

Abstract
Amyloid beta peptide (Aβ) causes neurodegeneration by several mechanisms including oxidative stress, which is known to induce DNA damage with the consequent activation of poly (ADP-ribose) polymerase (PARP-1). To elucidate the role of PARP-1 in the neurodegenerative process, SH-SY5Y neuroblastoma cells were treated with Aβ25–35 fragment in the presence or absence of MC2050, a new PARP-1 inhibitor. Aβ25–35 induces an enhancement of PARP activity which is prevented by cell pre-treatment with MC2050. These data were confirmed by measuring PARP-1 activity in CHO cells transfected with amylod precursor protein and in vivo in brains specimens of TgCRND8 transgenic mice overproducing the amyloid peptide. Following Aβ25–35 exposure a significant increase in intracellular ROS was observed. These data were supported by the finding that Aβ25–35 induces DNA damage which in turn activates PARP-1. Challenge with Aβ25–35 is also able to activate NF-kB via PARP-1, as demonstrated by NF-kB impairment upon MC2050 treatment. Moreover, Aβ25–35 via PARP-1 induces a significant increase in the p53 protein level and a parallel decrease in the anti-apoptotic Bcl-2 protein. These overall data support the hypothesis of PARP-1 involvment in cellular responses induced by Aβ and hence a possible rationale for the implication of PARP-1 in neurodegeneration is discussed.
Introduction
Free radical damage, which occurs during oxidative stress, is associated with neurodegenerative disorders, such as Alzheimer's disease (AD) and Parkinson's disease (PD) [1], [2]. The major cause of free radicals overproduction seems to be related to the accumulation of misfolded protein aggregates in brain tissues. In AD, the major component of these protein aggregates present in senile plaques, is the amyloid beta (Aβ), a peptide of 39–42 amino acid residues which derives from the sequential proteolytic processing of the amyloid precursor protein (APP) by beta- and gamma-secretases. When an unbalance between Aβ production and clearance due to genetic and/or environmental factors occurs, Aβ oligomerization takes places producing different species of soluble supramolecular assemblies and some of them finally converge towards fibrillar formation [3], [4]. Aβ plays a central role in the pathogenesis of AD, by causing neurodegeneration and disrupting the cognitive function although the molecular pathways leading to neuronal impairment are not yet fully elucidated. It has been shown that early formed pre-fibrillar aggregates of Aβ are mainly endowed with cytotoxicity, whereas mature fibrils are much less toxic or even harmless [5]. In particular, soluble Aβ oligomers are associated with the generation of free radicals via direct and indirect mechanisms: in the direct one, Aβ binds to transition metals ions, acquiring an oxidase activity leading to hydrogen peroxide production [6]. In the indirect mechanism neurons or microglia stimulated by Aβ oligomers produce free oxygen radicals by activation of NADPH oxidase [7].
Free radical injury may be responsible for neuronal loss by inducing DNA damage that in turn activates poly (ADP-ribose) polymerase enzyme (PARP-1).
PARP-1 is a 116 kDa zinc-binding nuclear enzyme consisting of three main domains: the N-terminal DNA-binding domain containing two zinc fingers motifs, the automodification domain, and the C-terminal catalytic domain. This enzyme catalyzes the covalent addition of the ADP-ribose moiety of nicotinamide adenine dinucleotide (NAD+) to nuclear proteins including histones, transcription factors and PARP-1 itself, and the subsequent elongation of the polymer. PARP-1 is involved in many physiological processes such as gene expression, maintenance of genomic stability and cell death and differentiation [8] [9].
Extensive PARP-1 activation by DNA damage contributes to the development and progression of various chronic diseases including diabetes, cancer, viral infections and neurodegenerative diseases [10]–[15]. In particular, the findings that parkinsonian neurotoxins and Aβ activate PARP-1 in dopaminergic neurons and hippocampal slices respectively, suggest a relationship between PARP-1 and neurodegeneration [16]–[18].
In the present study we focused on the comprehension of the molecular mechanisms that lead to PARP-1 activation by Aβ in SH-SY5Y neuroblastoma derived cells and in transgenic mice TgCRND8, an early onset model of AD and to the downstream ways activated by PARP-1. Since the suppression of over-activated PARP-1 by specific inhibitors might represent a useful tool to prevent neurotoxicity, we also analyzed the protection of SH-SY5Y cells from Aβ harmful effects by a newly synthesized PARP-1 inhibitor, MC2050 [19]. Our data show that challenge of SH-SY5Y cells with Aβ significantly increased PARP-1 activity following ROS generation and DNA damage and PARP-1 activated NF-kB and modulates pro-apoptotic proteins. These effects were significantly decreased in the presence of MC2050 suggesting a potential therapeutic application for this compound in neurodegenerative disease.
Materials and Methods
Preparation of the test substances
MC2050 (2-[2-(4-(2-pyridyl)-1-piperazinyl) ethylsulfanyl]-3H-quinazolin-4-one) dihydrochloride, a new PARP inhibitor was dissolved in water at an initial concentration of 50 mM and then diluted to a final concentration of 50 µM in PBS or culture medium. Two other PARP inhibitors were used in this work: 3-ABA (3-aminobenzamide) and PJ34. 3-ABA was dissolved at a concentration of 4 M in DMSO and then diluted to a final concentration of 4 mM in culture medium; PJ34 was dissolved at a concentration of 50 mM in H2O and then diluted to a final concentration of 5 µM in culture medium. Amyloid beta peptide 25–35 fragment (Aβ25–35) synthesized by conventional solid phase chemistry [20], was aggregated overnight at 37°C in phosphate buffered saline at a concentration of 1mM.
Animals
TgCRND8 mice (TgCRND8–129Sv), carrying the double mutant form APP695 (KM670/671NL+V717F), were obtained from Dr. David Westaway – Center of Research on Neurodegenerative Disease – University of Toronto and maintained in the heterozygous status (TgCRND8+/−) by mating male Tg with female WT (TgCRND8−/−) 129Sv mice (Charles River Laboratories, Wilmington MA, USA). These mice show amyloid peptide deposition at 3 months, with dense-cored plaques and neuritic pathology evident from 5 months of age [21].
Mice treatment
Biochemical analyses were carried out on a total of 16 animals (8 WT and 8 Tg). At the third month of life mice were anesthetized and sacrificed to obtain brains. Brains were perfused with PBS, removed and bisected on the sagittal plane. Hippocampus and entorhinal cortex, sites of major pathological changes in AD, were stored at −80°C and used for the biochemical and molecular analyses.
All experiments were performed in order to sacrifice the minimum number of animals required and were previously approved by author's Institution according to guidelines of the Italian Ministry of Health (D.L. 92/116) and of the EC directives 86/609/EEC and 2010/63/EU on the protection of animals used for experimental and other scientific purposes. According to Italian laws on animal care, the experimental protocol for the experiments described in this paper has been “communicated” to the Ministry of Health (IACUC) without necessity of any formal approval number.
Cell treatment
The human dopaminergic neuroblastoma cell line SH-SY5Y was obtained by ICLC (Genova, Italy) and maintained in DMEM/F12 supplemented with 10% heat-inactivated foetal bovine serum and 2 mM glutamine, at 37°C and under 5% CO2 in a humidified incubator. SH-SY5Y, an extremely versatile neuronal cell line, has been extensively utilized as an experimental model for the study of neuronal death processes induced by several agents [22]. Cells were treated with 10 µM Aβ25–35 in the presence or absence of the PARP-1 inhibitors. The inhibitors were added before Aβ25–35 treatment. In some experiments SH-SY5Y were also pre-treated with 5 µM quercetin, a natural antioxidant agent.
7PA2, a Chinese hamster ovary (CHO) cell line stably transfected with a complementary DNA coding for APP751 containing the Val717Phe familial Alzheimer's disease mutation that leads to Aβ overproduction, was a kind gift of Prof. D. Walsh, USA. CHO cells were used as a control. Both cell lines were maintained in DMEM/F12 supplemented with 10% heat-inactivated foetal bovine serum and 2 mM glutamine, at 37°C and under 5% CO2 in a humidified incubator.
PARP-1 activity assay
PARP-1 activity was tested in SH-SY5Y cells, in 7PA2 cells and in mice with a colorimetric PARP assay Kit from Trevigen. Briefly, the assay measures the incorporation of biotinylated poly (ADP-ribose) onto H1 histone in a 96 well plate. Enzyme assay was performed by adding biotinylated NAD+ to a reaction mixture containing lysates (20 µg protein). Cultured cells and homogenized tissue from hippocampus and entorhinal cortex were collected and centrifuged at 400 g for 10 min at 4°C and then resuspended in 100 μl of PARP lysis buffer (PARP buffer, 0.4M NaCl, 1% NP-40, 0.4 M PMSF, protease inhibitor). The lysates were incubated on ice for 30 min and centrifuged at 10000 g for 10 min at 4°C. The collected supernatants were subjected to protein determination by Bradford Assay (BioRAD). PARP Cocktail (containing biotinylated NAD+) was added to each wells and incubated for 60 min. Then, diluted Strep-HRP was added to each well, incubated for 20 min at room temperature and followed by TACS-Sapphire incubation for additional 30 min in the dark. The reaction was stopped by adding 0.2 M HCl and then the plate was read at 450 nm.
Cell viability assay
Cell viability was assessed by using the dye [4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT). The assay is based on the ability of living cells to convert MTT into an insoluble purple-coloured formazan, whose amount is proportional to the number of living cells. Cells seeded in 96 wells plates at a density of 15000 cells/well were exposed to Aβ25–35 at a final concentration of 10 µM. Cell viability was assessed at 24, 48 and 72 h. After the indicated time intervals, cells were treated with 20 µl of a 5 mg/ml solution of MTT in PBS and incubated at 37°C for 2 h. After discarding the medium, the formazan was extracted with DMSO, and absorbance read at 577 nm with a reference at 690 nm.
Cell Cycle
The SH-SY5Y cells were treated with Aβ25–35 (10 μM) for 24 and 48 h. The cells were then washed with PBS and centrifuged at 900 g for 10 min, resuspended in 70% ethanol and then centrifuged. The pellet was resuspended at 37°C for 30 min in 1 ml of PBS containing 100 μg of propidium iodide and 100 μg of RNAse. The samples were then analyzed by EPICS XL-MCL Flow cytometer (Coulter).
Permeation of the membrane
To assess the ability of amyloid peptide to alter the permeability of SH-SY5Y membrane, cells seeded in 96 wells plates at a density of 13000 cells/well were treated with Aβ25–3510 μM, in the presence or absence of MC2050. At different times the medium was removed and the cells were resuspended in 100 μl of PBS containing 1 μM Sytox Green. After Aβ25–35 addition, the increase in fluorescence, due to the binding of the dye to intracellular DNA, was measured at 37°C by a Polarstar Galaxy microplate reader (BMG Labotechnologies, Offenburg, Germany) using 485 and 520 nm filters for excitation and emission, respectively.
ROS detection
Reactive oxygen species formation in SH-SY5Y cells treated with Aβ25–35 was assayed by flow cytometry using the dye DCF-DA (dichlorofluoresceine-diacetate) and following standard methods. Briefly, DCF-DA (final concentration 10 µM) was added to cells grown on 6 wells plates for 15 min at 37°C in a humidified incubator and then cells were treated with the indicated amount of Aβ25–35, in the presence or absence of 5 µM quercetin, at different times. Cells were then scraped, washed in PBS and analyzed by Epics XL-MCL flow cytometer (Coulter) equipped with an Argon laser at 488 nm. Cells were gated on the basis of forward-angle light-scatter (FS) and 90° light-scatter parameters (SS). For every histogram, a minimum of 10000 events were counted. The mean fluorescence intensity was analyzed and expressed as percentage of relative ROS level vs. control cells.
Comet assay
Oxidative DNA damage was studied by electrophoresis of single cell in agarose gel.
SH-SY5Y (30000 cells) were resuspended in a 0,65% of low melting point agarose (LMPA) and left in a water bath (37°C) and then smeared on slides, which were earlier coated with a thin layer of 0.65% LMPA and left to dry out. After 10 min the slides were placed in the lysis buffer (2.5 M NaCl, 100 mM EDTA, 10 mM Tris, pH 10, 1% Triton X–100 and 10% DMSO) for 1 h at 4°C in dark. The slides were left for 40 min in a buffer for electrophoresis (1 mM EDTA, 300 mM NaOH pH >13) at 4°C in order to lose nuclear super-coiled DNA. Then, they were subjected to low-voltage electrophoresis at 300 mA, for 30 min at 4°C, and washed three times with neutral cold buffer (0.4 M Tris/HCl, pH 7.5). The slides were finally submerged for 5 min in distilled water and stained with propidium iodide at concentration of 2.5 μg/ml. DNA damage was visualized by an epifluorescence microscope (Olympus IX-50) equipped with a fluorescence source. The analysis of oxidative DNA damages was determined by olive moment parameter using the COMET PLUS 2.9 software (Comet Plus, Theta System Gmbh, Germany).
Electrophoretic mobility shift assay (EMSA)
SH-SY5Y cells were resuspended in high saline buffer (50 mM Tris-HCl, 400 nM NaCl, 1 mM EDTA, 1% Triton, 0.5% NP-40, 10% Glycerol, 2 mM DTT), protease and phosphatases inhibitors. The resulting cell lysates were centrifuged for 5 min at 15000 g at 4°C and 20 μg of protein extract were incubated with binding buffer 2X (20 mM Tris-HCl, 2 mM EDTA, 10% glycerol), 1mg/mL BSA, 1mg/mL Poly-d (I-C), 15 fmol/µL DIG-labeled (DIG oligonucleotide 3′ end-labeling kit, Roche Applied Science) NF–kB DNA probe [23]. The sequence of the “KB” oligonucleotide used was as follows:
5′- AGCTTCAGAGGGGACTTTCCGAGAGG –3′
3′- AGTCTCCCCTGAAAGGCTCTCCAGCT –5′
The DNA-protein complex formed was separated on 4% native polyacrylamide gel and transferred onto nitrocellulose membrane o/n. Detection was performed by DIG Gel Shift Kit (Roche®). Supershift assay using NF-κB p65 and p50 antibodies (Millipore) was also carried out to confirm the specificity of NF-κB DNA-binding activity. 30 μg of proteins from each sample were subjected to Western blot to verify equal loading.
Subcellular fractionation
SH-SY5Y cells were suspended in hypotonic buffer (10 mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 1 mM PMSF, protease inhibitors, 10 mM DTT, 0.1% NP-40) and incubated for 30 min on ice. The cell lysate was centrifuged for 5 min at 4500 g at 4°C, and the supernatant collected (cytosolic fraction). The nuclear fraction was obtained by sonication of the pellet in hypertonic buffer (5 mM HEPES, 1,5 mM MgCl2, 0,2 mM EDTA, 0,5 mM DTT, 26% glycerol, 1 mM PMSF, protease inhibitors), mixed quickly and incubated for 1 h on ice. After centrifugation for 20 min at 10000 g the supernatant was collected. Hence, cytosolic and nuclear fractions were submitted to western blotting analyses.
Western blotting
SH-SY5Y cells and mice brain tissues were lysed in 80 μl RIPA buffer (50 mM Tris-HCl pH 7.4, 150 mM NaCl, 0,1% NP-40, 0.1% SDS, 1 mM EDTA, 1 mM Na3VO4, 1 mM PMSF, 0,5 NaDOC, protease inhibitors). The lysates were incubated on ice for 30 min and centrifuged at 10000 g for 10 min at 4°C. The supernatants were collected and proteins quantification were performed using a Bradford Assay (BIO-RAD). Equal amounts of protein were separated on 12% SDS-PAGE Criterion (BIO-RAD) and subsequently transferred to nitrocellulose membrane (GE Healthcare). The membrane was probed with the primary anti-PAR (1∶5000) (Alexis), anti-PARP (1∶1000) (Alexis), anti-p53 (1∶400) (Sigma), anti-Bcl-2 (1:500) (Sigma), anti-p50 (1∶3000) (Millipore), anti-p65 (1∶1000) (Millipore), anti-actin (1∶5000) (Alexis), anti-vinculin (1∶500) (Santa Cruz), anti-lamin A/C (1∶1000) (Millipore) in PBS at 4°C overnight. The subsequent steps were performed with innovative SNAP i. d. system (Millipore). The bands were visualized by ECL system (Millipore) according to the manufacture's instructions. Densitometric analyses were performed with ImageJ software, and normalized to a reference protein.
Statistical analysis
Results are expressed as mean ± SD for at least three separate experiments. Graphics and data analysis were performed using GraphPAD prism 4 software. Statistical analyses were performed using the ANOVA and Bonferroni post hoc test. P<0.05 was deemed significant.
Results
Aβ affects PARP activity
To investigate whether Aβ could induce an increase in PARP-1 activity, SH-SY5Y cells were treated Aβ25–35, a toxic fragment of the full length Aβ1–42, for 2, 4, 8 and 24 h and enzymatic activity was assayed by a dedicated colorimetric kit. Results, reported in Fig. 1A, show that the amyloid peptide increased PARP-1 activity in a time-dependent manner. After 24 h, the activity in treated cells was 65% higher than in control. This activity level was maintained even when the stimulation was prolonged up to 48 h. Cells pre-treatment with 50 µM MC2050 prevented Aβ25–35 induced PARP-1 activation. The same results were obtained using 3 mM 3-ABA, a well known PARP inhibitor (data not shown). To assess whether the full length fragment Aβ1–42 is able to exert PARP-1 activation, 7PA2, CHO cells transfected with a mutated APP that lead to amyloid peptide overproduction, were assayed for PARP-1 activity. A 40% increase was observed in 7PA2 with respect to control CHO cells (Fig. 1B).
Figure 1. Aβ induces PARP-1 activation.
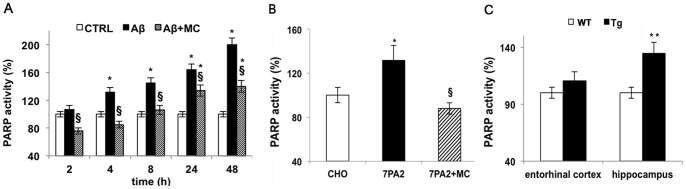
PARP-1 activity was determined in cells and tissues homogenates by the use of a dedicated colorimetric Kit from Trevigen, following manufacturer instructions. A) SH-SY5Y cells treated with 10 µM Aβ25–35 in the presence or absence of 50 µM MC2050 for the indicated times (white bar = control cells; black bar = Aβ25–35 treated cells; stripped bar = cells treated with Aβ25–35 in the presence of MC2050); B) PARP-1 activity in 7PA2 cells in the presence or absence of 50 µM MC2050. CHO cells were used as control (white bar = control cells; black bar = 7PA2 cells; stripped bar = 7PA2 cells in the presence of MC2050); C) Entorhinal cortex and hippocampus of transgenic mice TgCRND8 (Tg = black bar) compared to wild type mice 129Sv (WT = white bar). The values are the mean ± S.D. of at least three independent experiments. *p<0.05 vs. control cells or WT; § p<0.05 vs. Aβ25–35-treated cells or 7PA2 cells.
This demonstrates that a constant activation of PARP-1 takes place in the presence of both amyloid peptide species allowing us to use Aβ25–35 for the subsequent set of experiments since it is more easy-to-handle, cheaper and the results are more reproducible.
We confirmed a tight correlation between Aβ25–35 accumulation and PARP-1 activity by in vivo experiments using heterozygous transgenic mice. After 3 months, when early amyloid deposit occurs, mice were sacrificed and PARP-1 activity was assayed in hippocampus and in cortex compartments of both wild type and mutant mice. We observed an increase of more than 30% activity in the hippocampus, while a slight increase was recorded in the cortex region according to the direction of the spread of amyloid accumulation in AD (Fig. 1C). Moreover, immunoblot with anti-PAR antibodies showed an increase in PARylated proteins both in cells treated with Aβ25–35 up to 4 h, and in TgCRND8 mice. The pre-treatment of cells with MC2050 significantly reversed PARP-1 activation [Fig. 2 A, B].
Figure 2. Aβ induces protein PARylation.
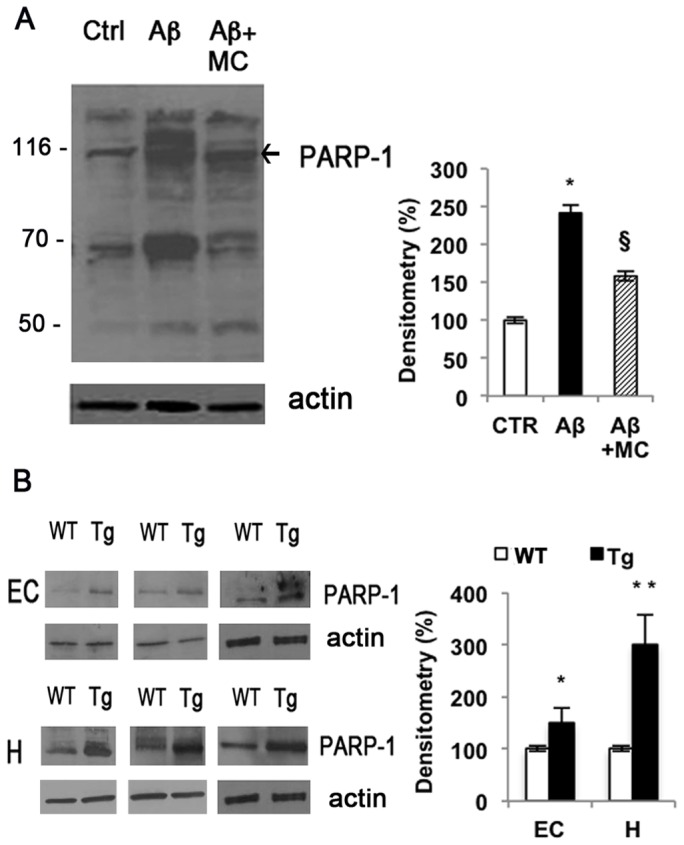
Equal concentrations of proteins (30 µg) from SH-SY5Y cell lysates treated with 10µM Aβ25–35 for 4 h in the presence or absence of 50 µM MC2050 (A) (white bar = control cells; black bar = Aβ25–35 treated cells; stripped bar = cells treated with Aβ25–35 in the presence of MC2050) and from entorhinal cortex and hippocampus of wild type and transgenic mice (B) (white bar = Wild Type mice; black bar = Transgenic mice) were subjected to 12% SDS-PAGE, and immunoblotted with anti-PAR polymer (1∶5000 Alexis). Densitometric analyses were performed with ImageJ software, and normalized to β-actin. Data are reported as percentage of control values and are the mean ± S.D. of at least three independent experiments. *p<0.05 vs. control cells or WT; **p<0.01 vs. WT; § p<0.05 vs. Aβ25–35-treated cells.
These in vitro and in vivo results drove us to understand the molecular causes of this enhancement of PARP-1 activity in injured SH-SY5Y cells.
Aβ25–35 generates membrane perturbation, ROS production and DNA damage
Since several works demonstrated that the toxicity of amyloid oligomers and larger fibrillar aggregates is related to a perturbing effect on the biological function of the adjacent cellular membrane [24], [25], firstly we assayed the Aβ25–35 capability to induce membrane damage. Obtained data showed that a perturbation occurred when cells were treated with 10 μM Aβ up to 4 h (Fig. 3A) and that the extent of membrane perturbation is correlated with the amyloid concentration (Fig. 3B). We also treated the cells with amyloid peptide in the presence or absence of MC2050 to demonstrate whether this compound was able to prevent membrane perturbation. MC2050 did not influence the process, indicating that membrane damage was not mediated by PARP-1 activity (Fig. 3A).
Figure 3. Aβ25–35 induces membrane perturbation in SH-SY5Y cells.
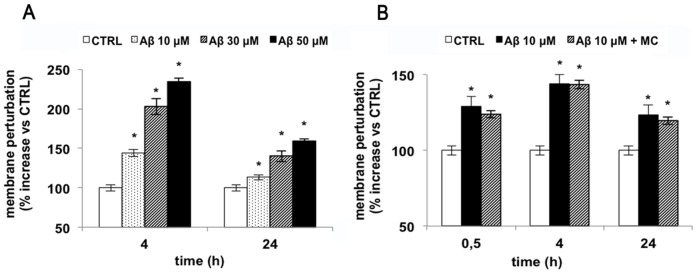
13000 cells were seeded in 96 well plates and treated with various amount of Aβ25–35 were incubated with 1 μM SYTOX™ Green in PBS. Changes in fluorescence were monitored (λex = 485 nm, λem = 520 nm) and plotted as the percentage of fluorescence relative to the control cells. A) Time-course of cells treated with 10 µM Aβ25–35 (white bar = control cells; black bar = Aβ25–35 treated cells; stripped bar = cells treated with Aβ25–35 in the presence of MC2050); B) Dose-dependent effect of Aβ25–35 on membrane perturbation (white bar = control cells; dotted bar = 10 µM Aβ25–35; stripped bar = 30 µM Aβ25–35; black bar = 50 µM Aβ25–35). Results are the mean of at least three independent experiments ± S.D. *p<0.05 vs. control cells.
In addition, to assess the cytotoxic effects exerted by Aβ25–35 on cells, viability assays were performed and the results revealed that the challenge with Aβ25–35 reduced cell viability in a time-dependent manner (Fig. 4A). To confirm these data we further checked cell toxicity by evaluating cell cycle after Aβ25–35 treatment. Results showed that the subG0 peak is significantly increased compared to control cells, raising from a basal value of about 4% to 20% within 48 h (Fig. 4B). Preliminary experiments on primary neuron treated with amyloid peptide showed a similar toxic effect (data not shown).
Figure 4. Effect of Aβ25–35 on SH-SY5Y neuroblastoma cell line viability.
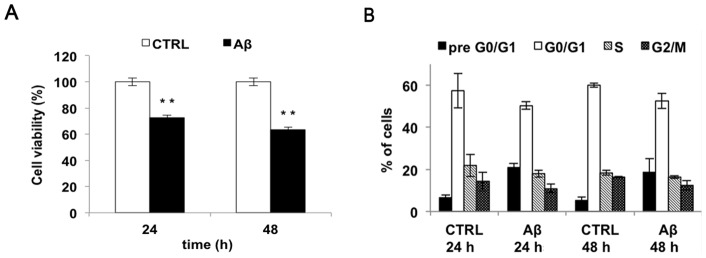
A) Cell viability of SH-SY5Y cells treated with 10 µM Aβ25–35 for 24, 48 h was monitored using MTT assay (white bar = control cells; black bar = Aβ25–35 treated cells). B) Cell cycle phases distribution of SH-SY5Y cells, as determined by FACS analysis Results are the mean of at least three independent experiments ± S.D. **p<0.01 vs. control cells.
Finally, since a well-known potent inducer of PARP-1 activation is DNA damage caused by ROS, intracellular free radical production and DNA damage in SH-SY5Y cells challenged by Aβ25–35 were monitored. ROS production, determined by flow-cytometric analysis, was markedly increased up to 70–80% upon treatment with Aβ25–35 within the first 2 h with respect to the control and then gradually decreased to 40–50% at 24 h (Fig. 5 A). DNA damage was evaluated by Comet assay analysis (expressed as Olive moment), in SH-SY5Y cells treated for 24 h and 48 h with Aβ25–35 (Fig. 5 B). Our results showed that as a consequence of treatment with the amyloidogenic peptide, the increase of DNA damage was observed in SH-SY5Y cells.
Figure 5. Aβ25–35 induces oxidative stress and DNA damage.
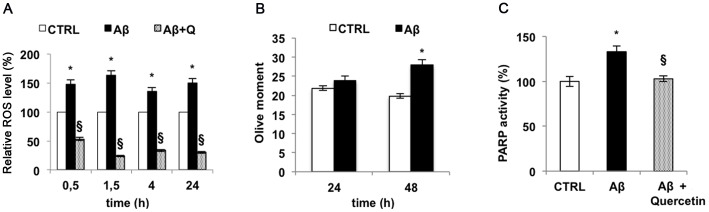
A) ROS produced by SH-SY5Y cells treated with 10 µM Aβ25–35 at different times were revealed by flow cytometry using DCF-DA (white bar = control cells; black bar = Aβ25–35 treated cells; fine dotted bar = cells treated with Aβ25–35 in the presence of quercetin). B) DNA damage was evaluated by Comet assay and expressed as Olive moment in SH-SY5Y treated with 10 µM Aβ25–35 for 24 and 48 h (white bar = control cells; black bar = Aβ25–35 treated cells). C) PARP-1 activity in SH-SY5Y treated with 10 µM Aβ25–35 in the presence or absence of 5 µM quercetin by the use of a dedicated colorimetric Kit from Trevigen (white bar = control cells; black bar = Aβ25–35 treated cells; fine dotted bar = cells treated with Aβ25–35 in the presence of quercetin). Results are the mean of at least three independent experiments ± S.D. *p<0.05 vs. control cells; § p<0.05 vs. Aβ25–35-treated cells.
Our data confirm the negative effect of Aβ25–35 on DNA integrity via ROS production and strongly suggest that PARP-1 activation depends on this event. To further strengthen this hypothesis, we used quercetin, a plant derived flavonoid, endowed with strong antioxidant capacity in order to blunt Aβ25–35 induced ROS production. When quercetin was added to the medium of SH-SY5Y cell cultures in the presence of Aβ25–35 the overall amount of free radicals dropped below the control level and PARP-1 activity did not show any variation with respect to the control (Fig. 5 A, C) strongly indicating that PARP-1 activation observed in Aβ25–35-treated cells depended on DNA damage caused by redox unbalance.
Then we moved on to investigate whether PARP-1 activation could affect cell signalling and cell death.
MC2050 prevents activation of NF-κB induced by Aβ25-35
Some authors have shown that Aβ can activate NF-kB in neuronal cells, suggesting that this molecular pathway may be responsible of the progression and of the pathogenesis of AD [26], [27]. Since Hassa and Hottinger [28] demostrated that PARP-1 regulates the function of NF-kB, we verified whether in our experimental conditions, Aβ25–35 modulates NF-kB activation through a PARP-1-dependent mechanism. Gel retardation assay (EMSA) showed a progressive increase of NF-kB activity during incubation with Aβ25–35. The o/n pre-treatment of SH-SY5Y cells with MC2050 followed by incubation with Aβ25–35 led to a significant reduction of NF-kB activation at 4 h (Fig. 6 A). We obtained similar results when we used PJ34 a commercially available PARP-1 inhibitor (data not shown).
Figure 6. Aβ25-35 induces NF-kB activation via PARP.
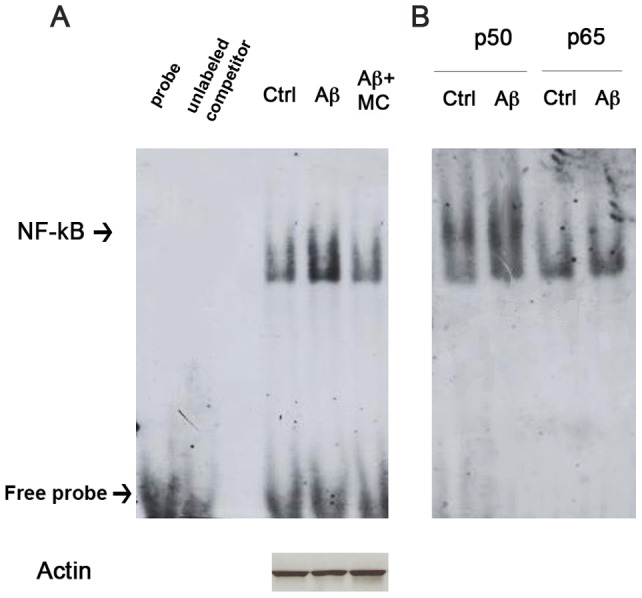
Cell lysates obtained from SH-SY5Y treated with 10 µM Aβ25–35 for 4 h, in the presence or absence of 50 µM MC2050, were incubated with a specific DNA probe for NF-kB, labeled with digoxigenin. A) The DNA-protein complexes separated on 4% native gels and transferred to nitrocellulose membrane were detected by chemiluminescence after hybridization with anti-DIG. B) Supershift of NF-kB detected by EMSA. The same cell lysates were incubated with the DNA probe for NF-kB in the presence of digoxigenin-labeled antibodies against p50 and p65 subunits.
To clarify which NF-kB subunit is responsible of the DNA binding activity, the cell extracts pre-treated or not with MC2050 were incubated with Aβ25–35 and antibodies against either p50 or p65. Figure 6B shows a slower electrophoretic migration of the complex in the presence of anti-p50, indicating the involvement of the p50 subunit in targeting the specific DNA sequence.
MC2050 prevents NF-kB translocation from cytoplasm to nucleus in SH-SY5Y
Several authors have shown that poly (ADP-ribosylation) plays a role as an intracellular signal involved in the transport of transcription factors across the nuclear envelope [29], [30]. To verify whether PARP-1 plays a role in the nuclear translocation of NF-kB, the cytosolic and nuclear lysates, obtained from SH-SY5Y cells incubated with Aβ25–35 and pretreated in the presence or absence of MC2050 were analyzed by Western blot.
The results show that p50 subunit translocated in the nuclei after 30 min of treatment, on the contrary p65 localized in the nuclei after 4 h of treatment. We observed a 50% increase in both protein levels in the cytosol and nuclei within 4 h compared to control ones (Fig. 7 A, B). MC2050 pre-treatment induced a decrease of 20% and 50% in protein level of p50 and p65 subunits at 4 h, respectively.
Figure 7. Effect of PARP-1 on NF-kB translocation and protein levels in SH-SY5Y treated with Aβ25-35.

A) Cell lysates obtained at 30 min and 4 h from SH-SY5Y cells treated with 10 µM Aβ25–35 in the presence or absence of 50 µM MC2050, were for subjected to 12% SDS-PAGE, and immunoblotted with antibodies against p50 and p65. Proteins were detected through ECL (Millipore). B) Densitometric analyses were performed with ImageJ software, β-actin and lamin A/C levels were used as cytosolic and nuclear protein loading controls, respectively (white bar = control cells; black bar = Aβ25–35 treated cells; stripped bar = cells treated with Aβ25-35 in the presence of MC2050). Results are the mean of at least three independent experiments ± S.D. *p<0.05 vs. control cells; § p<0.05 vs. Aβ25–35-treated cells.
Aβ25–35 regulates p53 and Bcl-2 protein levels in mice and in SH-SY5Y
In TgCRND8 mice, p53 protein levels increased by about 50% in the entorhinal cortex and 100% in hippocampus, compared to wild type mice, while the anti-apoptotic protein Bcl-2 decreased by about 50% and 75% in the same regions, respectively (Fig. 8 A, B). Thus, we assayed the effect of the amyloid peptide on these two apoptosis-related proteins in SH-SY5Y cell. Our data indicate that p53 protein levels increased by about 35% at 24 h while Bcl-2 levels decreased by about 30% within 48 h in cells treated with amyloid peptide compared to control. The pre-treatment of cells with the PARP-1 inhibitor MC2050 counteracted the apoptotic effect of Aβ25–35, restoring the basal protein levels (Fig. 9 A, B).
Figure 8. Aβ affects p53 and Bcl-2 protein levels in transgenic mice.
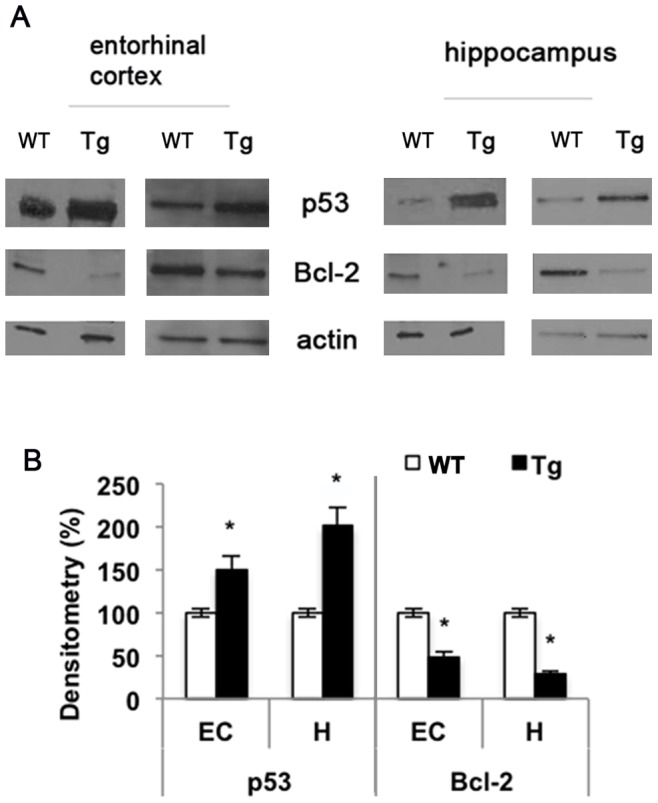
A) Entorhinal cortex and hippocampus homogenates were subjected to 12% SDS-PAGE and immunoblotted with antibodies against p53 and Bcl-2. Proteins were detected through ECL (Millipore). B) Densitometric analysis was performed with ImageJ software and normalized to β-actin (white bar = Wild Type mice; black bar = Transgenic mice). Results are the mean of at least three independent experiments ± S.D. *p<0.05 vs. WT.
Figure 9. Aβ affects p53 and Bcl-2 protein levels in SH-SY5Y via PARP-1.
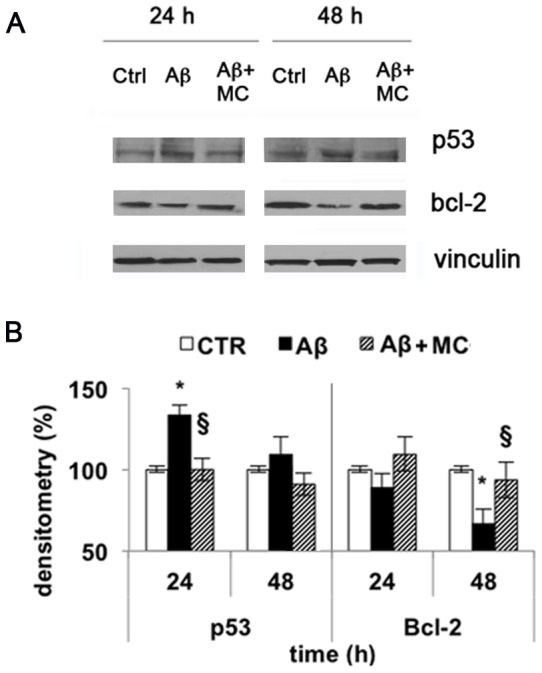
A) Cells treated with 10 µM Aβ25-35 for 24 and 48 h were lysed, subjected to 12% SDS-PAGE and immunoblotted with antibodies against p53 and Bcl-2. Proteins were detected through ECL (Millipore). B) Densitometric analysis performed with ImageJ software, and normalized to vinculin (white bar = control cells; black bar = Aβ25–35 treated cells; stripped bar = cells treated with Aβ25–35 in the presence of MC2050). Results are the mean of at least three independent experiments ± S.D. *p<0.05 vs. control; § p<0.05 vs. Aβ25–35-treated cells.
Discussion
PARP-1 is the most abundant member of the family of PARP enzymes and plays a critical role in the maintenance of DNA integrity by signalling DNA damage to the DNA repair machinery [31], [32]. When high levels of DNA damage occur, such as during oxidative stress insult, PARP-1 activity increases considerably causing a drastic reduction of NAD+ levels, with consequences on the ATP production and impairment of cell functions [33], [34]. Several studies have shown that pharmacological inhibition or gene deletion of PARP-1 could prevent tissue damage induced by oxidative stress associated with several diseases like stroke, diabetes, myocardial ischemia-reperfusion and Parkinson's disease [35]–[37].
In patients affected by AD, Love et al. [16] observed high levels of poly (ADP-ribosylated) proteins in the brain and recently two groups reported high level of PARP-1 activity in astrocytes and hippocampal brain slices [38], [39]. Since oxidative stress induced by the amyloid peptide, is implicated in the pathogenesis of AD [40], [41] here we explored how the crosstalk between PARP-1, ROS and DNA damage contributes to the neurodegenerative phenotype in AD.
To reach our goal, SH-SY5Y cells incubated with the fragment Aβ25–35, in the presence or absence of a new PARP inhibitor, a 4-quinazoline derivative, MC2050 and 7PA2 cells overproducing Aβ1–42 were used as cellular models. Experiments were also carried out on transgenic mice carrying the double mutant form APP695, showing amyloid peptide deposition at 3 months of age.
Our results showed an increase of about 60% of the enzyme activity over 24 h in the treated SH-SY5Y cells using Aβ25–35, a 40% increase in 7PA2 cells and 30% in hippocampal region of transgenic mice. The high level of PARylated proteins confirmed PARP-1 activation both in mice and in cells. The similar effect elicited by the two amyloidogenic species, confirms that the Aβ25–35 fragment is able to mimic the toxicity of Aβ1–40/42 and therefore is widely used to determine patterns of neurodegeneration. Millucci et al. [42] and Kaminsky et al. [43] reported the presence of the Aβ25–35 fragment in AD affected brains as the result of proteolysis of Aβ1–40 consequent to the racemization of serine 26. In comparative studies, Frozza et al. and Abramov et al. [44], [45] have shown that both Aβ1–42 and Aβ25–35 induce the same neuronal injury and toxic effects and Olivieri et al. [46] demonstrated the same effects on SH-SY5Y. Here we demonstrate that Aβ25–35 is able to induce PARP-1 activation in a similar manner to that reported for Aβ1–40/42 [31], [47]. Hence, all these researches support the use of Aβ25–35 as a suitable tool to study Alzheimer-type neurodegeneration.
The onset and development of AD are strictly associated with membrane integrity of neural cell. Indeed, oligomers and larger fibrillar aggregates made up by amyloid peptides seem to alter the biological function of the adjacent cellular membrane [24],[25]. Hence, first of all we assessed the ability of Aβ25–35 to interact with SH-SY5Y cell membrane. Data indicated that the Aβ25–35 targeted to cellular membrane inducing a 45% membrane perturbation after 4 h. An apparent decrease of membrane permeability was observed at 24 h, even if this phenomenon could be ascribed to cellular death. The neurotoxicity of Aβ25–35, assessed by the MTT assay, showed that the viability of cells treated with the amyloid peptide decrease of about 30% at 24 h, with a time-dependent trend, in agreement with the observations previously reported [48].
Many reports emphasized that in cells exposed to genotoxic insults, such as H2O2, PARP-1 is activated and modulates cell death [49], [50]. As shown by the cytofluorimetric analysis, Aβ25–35 induced oxidative stress and DNA damage as evaluated by comet assay that showed high DNA fragmentation in cells incubated with Aβ25–35 for 48 h. Then DNA damage led to PARP-1 activation that can be considered as one of the major cause of cell death probably depending on NAD depletion altering essential metabolic pathways as suggested by Abeti et al. in isolated astrocytes and in co-colture with hippocampal neurons [51].
Recently it has also been shown that PARP-1 plays an important role in the control of gene expression [52]. In particular, several studies explored the role of PARP-1 on the transcriptional activity of NF-kB, but the results are still ambiguous. Various authors have shown that the activation of NF-kB by PARP-1 is not due to its catalytic action, but to the interaction of PARP-1 with the two subunits of NF-kB [28], [53]. Conversely, other reports suggest a direct role of poly (ADP-ribosylation) in the co-activation of NF-kB [30], [47].
It is also known that NF-kB activation is strongly induced Aβ1–40, as well as Aβ1–42 in primary neurons or NT2N neuronal preparations, promoting the nuclear translocation of the subunits p65 and p50 and the expression of apoptotic genes [27]. Our data support the hypothesis that the activation of NF-kB caused by Aβ25–35 is mediated by PARP-1. In fact, by EMSA we show an increase of NF-kB activation in SH-SY5Y cells treated with Aβ25–35, which is prevented when the cells are pre-incubated with the PARP inhibitor MC2050. In addition, the “supershift” assay indicates p50 more than p65 as a component of the complex. Our data emphasize the role of PARP-1 as a trans-activator of NF-kB in the cells treated with the amyloid peptide, in agreement with data obtained by Chiarugi and Moskowitz [47], who used microglia and astrocytes as experimental model. Moreover, the specific involvement of the p50 subunit was also observed in vitro by Chang et al. [53], who demonstrated that NF-kappaB-p50 DNA binding was dependent on the presence of β-NAD+, and inhibited by the presence of PARP inhibitors.
Since activation of NF-kB is followed by its translocation from cytosol to nuclei as reported by Gilmore [54] and Perkins [55], we analysed the cellular localization of NF-kB. In SH-SY5Y cells treated with Aβ25–35 an initial translocation of p50 protein from the cytosol to the nucleus occurs within 30 min, whereas it seems that the p65 subunit is not implicated in this process. This data, together with the EMSA results, suggest that the p50 subunit is strictly involved in Aβ signal transduction. This hypothesis is supported also by data of Patel et al. [56] who observed p50 translocation in rat neurons following Aβ25–35 treatment.
We also tested whether the increase in NF-kB activity corresponds to an increase in NF-kB protein levels. We observed that Aβ25–35 affected the levels of p50 and p65 subunits in SH-SY5Y cells both in the nuclei and in the cytosol. Indeed after 4 h of treatment the levels of the two subunits significantly increase both in the cytosol and in the nuclei compared to control cells, indicating a positive regulation of NF-kB in cells exposed to Aβ. Data obtained from SH-SY5Y cells incubated with Aβ25–35 and pre-treated with MC2050 showed a decrease in protein levels of both subunits p50 and p65, suggesting that the significant p65 reduction in the nuclei could also be ascribed to a lack of p65 PARylation. Indeed, the translocation of NF-kB can be also modulated by PARP-1 through the poly (ADP-ribosylation) of the p65 subunit. The modified protein can no longer interact with the exportin Crm1, being retained in the nucleus [29].
Genotoxic insult such as oxidative stress and hypoxia cause DNA damage and trigger p53 activity. Misregulation in p53 activity can lead to cancer development, as well as to cardiovascular diseases, metabolic disorders and neurodegeneration.
We observed a significant increase in p53 protein levels in entorhinal cortex and hippocampus of transgenic mice and in SH-SY5Y cells treated with Aβ25–35. The observation that p53 is induced in cortex and brain of TgCRND8 mice point out the possible involvement of transcriptional regulation mechanisms. As a matter of fact, it was demonstrated that APP processing modulates p53 transcription via the signaling involving nuclear translocation of the AICD (APP intracellular domain) fragment generated by APP cleavage. Since mutant APP (more prone to gamma- and beta-secretase processing) is over-expressed in TgCRND8 mice, we can hypothesize that increased AICD is responsible for p53 up-regulation [57].
It is also well known that p53 is degraded rapidly in a ubiquitination-dependent proteasomal pathway and that the elevation in p53 levels occurs through stabilization and prevention of degradation [58]. Amyloid peptide can impair the proteasome, whose activity was found to be lower in AD brains than in age-matched controls [59], hence the altered levels of p53 in our experimental model could be also due to proteasomal impairment.
Once activated, p53 undergoes a large number of post-translational modifications including poly (ADP-ribosylation) that can stabilize the protein [60]. Several reports suggest a close relationship between PARP-1 and p53 [61] [62]. In experimental models of MPTP-induced parkinsonism, an increased level of p53 sustained by its the poly (ADP-ribosylation) prones neurons to cell death [17]. In our experimental model the pre-treatment of cells with MC2050 is able to counteract the toxic effects of Aβ25–35 thus restoring the basal p53 protein level.
Since Bcl-2 and p53 participate in the modulation and execution of cell death and since the increased expression of Bcl-2 protects PC12 cells from cell death induced by Aβ [63], we analyzed Bcl-2 protein levels in transgenic mice and SH-SY5Y cells treated with Aβ25–35. Our data indicated that the Bcl-2 protein level decreased markedly in both experimental models and that the pre-treatment of cells with MC2050 nullified the effect of amyloid treatment. These data are in agreement with Paradis et al. [64] who have shown that Aβ1–40 and Aβ1–42 induced a progressive decrease of Bcl-2 with a corresponding increase of the anti-apoptotic protein Bax in neuron primary cultures.
Since gene regulation occurs at different levels it is possible that PARP-1 affects Bcl-2 expression both at transcriptional or post-transcriptional level [65]. Recently, Dutta et al. [66] have demonstrated that Bcl-2 may influence PARP activity both in vitro and in cancer cells by a direct interaction with PARP-1. The overall relationship between PARP-1 and Bcl-2 may depend on the various inner environment of different cell lines and on the pathological condition.
In conclusion, our data show that PARP-1 has a prominent role of in the molecular mechanisms induced by Aβ that lead to cell death and neurodegeneration, through activation of NF-kB signalling and the modulation of p53 and Bcl-2.
Acknowledgments
The authors wish to thank Prof. A. Mai for critical reading of the manuscript and Prof. M.L. Mangoni and dr. A. De Leo for assistance with membrane perturbation and EMSA experiments. This work was supported by funds from MiUR and from Pasteur Institute, Cenci Bolognetti Foundation, Italy.
Funding Statement
This work was supported by funds from MIUR and from Istituto Pasteur, Fondazione Cenci Bolognetti, Roma, Italy. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Butterfield DA (2002) Amyloid beta-peptide (1–42)-induced oxidative stress and neurotoxicity: implications for neurodegeneration in Alzheimer's disease brain. A review. Free Radic Res 36: 1307–13. [DOI] [PubMed] [Google Scholar]
- 2. Hayashi M (2009) Oxidative stress in developmental brain disorders. Neuropathology 29: 1–8. [DOI] [PubMed] [Google Scholar]
- 3. Harper JD, Wong SS, Lieber CM, Lansbury PT (1999) Assembly of Abeta amyloid protofibrils: an in vitro model for a possible early event in Alzheimer's disease. Biochemistry 38: 8972–80. [DOI] [PubMed] [Google Scholar]
- 4. Larson ME, Lesné SE (2012) Soluble Aβ oligomer production and toxicity. J Neurochem 120 Suppl 1125–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Barnham KJ, Masters CL, Bush AI (2004) Neurodegenerative diseases and oxidative stress. Nat Rev Drug Discov 3: 205–14. [DOI] [PubMed] [Google Scholar]
- 6. Smith DG, Cappai R, Barnham KJ (2007) The redox chemistry of the Alzheimer's disease amyloid beta peptide. Biochim Biophys Acta 1768: 1976–90. [DOI] [PubMed] [Google Scholar]
- 7. Wilkinson BL, Landreth GE (2006) The microglial NADPH oxidase complex as a source of oxidative stress in Alzheimer's disease. J Neuroinflamm 3: 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Ji Y, Tulin AV (2010) The roles of PARP1 in gene control and cell differentiation. Curr Opin Genet Dev 20: 512–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. D'Amours D, Desnoyers S, D'Silva I, Poirier GG (1999) Poly (ADP-ribosyl) ation reactions in the regulation of nuclear functions. Biochem J 342: 249–68. [PMC free article] [PubMed] [Google Scholar]
- 10. de la Lastra CA, Villegas I, Sánchez-Fidalgo S (2007) Poly (ADP-ribose) polymerase inhibitors: new pharmacological functions and potential clinical implications. Curr Pharm Des 13: 933–62. [DOI] [PubMed] [Google Scholar]
- 11. Kauppinen TM, Swanson RA (2007) The role of poly (ADP-ribose) polymerase-1 in CNS disease. Neuroscience 145: 1267–72. [DOI] [PubMed] [Google Scholar]
- 12. Pacher P, Szabó C (2005) Role of poly (ADP-ribose) polymerase-1 activation in the pathogenesis of diabetic complications: endothelial dysfunction, as a common underlying theme. Antiox Redox Signal 7: 1568–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Peralta-Leal A, Rodríguez-Vargas JM, Aguilar-Quesada R, Rodríguez MI, Linares JL, et al. (2009) PARP inhibitors: new partners in the therapy of cancer and inflammatory diseases. Free Radic Biol Med 47: 13–26. [DOI] [PubMed] [Google Scholar]
- 14. Strosznajder RP, Jesko H, Zambrzycka A (2005) Poly (ADP-ribose) polymerase: the nuclear target in signal transduction and its role in brain ischemia-reperfusion injury. Mol Neurobiol 31: 149–67. [DOI] [PubMed] [Google Scholar]
- 15. Tempera I, Deng Z, Atanasiu C, Chen CJ, D'Erme M, et al. (2010) Regulation of Epstein-Barr virus OriP replication by poly (ADP-ribose) polymerase 1. J Virol 84: 4988–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Love S, Barber R, Wilcock GK (1999) Increased poly (ADP-ribosyl) ation of nuclear proteins in Alzheimer's disease. Brain 122: 247–53. [DOI] [PubMed] [Google Scholar]
- 17. Mandir AS, Przedborski S, Jackson-Lewis V, Wang ZQ, Simbulan-Rosenthal CM, et al. (1999) Poly (ADP-ribose) polymerase activation mediates 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced parkinsonism. Proc Natl Acad Sci USA 96: 5774–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Strosznajder JB, Jeśko H, Strosznajder RP (2000) Effect of amyloid beta peptide on poly (ADP-ribose) polymerase activity in adult and aged rat hippocampus. Acta Biochim Pol 47: 847–54. [PubMed] [Google Scholar]
- 19. Mosca L, Rotili D, Tempera I, Masci A, Fontana M, et al. (2011) Biological effects of MC2050, a quinazoline-based PARP-1 inhibitor, in human neuroblastoma and EBV-positive Burkitt's lymphoma cells. ChemMedChem 6: 606–11. [DOI] [PubMed] [Google Scholar]
- 20.Atherton E, Sheppard RC (1989) Solid Phase Synthesis – A Practical Approach. IRL Press at Oxford University Press, USA. p. 203.
- 21. Chishti MA, Yang DS, Janus C, Phinney AL, Horne P, et al. (2001) Early-onset amyloid deposition and cognitive deficits in transgenic mice expressing a double mutant form of amyloid precursor protein 695. J Biol Chem 276: 21562–70. [DOI] [PubMed] [Google Scholar]
- 22. Xie HR, Hu LS, Li GY (2010) SH-SY5Y human neuroblastoma cell line: in vitro cell model of dopaminergic neurons in Parkinson's disease. Chin Med J 123: 1086–92. [PubMed] [Google Scholar]
- 23. Zabel U, Schreck R, Baeuerle PA (1991) DNA binding of purified transcription factor NF-kappa B. Affinity, specificity, Zn2+ dependence, and differential half-site recognition. J Biol Chem 266: 252–60. [PubMed] [Google Scholar]
- 24. Poojari C, Kukol A, Strodel B (2012) How the amyloid-β peptide and membranes affect each other: An extensive simulation study. Biochim Biophys Acta 1828: 327–39. [DOI] [PubMed] [Google Scholar]
- 25. Tofoleanu F, Buchete NV (2012) Molecular interactions of Alzheimer's Aβ protofilaments with lipid membranes. J Mol Biol 421: 572–86. [DOI] [PubMed] [Google Scholar]
- 26. Kuner P, Schubenel R, Hertel C (1998) Beta-amyloid binds to p57NTR and activates NFkappaB in human neuroblastoma cells. J Neurosci Res 54: 798–804. [DOI] [PubMed] [Google Scholar]
- 27. Valerio A, Boroni F, Benarese M, Sarnico I, Ghisi V, et al. (2006) NF-kappaB pathway: a target for preventing beta-amyloid (Abeta)-induced neuronal damage and Abeta42 production. Eur J Neurosci 23: 1711–20. [DOI] [PubMed] [Google Scholar]
- 28. Hassa PO, Covic M, Hasan S, Imhof R, Hottiger MO (2001) The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J Biol Chem 276: 45588–97. [DOI] [PubMed] [Google Scholar]
- 29. Zerfaoui M, Errami Y, Naura AS, Suzuki Y, Kim H, et al. (2010) Poly (ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J Immunol 185: 1894–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Abd Elmageed ZY, Naura AS, Errami Y, Zerfaoui M (2012) The poly (ADP-ribose) polymerases (PARPs): new roles in intracellular transport. Cell Signal 24: 1–8. [DOI] [PubMed] [Google Scholar]
- 31. Bürkle A (2005) Poly (ADP-ribose). The most elaborate metabolite of NAD+ . FEBS J 272: 4576–89. [DOI] [PubMed] [Google Scholar]
- 32. Shall S, de Murcia G (2000) Poly (ADP-ribose) polymerase-1: what have we learned from the deficient mouse model? Mutat Res 460: 1–15. [DOI] [PubMed] [Google Scholar]
- 33. Alano CC, Garnier P, Ying W, Higashi Y, Kauppinen TM, et al. (2010) NAD+ depletion is necessary and sufficient for poly (ADP-ribose) polymerase-1-mediated neuronal death. J Neurosci 30: 2967–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mukhopadhyay P, Horváth B, Kechrid M, Tanchian G, Rajesh M, et al. (2011) Poly (ADP-ribose) polymerase-1 is a key mediator of cisplatin-induced kidney inflammation and injury. Free Radic Biol Med 51: 1774–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Czapski GA, Cakala M, Gajkowska B, Strosznajder JB (2006) Poly (ADP-ribose) polymerase-1 inhibition protects the brain against systemic inflammation. Neurochem Int 49: 751–5. [DOI] [PubMed] [Google Scholar]
- 36. d'Avila JC, Lam TI, Bingham D, Shi J, Won SJ, et al. (2012) Microglial activation induced by brain trauma is suppressed by post-injury treatment with a PARP inhibitor. J Neuroinflamm 15: 9–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pellicciari R, Camaioni E, Costantino G (2004) Life or death decisions: the cast of poly (ADP-ribose) polymerase (PARP) as a therapeutic target for brain ischaemia. Prog Med Chem 42: 125–69. [DOI] [PubMed] [Google Scholar]
- 38. Abeti R, Duchen MR (2012) Activation of PARP by oxidative stress induced by β-Amyloid: implications for Alzheimer's Disease. Neurochem Res 37: 2589–96. [DOI] [PubMed] [Google Scholar]
- 39. Strosznajder JB, Czapski GA, Adamczyk A, Strosznajder RP (2012) Poly (ADP-ribose) polymerase-1 in amyloid beta toxicity and Alzheimer's disease. Mol Neurobiol 46: 78–84. [DOI] [PubMed] [Google Scholar]
- 40. Atwood CS, Moir RD, Huang X, Scarpa RC, Bacarra NM, et al. (1998) Dramatic aggregation of Alzheimer abeta by Cu (II) is induced by conditions representing physiological acidosis. J Biol Chem 273: 12817–26. [DOI] [PubMed] [Google Scholar]
- 41. Cai Z, Zhao B, Ratka A (2011) Oxidative stress and β-amyloid protein in Alzheimer's disease. Neuromol Med 13: 223–50. [DOI] [PubMed] [Google Scholar]
- 42. Millucci L, Raggiaschi R, Franceschini D, Terstappen G, Santucci A (2009) Rapid aggregation and assembly in aqueous solution of A beta (25–35) peptide. J Biosci 34: 293–303. [DOI] [PubMed] [Google Scholar]
- 43. Kaminsky YG, Marlatt MW, Smith MA, Kosenko EA (2010) Subcellular and metabolic examination of amyloid-beta peptides in Alzheimer disease pathogenesis: evidence for Abeta (25–35). Exp Neurol 221: 26–37. [DOI] [PubMed] [Google Scholar]
- 44. Frozza RL, Horn AP, Hoppe JB, Simão F, Gerhardt D, et al. (2009) A comparative study of beta-amyloid peptides Abeta1-42 and Abeta25–35 toxicity in organotypic hippocampal slice cultures. Neurochem Res 34: 295–303. [DOI] [PubMed] [Google Scholar]
- 45. Abramov AY, Canevari L, Duchen MR (2004) Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci 24: 565–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Olivieri G, Baysang G, Meier F, Muller-Spahn F, Stahelin HB, et al. (2001) N-acetyl-L-cysteine protects SHSY5Y neuroblastoma cells from oxidative stress and cell cytotoxicity: effects on beta-amyloid secretion and tau phosphorylation. J Neurochem 76: 224–33. [DOI] [PubMed] [Google Scholar]
- 47. Chiarugi A, Moskowitz MA (2003) Poly (ADP-ribose) polymerase-1 activity promotes NF-kappaB-driven transcription and microglial activation: implication for neurodegenerative disorders. J Neurochem 85: 306–17. [DOI] [PubMed] [Google Scholar]
- 48. Datki Z, Juhász A, Gálfi M, Soós K, Papp R, et al. (2003) Method for measuring neurotoxicity of aggregating polypeptides with the MTT assay on differentiated neuroblastoma cells. Brain Res Bull 62: 223–9. [DOI] [PubMed] [Google Scholar]
- 49. Kang YH, Yi MJ, Kim MJ, Park MT, Bae S, et al. (2004) Caspase-independent cell death by arsenic trioxide in human cervical cancer cells: reactive oxygen species-mediated poly (ADP-ribose) polymerase-1 activation signals apoptosis-inducing factor release from mitochondria. Cancer Res 64: 8960–7. [DOI] [PubMed] [Google Scholar]
- 50. Szabó C (1996) The pathophysiological role of peroxynitrite in shock, inflammation, and ischemia-reperfusion injury. Shock 6: 79–88. [DOI] [PubMed] [Google Scholar]
- 51. Abeti R, Abramov AY, Duchen MR (2011) Beta-amyloid activates PARP causing astrocytic metabolic failure and neuronal death. Brain 134: 1658–72. [DOI] [PubMed] [Google Scholar]
- 52. Beneke S (2012) Regulation of chromatin structure by poly (ADP-ribosyl) ation. Front Genet 3: 169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Chang WJ, Alvarez-Gonzalez R (2001) The sequence-specific DNA binding of NF-kappa B is reversibly regulated by the automodification reaction of poly (ADP-ribose) polymerase 1. J Biol Chem 276: 47664–70. [DOI] [PubMed] [Google Scholar]
- 54. Gilmore TD (1999) The Rel/NF-kappaB signal transduction pathway: introduction. Oncogene 18: 6842–4. [DOI] [PubMed] [Google Scholar]
- 55. Perkins N (2006) Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 25: 6717–30. [DOI] [PubMed] [Google Scholar]
- 56. Patel JR, Brewer GJ (2008) Age-related differences in NFkappaB translocation and Bcl-2/Bax ratio caused by TNFalpha and Abeta42 promote survival in middle-age neurons and death in old neurons. Exp Neurol 213: 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Checler F, Sunyach C, Pardossi-Piquard R, Sevalle J, Vincent B, et al. (2007) The gamma/epsilon-secretase-derived APP intracellular domain fragments regulate p53. Curr Alzheimer Res 4: 423–6. [DOI] [PubMed] [Google Scholar]
- 58. Maki CG, Huibregtse JM, Howley PM (1996) In vivo ubiquitination and proteasome-mediated degradation of p53 (1). Cancer Res 56: 2649–54. [PubMed] [Google Scholar]
- 59. Chen JJ, Lin F, Qin ZH (2008) The roles of the proteasome pathway in signal transduction and neurodegenerative diseases. Neurosci Bull 24: 183–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wesierska-Gadek J, Schmid G (2001) Poly (ADP-ribose) polymerase-1 regulates the stability of the wild-type p53 protein. Cell Mol Biol Lett 6: 117–40. [PubMed] [Google Scholar]
- 61. Wieler S, Gagné JP, Vaziri H, Poirier GG, Benchimol S (2003) Poly (ADP-ribose) polymerase-1 is a positive regulator of the p53-mediated G1 arrest response following ionizing radiation. J Biol Chem 278: 18914–21. [DOI] [PubMed] [Google Scholar]
- 62. Valenzuela MT, Guerrero R, Núñez MI, Ruiz De Almodóvar JM, Sarker M, et al. (2002) PARP-1 modifies the effectiveness of p53-mediated DNA damage response. Oncogene 21: 1108–16. [DOI] [PubMed] [Google Scholar]
- 63. Song YS, Park HJ, Kim SY, Lee SH, Yoo HS, et al. (2004) Protective role of Bcl-2 on beta-amyloid-induced cell death of differentiated PC12 cells: reduction of NF-kappaB and p38 MAP kinase activation. Neurosci Res 49: 69–80. [DOI] [PubMed] [Google Scholar]
- 64. Paradis E, Douillard H, Koutroumanis M, Goodyer C, LeBlanc A (1996) Amyloid beta peptide of Alzheimer's disease downregulates Bcl-2 and upregulates bax expression in human neurons. J Neurosci 16: 7533–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Yang N, Gong F, Sun L, Yang D, Han X, et al. (2010) Poly (ADP-ribose) polymerase-1 binds to BCL2 major breakpoint region and regulates BCL2 expression. J Cell Biochem 110: 1208–18. [DOI] [PubMed] [Google Scholar]
- 66. Dutta C, Day T, Kopp N, van Bodegom D, Davids MS, et al. (2012) BCL2 suppresses PARP1 function and nonapoptotic cell death. Cancer Res 72: 4193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]