

Abstract
The ability of heterologous prime-boost vaccination to elicit robust CD8+ T cell responses has been well documented. In contrast, relatively little is known about how this immunotherapeutic strategy impacts the functional qualities of expanded T cells in the course of effector and memory responses. Using vesicular stomatitis virus (VSV) as a boosting vector in mice, we demonstrate that a massive secondary expansion of CD8+ T cells can be achieved shortly after priming with recombinant adenoviral vectors. Importantly, VSV-boosted CD8+ T cells were more potent than those primed by adenoviruses only, as measured by cytokine production, granzyme B expression, and functional avidity. Upon adoptive transfer, equivalent numbers of VSV-expanded CD8+ T cells were more effective (on a per-cell basis) in mediating antitumor and antiviral immunity than T cells only primed with adenoviruses. Furthermore, VSV boosting accelerated the progression of expanded CD8+ T lymphocytes to a central memory phenotype, thereby altering the effector memory profile typically associated with adenoviral vaccination. Finally, the functional superiority of VSV-expanded T cells remained evident 100 d after boosting, suggesting that VSV-driven immunological responses are of sufficient duration for therapeutic applications. Our data strongly support the choice of VSV as a boosting vector in prime-boost vaccination strategies, enabling a rapid amplification of CD8+ T cells and improving the quality of expanded T cells during both early and late immunological responses.
Keywords: adenovirus, CD8+ T cells, prime-boost, vaccination, vesicular stomatitis virus
Introduction
An outstanding challenge to successful immunotherapy is the development of antigen-specific vaccination regimens that can promote the generation of sufficient CD8+ T cell numbers for the treatment of infectious diseases and cancer. One promising approach to circumvent this problem involves prime-boost regimens using heterologous viral vectors that express the vaccination target. Indeed, numerous prior studies have shown that heterologous prime-boosting approaches generate larger amounts of antigen-specific CD8+ T cells than one-component vaccines and homologous boosting strategies.1-3 However, there is a limited availability of viral vectors that can be effectively used to boost viral vaccines to achieve an optimal expansion of CD8+ T cells. Vectors that can rapidly elicit strong primary and secondary immunological responses to allow for optimal control of progressing diseases are thus highly desirable for clinical application, yet remain poorly characterized.
Replication-incompetent, E1- and E3-deleted recombinant human type 5 adenovirus-based vectors are promising vaccines currently undergoing clinical evaluation. Recombinant adenoviral vectors are indeed highly effective in priming naïve T cells against transgenes, resulting in robust and antigen-specific protection against tumor or viral challenges.4-6 More importantly, adenoviral vectors have been shown to elicit effective immune responses even in individuals with pre-existing adenovirus-specific immunity, making them promising immunizing agents for the development of vaccines that are currently unavailable or unsatisfactory.7-10
A significant limitation of immunotherapeutic adenoviral vectors is that they elicit prolonged effector T (TEFF) cell and effector memory T (TEM) cell responses11-13 that require long intervals for an efficient boost. This is mainly due to the killing of migratory antigen-presenting dendritic cells (DCs) by cytolytic TEFF cells in the periphery, which prevents their engagement with central memory T (TCM) cells.14-16 Seeking to enhance the duration and potency of adenovirus-elicited immune responses, we have recently demonstrated that the intravenous administration of recombinant vesicular stomatitis virus (VSV) can provoke massive secondary expansion at the height of the adenovirus-primed CD8+ T-cell response.17 Furthermore, in an aggressive melanoma tumor model (B16), we showed that a strong primary immune response to adenovirus-based vaccines was essential for delaying tumor growth, creating a sufficient window to allow VSV boosting to further reduce the growth of—or even eradicate—neoplastic lesions.17 Since rapid boosting can be achieved without the need to compromise the TEFF population induced by primary immunization, our data suggest that recombinant adenoviruses and VSV can be combined to create a powerful therapeutic prime-boost vaccine.
Although VSV is an effective immunizing agent, it is also oncolytic agent. Thus, the relative contribution of these distinct biological properties towards the overall activity of VSV-based vaccines is difficult to discern, especially in the context of therapeutic (as opposed to prophylactic) settings.18 A better understanding of how VSV boosting impacts the functionality of expanded T cells may therefore be gained by investigating this approach in models of infectious disease, which may also broaden its clinical applicability. In the present study, we performed a detailed analysis of T-cell phenotype, cytokine secretion and immunological memory responses in tumor-free animals immunized with adenovirus-based vectors alone or in combination with VSV boosting. Furthermore, we compared the ability of these T cells (on a per cell basis) to protect animals against tumor or viral challenges. We report here that CD8+ T-cell responses from VSV-boosted mice are more potent than in mice that received adenovirus alone. More importantly, the superiority of VSV-expanded T cells was evident not only at the peak of secondary responses but was also 70 d after boosting, suggesting that the majority of antigen-specific CD8+ T cells elicited by VSV-boosting display a memory phenotype. Our data strongly support that the therapeutic use of recombinant VSV as a boosting vector in prime-boost vaccination strategies enables rapid and massive amplification of CD8+ T cells while improving their functional qualities at both early and late time points.
Results
Quantitative assessment of adenovirus-primed and VSV-boosted CD8+ T-cell responses
To compare adenovirus-primed and VSV-boosted CD8+ T cells, we reasoned that it would be best to coordinate the vaccinations so that peak expansion of primary and secondary epitope-specific CD8+ T-cell responses would coincide. To determine when these peaks occurred, mice were either primed with adenoviral vectors coding for human dopachrome tautomerase (Ad-hDCT) only, or primed with Ad-hDCT and then boosted (14 d later) with hDCT-coding VSV (VSV-hDCT). The dose and injection route of these vectors (i.e., 108 plaque-forming units (PFUs) i.m. for Ad-hDCT, and 109 PFUs i.v. for VSV-hDCT) had been previously optimized.17 As measured by the percentage of interferon γ (IFNγ)-expressing CD8+ T cells, kinetic analyses revealed that the peaks of the primary and secondary responses against the DCT180–188 epitope occurred about 11 d upon administration of Ad-hDCT (Fig. 1A) and 5 d upon that of VSV-hDCT (Fig. 1B), respectively. Of note, VSV-boosting significantly increased the magnitude of the response (~4.2% vs. ~25% IFNγ-expressing CD8+ T cells, Figure 1A and B).
Figure 1.

Quantitative assessment of adenovirus-primed and VSV-boosted CD8+ T cell responses. (A) C57BL/6 mice were primed by intramuscular injection of 1 × 108 plaque-forming units (PFUs) of Ad-hDCT. Blood samples were drawn on various days post-priming to quantify CD8+ T-cell response to the immunodominant epitope DCT180–188 by cytofluorometric detection of intracellular interferon γ (IFNγ) upon in vitro stimulation with the cognate peptide in the presence of brefeldin A. Data were pooled to plot the kinetics of the response. (B) Mice primed with Ad-hDCT were boosted after a 14-d interval by intravenous injection of 1 × 109 PFUs of VSV-hDCT. Transgene-specific CD8+ T-cell responses were measured in the blood at various days post-boosting to establish the kinetics of secondary responses. (C and D) Mice were primed by intramuscular injection of 1 × 108 PFUs of Ad-hDCT or Ad-SIINFEKL. Half of these mice were then boosted by intravenous injection of 1 × 109 PFUs of VSV-hDCT or VSV-SIINFEKL. The vaccinations were offset such that the peak of primary transgene-specific CD8+ T-cell responses in mice subjected to priming only (11 d post-priming) coincided with the peak of secondary responses in VSV-boosted animals (5 d post-boosting). The frequency of circulating CD8+ T cells specific for DCT180–188 (C) and SIINFEKL (D) was quantified by flow cytometry in terms of IFNγ-expressing cells upon in vitro antigenic stimulation. In all cases, n = 5 animals/group; data are reported as means ± SEM and are representative of two experiments. ν adenovirus-induced primary response; ϒ adenovirus-primed, VSV-boosted secondary response. Statistical significance was determined by two-way ANOVA: **p < 0.01, ***p < 0.001.
To evaluate the next phase of T cell-mediated immunity, we extended our observations for 2 weeks after the peak response (designated as “day 0”), confirming our previous finding that adenoviruses typically produce a CD8+ T-cell response with a protracted contraction phase (Fig. 1C). Although the frequency of VSV-expanded CD8+ T cells remained significantly higher than that of T cells developing in mice receiving Ad-hDCT only with during this 2-week time frame, VSV boosting did not appear to alter the occurrence or duration of the contraction phase (Fig. 1C). To determine if this phenomenon was unique to self-antigens, like hDCT, we repeated the experiment by substituting the transgene in both viral vectors with an immunodominant epitope derived from chicken ovalbumin (OVA), SIINFEKL (OVA257–264). Notably, SIINFEKL-specific T cells accounted for more than 22% of total circulating CD8+ T cells at the peak of adenoviral priming, a primary response that was increased to more than 80% by VSV boosting (Fig. 1C). Irrespective of both the enhanced magnitude of the response and the nature of the antigen, the protracted nature of the contraction phase remained unaltered (Fig. 1D).
To determine whether VSV could amplify CD8+ T-cell responses even at early time points, we boosted mice over shorter intervals with VSV-hDCT from 4 to 14 d after Ad-hDCT priming. In this setting, the frequency of transgene-specific CD8+ T cells was remarkably enhanced by boosting with VSV within 4 d of adenoviral priming (Fig. 2A). Also, boosting 7 d after priming was as effective as doing so 14 d thereafter, demonstrating the unique capacity of VSV to accelerate the kinetics of CD8+ T-cell responses. We also observed that VSV boosting was effective in the same time frame (i.e., 14 d after primary immunization) in mice primed with hDCT180–188-pulsed DCs (DC/SVY) or vaccinia viruses (VVs) expressing SIINFEKL (VV/SIIN) (Fig. 2B and C), indicating that the remarkable boosting potency of VSV is not limited to specific antigens or priming methods. Interestingly, however, the magnitude of boosting correlated with the intensity of primary responses (please compare Figure 2A with 2B and Figure 1D with 2C), indicating that the combination of efficient adenoviral vectors with VSV represents a robust prime-boost strategy to achieve therapeutically relevant immune responses.

Figure 2. VSV enables ultra-rapid boosting that is not limited to a single antigen or priming method. (A-C) C57BL/6 mice were primed by intramuscular injection of 1 × 108 plaque-forming units (PFUs) of Ad-hDCT (A) subcutaneous injection of 1 × 106 dendritic cells pulsed with the DCT180–188 peptide (DC/SVY) (B), or subcutaneous injection of 1 × 108 PFUs of VV-SIINFEKL (VV-SIIN). Four to 14 (A) or 14 (B and C) days later, mice were boosted by intravenous injection of 1 × 109 PFUs of VSV-hDCT (A and B) or VSV-SIINFEKL (VSV-SIIN) (C). Notes: In all cases, n = 5 animals/group; data are reported as means ± SEM and are representative of two experiments.
VSV-boosted CD8+ T cells produce more cytokines and granzyme B than those primed with adenoviruses
Although the ability of VSV to rapidly and quantitatively amplify TEFF responses elicited by adenoviral vaccines was highly attractive and unique, its impact on the qualitative function and memory development of adenovirus-primed T cells remained to be determined. As an initial approach to these incognita, we assessed the cytokine secretion profile of hDCT180–188- and SIINFEKL-specific cells developing in mice that had been primed with adenovirus as a standalone immunotherapeutic intervention or in combination with VSV boosting at various time points. Compared with primary T cells (i.e., cells developing in response to adenoviral priming), a higher frequency of secondary T cells (i.e., cell developing upon adenoviral priming and VSV boosting) could simultaneously produce IFNγ and tumor necrosis factor α (TNFα) (Fig. 3A), regardless of the nature of the immunizing epitope. VSV-boosted cells also produced more IFNγ (Fig. 3B) and TNFα (Fig. 3C) than adenovirus-primed cells on a per-cell basis. The dynamics of the response specifically showed that the amount of IFNγ produced by VSV-boosted T cells generally plateaued starting from 1-week post-peak (Fig. 3B), but the proportion of cells producing TNFα (Fig. 3A) and the amount of TNFα per cell (Fig. 3C) were still increasing at the final 2-week time point. In addition to enhanced cytokine production, CD8+ T cells from VSV-boosted mice also exhibited substantially increased expression levels of granzyme B than those from mice subjected to adenoviral priming only, particularly at the peak of response (Fig. 3D).

Figure 3. VSV-boosted CD8+ T cells produce more cytokines and granzyme B than those primed with adenoviruses only. (A-D) C57BL/6 mice were primed by intramuscular injection of 1 × 108 plaque-forming units (PFUs) of Ad-hDCT or Ad-SIINFEKL, and part of them boosted 14 d later by intravenous injection of 1 × 109 PFUs of VSV-hDCT or VSV-SIINFEKL. Immunizations were scheduled so that the peaks of primary or secondary transgene-specific CD8+ T-cell responses coincided. Circulating antigen-specific T cells were identified by flow cytometry upon stating of intracellular interferon γ (IFNγ) and tumor necrosis factor α (TNFα) after in vitro stimulation with cognate peptides in the presence of brefeldin A. (A) Multi-cytokine producing transgene-specific CD8+ T cells were defined as those capable of simultaneously producing IFNγ and TNFα. The amount of IFNγ (B) and TNFα (C) produced on a per cell basis was also evaluated. (D) Primary and secondary SIINFEKL-specific CD8+ T cells identified by tetramer staining were fixed, permeabilized and then assessed for granzyme B expression by intracellular immunostaining followed by cytofluorometric analysis. In all cases, n = 5 animals/group; data are reported as means ± SEM and are representative of two experiments. ○ adenovirus-primed, VSV-boosted secondary response. Statistical significance was determined by two-way ANOVA: *p < 0.05, **p < 0.01, ***p < 0.001.
VSV boosting enhances the functional avidity of CD8+ T cells
In light of our observations that secondary T cells had a superior cytokine secretion profile, we hypothesized that VSV boosting selectively induces the expansion of T-cell subsets that exhibit the highest avidity within the adenovirus-primed cell pool. To test this hypothesis, we assayed the functional avidity of CD8+ cells isolated from mice that were primed with either Ad-hDCT or Ad-SIINFEKL alone or in combination with a heterologous boost VSV-hDCT or VSV-SIINFEKL, respectively. At multiple time points, circulating lymphocytes were harvested and stimulated with serial dilutions of cognate peptides in the presence of brefeldin A (to trap cytokines in the cytoplasm of responding T cells). T-cell responses to each concentration of peptide were monitored by flow cytometry in terms of the percentage of IFNγ+ CD8+ T cells relative to that induced by the highest peptide concentration for each immunogen. In this assay, high T-cell avidities are defined by a large proportion of T cells that can respond to target peptides at low concentrations. Both at the peak of the immune response (Figure 4A) and 1-week later (Fig. 4B), VSV-boosted T cells were of higher avidity than adenovirus-primed cells upon exposure to both hDCT180–188 (Fig. 4, left panels) and SIINFEKL (Fig. 4, right panels). Interestingly, however, by 2-weeks post-peak, the average avidity of primary and secondary T cells was equivalent (Fig. 4C).

Figure 4. Boosting with VSV enhances the functional avidity of CD8+ T cells. (A-C) C57BL/6 mice were primed by intramuscular injection of 1 × 108 plaque-forming units (PFUs) of Ad-hDCT or Ad-SIINFEKL, and part of them boosted 14 d later by intravenous injection of 1 × 109 PFUs of VSV-hDCT (left panels) or VSV-SIINFEKL (right panels). Immunizations were scheduled so that the peaks of primary or secondary transgene-specific CD8+ T-cell responses coincided. Circulating transgene-specific T cells were evaluated over time upon the cytofluorometric detection of intracellular interferon γ (IFNγ) after in vitro stimulation with serial dilutions of cognate peptides in the presence of brefeldin A. (A) Peak response. (B) One-week post-peak response. (C) Two-weeks post-peak response. In all cases, n = 5 animals/group; data are reported as means ± SEM and are representative of two experiments. ■ adenovirus-induced primary response; ○ adenovirus-primed, VSV-boosted secondary response. Statistical significance was determined by two-way ANOVA: *p < 0.05.
VSV-boosted CD8+ T cells are functionally superior in vivo to those primed by adenoviruses only
Having determined that VSV-boosted CD8+ T cells display higher avidity and produce more cytokines than adenovirus-primed cells, we sought to determine if these characteristics translated into enhanced immunotherapeutic activity in vivo, in appropriate tumor and viral challenge models. We selected SIINFEKL as the target epitope for this experiment because an almost 14-fold increase in transgene-specific T cells can be achieved with SIINFEKL as compared with DCT180–188 (Fig. 1), significantly reducing the number of donor animals required. Also, we excluded quantitative differences as a confounding factor by using flow cytometry to sort transgene-specific CD8+ T cells from mice primed with either Ad-SIINFEKL alone or subjected to a prime-boost regimen involving Ad-SIINFEKL and VSV-SIINFEKL. CD8+ T cells purified from each group of mice were then adoptively transferred into naïve recipients in equal amounts (4.5 × 105 cells/mouse). Negative control animals received the same number of CD8+ T cells obtained from mice that had been sequentially treated with empty adenoviral (Ad-BHG) and VSV (VSV-MT) vectors. Transferred SIINFEKL-specific CD8+ T cells were allowed to home to their target tissues for 24 h prior to the administration of the immunogen.
In order to test the utility of heterologous VSV boosting against both malignant and infectious diseases, we chose to test two different challenge models. Mice first received 5 × 105 OVA-expressing B16 (B16-OVA) murine melanoma cells. Twenty-one days after challenge, mice were euthanized and lung metastases were enumerated. Transferring SIINFEKL-targeting CD8+ T cells from Ad-SIIN-primed donor mice effectively decreased the frequency of metastases (relative to CD8+ T cells derived from Ad-BHG-primed donor mice), confirming that adenoviruses efficiently prime protective CD8+ T-cell responses. As expected, VSV-boosted T cells reduced the proportion of metastases even further (Fig. 5A; p < 0.0001). These observations provide compelling evidence that VSV boosting indeed improves the cytotoxic function of adenovirus-primed CD8+ lymphocytes, at least in the context of antitumor immunity. To extend the relevancy of our findings to the treatment of infectious diseases, we assayed the relative efficacy of adenovirus-primed vs. adenovirus-primed and VSV-boosted CD8+ T cells in a model of viral challenge. Following adoptive T-cell transfer, mice were challenged i.p. with 1 × 106 PRUs of a recombinant VV engineered to express SIINFEKL (VV-SIINFEKL). Three days after challenge, mice were euthanized and VV titers were quantified in preparations derived from the ovaries, a tissue for which VVs have a natural tropism. VSV-boosted CD8+ T cells were superior to an equal number of primary (adenovirus-primed only) cells in controlling VV infection, as measured by reduced viral titers (Fig. 5B, p = 0.027).
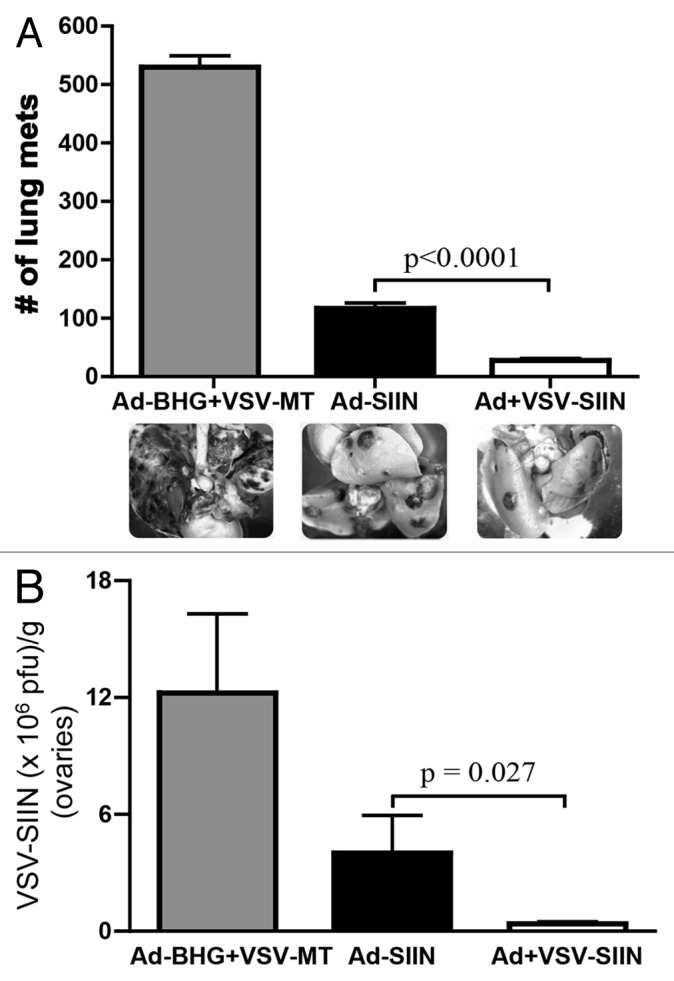
Figure 5. VSV-boosted CD8+ T cells are functionally superior to T cells subjected to adenoviral priming only. (A-B) Donor C57BL/6 mice were primed by intramuscular injection of 1 × 108 plaque-forming units (PFUs) of Ad-SIINFEKL (Ad-SIIN), or primed and then boosted 14 d later by intravenous injection of 1 × 109 PFUs of VSV-SIINFEKL (VSV-SIIN). Immunizations were scheduled so that the peaks of primary or secondary transgene-specific CD8+ T-cell responses coincided. At the peak of these responses, 4.5 × 105 of either primary or secondary SIINFEKL-specific CD8+ T cells (4.5 × 105) was adoptively transferred into naïve C57BL/6 hosts. Control animals received an equal number of CD8+ T cells obtained from mice that had been treated with 1 x 108 PFUs of Ad-BHG i.m. followed by 1 × 109 PFUs of VSV-MT i.v. (both are empty vectors). (A) Twenty-four hours after adoptive cell transfer, mice were challenged with 5 × 105 B16-OVA cells i.v.. Twenty-one days later, lung metastases, and representative images are shown. (B) Twenty-four hours after adoptive cell transfer, mice were challenged with 1 × 106 PFUs of VV-SIINFEKL (VV-SIIN) i.p.. Three days later, VV titers were quantified in ovaries. Both these challenge experiments were repeated twice with n = 3–6 mice/group and yielded similar results. In all cases, data are reported as means ± SEM . Statistical significance was determined by one-way ANOVA.
VSV-boosted CD8+ T cells exhibit enhanced functional memory
The quantitative and qualitative comparisons of CD8+ T cells developing in adenovirus-primed and VSV-boosted recipients had been, so far, limited to early time points (ranging from the peak of the response to 2-weeks later). Tumor and viral challenges were performed with adoptively transferred CD8+ T lymphocytes that had been purified from mice at the peak of primary and secondary responses (i.e., during the effector phase). To assay the impact of VSV boosting on the induction of immunological memory and to determine whether functional differences in the resulting CD8+ T cells persist over time, we repeated our assessments 70 d after the peak of adenovirus-primed or VSV-boosted responses against hDCT and OVA. As shown by enhanced tetramer staining and increased percentages of IFNγ-expressing CD8+ T cells, the frequency of transgene-specific T cells remained significantly higher in VSV-boosted mice than in animals subjected to adenoviral priming only (Fig. 6A). We also monitored the development of memory responses by using an antibody specific for CD127, the interleukin (IL)-7 receptor associated with memory cells. We observed a dominance shift from effector (TEFF) to memory (comprising TEM and TCM) cells 70 d after the peak of responses (Fig. 6B). Of particular interest, VSV boosting exaggerated the predominance of CD127+ memory T cells as compared with adenovirus priming only (Fig. 6B). We further analyzed DCT-specific CD8+ T cells 113 d post-peak to elucidate the relative contribution of TEM and TCM cells towards the increased abundance of memory T lymphocytes. Consistent with previous observations,6,11 adenoviral immunization mainly resulted in the generation of TEM (tetramer+CD127+CD62L-) cells (Fig. 6C). By contrast, the vast majority of the memory compartment in VSV-boosted animals (at this time point) displayed a central memory phenotype (tetramer+CD127+CD62L+), suggesting that VSV boosting does not only increase the abundance and promote the early cytotoxic functions of adenovirus-primed TEM cells, but also influences the frequency and phenotype of memory responses to priming.

Figure 6. VSV-boosted CD8+ T cells exhibit enhanced functional memory. (A-C) C57BL/6 mice were primed by intramuscular injection of 1 × 108 plaque-forming units (PFUs) of Ad-hDCT or Ad-SIINFEKL, or primed and then boosted 14 d later by intravenous injection 1 × 109 PFUs of VSV-hDCT or VSV-SIINFEKL. Immunizations were scheduled so that the peaks of primary or secondary transgene-specific CD8+ T-cell responses coincided. Antigen-specific T cells were identified in the blood by tetramer staining or cytofluorometric detection of intracellular interferon γ (IFNγ) upon in vitro stimulation with cognate peptides (DCT180–188 or SIINFEKL) in the presence of brefeldin A. (A) Frequencies of transgene-specific cells on day 70 post-peak as measured by tetramer and intracellular cytokine staining (ICS). (B) Kinetics of CD127 expression on transgene-specific cells as measured by tetramer staining. (C) Proportion of DCT180–188-tetramer-positive CD8+ T cells with effector (TEFF, CD127-), effector memory (TEM; CD127+CD62L-) and central memory (TCM; CD127+ CD62L+) phenotypes at day 113 post-peak. Data are representative of two experiments. ○ adenovirus-primed, + VSV-boosted secondary response. Statistical significance in (A) and (C) was determined by one-way ANOVA, p values are reported. Statistical significance in (B) was determined by two-way ANOVA: *p < 0.05, **p < 0.01, ***p < 0.001.
Finally, to directly determine the functionality of adenovirus-primed or VSV-boosted T cells well beyond their peak responses, we purified primary or secondary T cells from SIINFEKL-immunized mice 70 d post-peak and adoptively transferred them in equal amounts (4.5 × 105 cells/mouse) into distinct naïve hosts. These mice were intravenously challenged with 5 × 105 B16-OVA cells 24 h after T-cell transfer. Similar to what observed when T cells derived from peak immune responses were transferred (Fig. 5A) and in line with our previous findings,6,11 fewer metastases developed in mice receiving adenovirus-primed T cells than in animals treated with control lymphocytes, suggesting that the therapeutic potential of adenoviral priming persists even 70 d after the peak response. In addition, VSV-boosted cells were significantly superior to adenovirus-primed cells in attenuating the metastatic spread of B16-OVA cells to the lung (Fig. 7; p = 0.0001). This provides firm experimental evidence indicating that the functional improvement of T cells achieved by VSV boosting is long lasting, presumably due to enhanced immunological memory.
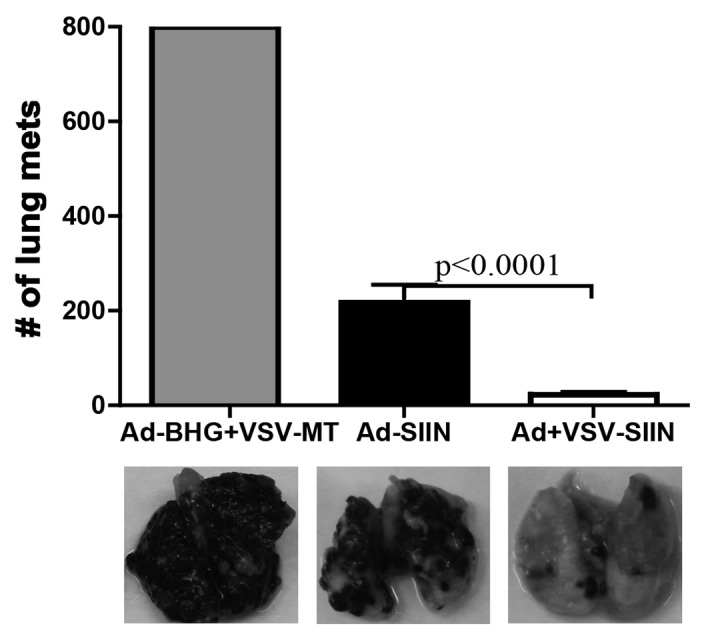
Figure 7. VSV-boosted CD8+ T cells are functionally superior at the memory stage. C57BL/6 mice were primed by intramuscular injection of 1 × 108 plaque-forming units (PFUs) of Ad-SIINFEKL (Ad-SIIN), or primed and then boosted 14 d later by intravenous injection of 1 × 109 PFUs of VSV-SIINFEKL (VSV-SIIN). Immunizations were scheduled so that the peaks of primary or secondary transgene-specific CD8+ T-cell responses coincided. At 70 d post-peak, 4.5 × 105 primary or secondary SIINFEKL-specific CD8+ T cells were adoptively transferred into naïve C57BL/6 hosts. Control animals received an equal number of CD8+ T cells from obtained from mice that had been treated with 1 × 108 PFUs of Ad-BHG i.m. followed by 1 × 109 PFUs of VSV-MT i.v. (both are empty vectors). Twenty-four hours after transfer, mice were challenged with 5 × 105 B16-OVA cells i.v.. Twenty-one days after challenge, lung metastases were enumerated. In all cases, n = 5 animals/group; data are reported as means ± SEM and are representative of two experiments. Statistical significance was determined by one-way ANOVA, p values are reported.
Discussion
The success of immunotherapeutic vaccines is dependent upon sufficient elicitation of antigen-specific cytotoxic T lymphocyte-mediated cell death and immune responses that persist over time. Here, we report that the magnitude, cytotoxic function and memory phenotype of antigen-specific CD8+ T lymphocytes produced by adenoviral vaccines can be reshaped by VSV boosting, leading to dramatically enhanced CD8+ T-cell immunity in mouse models of cancer and viral infection. Combining adenoviruses and VSV into a prime-boost regimen can rapidly stimulate increased numbers of both effector and memory CD8+ T cells, which are both essential for improved clinical outcomes. These data have broad implications for the treatment of both cancer and infectious diseases, especially in therapeutic (as opposed to prophylactic) settings.
It has been previously reported that recombinant adenoviruses are effective vectors that can consistently foster robust CD8+ T cell immunity in multiple infection models as well as in patients affected by various diseases.7,19-22 CD8+ T cells induced by adenovirus-based vaccines exhibit unique characteristics amenable to therapeutic use, including the preferential accumulation of TEM cells and their persistence at high numbers.6,11,12 These clinically relevant responses may potentiate immune protection against peripheral (e.g., skin, mucosal) infections and, by similar mechanisms, against cancer.23-26 Our results have confirmed this adenovirus-induced CD8+ T cell phenotype. Moreover, by adoptive cell transfer experiments, we have further evinced the robust immunotherapeutic functions of adenovirus-primed cells at both the early and late stages of the immune response.
Remarkably, VSV boosting can further magnify the high number of effector T cells that develop in response to adenoviral priming. Strikingly, this immunologically advantageous boosting effect can be achieved over a short interval (within 4–14 d upon priming) and offers an effective vaccination strategy for circumstances in which immediate protection is essential (e.g., malignancy or potentially pandemic infections). The ability of VSV to accelerate the secondary expansion of antigen-specific CD8+ T cells is a result of its unique intrinsic properties, as VV systems as well as DC-based vaccines failed to boost T-cell responses to a similar extent within the days following immunization (ref. 16 and unpublished data). Indeed, we have recently demonstrated that the intravenous delivery of VSV-based vaccines results in the direct infection of follicular B cells in the spleen, leading to antigen presentation by neighboring DCs in the follicular region (Bridle and Wan, manuscript in preparation). Since TCM cells are also located within B-cell follicles and TEFF cells cannot traffic through the same areas, antigen-loaded follicular antigen-presenting cells (APCs) might, at least conceptually, efficiently engage TCM cells while escaping TEFF cell-dependent killing, even at the peak of primary responses.27-29
Interestingly, VSV does not only increase the magnitude of adenovirus-primed CD8+ T-cell responses but also enhances the cytotoxic activity of these cells, as measured by increased cytokine production, granzyme B expression, and avidity for their targets. We observed that VSV-boosted T cells are functionally superior—on a per-cell basis (i.e., in equal amounts)—to adenovirus-primed T cells in mediating immune responses against either viral or tumor challenges. Improving the quality of T cells has been a problem in particular for the treatment of cancer or chronic infections, two pathological conditions in which T cells either intrinsically exhibit a low avidity for cognate antigens or, alternatively, become functionally exhausted upon prolonged antigenic stimulation in the context of an inflammatory milieu, such as it occurs in the tumor environment.30-32 Combining adenoviruses with VSV may be able to potentiate the magnitude as well as the quality of immune responses, both of which are essential for the successful outcome of vaccination in patients. The precise molecular mechanism by which VSV boosting enhances the quality of expanded CD8+ T lymphocytes remains unclear; however, we postulate that it may relate to antigen cross-presentation by DCs, which has been linked to the immunological profile of CD8+ T cells.33-35 In our experience, VSV appears to primarily infect follicular B cells while antigen presentation in this setting relies on DCs (Bridle and Wan, manuscript in preparation), suggesting that cross-presentation may constitute the predominant mechanism which mediates the increased immunological efficacy of VSV-boosted CD8+ T cells that we report here. VSV vectors possess self-replicating, cytolytic and oncolytic properties that may facilitate the cross-presentation of antigens released in the follicles upon cell death, leading to superior CD8+ T-cell responses. Furthermore, like other viral vectors, VSV generates a variety of “danger signals” that engage host Toll-like receptors (TLRs) and other pattern recognition receptors to activate the innate immune response.36-38 Specifically, it has been reported that ligation of TLR4 and/or TLR7/TLR8 on APCs can influence the phenotype of T cells, enhancing the generation of polyfunctional CD8+ T lymphocytes.39,40 We and others have previously shown that VSV infection stimulates DCs to produce large amounts of IFNα/β, IL-12 and IL-15, APC responses that may contribute to the functional improvement of expanding CD8+ T cells.41-44
It has been previously shown that adenoviral immunization favors the commitment of antigen-specific CD8+ T cells to the TEM lineage, mainly due to prolonged antigenic stimulation.11,12,45 In our hands, prolonged antigen presentation upon adenoviral vaccination is exclusively mediated by non-hematopoietic APCs that persistently stimulate existing memory cells.6 We surmise that the rapid VSV boosting of effector CD8+ T cells with a high functional avidity may clear such non-hematopoietic APCs, resulting in shortened antigen presentation, a situation that resembles the outcome of extinction of transgene expression.45 On the contrary, boosting with VSV over a short interval may prolong antigen presentation by conventional hematopoietic APCs, effectively accelerating CD8+ T-cell responses and the acquisition of a TCM phenotype.
Whether the quality or the magnitude of the immune response is a better predictor of the clinical efficacy of vaccination is an important and hitherto unresolved question for the field.26,46-48 It is likely that an optimal protection against individual pathogens relies on T cells with a distinct functional profile, while both the magnitude and quality of CD8+ T cells are required to effectively control neoplastic diseases.49 Accumulating data highlight the potential efficacy of recombinant adenoviruses as highly versatile vectors that are capable of generating high numbers of CD8+ effector-like T cells in peripheral tissues to provide immediate immunological protection.20,48 Heterologous boosting with VSV offers a new strategy not only to improve the already desirable properties of memory T cells elicited by adenoviruses (notably their persistence at high levels and their TEM phenotype) but also to increase the amounts of TCM cells, a subset of CD8+ T cells that may be preferable in the context of certain pathogens. Thus, the administration of recombinant adenoviral vectors (priming) followed by that of VSV expressing the same antigenic epitope (boosting) represents a novel combinatorial approach to vaccination that elicits CD8+ T cells with optimal magnitude and phenotype at both effector and memory stages.
Materials and Methods
Mice and cell cultures
Age-matched (8–10 weeks old at initiation of each experiment) female C57BL/6 (haplotype H-2b) mice (Charles River Laboratories) were housed in a dedicated pathogen-free facility. Animal studies complied with Canadian Council on Animal Care guidelines and were approved by McMaster University’s Animal Research Ethics Board. B16-OVA cells, a murine melanoma line stably transfected with an expression vector encoding full-length OVA, were cultured in MEM-F11 medium supplemented with 10% fetal bovine serum (FBS), 2 mM l-glutamine, 55 µM 2-mercaptoethanol, 1× vitamin solution and antibiotics. G418 (at the dose of 800 μg/mL) was used to maintain OVA expression. Vero cells were cultured in Eagle’s minimum essential medium containing Earle’s salts supplemented with 10% FBS, 2 mM l-glutamine and antibiotics. All cell culture reagents were from Life Technologies.
Viruses
The E1/E3-deleted, replication-deficient, recombinant human serotype 5 adenovirus and the replication-competent, recombinant vesicular stomatitis virus (VSV) with a methionine residue deleted at position 51 of the matrix protein (to abrogate its ability to inhibit Type I IFN responses) have been described previously.6,41 Ad-hDCT and VSV-hDCT vectors express the human melanoma-associated antigen dopachrome tautomerase (hDCT); Ad-SIINFEKL and VSV-SIINFEKL express the H-2b-dominant CD8+ T-cell epitope OVA257–264 (SIINFEKL). Recombinant VVs expressing SIINFEKL (VV-SIINFEKL) has previously been described.6,50 Ad-BHG and VSV-MT were empty control vectors.
Peptides
The immunodominant peptide from DCT that binds to H-2Kb (DCT180–188, SVYDFFVWL; shared by human and murine DCT) was synthesized by PepScan Systems (Lelystad). The H-2Kb-restricted OVA-derived SIINFEKL peptide was synthesized by Biomer Technologies.
Dendritic cell-based vaccine
Murine bone marrow-derived DCs were generated in the presence of 40 ng/mL recombinant murine granulocyte macrophage colony-stimulating factor (GM-CSF; from PeproTech) for 7 d, as previously described,42 and then loaded with 1 μg/mL DCT180–188 for 4 h in the presence of 2 μg/mL lipopolysaccharide LPS (Sigma-Aldrich). To vaccinate mice, 5 × 105 peptide-pulsed DCs were injected s.c. into each hind footpad (total dose = 1 × 106 cells).
Administration of viral vaccines
Anesthetized mice were immunized by injection of 1 × 108 PFUs of adenoviral vectors in 100 μL PBS (50 μL/hamstring) i.m., or 1 × 108 PFUs of VV vectors in 200 μL PBS i.p.. When appropriate, boosting was performed by injection of 1 × 109 PFUs of VSV in 200 μL PBS i.v., into the tail vein.
Antibodies and tetramers
The following monoclonal antibodies were used in flow cytometry assays. Anti-CD16/CD32 (clone 2.4G2) antibodies were employed to block FC receptors; anti-CD8 (clone 53–6.7), anti-CD62L (clone MEL-14) and anti-CD127 (clone SB/199) antibodies were used for cell-surface staining; and anti-IFNγ (clone XMG1.2), anti-TNFα (clone MP6-XT22) and anti-granzyme B (clone GB11) antibodies were employed for intracellular staining. All antibodies were from BD Biosciences. For the quantification of antigen-specific T cells, the following allophycocyanin (APC)-conjugated tetramers were used: Kb-DCT180–188-APC and Kb-OVA257–264-APC (MHC Tetramer Lab, Baylor College of Medicine).
Intracellular cytokine staining
To assess antigen-specific T-cell responses, blood was collected from the peri-orbital sinus into heparinized tubes and red blood cells were lysed. Cells were kept on ice during handling and the time from sample collection to the end of processing was less than 2 h. Only fresh cells were used in cytofluorometric assays. To this aim, cells were counted on an improved Neubauer hemocytometer and cell viability was ensured consistently greater than 90% (as assessed based on the exclusion of trypan blue). Mononuclear cells were stimulated with 1 µg/mL peptides (controls were exposed to irrelevant peptides at the same concentration) in RPMI medium supplemented with 10% FBS, 2 mM l-glutamine, antibiotics and 1 µg/mL brefeldin A (GolgiPlug, BD Biosciences, added after 1 h of incubation) . During the 5 h total incubation time, FC receptors were blocked with anti-CD16/CD32 antibodies and then cells were stained with fluorescent anti-CD8 antibody in PBS supplemented with 5% bovine serum albumin (BSA). Cells were then fixed/permeabilized with Cytofix/Cytoperm (BD Biosciences) and stained for the detection of intracellular cytokines. Data were acquired using a FACSCanto flow cytometer with the FACSDiva v.5.0.2 software (BD Biosciences) and analyzed with the FlowJo software (Tree Star).
Functional avidity assays
The functional avidity of antigen-specific CD8+ T cells was determined by intracellular cytokine staining, as described above, following stimulation with serial log-dilutions of peptides in vitro. Peptide concentration varied from 1,000 to 0.1 ng/mL. Data are expressed as percentages of the response to the maximal peptide concentration, calculated as follows: (% of CD8+ cells responding to a given concentration of peptides / % of CD8+ cells responding to the highest concentration of peptides) × 100.
Tetramer staining
The flow cytometry-assisted phenotyping of antigen-specific T cells for the expression of memory markers was accomplished using circulating lymphocytes stained with fluorochrome-conjugated tetramers and anti-CD8, anti-CD62L and anti-CD127 antibodies. The cytolytic potential of antigen-specific T cells was assessed by surface-staining with anti-CD8 antibodies and tetramers followed by fixation, permeabilization and intracellular staining for granzyme B.
Gating strategy for analyzing cytofluorometric data
Using forward vs. side scatter-width parameters (FSC vs. SSC), doublets were excluded from analyses and single lymphocytes were gated on. Single CD8+ cells were subsequently gated on using a histogram so that their staining with tetramers or intracellular cytokine-specific antibodies could be queried.
Adoptive T-cell transfer
Equal numbers of SIINFEKL-specific T cells were transferred into naïve recipient mice. To accomplish this, spleens from donor mice that has been previously immunized with adenoviral vectors alone or coupled to VSV boosting were harvested either at the peak of primary (i.e., day 11 post-priming) or secondary (i.e., day 5 post-boosting) responses or 70 d post-peak. Aliquots from single splenocyte suspensions were labeled with tetramers and anti-CD8 antibodies so that the frequency of antigen-specific T cells could be quantified by flow cytometry. Equal doses of SIINFEKL-specific adenovirus-primed and VSV-boosted T cells were then prepared. The total amounts of splenocytes within each dose was equalized equal by topping up VSV-boosted T cells with splenocytes derived from naïve mice. Preparations containing equal numbers of both antigen-specific T cells and total splenocytes were then injected i.v. into recipients. Controls received splenocytes from mice receiving Ad-BHG followed by VSV-MT.
Tumor and virus challenge models
Mice into which SIINFEKL-specific T cells had been adoptively transferred (see above) were assessed for their ability to clear OVA-expressing tumor cells and viruses. In both cases, adoptively transferred T cells were given 24 h to achieve homeostatic trafficking patterns. In the lung metastasis model, mice received 5 × 105 B16-OVA cells i.v. After 21 d, mice were euthanized and tumor nodules growing on the surface of the lungs were counted. When too many tumor nodules were present to obtain a reliable count, a value of 800 was assigned based on a semi-quantitative estimation. In the infection model, 1 × 106 PFU of VV-SIINFEKL were injected i.v.. After 3 d, mice were euthanized and VV titers in homogenized ovaries were determined by a plaque assay on Vero cells.
Statistical analyses
GraphPad Prism version 4.00 for Windows (GraphPad Software) was used for all graphing and statistical analyses. Differences in T-cell responses were analyzed for statistical significance by two-way ANOVA. Data from tumor and viral challenge experiments were analyzed for statistical significance by one-way ANOVA. If required, data were normalized by log transformation prior to statistical analyses. Differences between means were considered statistically significant when p ≤ 0.05. Data are reported as means ± SEM.
Acknowledgments
This work was supported by grants to Y.W. from the Canadian Institutes of Health Research [MOP-67066]. The authors thank Natasha Khazdan for producing the VSV vectors.
Glossary
Abbreviations:
- APC
antigen-presenting cell
- DC
dendritic cell
- DCT
dopachrome tautomerase
- IFN
interferon γ
- IL
interleukin
- OVA
ovalbumin
- PFU
plaque forming unit
- TCM
central memory T
- TEFF
effector T
- TEM
effector memory T
- TLR
Toll-like receptor
- TNFα
tumor necrosis factor α
- VSV
vesicular stomatitis virus
- VV
vaccinia virus
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/oncoimmunology/article/26013
References
- 1.Ramshaw IA, Ramsay AJ. The prime-boost strategy: exciting prospects for improved vaccination. Immunol Today. 2000;21:163–5. doi: 10.1016/S0167-5699(00)01612-1. [DOI] [PubMed] [Google Scholar]
- 2.Irvine KR, Chamberlain RS, Shulman EP, Surman DR, Rosenberg SA, Restifo NP. Enhancing efficacy of recombinant anticancer vaccines with prime/boost regimens that use two different vectors. J Natl Cancer Inst. 1997;89:1595–601. doi: 10.1093/jnci/89.21.1595. [DOI] [PubMed] [Google Scholar]
- 3.Woodland DL. Jump-starting the immune system: prime-boosting comes of age. Trends Immunol. 2004;25:98–104. doi: 10.1016/j.it.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 4.Rein DT, Breidenbach M, Curiel DT. Current developments in adenovirus-based cancer gene therapy. Future Oncol. 2006;2:137–43. doi: 10.2217/14796694.2.1.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lasaro MO, Ertl HC. New insights on adenovirus as vaccine vectors. Mol Ther. 2009;17:1333–9. doi: 10.1038/mt.2009.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bassett JD, Yang TC, Bernard D, Millar JB, Swift SL, McGray AJ, VanSeggelen H, Boudreau JE, Finn JD, Parsons R, et al. CD8+ T-cell expansion and maintenance after recombinant adenovirus immunization rely upon cooperation between hematopoietic and nonhematopoietic antigen-presenting cells. Blood. 2011;117:1146–55. doi: 10.1182/blood-2010-03-272336. [DOI] [PubMed] [Google Scholar]
- 7.Priddy FH, Brown D, Kublin J, Monahan K, Wright DP, Lalezari J, Santiago S, Marmor M, Lally M, Novak RM, et al. Merck V520-016 Study Group Safety and immunogenicity of a replication-incompetent adenovirus type 5 HIV-1 clade B gag/pol/nef vaccine in healthy adults. Clin Infect Dis. 2008;46:1769–81. doi: 10.1086/587993. [DOI] [PubMed] [Google Scholar]
- 8.Richardson JS, Pillet S, Bello AJ, Kobinger GP. Airway delivery of an adenovirus-based Ebola virus vaccine bypasses existing immunity to homologous adenovirus in nonhuman primates. J Virol. 2013;87:3668–77. doi: 10.1128/JVI.02864-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buchbinder SP, Mehrotra DV, Duerr A, Fitzgerald DW, Mogg R, Li D, Gilbert PB, Lama JR, Marmor M, Del Rio C, et al. Step Study Protocol Team Efficacy assessment of a cell-mediated immunity HIV-1 vaccine (the Step Study): a double-blind, randomised, placebo-controlled, test-of-concept trial. Lancet. 2008;372:1881–93. doi: 10.1016/S0140-6736(08)61591-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gabitzsch ES, Balint-Junior JP, Xu Y, Balcaitis S, Sanders-Beer B, Karl J, Weinhold KJ, Paessler S, Jones FR. Control of SIV infection and subsequent induction of pandemic H1N1 immunity in rhesus macaques using an Ad5 [E1-, E2b-] vector platform. Vaccine. 2012;30:7265–70. doi: 10.1016/j.vaccine.2012.09.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang TC, Millar J, Groves T, Grinshtein N, Parsons R, Takenaka S, Wan Y, Bramson JL. The CD8+ T cell population elicited by recombinant adenovirus displays a novel partially exhausted phenotype associated with prolonged antigen presentation that nonetheless provides long-term immunity. J Immunol. 2006;176:200–10. doi: 10.4049/jimmunol.176.1.200. [DOI] [PubMed] [Google Scholar]
- 12.Tatsis N, Fitzgerald JC, Reyes-Sandoval A, Harris-McCoy KC, Hensley SE, Zhou D, Lin SW, Bian A, Xiang ZQ, Iparraguirre A, et al. Adenoviral vectors persist in vivo and maintain activated CD8+ T cells: implications for their use as vaccines. Blood. 2007;110:1916–23. doi: 10.1182/blood-2007-02-062117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yang TC, Dayball K, Wan YH, Bramson J. Detailed analysis of the CD8+ T-cell response following adenovirus vaccination. J Virol. 2003;77:13407–11. doi: 10.1128/JVI.77.24.13407-13411.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wong P, Pamer EG. Feedback regulation of pathogen-specific T cell priming. Immunity. 2003;18:499–511. doi: 10.1016/S1074-7613(03)00081-5. [DOI] [PubMed] [Google Scholar]
- 15.Ronchese F, Hermans IF. Killing of dendritic cells: a life cut short or a purposeful death? J Exp Med. 2001;194:F23–6. doi: 10.1084/jem.194.5.F23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luketic L, Delanghe J, Sobol PT, Yang P, Frotten E, Mossman KL, Gauldie J, Bramson J, Wan Y. Antigen presentation by exosomes released from peptide-pulsed dendritic cells is not suppressed by the presence of active CTL. J Immunol. 2007;179:5024–32. doi: 10.4049/jimmunol.179.8.5024. [DOI] [PubMed] [Google Scholar]
- 17.Bridle BW, Stephenson KB, Boudreau JE, Koshy S, Kazdhan N, Pullenayegum E, Brunellière J, Bramson JL, Lichty BD, Wan Y. Potentiating cancer immunotherapy using an oncolytic virus. Mol Ther. 2010;18:1430–9. doi: 10.1038/mt.2010.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stojdl DF, Lichty B, Knowles S, Marius R, Atkins H, Sonenberg N, Bell JC. Exploiting tumor-specific defects in the interferon pathway with a previously unknown oncolytic virus. Nat Med. 2000;6:821–5. doi: 10.1038/77558. [DOI] [PubMed] [Google Scholar]
- 19.Ledgerwood JE, Costner P, Desai N, Holman L, Enama ME, Yamshchikov G, Mulangu S, Hu Z, Andrews CA, Sheets RA, et al. VRC 205 Study Team A replication defective recombinant Ad5 vaccine expressing Ebola virus GP is safe and immunogenic in healthy adults. Vaccine. 2010;29:304–13. doi: 10.1016/j.vaccine.2010.10.037. [DOI] [PubMed] [Google Scholar]
- 20.Draper SJ, Heeney JL. Viruses as vaccine vectors for infectious diseases and cancer. Nat Rev Microbiol. 2010;8:62–73. doi: 10.1038/nrmicro2240. [DOI] [PubMed] [Google Scholar]
- 21.Sullivan NJ, Hensley L, Asiedu C, Geisbert TW, Stanley D, Johnson J, Honko A, Olinger G, Bailey M, Geisbert JB, et al. CD8+ cellular immunity mediates rAd5 vaccine protection against Ebola virus infection of nonhuman primates. Nat Med. 2011;17:1128–31. doi: 10.1038/nm.2447. [DOI] [PubMed] [Google Scholar]
- 22.Sullivan NJ, Geisbert TW, Geisbert JB, Shedlock DJ, Xu L, Lamoreaux L, Custers JH, Popernack PM, Yang ZY, Pau MG, et al. Immune protection of nonhuman primates against Ebola virus with single low-dose adenovirus vectors encoding modified GPs. PLoS Med. 2006;3:e177. doi: 10.1371/journal.pmed.0030177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jeyanathan M, Mu J, McCormick S, Damjanovic D, Small CL, Shaler CR, Kugathasan K, Xing Z. Murine airway luminal antituberculosis memory CD8 T cells by mucosal immunization are maintained via antigen-driven in situ proliferation, independent of peripheral T cell recruitment. Am J Respir Crit Care Med. 2010;181:862–72. doi: 10.1164/rccm.200910-1583OC. [DOI] [PubMed] [Google Scholar]
- 24.Hansen SG, Ford JC, Lewis MS, Ventura AB, Hughes CM, Coyne-Johnson L, Whizin N, Oswald K, Shoemaker R, Swanson T, et al. Profound early control of highly pathogenic SIV by an effector memory T-cell vaccine. Nature. 2011;473:523–7. doi: 10.1038/nature10003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huster KM, Koffler M, Stemberger C, Schiemann M, Wagner H, Busch DH. Unidirectional development of CD8+ central memory T cells into protective Listeria-specific effector memory T cells. Eur J Immunol. 2006;36:1453–64. doi: 10.1002/eji.200635874. [DOI] [PubMed] [Google Scholar]
- 26.Stock AT, Jones CM, Heath WR, Carbone FR. Cutting edge: central memory T cells do not show accelerated proliferation or tissue infiltration in response to localized herpes simplex virus-1 infection. J Immunol. 2006;177:1411–5. doi: 10.4049/jimmunol.177.3.1411. [DOI] [PubMed] [Google Scholar]
- 27.Khanna KM, McNamara JT, Lefrançois L. In situ imaging of the endogenous CD8 T cell response to infection. Science. 2007;318:116–20. doi: 10.1126/science.1146291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dauner JG, Williams IR, Jacob J. Differential microenvironment localization of effector and memory CD8 T cells. J Immunol. 2008;180:291–9. doi: 10.4049/jimmunol.180.1.291. [DOI] [PubMed] [Google Scholar]
- 29.Weninger W, Crowley MA, Manjunath N, von Andrian UH. Migratory properties of naive, effector, and memory CD8(+) T cells. J Exp Med. 2001;194:953–66. doi: 10.1084/jem.194.7.953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lyman MA, Nugent CT, Marquardt KL, Biggs JA, Pamer EG, Sherman LA. The fate of low affinity tumor-specific CD8+ T cells in tumor-bearing mice. J Immunol. 2005;174:2563–72. doi: 10.4049/jimmunol.174.5.2563. [DOI] [PubMed] [Google Scholar]
- 31.Baitsch L, Fuertes-Marraco SA, Legat A, Meyer C, Speiser DE. The three main stumbling blocks for anticancer T cells. Trends Immunol. 2012;33:364–72. doi: 10.1016/j.it.2012.02.006. [DOI] [PubMed] [Google Scholar]
- 32.McGray AJ, Bernard D, Hallett R, Kelly R, Jha M, Gregory C, Bassett JD, Hassell JA, Pare G, Wan Y, et al. Combined vaccination and immunostimulatory antibodies provides durable cure of murine melanoma and induces transcriptional changes associated with positive outcome in human melanoma patients. Oncoimmunology. 2012;1:419–31. doi: 10.4161/onci.19534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gasteiger G, Kastenmuller W, Ljapoci R, Sutter G, Drexler I. Cross-priming of cytotoxic T cells dictates antigen requisites for modified vaccinia virus Ankara vector vaccines. J Virol. 2007;81:11925–36. doi: 10.1128/JVI.00903-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pham NL, Pewe LL, Fleenor CJ, Langlois RA, Legge KL, Badovinac VP, Harty JT. Exploiting cross-priming to generate protective CD8 T-cell immunity rapidly. Proc Natl Acad Sci U S A. 2010;107:12198–203. doi: 10.1073/pnas.1004661107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Abadie V, Bonduelle O, Duffy D, Parizot C, Verrier B, Combadière B. Original encounter with antigen determines antigen-presenting cell imprinting of the quality of the immune response in mice. PLoS One. 2009;4:e8159. doi: 10.1371/journal.pone.0008159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–84. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 37.Georgel P, Jiang Z, Kunz S, Janssen E, Mols J, Hoebe K, Bahram S, Oldstone MB, Beutler B. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology. 2007;362:304–13. doi: 10.1016/j.virol.2006.12.032. [DOI] [PubMed] [Google Scholar]
- 38.Lund JM, Alexopoulou L, Sato A, Karow M, Adams NC, Gale NW, Iwasaki A, Flavell RA. Recognition of single-stranded RNA viruses by Toll-like receptor 7. Proc Natl Acad Sci U S A. 2004;101:5598–603. doi: 10.1073/pnas.0400937101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pufnock JS, Cigal M, Rolczynski LS, Andersen-Nissen E, Wolfl M, McElrath MJ, Greenberg PD. Priming CD8+ T cells with dendritic cells matured using TLR4 and TLR7/8 ligands together enhances generation of CD8+ T cells retaining CD28. Blood. 2011;117:6542–51. doi: 10.1182/blood-2010-11-317966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wille-Reece U, Flynn BJ, Loré K, Koup RA, Miles AP, Saul A, Kedl RM, Mattapallil JJ, Weiss WR, Roederer M, et al. Toll-like receptor agonists influence the magnitude and quality of memory T cell responses after prime-boost immunization in nonhuman primates. J Exp Med. 2006;203:1249–58. doi: 10.1084/jem.20052433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Boudreau JE, Bridle BW, Stephenson KB, Jenkins KM, Brunellière J, Bramson JL, Lichty BD, Wan Y. Recombinant vesicular stomatitis virus transduction of dendritic cells enhances their ability to prime innate and adaptive antitumor immunity. Mol Ther. 2009;17:1465–72. doi: 10.1038/mt.2009.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Boudreau JE, Stephenson KB, Wang F, Ashkar AA, Mossman KL, Lenz LL, Rosenthal KL, Bramson JL, Lichty BD, Wan Y. IL-15 and type I interferon are required for activation of tumoricidal NK cells by virus-infected dendritic cells. Cancer Res. 2011;71:2497–506. doi: 10.1158/0008-5472.CAN-10-3025. [DOI] [PubMed] [Google Scholar]
- 43.Ahmed M, Mitchell LM, Puckett S, Brzoza-Lewis KL, Lyles DS, Hiltbold EM. Vesicular stomatitis virus M protein mutant stimulates maturation of Toll-like receptor 7 (TLR7)-positive dendritic cells through TLR-dependent and -independent mechanisms. J Virol. 2009;83:2962–75. doi: 10.1128/JVI.02030-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ahmed M, Puckett S, Arimilli S, Braxton CL, Mizel SB, Lyles DS. Stimulation of human dendritic cells by wild-type and M protein mutant vesicular stomatitis viruses engineered to express bacterial flagellin. J Virol. 2010;84:12093–8. doi: 10.1128/JVI.00406-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Finn JD, Bassett J, Millar JB, Grinshtein N, Yang TC, Parsons R, Evelegh C, Wan Y, Parks RJ, Bramson JL. Persistence of transgene expression influences CD8+ T-cell expansion and maintenance following immunization with recombinant adenovirus. J Virol. 2009;83:12027–36. doi: 10.1128/JVI.00593-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ahlers JD, Belyakov IM. Memories that last forever: strategies for optimizing vaccine T-cell memory. Blood. 2010;115:1678–89. doi: 10.1182/blood-2009-06-227546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Seder RA, Darrah PA, Roederer M. T-cell quality in memory and protection: implications for vaccine design. Nat Rev Immunol. 2008;8:247–58. doi: 10.1038/nri2274. [DOI] [PubMed] [Google Scholar]
- 48.Bassett JD, Swift SL, Bramson JL. Optimizing vaccine-induced CD8(+) T-cell immunity: focus on recombinant adenovirus vectors. Expert Rev Vaccines. 2011;10:1307–19. doi: 10.1586/erv.11.88. [DOI] [PubMed] [Google Scholar]
- 49.Bridle BW, Chen L, Lemay CG, Diallo JS, Pol J, Nguyen A, Capretta A, He R, Bramson JL, Bell JC, et al. HDAC inhibition suppresses primary immune responses, enhances secondary immune responses, and abrogates autoimmunity during tumor immunotherapy. Mol Ther. 2013;21:887–94. doi: 10.1038/mt.2012.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Oldstone MB, Tishon A, Eddleston M, de la Torre JC, McKee T, Whitton JL. Vaccination to prevent persistent viral infection. J Virol. 1993;67:4372–8. doi: 10.1128/jvi.67.7.4372-4378.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]