

Abstract
The RNA-programmable Cas9 endonuclease cleaves double-stranded DNA at sites complementary to a 20-base-pair guide RNA. The Cas9 system has been used to modify genomes in multiple cells and organisms, demonstrating its potential as a facile genome-engineering tool. We used in vitro selection and high-throughput sequencing to determine the propensity of eight Cas9:guide RNA complexes to cleave each of 10^12 potential off-target DNA sequences. The selection results predicted five off-target sites in the human genome that were confirmed to undergo genome cleavage in HEK293T cells upon expression of one of two Cas9:guide RNA complexes. In contrast to previous models, our results show that Cas9:guide RNA specificity extends past a 7- to 12-base pair seed sequence. Our results also suggest a tradeoff between activity and specificity both in vitro and in cells as a shorter, less-active guide RNA is more specific then a longer, more-active guide RNA. High concentrations of Cas9:guide RNA complexes can cleave off-target sites containing mutations near or within the PAM that are not cleaved when enzyme concentrations are limiting.
Sequence-specific endonucleases including zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) have become important tools to modify genes in induced pluripotent stem cells (iPSCs),1-3 in multi-cellular organisms,4-8 and in ex vivo gene therapy clinical trials9, 10. Although ZFNs and TALENs have proved effective for such genetic manipulation, a new ZFN or TALEN protein must be generated for each DNA target site. By contrast, the RNA-guided Cas9 endonuclease uses RNA:DNA hybridization to find target DNA cleavage sites, enabling a single monomeric protein to cleave, in principle, any sequence specified by the guide RNA.11
Previous studies12-17 demonstrated that Cas9 mediates genome editing at sites complementary to a 20-nucleotide sequence in a bound guide RNA. In addition, target sites must include a protospacer adjacent motif (PAM) at the 3′ end adjacent to the 20-base pair target site; for Streptococcus pyogenes Cas9, the PAM sequence is NGG. Cas9-mediated DNA cleavage specificity both in vitro and in cells has been inferred previously based on assays against small collections of potential single-mutation off-target sites. These studies suggested that perfect complementarity between guide RNA and target DNA is required in the 7-12 base pairs adjacent to the PAM end of the target site (3′ end of the guide RNA) and mismatches are tolerated at the non-PAM end (5′ end of the guide RNA).11, 12, 17-19
Although such a limited number of nucleotides specifying Cas9:guide RNA target recognition would predict multiple sites of DNA cleavage in genomes of moderate to large size (> ~107 bp), Cas9:guide RNA complexes have been successfully used to modify both cells12, 13, 15 and organisms.14 A study using Cas9:guide RNA complexes to modify zebrafish embryos observed toxicity at a rate similar to that of ZFNs and TALENs.14 A recent, broad study of the specificity of DNA binding (transcriptional repression) in E. coli of a catalytically inactive Cas9 mutant using high-throughput sequencing found no detectable off-target transcriptional repression in the relatively small E. coli transcriptome.20 Although these studies have substantially advanced our basic understanding of Cas9, a systematic and comprehensive profile of Cas9:guide RNA-mediated DNA cleavage specificity generated from measurements of Cas9 cleavage on a large number of related mutant target sites has not been described. Such a specificity profile is needed to understand and improve the potential of Cas9:guide RNA complexes as research tools and future therapeutic agents.
To determine the off-target DNA cleavage profiles of Cas9:single guide RNA (sgRNA)11 complexes, we modified our previously published in vitro selection protocol21 to process the blunt-ended cleavage products produced by Cas9 instead of the overhang-containing products of ZFN cleavage. Each selection experiment used DNA substrate libraries containing ~1012 sequences, a size sufficiently large to include ten-fold coverage of all sequences with eight or fewer mutations relative to each 22-base pair target sequence (including the two-base pair PAM) (Figure 1). We used partially randomized nucleotide mixtures at all 22 target-site base pairs to create a binomially distributed library of mutant target sites with an expected mean of 4.62 mutations per target site. In addition, target site library members were flanked by four fully randomized base pairs on each side to test for specificity patterns beyond those imposed by the canonical 20-base pair target site and PAM.
Figure 1. In vitro selection overview.
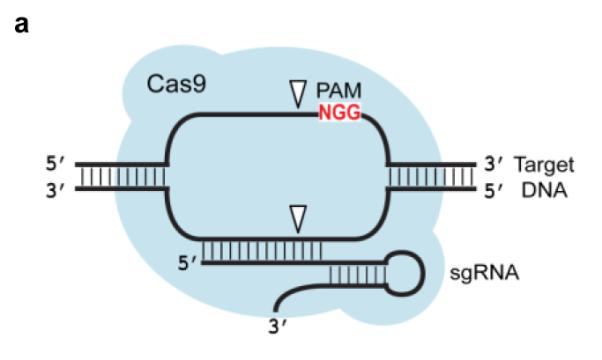

(a) Cas9 complexed with a short guide RNA (sgRNA) recognizes ~20 bases of a target DNA substrate that is complementary to the sgRNA sequence and cleaves both DNA strands. The white triangles represent cleavage locations. (b) A modified version of our previously described in vitro selection21 was used to comprehensively profile Cas9 specificity. A concatemeric pre-selection DNA library in which each molecule contains one of 10^12 distinct variants of a target DNA sequence (white rectangles) was generated from synthetic DNA oligonucleotides by ligation and rolling-circle amplification. This library was incubated with a Cas9:sgRNA complex of interest. Cleaved library members contain 5′ phosphate groups (green circles) and therefore are substrates for adapter ligation and PCR. The resulting amplicons were subjected to high-throughput DNA sequencing and computational analysis.
Pre-selection libraries of 1012 individual potential off-target sites were generated for each of four different target sequences in the human clathrin light chain A (CLTA) gene (Supplementary Figure S1). Synthetic 5′-phosphorylated 53-base oligonucleotides were self-ligated into circular single-stranded DNA in vitro and then converted into concatemeric 53-base pair repeats through rolling-circle amplification. The resulting pre-selection libraries were incubated with their corresponding Cas9:sgRNA complexes. Cleaved library members containing free 5′ phosphates were separated from intact library members through the 5′ phosphate-dependent ligation of non-phosphorylated double-stranded sequencing adapters. The ligation-tagged post-selection libraries were amplified by PCR. The PCR step generated a mixture of post-selection DNA fragments containing 0.5, 1.5, or 2.5, etc. repeats of library members cleaved by Cas9, resulting from amplification of an adapter-ligated cut half-site with or without one or more adjacent corresponding full sites (Figure 1). Post-selection library members with 1.5 target-sequence repeats were isolated by gel purification and analyzed by high-throughput sequencing. In a final computational selection step to minimize the impact of errors during DNA amplification or sequencing, only sequences with two identical copies of the repeated cut half-site were analyzed.
Pre-selection libraries were incubated under enzyme-limiting conditions (200 nM target site library, 100 nM Cas9:sgRNA v2.1) or enzyme-excess conditions (200 nM target site library, 1000 nM Cas9:sgRNA v2.1) for each of the four guide RNAs targets tested (CLTA1, CLTA2, CLTA3, and CLTA4) (Supplementary Figure S1c and S1d). A second guide RNA construct, sgRNA v1.0, which is less active than sgRNA v2.1, was assayed under enzyme-excess conditions alone for each of the four guide RNA targets tested (200 nM target site library, 1000 nM Cas9:sgRNA v1.0). The two guide RNA constructs differ in their length (Supplementary Figure S1) and in their DNA cleavage activity level under the selection conditions, consistent with previous reports15 (Supplementary Figure S2). Both pre-selection and post-selection libraries were characterized by high-throughput DNA sequencing and computational analysis. As expected, library members with fewer mutations were significantly enriched in post-selection libraries relative to pre-selection libraries (Supplementary Text and Supplementary Figure S3).
We calculated specificity scores to quantify the enrichment level of each base pair at each position in the post-selection library relative to the pre-selection library, normalized to the maximum possible enrichment of that base pair. Positive specificity scores indicate base pairs that were enriched in the post-selection library and negative specificity scores indicate base pairs that were de-enriched in the post-selection library. For example, a score of +0.5 indicates that a base pair is enriched to 50% of the maximum enrichment value, whereas a score of −0.5 indicates that a base pair is de-enriched to 50% of the maximum de-enrichment value.
In addition to the two base pairs specified by the PAM, all 20 base pairs targeted by the guide RNA were enriched in the sequences from the CLTA1 and CLTA2 selections (Figure 2, Supplementary Figures S4 and S7, and Supplementary Table S1). For the CLTA3 and CLTA4 selections (Supplementary Figures S5 and S6, and Supplementary Table S1), guide RNA-specified base pairs were enriched at all positions except for the one or two most distal base pairs from the PAM (5′ end of the guide RNA), respectively. At these non-specified positions farthest from the PAM, at least two of the three alternate base pairs were nearly as enriched as the specified base pair. Our finding that the entire 20 base-pair target site and two base pair PAM can contribute to Cas9:sgRNA DNA cleavage specificity contrasts with the results from previous single-substrate assays suggesting that only 7-12 base pairs and two base pair PAM are specified11, 12, 15.
Figure 2. In vitro selection results for Cas9:CLTA1 sgRNA.

Heat maps21 show the specificity profiles of Cas9:CLTA1 sgRNA v2.1 under enzyme-limiting conditions (a, b), Cas9:CLTA1 sgRNA v1.0 under enzyme-excess conditions (c, d), and Cas9:CLTA1 sgRNA v2.1 under enzyme-excess conditions (e, f). Heat maps show all post-selection sequences (a, c, e) or only those sequences containing a single mutation in the 20-base pair sgRNA-specified target site and two-base pair PAM (b, d, f). Specificity scores of 1.0 (dark blue) and -1.0 (dark red) corresponds to 100% enrichment for and against, respectively, a particular base pair at a particular position. Black boxes denote the intended target nucleotides. (g) Effect of Cas9:sgRNA concentration on specificity. Positional specificity changes between enzyme-limiting (200 nM DNA, 100 nM Cas9:sgRNA v2.1) and enzyme-excess (200 nM DNA, 1000 nM Cas9:sgRNA v2.1) conditions are shown for CLTA1. Red lines indicate the maximum possible change in positional specificity for a given position. (h) Effect of sgRNA architecture on specificity. Positional specificity changes between sgRNA v1.0 and sgRNA v2.1 under enzyme-excess conditions are shown for CLTA1. Red lines indicate the maximum possible change in positional specificity for a given position. See Supplementary Figures S4-S6, S23, and S24 for corresponding data for CLTA2, CLTA3, and CLTA4.
All single-mutant pre-selection (n ≥ 14,569) and post-selection library members (n ≥ 103,660) were computationally analyzed to provide a selection enrichment value for every possible single-mutant sequence. The results of this analysis (Figure 2 and Supplementary Figures S4-S6) show that when only single-mutant sequences are considered, the six to eight base pairs closest to the PAM are generally highly specified and the non-PAM end is poorly specified under enzyme-limiting conditions, consistent with previous findings11, 12, 17-19. Under enzyme-excess conditions, however, single mutations even in the six to eight base pairs most proximal to the PAM are tolerated, suggesting that the high specificity at the PAM end of the DNA target site can be compromised when enzyme concentrations are high relative to substrate (Figure 2). The observation of strong discrimination against single mutations close to the PAM only applies to sequences with a single mutation, and the selection results do not support a model in which any combination of mutations is tolerated in the region of the target site farthest from the PAM (Supplementary Text and Supplementary Figures S8-S15). See the Supplementary Text for analyses of pre- and post-selection library composition, position-dependent specificity patterns (Supplementary Figures S16-S18), PAM nucleotide specificity (Supplementary Figures S19-S22), and more detailed effects of Cas9:sgRNA concentration on specificity (Figure 2g and Supplementary Figure S23).
The selection results also reveal that the choice of guide RNA structure affects cleavage of off-target sites. The shorter, less-active sgRNA v1.0 constructs are less tolerant of mutations than the longer, more-active sgRNA v2.1 constructs when assayed under identical, enzyme-excess conditions that reflect a cellular context of excess of enzyme relative to substrate (Figure 2 and Supplementary Figures S3-S6). The difference in off-target activity between sgRNA v1.0 and sgRNA v2.1 is greater for CLTA1 and CLTA2 (~40-90% difference) than for CLTA3 and CLTA4 (< 40% difference). The differences in tolerated off-target mutations between sgRNA constructs are localized to different regions of the target site for each target sequence (Figure 2h and Supplementary Figure S24). Collectively, these results indicate that different guide RNA architectures result in different off-target DNA cleavage activities, and that guide RNA-dependent changes in specificity do not affect all positions in the target site equally. Given the inverse relationship between Cas9:sgRNA concentration and specificity described above, we speculate that the differences in off-target activities between guide RNA architectures arises from differences in their overall level of DNA-cleavage activities.
To confirm that the in vitro selection results accurately reflect the cleavage behavior of Cas9 in vitro, we performed discrete cleavage assays of six CLTA4 off-target substrates containing one to three mutations in the target site. We calculated enrichment values for all sequences in the post-selection libraries for the Cas9:CLTA4 v2.1 sgRNA under enzyme-excess conditions by dividing the abundance of each sequence in the post-selection library by the calculated abundance in the pre-selection library. Under enzyme-excess conditions, the single one, two, and three mutation sequences with the highest enrichment values (27.5, 43.9, and 95.9) were cleaved to ≥ 72% completion (Supplementary Figure S25). A two-mutation sequence with an enrichment value of 1.0 was cleaved to 35%, and a two-mutation sequence with an enrichment value near zero (0.064) was not cleaved. The three-mutation sequence, which was cleaved to 77% by CLTA4 v2.1 sgRNA, was cleaved to a lower efficiency of 53% by CLTA4 v1.0 sgRNA (Supplementary Figure S26). These results indicate that the selection enrichment values of individual sequences are predictive of in vitro cleavage efficiencies.
To determine if results of the in vitro selection and in vitro cleavage assays pertain to Cas9:guide RNA activity in human cells, we identified 51 off-target sites (19 for CLTA1 and 32 for CLTA4) containing up to eight mutations that were both enriched in the in vitro selection and present in the human genome (Supplementary Tables S2-S4). We expressed Cas9:CLTA1 sgRNA v1.0, Cas9:CLTA1 sgRNA v2.1, Cas9:CLTA4 sgRNA v1.0, Cas9:CLTA4 sgRNA v2.1, or Cas9 without sgRNA in HEK293T cells by transient transfection and used genomic PCR and high-throughput DNA sequencing to look for evidence of Cas9:sgRNA modification at 46 of the 51 off-target sites as well as at the on-target loci; no specific amplified DNA was obtained for five of the 51 predicted off-target sites (three for CLTA1 and two for CLTA4).
Deep sequencing of genomic DNA isolated from HEK293T cells treated with Cas9:CLTA1 sgRNA or Cas9:CLTA4 sgRNA identified sequences evident of non-homologous end-joining (NHEJ) at the on-target sites and at five of the 49 tested off-target sites (CLTA1-1-1, CLTA1-2-2, CLTA4-3-1, CLTA4-3-3, and CLTA4-4-8) (Table 1 and Supplementary Tables S5-S7). The CLTA4 target site was modified by Cas9:CLTA4 v2.1 sgRNA at a frequency of 76%, whereas off-target sites, CLTA4-3-1 CLTA4-3-3, and CLTA4-4-8, were modified at frequencies of 24%, 0.47% and 0.73%, respectively. The CLTA1 target site was modified by Cas9:CLTA1 v2.1 sgRNA at a frequency of 0.34%, whereas off-target sites, CLTA1-1-1 and CLTA1-2-2, were modified at frequencies of 0.09% and 0.15%, respectively.
Table 1. Cellular modification induced by Cas9:CLTA4 sgRNA.
33 human genomic DNA sequences were identified that were enriched in the Cas9:CLTA4 v2.1 sgRNA in vitro selections under enzyme-limiting or enzyme-excess conditions. Sites shown in red contain insertions or deletions (indels) that are consistent with significant Cas9:sgRNA-mediated modification in HEK293T cells. In vitro enrichment values for selections with Cas9:CLTA4 v1.0 sgRNA or Cas9:CLTA4 v2.1 sgRNA are shown for sequences with three or fewer mutations. Enrichment values were not calculated for sequences with four or more mutations due to low numbers of in vitro selection sequence counts. Modification frequencies (number of sequences with indels divided by total number of sequences) in HEK293T cells treated with Cas9 without sgRNA (“no sgRNA”), Cas9 with CLTA4 v1.0 sgRNA, or Cas9 with CLTA4 v2.1 sgRNA. P-values are listed for those sites that show significant modification in v1.0 sgRNA- or v2.1 sgRNA-treated cells compared to cells treated with Cas9 without sgRNA. P-values were calculated using a one-sided Fisher exact text. “Not tested (n.t.)” indicates that PCR of the genomic sequence failed to provide specific amplification products.
|
in vitro enrichment |
modification frequency in HEK293T cells |
P-value | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| # of mutations |
sequence | gene | v1.0 | v2.1 | no sgRNA |
v1.0 | v2.1 | v1.0 | v2.1 | |
| CLTA4-0-1 | 0 | GCAGATGTAGTGTTTCCACAGGG | CLTA | 20 | 7.95 | 0.021% | 11% | 76% | <1E-55 | <1E-55 |
| CLTA4-3-1 | 3 | aCAtATGTAGTaTTTCCACAGGG | 16.5 | 12.5 | 0.006% | 0.055% | 24% | 6.0E-04 | <1E-55 | |
| CLTA4-3-2 | 3 | GCAtATGTAGTGTTTCCAaATGt | 2.99 | 6.97 | 0.017% | 0% | 0.014% | |||
| CLTA4-3-3 | 3 | cCAGATGTAGTaTTcCCACAGGG | CELF1 | 1.00 | 4.95 | 0% | 0% | 0.469% | 2.5E-21 | |
| CLTA4-3-4 | 3 | GCAGtTtTAGTGTTTtCACAGGG | BC073807 | 0.79 | 3.12 | 0% | 0% | 0% | ||
| CLTA4-3-5 | 3 | GCAGAgtTAGTGTTTCCACACaG | MPPED2 | 0 | 1.22 | 0.005% | 0.015% | 0.018% | ||
| CLTA4-3-6 | 3 | GCAGATGgAGgGTTTtCACAGGG | DCHS2 | 1.57 | 1.17 | 0.015% | 0.023% | 0.021% | ||
| CLTA4-3-7 | 3 | GgAaATtTAGTGTTTCCACAGGG | 0.43 | 0.42 | 0.005% | 0.012% | 0.003% | |||
| CLTA4-4-1 | 4 | aaAGAaGTAGTaTTTCCACATGG | n.t. | n.t. | n.t. | |||||
| CLTA4-4-2 | 4 | aaAGATGTAGTcaTTCCACAAGG | 0.004% | 0% | 0.005% | |||||
| CLTA4-4-3 | 4 | aaAtATGTAGTcTTTCCACAGGG | 0.004% | 0.009% | 0% | |||||
| CLTA4-4-4 | 4 | atAGATGTAGTGTTTCCAaAGGa | NR1H4 | 0.032% | 0.006% | 0.052% | ||||
| CLTA4-4-5 | 4 | cCAGAgGTAGTGcTcCCACAGGG | 0.005% | 0.006% | 0.007% | |||||
| CLTA4-4-6 | 4 | cCAGATGTgagGTTTCCACAAGG | XKR6 | 0.018% | 0% | 0.007% | ||||
| CLTA4-4-7 | 4 | ctAcATGTAGTGTTTCCAtATGG | HKR1 | 0.006% | 0% | 0.008% | ||||
| CLTA4-4-8 | 4 | ctAGATGaAGTGcTTCCACATGG | CDK8 | 0.009% | 0.013% | 0.730% | 9.70E-21 | |||
| CLTA4-4-9 | 4 | GaAaATGgAGTGTTTaCACATGG | 0% | 0% | 0.004% | |||||
| CLTA4-4-10 | 4 | GCAaATGaAGTGTcaCCACAAGG | 0.004% | 0% | 0% | |||||
| CLTA4-4-11 | 4 | GCAaATGTAtTaTTTCCACtAGG | NOV | 0% | 0.00% | 0% | ||||
| CLTA4-4-12 | 4 | GCAGATGTAGctTTTgtACATGG | 0% | 0.00% | 0% | |||||
| CLTA4-4-13 | 4 | GCAGcTtaAGTGTTTtCACATGG | GRHL2 | 0.020% | 0.02% | 0.030% | ||||
| CLTA4-4-14 | 4 | ttAcATGTAGTGTTTaCACACGG | LINC00535 | n.t. | n.t. | n.t. | ||||
| CLTA4-5-1 | 5 | GaAGAgGaAGTGTTTgCcCAGGG | RNH1 | 0.004% | 0.01% | 0.006% | ||||
| CLTA4-5-2 | 5 | GaAGATGTgGaGTTgaCACATGG | FZD3 | 0.004% | 0.00% | 0% | ||||
| CLTA4-5-3 | 5 | GCAGAaGTAcTGTTgttACAAGG | 0.002% | 0.00% | 0.003% | |||||
| CLTA4-5-4 | 5 | GCAGATGTgGaaTTaCaACAGGG | SLC9A2 | 0% | 0.00% | 0% | ||||
| CLTA4-5-5 | 5 | GCAGtcaTAGTGTaTaCACATGG | 0.004% | 0.00% | 0.005% | |||||
| CLTA4-5-6 | 5 | taAGATGTAGTaTTTCCAaAAGt | 0.007% | 0.01% | 0% | |||||
| CLTA4-6-1 | 6 | GCAGcTGgcaTtTcTCCACACGG | n.t. | n.t. | n.t. | |||||
| CLTA4-6-2 | 6 | GgAGATcTgaTGgTTCtACAAGG | 0.007% | 0.00% | 0.009% | |||||
| CLTA4-6-3 | 6 | taAaATGcAGTGTaTCCAtATGG | SMA4 | 0.015% | 0.00% | 0% | ||||
| CLTA4-7-1 | 7 | GCcagaaTAGTtTTTCaACAAGG | SEPHS2 | 0% | 0.00% | 0.007% | ||||
| CLTA4-7-2 | 8 | ttgtATtTAGaGaTTgCACAAGG | RORB | 0% | 0.00% | 0% | ||||
Under enzyme-excess conditions with the v2.1 sgRNA, the two verified CLTA1 off-target sites, CLTA1-1-1 and CLTA1-2-2, were two of the three most highly enriched sequences identified in the in vitro selection. CLTA4-3-1 and CLTA4-3-3 were the highest and third-highest enriched sequences of the seven CLTA4 three-mutation sequences enriched in the in vitro selection that are also present in the genome. The in vitro selection enrichment values of the four-mutation sequences were not calculated, because 12 out of the 14 CLTA4 sequences in the genome containing four mutations, including CLTA4-4-8, were observed at a level of only one sequence count in the post-selection library. Taken together, these results confirm that several of the off-target substrates identified in the in vitro selection that are present in the human genome are indeed cleaved by Cas9:sgRNA complexes in human cells and also suggest that the most highly enriched genomic off-target sequences in the selection are modified in cells to the greatest extent.
The off-target sites we identified in cells were among the most-highly enriched in our in vitro selection and contain up to four mutations relative to the intended target sites. Although it is possible that heterochromatin or covalent DNA modifications could diminish the ability of a Cas9:guide RNA complex to access genomic off-target sites in cells, the identification of five out of 49 tested cellular off-target sites in this study, rather than zero or many, strongly suggests that Cas9-mediated DNA cleavage is not limited to specific targeting of only a 7-12-base pair target sequence, as suggested in recent studies.11, 12, 19
The cellular genome modification data are also consistent with the tradeoff between activity and specificity of sgRNA v1.0 compared to sgRNA v2.1 observed in the in vitro selection data and discrete assays (Table 1 and Supplementary Table S5). The on-target CLTA4-0-1 site had a modification frequency that was seven-fold lower (11% vs. 76%) in cells expressing Cas9:sgRNA v1.0 compared to cells expressing Cas9:sgRNA v2.1. Although the CLTA4-3-3 and CLTA4-4-8 sites were modified by the Cas9-sgRNA v2.1 complexes, no evidence of modification at any of these three sites was detected in Cas9:sgRNA v1.0-treated cells. The CLTA4-3-1 site, which was modified at 32% of the frequency of on-target CLTA4 site modification in Cas9:v2.1 sgRNA-treated cells, was modified at only 0.5% of the on-target modification frequency in v1.0 sgRNA-treated cells, representing a 62-fold change in selectivity. Taken together, these results suggest that guide RNA architecture can have a significant influence on both Cas9 activity and specificity in cells. Our specificity profiling findings present a potential caveat to recent and ongoing efforts to improve the overall DNA modification activity of Cas9:guide RNA complexes through guide RNA engineering.11, 15
Overall, the off-target DNA cleavage profiling of Cas9 and subsequent analyses show that (i) Cas9:guide RNA recognition extends to 18-20 specified target site base pairs and a two-base pair PAM for the four target sites tested; (ii) increasing Cas9:guide RNA concentrations can decrease DNA-cleaving specificity in vitro; (iii) using more active sgRNA architectures can increase DNA-cleavage activity both in vitro and in cells but also can increase cleavage of off-target sites both in vitro and in cells; and (iv) as predicted by our in vitro results, Cas9:guide RNA can modify off-target sites in cells, some of which contain four mutations relative to the on-target site. Our findings provide insights into RNA-programmed Cas9 specificity and reveal a previously unknown role for sgRNA architecture in DNA-cleavage specificity. The principles revealed in this study may also apply to Cas9-based effectors engineered to mediate functions beyond DNA cleavage.
METHODS
Oligonucleotides
All oligonucleotides used in this study were purchased from Integrated DNA Technologies. Oligonucleotide sequences are listed in Supplementary Table S8.
Expression and Purification of S. pyogenes Cas9
E. coli Rosetta (DE3) cells were transformed with plasmid pMJ80611, encoding the S. pyogenes cas9 gene fused to an N-terminal 6xHis-tag/maltose binding protein. The resulting expression strain was inoculated in Luria-Bertani (LB) broth containing 100 μg/mL of ampicillin and 30 μg/mL of chloramphenicol at 37 °C overnight. The cells were diluted 1:100 into the same growth medium and grown at 37 °C to OD600 ~0.6. The culture was incubated at 18 °C for 30 min and isopropyl β-D-1-thiogalactopyranoside (IPTG) was added at 0.2 mM to induce Cas9 expression. After ~17 h, the cells were collected by centrifugation at 8,000 g and resuspended in lysis buffer (20 mM tris(hydroxymethyl)-aminomethane (Tris)-HCl, pH 8.0, 1 M KCl, 20 % glycerol, 1 mM tris (2-carboxyethyl)phosphine (TCEP)). The cells were lysed by sonication (10 sec pulse-on and 30 sec pulse-off for 10 min total at 6 W output) and the soluble lysate was obtained by centrifugation at 20,000 g for 30 min. The cell lysate was incubated with nickel-nitriloacetic acid (nickel-NTA) resin (Qiagen) at 4 °C for 20 min to capture His-tagged Cas9. The resin was transferred to a 20-mL column and washed with 20 column volumes of lysis buffer. Cas9 was eluted in 20 mM Tris-HCl (pH 8), 0.1 M KCl, 20 % glycerol, 1 mM TCEP and 250 mM imidazole and concentrated by Amicon ultra centrifugal filter (Millipore, 30-kDa molecular weight cut-off) to ~50 mg/mL. The 6xHis tag and maltose-binding protein were removed by TEV protease treatment at 4 °C for 20 h and captured by a second Ni-affinity purification step. The eluent, containing Cas9, was injected into a HiTrap SP FF column (GE Healthcare) in purification buffer containing 20 mM Tris-HCl (pH 8), 0.1 M KCl, 20 % glycerol and 1 mM TCEP. Cas9 was eluted with purification buffer containing a linear KCl gradient from 0.1 M to 1 M over five column volumes. The eluted Cas9 was further purified by a HiLoad Superdex 200 column in purification buffer, snap-frozen in liquid nitrogen and stored in aliquots at −80 °C.
In Vitro RNA Transcription
100 pmol CLTA(#) v2.1 fwd and v2.1 template rev were incubated at 95 °C and cooled at 0.1 °C/s to 37 °C in NEBuffer2 (50 mM sodium chloride, 10 mM Tris-HCl, 10 mM magnesium chloride, 1 mM dithiothreitol, pH 7.9) supplemented with 10 μM dNTP mix (Bio-Rad). 10 U of Klenow Fragment (3′→5′ exo−) (NEB) were added to the reaction mixture and a double-stranded CLTA(#) v2.1 template was obtained by overlap extension for 1 h at 37 °C. 200 nM CLTA(#) v2.1 template alone or 100 nM CLTA(#) template with 100 nM T7 promoter oligo was incubated overnight at 37 °C with 0.16 U/μL of T7 RNA Polymerase (NEB) in NEB RNAPol Buffer (40 mM Tris-HCl, pH 7.9, 6 mM magnesium chloride, 10 mM dithiothreitol, 2 mM spermidine) supplemented with 1 mM rNTP mix (1 mM rATP, 1 mM rCTP, 1 mM rGTP, 1 mM rUTP). In vitro transcribed RNA was precipitated with ethanol and purified by gel electrophoresis on a Criterion 10% polyacrylamide TBE-Urea gel (Bio-Rad). Gel-purified sgRNA was precipitated with ethanol and redissolved in water.
In Vitro Library Construction
10 pmol of CLTA(#) lib oligonucleotides were separately circularized by incubation with 100 units of CircLigase II ssDNA Ligase (Epicentre) in 1× CircLigase II Reaction Buffer (33 mM Tris-acetate, 66 mM potassium acetate, 0.5 mM dithiothreitol, pH 7.5) supplemented with 2.5 mM manganese chloride in a total reaction volume of 20 μL for 16 hours at 60 °C. The reaction mixture was incubated for 10 minutes at 85 °C to inactivate the enzyme. 5 μL (5 pmol) of the crude circular single-stranded DNA were converted into the concatemeric pre-selection libraries with the illustra TempliPhi Amplification Kit (GE Healthcare) according to the manufacturer’s protocol. Concatemeric pre-selection libraries were quantified with the Quant-it PicoGreen dsDNA Assay Kit (Invitrogen).
In Vitro Cleavage of On-Target and Off-Target Substrates
Plasmid templates for PCR were constructed by ligation of annealed oligonucleotides CLTA(#) site fwd/rev into HindIII/XbaI double-digested pUC19 (NEB). On-target substrate DNAs were generated by PCR with the plasmid templates and test fwd and test rev primers, then purified with the QIAquick PCR Purification Kit (Qiagen). Off-target substrate DNAs were generated by primer extension. 100 pmol off-target (#) fwd and off-target (#) rev primers were incubated at 95 °C and cooled at 0.1 °C/s to 37 °C in NEBuffer2 (50 mM sodium chloride, 10 mM Tris-HCl, 10 mM magnesium chloride, 1 mM dithiothreitol, pH 7.9) supplemented with 10 μM dNTP mix (Bio-Rad). 10 U of Klenow Fragment (3′→5′ exo-) (NEB) were added to the reaction mixture and double-stranded off-target templates were obtained by overlap extension for 1 h at 37 °C followed by enzyme inactivation for 20 min at 75 °C, then purified with the QIAquick PCR Purification Kit (Qiagen). 200 nM substrate DNAs were incubated with 100 nM Cas9 and 100 nM (v1.0 or v2.1) sgRNA or 1000 nM Cas9 and 1000 nM (v1.0 or v2.1) sgRNA in Cas9 cleavage buffer (200 mM HEPES, pH 7.5, 1.5 M potassium chloride, 100 mM magnesium chloride, 1 mM EDTA, 5 mM dithiothreitol) for 10 min at 37 °C. On-target cleavage reactions were purified with the QIAquick PCR Purification Kit (Qiagen) and off-target cleavage reactions were purified with the QIAquick Nucleotide Removal Kit (Qiagen) before electrophoresis in a Criterion 5% polyacrylamide TBE gel (Bio-Rad).
In Vitro Selection
200 nM concatemeric pre-selection libraries were incubated with 100 nM Cas9 and 100 nM sgRNA or 1000 nM Cas9 and 1000 nM sgRNA in Cas9 cleavage buffer (200 mM HEPES, pH 7.5, 1.5 M potassium chloride, 100 mM magnesium chloride, 1 mM EDTA, 5 mM dithiothreitol) for 10 min at 37 °C. Pre-selection libraries were also separately incubated with 2 U of BspMI restriction endonuclease (NEB) in NEBuffer 3 (100 mM NaCl, 50 mM Tris-HCl, 10 mM MgCl2, 1 mM dithiothreitol, pH 7.9) for 1 h at 37 °C. Blunt-ended post-selection library members or sticky-ended pre-selection library members were purified with the QIAQuick PCR Purification Kit (Qiagen) and ligated to 10 pmol adapter1/2(AACA) (Cas9:v2.1 sgRNA, 100 nM), adapter1/2(TTCA) (Cas9:v2.1 sgRNA, 1000 nM), adapter1/2 (Cas9:v2.1 sgRNA, 1000 nM), or lib adapter1/CLTA(#) lib adapter 2 (pre-selection) with 1,000 U of T4 DNA Ligase (NEB) in NEB T4 DNA Ligase Reaction Buffer (50 mM Tris-HCl, pH 7.5, 10 mM magnesium chloride, 1 mM ATP, 10 mM dithiothreitol) overnight (> 10 h) at room temperature. Adapter-ligated DNA was purified with the QIAquick PCR Purification Kit and PCR-amplified for 10-13 cycles with Phusion Hot Start Flex DNA Polymerase (NEB) in Buffer HF (NEB) and primers CLTA(#) sel PCR/PE2 short (post-selection) or CLTA(#) lib seq PCR/lib fwd PCR (pre-selection). Amplified DNAs were gel purified, quantified with the KAPA Library Quantification Kit-Illumina (KAPA Biosystems) and subjected to single-read sequencing on an Illumina MiSeq or Rapid Run single-read sequencing on an Illumina HiSeq 2500 (Harvard University FAS Center for Systems Biology Core facility, Cambridge, MA).
Selection Analysis
Pre-selection and post-selection sequencing data were analyzed as previously described21, with modification (Supplementary Algorithms) using scripts written in C++. Scripts are available upon request. Raw sequence data is available at the NCBI sequence read archive (SRA); see Supplementary Table S1 for a curated summary. Specificity scores were calculated with the formulae: positive specificity score = (frequency of base pair at position[post-selection] - frequency of base pair at position[pre-selection]) / (1 - frequency of base pair at position[pre-selection]) and negative specificity score = (frequency of base pair at position[post-selection] - frequency of base pair at position[pre-selection]) / (frequency of base pair at position[pre-selection]). Normalization for sequence logos was performed as previously described22.
Cellular Cleavage Assays
HEK293T cells were split at a density of 0.8×105 per well (6-well plate) before transcription and maintained in Dulbecco’s modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in a 37°C humidified incubator with 5% CO2. After 1 day, cells were transiently transfected using Lipofectamine 2000 (Invitrogen) following the manufacturer’s protocols. HEK293T cells were transfected at 70% confluency in each well of 6-well plate with 1.0 μg of the Cas9 expression plasmid (Cas9-HA-2xNLS-GFP-NLS) and 2.5 μg of the single-strand RNA expression plasmid pSilencer-CLTA (version 1.0 or 2.1). The transfection efficiencies were estimated to be ~70%, based on the fraction of GFP-positive cells observed by fluorescence microscopy. 48 h after transfection, cells were washed with phosphate buffered saline (PBS), pelleted and frozen at −80 °C. Genomic DNA was isolated from 200 μL cell lysate using the DNeasy Blood and Tissue Kit (Qiagen) according to the manufacturer’s protocol.
Off-Target Site Sequence Determination
100 ng genomic DNA isolated from cells treated with Cas9 expression plasmid and single-strand RNA expression plasmid (treated cells) or Cas9 expression plasmid alone (control cells) were amplified by PCR with 10 s 72°C extension for 35 cycles with primers CLTA(#)-(#)-(#) fwd and CLTA(#)-(#)-(#) rev and Phusion Hot Start Flex DNA Polymerase (NEB) in Buffer GC (NEB), supplemented with 3% DMSO. Relative amounts of crude PCR products were quantified by gel and Cas9-treated (control) and Cas9:sgRNA-treated PCRs were separately pooled in equimolar concentrations before purification with the QIAquick PCR Purification Kit (Qiagen). Purified DNA was amplified by PCR with primers PE1-barcode# and PE2-barcode# for 7 cycles with Phusion Hot Start Flex DNA Polymerase (NEB) in Buffer HF (NEB). Amplified control and treated DNA pools were purified with the QIAquick PCR Purification Kit (Qiagen), followed by purification with Agencourt AMPure XP (Beckman Coulter). Purified control and treated DNAs were quantified with the KAPA Library Quantification Kit-Illumina (KAPA Biosystems), pooled in a 1:1 ratio and subjected to paired-end sequencing on an Illumina MiSeq.
Statistical Analysis
Statistical analysis was performed as previously described21. P values in Supplementary Figure S3 were calculated for a one-sided test of the difference in the means of the number of mutations in all possible pairwise comparisons between pre- and post-selection libraries. The t-statistic was calculated as t = (x_bar1 − x_bar2) / sqrt(l • p_hat1 • (1 − p_hat1) / n1 + l • p_hat2 • (1 − p_hat2) / n2), where x_bar1 and x_bar2 are the means of the distributions being compared, l (= 22) is the target site length, p_hat1 and p_hat2 are the calculated probabilities of mutation (x_bar / l) for each library and n1 and n2 are the total number of sequences analyzed for each selection (Supplementary Text: Pre- and Post-Selection Library Composition). All pre- and post-selection libraries were assumed to be binomially distributed. P-values in Table 1 and Supplementary Table S5 were calculated for a one-sided Fisher exact test.
Supplementary Material
Acknowledgements
V.P., J.P.G. and D.R.L. were supported by DARPA HR0011-11-2-0003, DARPA N66001-12-C-4207, and the Howard Hughes Medical Institute. V.P. was supported by award Number T32GM007753 from the National Institute of General Medical Sciences. S.L and J.A.D. were supported by the Howard Hughes Medical Institute; E.M. was supported by NIH grant R01GM073794-05 to J.A.D.; J.A.D. and D.R.L. are HHMI Investigators.
Footnotes
Author Contributions V.P., S.L., J.P.G., and E.M. performed the experiments, designed the research, analyzed the data, and wrote the manuscript. J.A.D. and D.R.L. designed the research, analyzed the data, and wrote the manuscript.
Competing Financial Interests The co-authors have filed provisional patents related to this work.
References
- 1.Hockemeyer D, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nature Biotechnology. 2011;29:731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zou J, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5:97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hockemeyer D, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nature Biotechnology. 2009;27:851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Doyon Y, et al. Heritable targeted gene disruption in zebrafish using designed zinc-finger nucleases. Nature Biotechnology. 2008;26:702–708. doi: 10.1038/nbt1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Meng X, Noyes MB, Zhu LJ, Lawson ND, Wolfe SA. Targeted gene inactivation in zebrafish using engineered zinc-finger nucleases. Nature Biotechnology. 2008;26:695–701. doi: 10.1038/nbt1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sander JD, et al. Targeted gene disruption in somatic zebrafish cells using engineered TALENs. Nature Biotechnology. 2011;29:697–698. doi: 10.1038/nbt.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tesson L, et al. Knockout rats generated by embryo microinjection of TALENs. Nature Biotechnology. 2011;29:695–696. doi: 10.1038/nbt.1940. [DOI] [PubMed] [Google Scholar]
- 8.Cui X, et al. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nature Biotechnology. 2011;29:64–67. doi: 10.1038/nbt.1731. [DOI] [PubMed] [Google Scholar]
- 9.Perez EE, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nature Biotechnology. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.NCT00842634, NCT01044654, NCT01252641, NCT01082926.
- 11.Jinek M, et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hwang WY, et al. Efficient genome editing in zebrafish using a CRISPR-Cas system. Nature Biotechnology. 2013;31:227–229. doi: 10.1038/nbt.2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jinek M, et al. RNA-programmed genome editing in human cells. eLife. 2013;2:e00471. doi: 10.7554/eLife.00471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dicarlo JE, et al. Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systems. Nucleic Acids Research. 2013 doi: 10.1093/nar/gkt135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jiang W, Bikard D, Cox D, Zhang F, Marraffini LA. RNA-guided editing of bacterial genomes using CRISPR-Cas systems. Nature Biotechnology. 2013;31:233–239. doi: 10.1038/nbt.2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sapranauskas R, et al. The Streptococcus thermophilus CRISPR/Cas system provides immunity in Escherichia coli. Nucleic Acids Research. 2011;39:9275–9282. doi: 10.1093/nar/gkr606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Semenova E, et al. Interference by clustered regularly interspaced short palindromic repeat (CRISPR) RNA is governed by a seed sequence. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:10098–10103. doi: 10.1073/pnas.1104144108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qi LS, et al. Repurposing CRISPR as an RNA-Guided Platform for Sequence-Specific Control of Gene Expression. Cell. 2013;152:1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nature Methods. 2011;8:765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doyon JB, Pattanayak V, Meyer CB, Liu DR. Directed evolution and substrate specificity profile of homing endonuclease I-SceI. Journal of the American Chemical Society. 2006;128:2477–2484. doi: 10.1021/ja057519l. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.