs (2–30). In three patients, HPS was the initial presentation, and PAH developed after its resolution; in these three, HPS responded dramatically to ERT. In seven patients, sequencing of the coding regions of BMPR2 and ALK1 was undertaken: 3/7 were heterozygous for BMPR2 polymorphisms; none harbored ALK1 variants. With ERT (± adjuvant therapy), 5/14 improved dramatically, five remained stable, two worsened, and two died prematurely. In this largest series of GD/PAH-HPS patients, there is preponderance of females and N370S heteroallelic GBA1 genotype. Splenectomy appears essential to development of this phenotype. In some patients, HPS precedes PAH. BMPR2 and ALK1 appear not be modifier genes for this rare phenotype of GD. ERT +/− adjuvant therapy improves prognosis of this devastating GD phenotype.
Introduction
Inherited deficiency of acid β-glucosidase in Gaucher disease (GD) leads to a complex multisystemic phenotype that is attributed to widespread accumulation of glycolipid-engorged macrophages (Grabowski et al. 2006). Phenotypic expression is highly heterogenous and mutations in GBA1 gene that encodes acid β-glucosidase does not explain this extreme variability (Mistry et al. 2010a; Grabowski 2008). A number of manifestations of GD cannot be directly attributed to lysosomal accumulation of glycolipids, such as predilection to gallstones (Taddei et al. 2009), cancers (Taddei et al. 2009), pulmonary hypertension (Mistry et al. 2002; Elstein et al. 1998) and hepatopulmonary syndrome (Dawson et al. 1996). The latter two pulmonary vascular manifestations affect only a minority of patients but they result in life-threatening disease. In fact in the pre-enzyme replacement therapy with alglucerase/imiglucerase (ERT) era, these uncommon complications of GD were recognized to be a cause of premature deaths (Lee and Yousem 1988). The clinical spectrum, determinants of GD phenotype that exhibit symptomatic pulmonary vascular disease, and long-term outcomes of response to therapy are not known.
Pulmonary arterial hypertension (PAH) and hepatopulmonary syndrome (HPS) are associated with a number of other diseases and etiologies that result in significant morbidity and mortality. Primary PAH is due to mutations in BMPR2 gene. In another monogenic disease, hereditary hemorrhagic telangiectasia mutations in ALK1 gene lead to a broader phenotypic expression of pulmonary vascular disease ranging from predominantly PAH to predominantly intrapulmonary vascular dilatations. The penetrance of mutations in these two genes is low, and mutations lead to highly variable phenotypic expression of pulmonary vascular disease (Newman et al. 2008; Austin and Loyd 2007; Morse 2002). In GD, we reported that pathological lesions of PAH (plexogenic arteriopathy) and intrapulmonary shunting (intrapulmonary vascular dilatations) may occur simultaneously in the same patient, suggesting that a common pathophysiologic mechanism may underlie the extreme of the spectrum of pulmonary vascular disease (Mistry et al. 2002). Three distinct patterns of lung involvement in GD are recognized: (1) alveolar consolidation by Gaucher cells filling alveolar spaces, which usually occurs in types 2 and 3 GD, (2) interstitial infiltrates of Gaucher cells with associated fibrosis, which usually occurs in type 3 GD, and (3) PAH which usually occurs in type 1 GD (Lee and Yousem 1988). In addition, HPS has been described in GD patients with advanced liver involvement (Lachmann et al. 2000). We and others have reported a relatively high prevalence of mild PAH in GD1 by Doppler echocardiography (Elstein et al. 1998) that improves with ERT (Mistry et al. 2002). In addition, we found that severe, potentially life-threatening PAH occurred in ~1% of GD patients (compared to one in one million of the general population).
Herein, we report 14 consecutive patients evaluated at our center with GD/PAH-HPS phenotype, representing the largest case series ever described (Table 1). We examined GBA1 genotype, spleen status, GD Severity Score Index (SSI) and other clinical characteristics of patients as determinants of GD/PAH-HPS phenotype as well as long-term outcomes of enzyme replacement therapy (ERT) +/− adjuvant therapies. We examined the potential role of two candidate genes, BMPR2 and ALK1, as genetic modifiers that result in pulmonary vascular disease in GD1.
Table 1.
Characteristics of 14 patients with GD1 and Severe PAH/HPS
| Patient | GBA1 Genotype | Ethnicity (A/O) | Age at diagnosis of GD | Age at splenectomy | Age at diagnosis of Hepato-pulmonary syndrome | Age at diagnosis of Pulmonary HTN | SSI | Age at ERT | Adjuvant therapy-age initiated | PVR (Wood units) | PAP (mmHg) | Outcome | BMPR2 mutationsa | ALK1 mutationsa | Current Age |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | N370S/T3231 | O | ND | 13 | NA | 48 | 13 | 48 | Bosentan, 57 | 8–10 | 93/16 | Deceased, age 59 | NP | NP | Deceased |
| 2a | N370S/84GG | A | 5 | ~5–10 | 22 | 32 | 20 | 29 | Ø | ND | 49/16 | Resolved | None | None | 49 |
| 3 | N370S/L444P | O | 4 | 7 | NA | 24 | 7 | 16 | Prostacyclin, 24 | 19.2 | 95/45 | Deceased age 26; sepsis | NP | NP | Deceased |
| 4 | N370S/L444P | O | 5 | 16 | NA | 36 | 8 | 43 | Prostacyclin, 49; Bosentan, 52 | ND | 61/26, mean 38 | R & L sided CHF | NP | NP | 61 |
| 5 | N370S/84GG | ND | 2 | 7 | NA | 27 | 14 | 21 | Bosentan, ND | 3.4 | 55/20, mean 32 | Much improved | S775N (AGC→AAC) heterozygous | None | 38 |
| 6 | N370S/IVS2+1 | A | 14 | 14 | NA | 36 | 17 | 36 | Bosentan, 46; Sildenafil, 52 | 7.1 | ND | NYHA 1 | None | None | 54 |
| 7a | N370S/55 b.p. del. | O | 3 | 8 | 31 | 35 | 17 | 33 | Ø | ND | ND | Resolved | None | None | 50 |
| 8 | N370S/L444P | O | 5 | 11 | NA | 44 | 8 | 37 | Ø | ND | 77/35, mean 51 | Much improved | NP | NP | 30 |
| 9a | N370S/R257Q | O | 9 | 19 | ND | 63 | 20 | 50 | Ø | ND | 94/32, mean 57 | Minimally improved | R937R (AGG→AGA) heterozygous | None | 66 |
| 10 | N370S/R257Q | ND | 2 | 2 | NA | 39 | 16 | 39 | Bosentan, ND | ND | Systolic 60–70 | NYHA 2–3 | NP | NP | 57 |
| 11 | N370S/N370S | ND | 2 | 17 | NA | 49 | ND | 49 | Prostacyclin, ND | ND | Systolic 130 | Minimally improved | NP | NP | 67 |
| 12 | N370S/L444P | O | 19 | 19 | NA | 33 | 14 | 30 | Sildenafil, 34; Bosentan, 34 | 2.8 | ND | Much improved | R937R (AGG→AGA) heterozygous | None | 41 |
| 13 | ND | A | 3 | 3 | NA | 22 | ND | 22 | Prostacyclin, ND | ND | Systolic 85 | ND | NP | NP | 41 |
| 14 | N370S/N370S | A | 22 | 30 | NA | 63 | 17 | 53 | ND | ND | Systolic 77 | CHF, age 71 | None | None | 72 |
A Ashkenazi Jewish; O Other, i.e., non-Jewish; NA Not applicable; ND Not determined; NP Not performed; PVR Pulmonary vascular resistance; PAP Pulmonary arterial pressure
Patient first presented with HPS
Please see SI for PCR conditions and primer sequences
Materials and methods
We performed full phenotypic/genotypic characterization and assessed long-term outcomes of treatment in 14 consecutive patients with GD1 and severe PAH with or without HPS. These patients were drawn from a population of patients who presented to a tertiary GD referral clinic. The patients had confirmed diagnosis of GD by demonstration of low leukocyte acid β-glucosidase activity (<10% of normal), supplemented by GBA1 genotyping. All patients were clinically deemed to have type 1 GD; indeed all harbored at least one N370S mutant GBA1 allele. Patients with clinical symptoms of PAH or HPS were further evaluated. Patients were diagnosed with severe PAH defined by symptoms and initial Doppler echocardiographic estimate of pulmonary artery pressure >50 mmHg and right heart catheterization demonstrating all of the following: mean pulmonary pressure >25 mm Hg, pulmonary vascular resistance >120 dynes-sec-cm5, and pulmonary capillary wedge pressure <15 mm Hg. Patients were excluded if they had any of the following: presence of severe cardiac disease not related to GD, presence of severe COPD, presence of an active infectious disease, history of anorexin use, or history of alcoholism or drug abuse. Diagnosis of HPS was based on classical clinical criteria of hypoxemia on room air, digital clubbing, signs of advanced hepatic disease (massive hepatomegaly, impaired tests of liver function), and radionuclide scan evidence of intrapulmonary shunting.
PCR amplification of BMPR2 and ALK1 coding regions
In seven of the 14 patients, we examined BMPR2 and ALK1 genes as candidate modifier genes for GD/PAH-HPS phenotype. Coding regions together with consensus splice sequences of BMPR2 and ALK1 gene were amplified by PCR for DNA sequencing. Genomic DNA was extracted from peripheral blood of GD patients using the Puregene genomic DNA purification kit (Gentra Systems, Inc, Big Lake, MN) according to manufacturer’s protocol. DNA concentration and quality were determined on a Nanodrop 2000c spectrophotometer (Thermo Scientific) at 260 nm and 280 nm and 100–400 ng was used as primary PCR template. Normal genomic DNA was also collected as control.
All primers were synthesized at the scale of 40 nm and were used to amplify respective exon(s) of various lengths. GBA1 sequencing was performed as described previously (Tayebi et al. 1996).
Results
Patient characteristics are summarized in Table 2. There was a preponderance of females (10:4), non-Jewish patients (10:4) and N370S heteroallelic mutations compared to N370S homozygous (12:2, i.e., four N370S/L444P and two each of N370S/84GG and N370S/R257Q). The median age at diagnosis of PAH was 36, with a range of 22 to 63. Eleven patients had PAH as the sole overt pulmonary vascular abnormality, whereas three had both PAH and HPS. In the latter three patients, initial presentation was florid HPS and massive hepatomegaly that responded dramatically to ERT, unmasking PAH. Prior to diagnosis of PAH/HPS, all 14 patients had undergone splenectomy; the median age at splenectomy was 12 (range 2–30).
Table 2.
Characteristics of GD1 patients with severe PAH with or without preceding HPS (N=14)
| Gender | ||
| Males | 4 | |
| Females | 10 | |
| Age at diagnosis of GD | ||
| Median | 5 | |
| Range | 2–22 | |
| Ethnicity | ||
| Ashkenazi Jewish | 4 | |
| Other | 7 | |
| Genotype | ||
| N370S/L444P | 4 | |
| N370S/84GG | 2 | |
| N370S/R257Q | 2 | |
| N370S/N370S | 2 | |
| N370S/T3231 | 1 | |
| N370S/IVS2+1 | 1 | |
| N370S/55 b.p. del. | 1 | |
| Severity Score Index (SSI) | ||
| Median | 15 | |
| Range | 7–20 | |
| Splenectomy | ||
| Y | 14 | |
| N | 0 | |
| Age at splenectomy | ||
| Median | 12 | |
| Range | 2–30 | |
| Hepatopulmonary syndrome (HPS)/Pulmonary arterial hypertension (PAH) | ||
| HPS alone | 0 | |
| HPS/PAH | 3 | |
| PAH alone | 11 | |
| Age at ERT | ||
| Median | 36.5 | |
| Range | 16–53 | |
| Age at adjuvant therapy | ||
| Median | 36 | |
| Range | 24–57 | |
| Age at diagnosis of PHT | ||
| Median | 36 | |
| Range | 22–63 | |
| Current age | ||
| Median | 52 | |
| Range | 26–72 | |
| BMPR2 mutations | ||
| Homozygous | 0/7 | |
| Heterozygous | 3/7 | |
| ALK1 mutations | ||
| Homozygous | 0/7 | |
| Heterozygous | 0/7 | |
GD Severity Score Index (SSI), calculated at the time of PAH-HPS diagnosis, varied widely, with median value of 15, ranging from 7 to 20. SSI assigns four points for presence of pulmonary disease (Zimran et al. 1992). Therefore as depicted in Fig. 1, 3/14 patients had exceptionally low GD severity (SSI 7, 8, and 8) despite the presence of life-threatening pulmonary vascular disease. In contrast, the three patients who first presented with HPS and later exhibited PAH, had consistently high SSI. These patients with HPS had SSI’s of 17, 20, and 20, respectively. Therefore, there is a dichotomy in the relationship of SSI to pulmonary vascular disease, with patients presenting with pure PAH having low to high SSI, while those with HPS/PAH exhibit consistently high SSI.
Fig. 1.
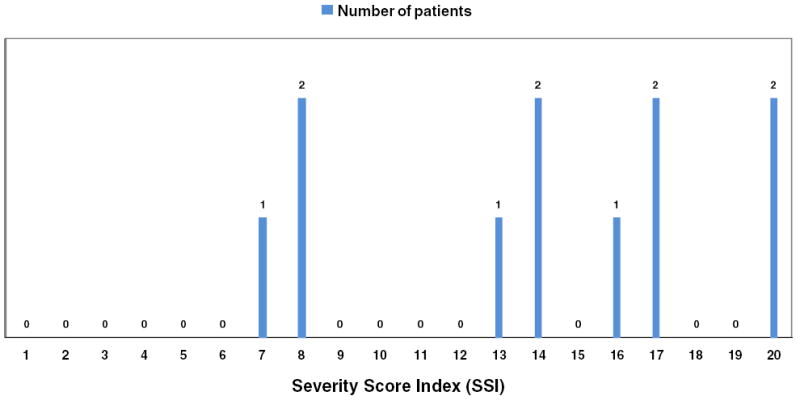
Distribution of Severity Score Index in GD1 patients with severe PAH. The numbers above each bar represent the number of patients with corresponding SSI on the X-axis
The median age at initiation of ERTwas 36.5 (range 16–53). The median age at initiation of adjuvant therapy was 36 (range 24–57). Figure 2 depicts the natural history of PAH in this series of patients. Of the seven patients tested for BMPR2 or ALK1 mutations, three were heterozygous for SNPs in BMPR2 gene (two synonymous and one nonsynonymous) but there were no variants in ALK1.
Fig. 2.

Natural history of PAH/HPS in GD1 derived from aggregate data of all 14 patients
Discussion
We report phenotypic associations and long-term outcomes of GD/PAH phenotype in 14 consecutively evaluated patients with this rare complication. Given our previous findings that the pathological lesions of PAH (plexogenic arteriopathy) and intrapulmonary shunting (intrapulmonary vascular dilatations) may occur simultaneously in GD patients, we hypothesized that common pathophysiologic mechanisms may underlie the extreme ends of the clinical spectrum of pulmonary vascular disorders (Mistry et al. 2002). Therefore, we examined ALK1 gene as a modifier of phenotype since mutations in this gene lead to pulmonary vascular disease of similar clinical spectrum; in addition we examined BMPR2 gene, which when mutated leads to primary PAH. In a minority of patients, the GD/PAH phenotype was associated with polymorphisms in BMPR2 gene (Table 1). However, no patient harbored polymorphisms or mutations in ALK1 gene (Table 1). Nevertheless, we found that severe PAH/HPS was not consistently related to GD severity, underscoring the likely significant role of hitherto unidentified genetic and other modifiers in the development of this highly specific phenotype. In our study we ruled out ALK1 and BMPR2 as candidate modifier genes. Identification of potential genetic modifiers underlying GD/pulmonary vascular disease phenotype might best be pursued using newer genomic approaches in carefully selected patients (Choi et al. 2009).
Pathophysiologic effects of GD in cell types other than macrophages have been attributed to increased extralysosomal accumulation of glucosylceramide and the effects of the minor water-soluble substrate glucosylsphingosine (Mistry et al. 2010b; Elleder 2006; Hůlková et al. 2010). Such mechanisms operating in endothelial cells in predisposed individuals may underlie the development of unusual GD phenotypes that exhibit pulmonary vascular disorders.
Another contributor to plexogenic arteriopathy in GD/PAH phenotype may be the tendency to cellular proliferation evidenced by high cancer risk in GD (Taddei et al. 2009; Lo et al. 2010). Interestingly, evidence for monoclonal proliferation of smooth muscle cells in plexiform lesions (Lee et al. 1998; Loyd et al. 1988) has led to speculation of a model of end-stage PAH similar to that of progression to cancer, with dysregulation of the cell-cycle and apoptosis as predominant features (Humbert et al. 2004; Yeager et al. 2001). Indeed, aberrant gene expression in cell cycle and apoptosis pathways has been described in a mouse model of GD1 (Mistry et al. 2010b).
As in other forms of PAH, in GD, it is likely that other genetic or environmental modifiers mediate the clinical expression of disease. Several possible mechanisms for PAH in GD1 have been suggested. Recently it has been proposed that megakaryocytes in the lungs due to extramedullary hematopoesis (EMH) might mediate PAH (Thachil 2009). Indeed megakaryocytes and EMH in the lungs has been reported in GD previously (Theise and Ursell 1990). We recently reported EMH in the liver and the spleen of a mouse model of GD1 (Mistry et al. 2010b). Other potential contributors to this phenotype include: coincidental unrelated disorders (e.g., micropulmonary emboli or idiopathic PAH concurrent with GD) and PAH as an intrinsic complication of GD. Capillary plugging by Gaucher cells was described by Ross et al., who recovered Gaucher cells from a sample of pulmonary capillary blood aspirated from a PA catheter (Ross et al. 1997). In another report, the pathologic findings were of plexogenic arteriopathy, similar to other forms of PAH (Theise and Ursell 1990).
Our patients were treated with ERT, adjuvant therapy (prostacyclin, bosentan and/or sildenafil) or a combination of both, and the majority exhibited improvement in their PAH by clinical as well as hemodynamic parameters. All 14 patients in our series were treated with imiglucerase ERT: we found that about one-third improved dramatically, one-third remained stable with long-term survival, and the remainder worsened or died prematurely. Numerous deaths in the latter group were not directly attributable to PAH but rather to intercurrent infections and overwhelming sepsis in the setting of asplenia. Therefore, the overall outcomes are clearly much better compared to non-GD patients with PAH/HPS treated with vasodilator therapies. In three of our 14 patients (~20%), the pattern of clinical diseases was initial development of HPS, which necessitated ERT, and subsequent unmasking of PAH as HPS reversed. In two of the three HPS patients, PAH ultimately resolved without requiring the initiation of adjuvant therapies, and in the third patient, it improved minimally with ERT alone. The dramatic reversal of HPS by imiglucerase ERT is unprecedented, since there are no effective medical therapies for the hepatopulmonary syndrome complicating end-stage liver disease, and liver transplantation is the only successful life-saving procedure (Rodriguez-Roisin and Krowka 1994).
It should be kept in mind that PAH was a recognized cause of premature death in GD long before the advent of ERT (Theise and Ursell 1990; Roberts and Fredrickson 1967; Smith et al. 1978). Improved therapeutic outcomes in our experience may be related to higher doses of imiglucerase ERT. The lung represents a sequestered site not readily accessible to ERT due to high uptake in the liver (Mistry et al. 1996), and therefore, a higher dose regimen may permit an adequate enzyme delivery to the lung parenchyma.
In our study, we found that severe PAH is not consistently related to GD severity, except in patients who initially presented with HPS. This is counterintuitive since life-threatening PAH would be expected to be associated with overall severe GD of commensurate degree. We found that several patients with severe PAH had low SSIs (3/14 patients had SSI ≤8), indicating that overall GD severity was mild. This finding further suggests a role for a significant effect of a genetic modifier that predisposes a minority of asplenic GD patients to life-threatening PAH.
Additionally, we observed that several (2/14 patients) of our GD1 patients with severe PAH had a normal baseline echo but their severe PAH was unmasked after exercise. Recently, genetic heterogeneity in primary PAH was revealed though stress Doppler echocardiography leading to identification of a specific sub-group of patients in whom severe PAH is only unmasked on exercise (Möller et al. 2010). Specific genetic basis of this type of PAH has not yet been identified; when this defect is revealed, it would be interesting to examine its occurrence in patients with GD1/PAH phenotype.
In keeping with the general literature on PAH, we found a significant preponderance of females amongst our GD1 patients with PAH (5:2). The basis of this sex predilection is not known. The fact that more women are affected may be caused by the loss of male fetuses from an unrecognized defect in embryologic development (Austin and Loyd 2007). In fact, an abnormal gender ratio among the offspring of obligate BMPR2 gene mutation carriers has been described (Loyd et al. 1998). The increased ratio of female live births suggests a difference in fertilization, or a possible selective demise of male fetuses (Loyd et al. 1998; Morse 2002).
Conclusions
Our study on PAH in GD1 highlights the importance of defining specific phenotypes within the vast clinical spectrum of type 1 GD. We further confirmed the invariable association of GD/PAH-HPS phenotype with asplenia. Our findings support the recommendation that splenectomy be avoided particularly in high-risk patients with GD1. Fortunately, the striking reduction of splenectomy rates because of the success of ERT (Cox et al. 2008) is likely to translate into elimination of the GD1/severe PAH phenotype in the new generation of patients. Additionally, we found compelling evidence that severe PAH is not consistently related to GD1 severity, specifically that mild GD1 patients may have life-threatening PAH. Conversely, we found that HPS patients who progressed to PAH demonstrated high SSIs as a rule. Our studies suggest that genetic modifiers not linked to the GBA1 locus may result in this severe phenotype. Although we were unable to implicate BMPR2 and ALK1 genes as modifier genes in our series, this question is worthy of further study using newer unbiased whole genomic approaches. Moreover, the favorable therapeutic outcomes of our patients support aggressive treatment with ERT+/− adjuvant therapy with sildenafil, bosentan, and anticoagulation in GD1 patients with PAH.
Acknowledgments
We would like to thank the patients for participating in these studies. We thank colleagues from around the US who have referred patients to us.
Contract grant sponsor NIH T32 post-doctoral training program in investigative hematology (T32-HL07262) supported SML. PKM is supported by NIDDK K24DK066306 mid-career clinical investigator award and research support from Genzyme Corporation. The study was supported in part by a pilot project grant from the Yale Liver Center (NIDDK 3P30DK034989).
Abbreviations
- ERT
Enzyme replacement therapy
- GD
Gaucher disease
- GD1
Type 1 Gaucher disease
- HPS
Hepatopulmonary syndrome
- PAH
Pulmonary arterial hypertension
- RVSP
Right ventricular systolic pressure
- SSI
Severity Score Index
Footnotes
Communicated by: Ed Wraith
Competing interest: None declared.
Contributor Information
Sarah Michelman Lo, Section of Hematology-Oncology, Department of Pediatrics, Yale University School of Medicine, New Haven, CT, USA.
Jun Liu, Department of Pediatrics, Section of Gastroenterology and Hepatology, Yale University School of Medicine, New Haven, CT, USA.
F. Chen, Section of Digestive Diseases, Department of Internal Medicine, Yale University School of Medicine, New Haven, CT, USA
G. M. Pastores, Department of Neurology, New York University School of Medicine, New York, NY, USA
J. Knowles, Department of Psychiatry, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA
M. Boxer, U.S. Oncology, Tucson, AZ, USA
Kirk Aleck, St. Joseph’s Hospital and Medical Center, Phoenix, AZ, USA.
Pramod K. Mistry, Departments of Pediatrics and Internal Medicine, Section of Pediatric Hepatology and Gastroenterology, Yale University School of Medicine, 333 Cedar Street, P.O. Box 208064, New Haven, CT 06520–8064, USA pramod.mistry@yale.edu
References
- Austin ED, Loyd JE. Genetics and mediators in pulmonary arterial hypertension. Clin Chest Med. 2007;28(1):43–57. vii–viii. doi: 10.1016/j.ccm.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi M, Scholl UI, Ji W, et al. Genetic diagnosis by whole exome capture and massively parallel DNA sequencing. Proc Natl Acad Sci USA. 2009;106(45):19096–19101. doi: 10.1073/pnas.0910672106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox TM, Aerts JM, Belmatoug N, et al. Management of non-neuronopathic Gaucher disease with special reference to pregnancy, splenectomy, bisphosphonate therapy, use of biomarkers and bone disease monitoring. J Inherit Metab Dis. 2008;31(3):319–36. doi: 10.1007/s10545-008-0779-z. Epub 2008 May 23. Review. [DOI] [PubMed] [Google Scholar]
- Dawson A, Elias DJ, Rubenson D, et al. Pulmonary hypertension developing after alglucerase therapy in two patients with type 1 Gaucher disease complicated by the hepatopulmonary syndrome. Ann Intern Med. 1996;125(11):901–904. doi: 10.7326/0003-4819-125-11-199612010-00005. [DOI] [PubMed] [Google Scholar]
- Elleder M. Glucosylceramide transfer from lysosomes–the missing link in molecular pathology of glucosylceramidase deficiency: a hypothesis based on existing data. J Inherit Metab Dis. 2006;29(6):707–715. doi: 10.1007/s10545-006-0411-z. [DOI] [PubMed] [Google Scholar]
- Elstein D, Klutstein MW, Lahad A, et al. Echocardiographic assessment of pulmonary hypertension in Gaucher’s disease. Lancet. 1998;351:1544–1546. doi: 10.1016/S0140-6736(98)10194-0. [DOI] [PubMed] [Google Scholar]
- Grabowski GA. Phenotype, diagnosis, and treatment of Gaucher’s disease. Lancet. 2008;372(9645):1263–1271. doi: 10.1016/S0140-6736(08)61522-6. [DOI] [PubMed] [Google Scholar]
- Grabowski GA, Kolodyny EH, Weinreb NJ, et al. Gaucher disease: phenotypic and genetic variation. In: Schriver CR, Beaudet AL, Sly WS, et al., editors. The Online Metabolic and Molecular Basis of Inherited Diseases. Ch 146.1. McGraw-Hill; New York: 2006. ww.ommbid.com. [Google Scholar]
- Hůlková H, Poupetová H, Harzer K, et al. Abnormal nonstoring capillary endothelium: a novel feature of Gaucher disease. Ultrastructural study of dermal capillaries. J Inherit Metab Dis. 2010;33(1):69–78. doi: 10.1007/s10545-009-9018-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humbert M, Morrell N, Archer S, et al. Cellular and molecular pathobiology of pulmonary arterial hypertension. J Am Coll Cardiol. 2004;43(12 Suppl S):13S–24S. doi: 10.1016/j.jacc.2004.02.029. [DOI] [PubMed] [Google Scholar]
- Lachmann RH, Wight DG, Lomas DJ, et al. Massive hepatic fibrosis in Gaucher’s disease: clinico-pathological and radiological features. QJM. 2000;93(4):237–244. doi: 10.1093/qjmed/93.4.237. [DOI] [PubMed] [Google Scholar]
- Lee RE, Yousem SA. The frequency and type of lung involvement in patients with Gaucher’s disease. Lab Invest. 1988;58:54A. [Google Scholar]
- Lee S, Shroyer K, Markham N, Cool C, Voelkel N, Tuder R. Monoclonal endothelial cell proliferation is present in primary but not secondary pulmonary hypertension. J Clin Invest. 1998;101:927–934. doi: 10.1172/JCI1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo SM, Stein P, Mullaly S, et al. Expanding spectrum of the association between Type 1 Gaucher disease and cancers: a series of patients with up to 3 sequential cancers of multiple types–correlation with genotype and phenotype. Am J Hematol. 2010;85(5):340–5. doi: 10.1002/ajh.21684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loyd JE, Atkinson JB, Pietra GG, et al. Heterogeneity of pathologic lesions in familial primary pulmonary hypertension. Am Rev Respir Dis. 1988;138(4):952–957. doi: 10.1164/ajrccm/138.4.952. [DOI] [PubMed] [Google Scholar]
- Loyd JE, Butler MD, Foroud TM, et al. Genetic anticipation and abnormal gender ratio at birth in familial primary pulmonary hypertension. Am J Respir Crit Care Med. 1998;152:93–97. doi: 10.1164/ajrccm.152.1.7599869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry PK, Wraight EP, Cox TM. Therapeutic delivery of proteins to macrophages: implications for treatment of Gaucher’s disease. Lancet. 1996;348(9041):1555–1559. doi: 10.1016/S0140-6736(96)04451-0. [DOI] [PubMed] [Google Scholar]
- Mistry PK, Sirrs S, Chan A, et al. Pulmonary hypertension in type 1 Gaucher’s disease: genetic and epigenetic determinants of phenotype and response to therapy. Mol Genet Metab. 2002;77:91–98. doi: 10.1016/s1096-7192(02)00122-1. [DOI] [PubMed] [Google Scholar]
- Mistry PK, Cappellini MD, Lukina E, et al. A reappraisal of Gaucher disease-diagnosis and disease management algorithms. Am J Hematol. 2010 doi: 10.1002/ajh.21888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry PK, Liu J, Yang M, et al. Glucocerebrosidase gene-deficient mouse recapitulates Gaucher disease displaying cellular and molecular dysregulation beyond the macrophage. Proc Natl Acad Sci USA. 2010b;107(45):19473–19478. doi: 10.1073/pnas.1003308107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möller T, Leren TP, Eiklid KL, et al. A novel BMPR2 gene mutation associated with exercise-induced pulmonary hypertension in septal defects. Scand Cardiovasc J. 2010;44(6):331–336. doi: 10.3109/14017431.2010.525747. [DOI] [PubMed] [Google Scholar]
- Morse JH. Bone morphogenetic protein receptor 2 mutations in pulmonary hypertension. Chest. 2002;121(3 Suppl):50S–53S. doi: 10.1378/chest.121.3_suppl.50s. [DOI] [PubMed] [Google Scholar]
- Newman JH, Phillips JA, 3rd, Loyd JE. Narrative review: the enigma of pulmonary arterial hypertension: new insights from genetic studies. Ann Intern Med. 2008;148(4):278–283. doi: 10.7326/0003-4819-148-4-200802190-00006. [DOI] [PubMed] [Google Scholar]
- Roberts WC, Fredrickson DS. Gaucher’s disease of the lung causing severe pulmonary hypertension with associated acute recurrent pericarditis. Circulation. 1967;35:783–789. doi: 10.1161/01.cir.35.4.783. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Roisin R, Krowka MJ. Is severe arterial hypoxaemia due to hepatic disease an indication for liver transplantation? A new therapeutic approach. Eur Respir J. 1994;7:839–842. [PubMed] [Google Scholar]
- Ross DJ, Spira S, Buchbinder NA. Gaucher Cells in pulmonary-capillary blood in association with pulmonary hypertension. N Engl J Med. 1997;336:379–381. doi: 10.1056/NEJM199701303360516. [DOI] [PubMed] [Google Scholar]
- Smith RL, Hutchins GM, Sack GH, Jr, Ridolfi RL. Unusual cardiac, renal and pulmonary involvement in Gaucher’s disease. Interstitial glucocerebroside accumulation, pulmonary hypertension and fatal bone marrow embolization. Am J Med. 1978;65:352–360. doi: 10.1016/0002-9343(78)90832-x. [DOI] [PubMed] [Google Scholar]
- Taddei TH, Kacena KA, Yang M, et al. The underrecognized progressive nature of N370S Gaucher disease and assessment of cancer risk. Am J Hematol. 2009;84(4):208–214. doi: 10.1002/ajh.21362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayebi N, Cushner S, Sidransky E. Differentiation of the glucocerebrosidase gene from pseudogene by long-template PCR: implications for Gaucher disease. Am J Hum Genet. 1996;59:740–741. [PMC free article] [PubMed] [Google Scholar]
- Thachil J. The enigma of pulmonary hypertension after splenectomy – does the megakaryocyte provide a clue? QJM. 2009;102(10):743–5. doi: 10.1093/qjmed/hcp092. Epub 2009 Jul 21. [DOI] [PubMed] [Google Scholar]
- Theise ND, Ursell PC. Pulmonary hypertension and Gaucher’s disease: logical association or mere coincidence? Am J Pediatr Hematol Oncol. 1990;12:74–76. doi: 10.1097/00043426-199021000-00014. [DOI] [PubMed] [Google Scholar]
- Yeager M, Halley G, Golpon H, Voelkel N, Tuder R. Microsatellite instability of endothelial cell growth and apoptosis genes within plexiform lesions in primary pulmonary hypertension. Circ Res. 2001;88:E2–E11. doi: 10.1161/01.res.88.1.e2. [DOI] [PubMed] [Google Scholar]
- Zimran A, Kay A, Gelbart T, et al. Gaucher disease: clinical, laboratory radiologic and genetic features of 53 patients. Medicine. 1992;71:337–353. [PubMed] [Google Scholar]