

Abstract
Brain-derived neurotrophic factor (BDNF) is a potent regulator of neuronal activity, neurogenesis, and depressive-like behaviors; however, downstream effectors by which BDNF exerts these varying actions remain to be determined. Here we reveal that BDNF induces long-lasting enhancements in the efficacy of synaptic inhibition by stabilizing γ2 subunit-containing GABAA receptors (GABAARs) at the cell surface, leading to persistent reductions in neuronal excitability. This effect is dependent upon enhanced phosphorylation of tyrosines 365 and 367 (Y365/7) in the GABAAR γ2 subunit as revealed using mice in which these residues have been mutated to phenyalanines (Y365/7F). Heterozygotes for this mutation exhibit an antidepressant-like phenotype, as shown using behavioral-despair models of depression. In addition, heterozygous Y365/7F mice show increased levels of hippocampal neurogenesis, which has been strongly connected with antidepressant action. Both the antidepressant phenotype and the increased neurogenesis seen in these mice are insensitive to further modulation by BDNF, which produces robust antidepressant-like activity and neurogenesis in wild-type mice. Collectively, our results suggest a critical role for GABAAR γ2 subunit Y365/7 phosphorylation and function in regulating the effects of BDNF.
Introduction
GABAA receptors (GABAARs) are ligand-gated Cl− channels that mediate the majority of fast synaptic inhibition in the brain (for review, see Sieghart, 2006). These receptors are heteropentamers assembled from four major subunit classes: α1–6, β1–3 γ1–3, and δ, with the majority of synaptic benzodiazepine-sensitive GABAARs being assembled from α1, β2, and γ2 subunits (for review, see Sieghart, 2006).
While deficits in the activity of GABAARs are accepted to contribute to epilepsy and anxiety, experimental evidence now suggests that GABAergic deficits are central to depressive disorders. Clinical studies from depressed patients show reductions in the level of GABAAR expression in the prefrontal cortex (PFC; Feighner et al., 1983; Petty et al., 1995; Earnheart et al., 2007; Kalueff and Nutt, 2007; Maguire and Mody, 2008; Klumpers et al., 2010). A key mechanistic insight into the role that GABAARs play in depression comes from the study of heterozygous γ2 subunit knock-out mice, which show a depressive-like phenotype and reduced levels of hippocampal neurogenesis (Earnheart et al., 2007; Luscher et al., 2011a; Song et al., 2012).
It is well established that the efficacy of synaptic inhibition relies upon the accumulation of γ2-containing GABAARs at postsynaptic sites, a process that is determined by rates of exocytosis and endocytosis (for review, see Vithlani et al., 2011). Tyrosine residues 365 and 367 (Y365/7) within the intracellular domain of the γ2 subunit play a key role in facilitating endocytosis of GABAARs as they bind with nanomolar affinity to the clathrin adaptor protein-2 (AP2), a central component in the formation of endocytic vesicles (Kittler et al., 2008; Tretter et al., 2009). Phosphorylation of Y365/7 by Src family kinases decreases affinity of the γ2 subunit for AP2 and slows receptor endocytosis, a process that can be mimicked by their mutation to phenylalanine residues (Jurd et al., 2010). Thus Y365/7 phosphorylation enhances the stability of GABAARs at the cell surface and the efficacy of synaptic inhibition (Kittler et al., 2008; Tretter et al., 2009).
The first messengers that regulate phosphorylation of Y365/7 remain to be identified. One attractive molecule is brain-derived neurotrophic factor (BDNF), which exerts pleiotrophic effects on neuronal activity mediated by multiple effectors downstream of TrKB receptors. BDNF is known to exert region-specific events on depressive-like behaviors in rodents with potent antidepressant actions being seen in the hippocampus, but prodepressant effects in the nucleus accumbens. In addition to regulating depressive-like behaviors, BDNF also exerts potent effects on appetite, anxiety, cognition, and reward (Huang and Reichardt, 2001; Waterhouse and Xu, 2009). Studies in culture and brain slices have revealed that GABAAR phosphorylation, cell-surface number, and miniature inhibitory synaptic currents (mIPSCs) are modulated by BDNF (Jacob et al., 2008). However, the significance of this modulation in determining neuronal excitability, depressive-like behaviors, and neurogenesis remains unknown.
Here we demonstrate that BDNF enhances the phosphorylation of Y365/7 in the γ2 subunit, leading to persistent increases in the cell-surface accumulation of GABAARs and the efficacy of synaptic inhibition. Using Y365/7F mice (mice in which tyrosine residues Y365/7 have been mutated to phenyalanines), we reveal that phosphorylation of Y365/7 determines the excitability of the PFC, increases hippocampal neurogenesis, reduces depressive-like behaviors, and blocks the therapeutic actions of BDNF. Our results suggest that phosphorylation of GABAAR γ2 mediates both the antidepressant efficacy of BDNF and its regulation of neurogenesis.
Materials and Methods
Housing and handling of animals.
All procedures were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by Tufts University and the Institutional Animal Care and Use Committee. Two-to-four-month-old male Y365/7F mice (Tretter et al., 2009) were group-housed with wild-type (WT) littermate controls in a temperature-controlled facility with a 12 h light/dark cycle and fed standard rodent chow and water ad libitum. Before any behavioral testing, animals were brought to the testing room and allowed to acclimatize for 30 min.
Acute PFC slices.
Preparation of acute PFC slices was performed as described previously (Svenningsson et al., 2002). Briefly, animals were deeply anesthetized with isoflurane and then decapitated at the second cervical vertebrae. Brains were carefully removed from the skull and rinsed in ice-cold ACSF equilibrated with 95% O2 and 5% CO2 to yield a pH of 7.4. The PFC was then cut into 350 μm slices using a Leica VT1000S vibratome and allowed to recover in ACSF under constant oxygenation for 1 h at 32°C before experimentation.
Biotinylation and immunoblotting.
Biotinylation of cell-surface proteins was performed as previously described (Terunuma et al., 2008). Briefly, PFC slices were either analyzed untreated or following treatment with 100 ng/ml BDNF (or vehicle) for 2 h at 32°C. Slices were chilled to 4°C and incubated for 30 min with 1 mg/ml sulfosuccinimidyl-2-(biotinamido)ethyl-1,3′-dithiopropionate (Pierce) to label surface fractions. Excess biotin was removed by washing three times in ice-cold ACSF and biotinylated proteins were pulled down by incubating detergent-soluble extracts with Neutravidin (Pierce) overnight at 4°C. Bound material was subjected to SDS-PAGE, followed by immunoblotting with antibodies and visualized by ECL (Pierce). Blots were then quantified using the CCD-based FujiFilm LAS 3000 system and MultiGauge software.
Electrophysiology.
Synaptic currents and spontaneous action potentials were recorded from layer V medial PFC pyramidal neurons from 2–4-month-old mice as described previously (Chen et al., 2006; Kittler et al., 2008; Tretter et al., 2009; Yuen et al., 2011). PFC slices were placed in a perfusion chamber attached to the fixed stage of an upright microscope (Olympus) and submerged in continuously flowing oxygenated ACSF containing the following (in mm): 130 NaCl, 26 NaHCO3, 3 KCl, 5 MgCl2, 1.25 NaH2PO4, 1 CaCl2, 10 glucose, pH 7.4, 300 mOsm. CNQX (20 μm) and d(-)-2-amino-5-phosphonopentanoic acid (d-APV; 50 μm) were added in IPSC recordings; bicuculline (10 μm) and d-APV were added in EPSC recordings. TTX (0.5 μm) was added in miniature synaptic current recordings. Cells were visualized with a 40× water-immersion lens and illuminated with near-infrared (IR) light, and the image was detected with an IR-sensitive CCD camera. A Multiclamp 700A amplifier was used for these recordings. Tight seals (2–10 GΩ) from visualized pyramidal neurons were obtained by applying negative pressure. The membrane was disrupted with additional suction, and the whole-cell configuration was obtained. Membrane potential was held at −70 mV during synaptic current recordings. Evoked currents were generated with a pulse from a stimulation isolation unit controlled by a S48 pulse generator (Astro Med). A bipolar stimulating electrode (FHC) was positioned ∼100 μm from the neuron under recording.
For eIPSC or mIPSC recording, internal solution contained the following (in mm): 100 CsCl, 30 N-methyl-d-glucamine, 4 NaCl, 10 HEPES, 1 MgCl2, 5 EGTA, 2.2 QX314, 12 phosphocreatine, 5 MgATP, 0.2 Na3GTP, 0.1 leupeptin. For mEPSC recording, internal solution contained the following (in mm): 130 Cs-methanesulfonate, 10 CsCl, 4 NaCl, 10 HEPES, 1 MgCl2, 5 EGTA, 2.2 QX-314, 12 phosphocreatine, 5 MgATP, 0.2 Na3GTP, 0.1 leupeptin, pH 7.2–7.3, 265–270 mOsm.
To record the spontaneous action potential (sAP), we used a modified ACSF that slightly elevated basal neuronal activity (in mm: 130 NaCl, 26 NaHCO3, 2 CaCl2, 0.5 MgCl2, 3.5 KCl, 10 glucose, 1.25 NaH2PO4), which more closely mimics the ionic composition of brain interstitial fluid in situ (1.0–1.2 mm Ca2+, 1 mm Mg2+, and 3.5 mm K+; Sanchez-Vives and McCormick, 2000; Maffei et al., 2004). sAP was induced and recorded in the whole-cell current-clamp mode with the internal solution containing the following (in mm): 20 KCl, 100 K-gluconate, 10 HEPES, 4 MgATP, 0.5 Na2GTP, and 10 phosphocreatine (Maffei and Turrigiano, 2008). The interspike membrane potential was −60 mV either without a holding current or with a small (<50 pA) adjusting current (positive DC).
Data analyses were performed with Clampfit (Axon) and Kaleidagraph (Albeck Software).
Cannula implantation.
Cannula implantation surgeries were performed to target the cerebral ventricles using one site [anteroposterior, −1.0 mm; mediolateral, +1.0 mm (left side); dorsoventral, −2.0 mm; Paxinos and Franklin, 2001]. Animals were deeply anesthetized via intraperitoneal injection with a mixture of a ketamine (120 mg/kg) and xylazine (10 mg/kg) and placed into a stereotaxic frame. A small burr hole was drilled for the cannula according to stereotaxic coordinates, along with an additional hole for an anchoring screw. Mice were singly housed following surgery and were allowed to recover for 1 week before infusion and testing. For the infusion, mice were scruffed so that the cannula could be inserted, and then allowed to move freely in a small enclosure while the infusion took place (0.1 μl/min for 10 min).
Forced-swim task.
The task arena consisted of a cylinder (25 cm high, 25 cm diameter) containing 20 cm (height) of water at a temperature of 23–25°C. Animals were placed in the arena and allowed to swim freely for 10 min. Animals typically display very fast swimming for the initial 1–2 min, followed by increasing immobility. The animals were video recorded and movements were digitized from above. Immobility was automatically scored using Ethovision (Noldus Information Technology). Immobile time used for statistical analysis was measured during the last 4 min of the task.
Tail-suspension test.
The apparatus for the tail suspension test was composed of a horizontal wooden bar located inside a white plastic box (40 × 46 × 40 cm) ∼35 cm above the floor. Each mouse was suspended by its tail from the bar by adhesive tape placed 1–1.5 cm from the tip of the tail. Each mouse was video recorded for 6 min (360 s) followed by digitizing and automated scoring using Ethovision (Noldus Information Technology). When mice where observed to climb their tails a total of 10% or more of the total time, they were eliminated from further analyses.
Open-field test.
The open-field apparatus was based on that used in the European Mouse Phenotyping Resource of Standardized Screens (Brown et al., 2005). Animals were assessed for 30 min and video recorded from above for scoring with Ethovision (Noldus Information Technology).
Immunohistochemistry.
Animals were transcardially perfused with a periodate-lysine-paraformaldehyde solution. Forty-micrometer-thick sections were stained free-floating with anti-doublecortin (anti-Dcx, rabbit; Cell Signaling Technology) and with DAPI for fluorescent staining experiments (Life Technologies, Invitrogen). Fluorescent images were taken with a 60× objective on a Nikon confocal microscope and were quantified using MetaMorph. For quantification of the total number of cells per granule cell layer (GCL), images were taken in a series of every eighth section (320 μm interval) spanning the entire hippocampus. The number of Dcx-positive cells was then estimated by multiplying the total number of cells by eight.
Statistical analysis.
Data expressed are mean ± SE and the statistical significance of differences in mean values was assessed by paired and unpaired t test, or two-way ANOVA with a post hoc Student–Newman–Keuls multiple-comparison test, as appropriate.
Results
BDNF enhances the cell-surface stability of GABAARs by stimulating phosphorylation of the γ2 subunit at Y365/7
To examine the role that BDNF plays in regulating the functional expression of GABAARs, our experiments focused on the PFC, the activity of which is strongly linked to depression in humans and depressive-like behaviors in rodents. Acute PFC slices prepared from WT mice were treated with 100 ng/ml BDNF (or vehicle) for 2 h at 32°C and the effects on γ2 subunit cell-surface expression measured via biotinylation coupled with immunoblotting. Under these conditions, BDNF significantly increased the cell-surface levels of γ2 subunit-containing GABAARs to 179.0 ± 17.5% of vehicle-treated control (p < 0.01, n = 6 mice; Fig. 1A). Cell-surface fractions were essentially free of actin, verifying the validity of the biotinylation assay. We have previously shown that the cell-surface stability of GABAARs and their accumulation at inhibitory synapses is enhanced via phosphorylation of Y365/7 within the intracellular domain of the γ2 subunit, which drastically reduces the affinity of GABAARs for the μ2 adaptin of the AP2 complex to slow receptor endocytosis (Kittler et al., 2008; Tretter et al., 2009). In line with this, BDNF-induced increase in the levels of γ2 subunit-containing GABAARs at the cell surface was accompanied by an increase in phosphorylation of the γ2 subunit at Y365/7 to 225.2 ± 18.5% of vehicle-treated control (p < 0.01, n = 6 mice; Fig. 1A), as measured using a phospho-specific antibody directed against tyrosine 367 (pY367; Brandon et al., 2001; Tretter et al., 2009).
Figure 1.

BDNF enhances GABAAR γ2 subunit Y365/7 phosphorylation and cell-surface stability. A, PFC slices from C57BL/6 mice treated with either vehicle (−) or 100 ng/ml BDNF (+) for 2 h at 32°C were labeled with 1 mg/ml sulfosuccinimidyl-2-(biotinamido)ethyl-1,3′-dithiopropionate and detergent-soluble extracts purified on avidin. Surface and 10% of input (Total) fractions were then analyzed by immunoblotting with γ2 and pY367 antibodies. In addition, membranes were probed with actin to assess the validity of the biotinylation assay. Surface and phosphorylated γ2 levels were normalized to input. B, PFC slices from WT and Y365/7F mice were subject to biotinylation and analyzed by immunoblotting with γ2 and GluR1 antibodies as above. Surface levels of GABAAR and AMPA-receptor subunits were normalized to input. C, PFC slices from Y365/7 mice were subject to BDNF treatment followed by biotinylation and then analyzed by immunoblotting as detailed above. Surface γ2 levels were normalized to input. D, PFC slices were subject to BDNF treatment as above and then SDS-soluble extracts immunoblotted with total and phospho-specific (pS408/9) β3 antibodies. Phosphorylated β3 levels were normalized to total levels. E, 350 μm hippocampal slices from WT and BDNFL/L/CamK-Cre mice were subject to biotinylation and then blotted with γ2 antibodies and pY367 antibodies. Statistical significance was determined using the Student's t test.
To further examine the role that BDNF plays in modulating plasma membrane levels of GABAARs we used mice in which these residues were mutated to phenylalanines (Y365/7F). In common with their phosphorylation, Y365/7F mutation reduces binding of the γ2 subunit to AP2 to slow GABAAR endocytosis (Tretter et al., 2009). While homozygotes for this mutation die in utero, heterozygotes for the Y365/7F mutation are viable. As determined using biotinylation, cell-surface levels of γ2 subunit-containing GABAARs was significantly increased in Y365/7F mice to 155.5 ± 17.5% of WT control (p < 0.01, n = 5–6 mice; Fig. 1B), while the cell-surface levels of GluR1 subunit-containing AMPA receptors were similar between genotypes (Fig. 1B). In contrast to WT mice, BDNF did not significantly modify the cell-surface expression of the γ2 subunit in Y365/7F mice (Fig. 1C). However, the ability of BDNF to modulate the phosphorylation of serines 408 and 409 (S408/9) in the GABAAR receptor β3 subunit, previously identified substrates of this neurotrophin (Jovanovic et al., 2004; Porcher et al., 2011), was not compromised upon mutation of Y365/7 (Fig. 1D). This suggests that Y365/7F mutation does not induce a generalized loss in the efficacy of BDNF-dependent signaling. As a means of independently assessing the significance of BDNF as a determinant of the phosphorylation and membrane trafficking of GABAARs, we used mice that have postnatal deficits in BDNF expression in the forebrain (BDNFL/L/CamKCre; Rios et al., 2006). Compared with WT mice, BDNFL/L/CamKCre mice exhibited a significant decrease in γ2 subunit cell-surface levels (67.5 ± 10.5% of WT control, p < 0.01, n = 7 mice) and phosphorylation at Y365/7 (45.6 ± 18.5% of WT control, p < 0.01, n = 7 mice) within the PFC (Fig. 1E).
Collectively, our results suggest that the ability of BDNF to modulate GABAAR cell-surface stability is dependent on Y365/7 in the receptor γ2 subunit.
BDNF enhances synaptic inhibition by blocking endocytosis of γ2 subunit-containing GABAARs
To examine the significance of our biochemical studies, we measured the effects of BDNF on mIPSCs in layer V of medial PFC pyramidal neurons. As shown in Figure 2, BDNF treatment (100 ng/ml, 2 h) significantly increased mIPSC amplitude (control: 27.3 ± 1.6 pA, n = 14 neurons; BDNF: 34.9 ± 1.7 pA, n = 14 neurons, p < 0.05) and frequency (control: 1.9 ± 0.1 Hz, n = 14 neurons; BDNF: 3.0 ± 0.3 Hz, n = 14 neurons; p < 0.05). This result suggests that BDNF may act to potentiate inhibitory transmission via both a postsynaptic mechanism affecting the number and/or activity of GABAARs (indicated by the increase in mIPSC amplitude) and a presynaptic mechanism that modifies GABA release (indicated by the increase in mIPSC frequency).
Figure 2.

BDNF enhances mIPSC amplitude by blocking endocytosis of γ2 subunit-containing GABAARs. A–C, E–G, Cumulative distribution plots and representative traces of mIPSCs in WT PFC pyramidal neurons untreated (control) or treated with 100 ng/ml BDNF for 2 h at 32°C in either the absence (A–C) or presence (E–G) of TAT-YECL peptide. Scale bars: 20 pA, 5 s. D, H, Bar graphs show mIPSC amplitude or frequency in control versus BDNF-treated PFC slices in either the absence (D) or presence (H) of TAT-YECL peptide (*p < 0.05).
As BDNF acted to increase phosphorylation of Y365/7 in the GABAAR γ2 subunit (Fig. 1A), a post-translation covalent modification accepted to slow receptor endocytosis (Kittler et al., 2008), we sought to determine whether the effects of BDNF on mIPSC amplitude are mediated via alterations in GABAAR endocytosis. To do so, we treated PFC slices from WT mice with membrane-permeable YECL peptide (TAT-YECL) that blocks binding of the AP2-μ2 adaptin to the GABAAR γ2 subunit, and has been established to specifically modulate receptor endocytosis and mIPSC but not mEPSC amplitude (Kittler et al., 2008). Thus we compared the effects of TAT-YECL on mIPSC properties when applied to PFC slices alone and in combination with BDNF. TAT-YECL-pep (20 μm, 1 h) caused a significant increase (p < 0.05) in mIPSC amplitude similar to that seen with BDNF (Fig. 2; TAT-YECL, 33.8 ± 2.1 pA, n = 6 neurons). However TAT-YECL occluded the effects of BDNF on mIPSC amplitude (Fig. 2; TAT-YECL plus BDNF, 35.2 ± 2.3 pA, n = 6 neurons). In contrast to this, TAT-YECL had minimal effects on mIPSC frequency when applied alone and BDNF significantly increased (p < 0.05) mIPSC frequency in the presence of this peptide (Fig. 2; TAT-YECL, 2.26 ± 0.2 Hz, n = 6; TAT-YECL plus BDNF, 3.29 ± 0.3 Hz, n = 6 neurons).
Together these results suggest that the ability of BDNF to increase the amplitude of mIPSCs is mediated via a reduction in the endocytosis of γ2 subunit-containing GABAAR subtypes.
BDNF modulation of mIPSC amplitude is dependent on γ2 Y365/7
Given that the ability of BDNF to modulate the cell-surface stability of GABAARs is dependent on Y365/7 in the receptor γ2 subunit, we assessed the role that these residues play in mediating the effects of BDNF on mIPSCs. To do so, we compared the properties of mIPSCs in WT and Y365/7F mice. The amplitude of mIPSCs was selectively enhanced (p < 0.05) in the PFC of Y365/7F mice (Fig. 3; WT: 21.2 ± 1.2 pA, n = 11 neurons; Y365/7F: 27.0 ± 0.9 pA, n = 10 neurons), while mIPSC frequency was similar (Fig. 3; WT: 1.8 ± 0.3 Hz, n = 11 neurons; Y365/7F: 2.1 ± 0.2 Hz, n = 10 neurons). These results are similar to observations published in CA3 pyramidal neurons from Y365/7F male mice that have elevated mIPSC amplitudes, which correlate with increases in the size and number of inhibitory synapses (Tretter et al., 2009).
Figure 3.

mIPSC amplitude is enhanced in Y365/7F mice. A–C, Cumulative distribution plots and representative traces of mIPSCs in PFC pyramidal neurons from WT versus Y365/7F mice. Scale bars: 20 pA, 5 s. D, Bar graphs showing mIPSC amplitude or frequency in WT versus Y365/7F mice (*p < 0.05).
Next, the ability of BDNF to modulate mIPSC properties was compared between genotypes. This revealed that the ability of BDNF to increase mIPSC amplitude was significantly impaired (p < 0.05) in Y365/7F mice (Fig. 4; WT: 27.6 ± 6.6% increase, n = 8 neurons; Y365/7F: 5.8 ± 5.1% increase, n = 8 neurons). In contrast, the ability of BDNF to increase mIPSC frequency was not influenced by mutation of Y365/7 (Fig. 4; WT: 84.8 ± 28.2% increase, n = 8 neurons; Y365/7F: 88.7 ± 28.5% increase, n = 8 neurons).
Figure 4.

BDNF modulation of mIPSC amplitude is lost in Y365/7 mice. A–D, Cumulative distribution plots and representative traces of mIPSCs in PFC neurons from WT versus Y365/7F mice untreated (control) or treated with 100 ng/ml BDNF for 2 h at 32°C. Scale bars: 20 pA, 5 s. E, Bar graphs show the mIPSC amplitude or frequency in control versus BDNF-treated PFC pyramidal neurons from either WT or Y365/7F mice. F, Bar graph shows percentage change induced by BDNF in mIPSC amplitude and frequency between genotypes.
To ascertain whether excitatory synaptic transmission was modified in Y365/7F mice, the properties of mEPSCs under basal conditions and in the presence of BDNF were compared between genotypes. mEPSC amplitude and frequency were comparable in WT and Y365/7F mice (Fig. 5; WT: 12.6 ± 0.6 pA, 2.3 ± 0.2 Hz, n = 5 neurons; Y365/7F: 11.7 ± 0.4 pA, 2.0 ± 0.2 Hz, n = 5 neurons). Moreover, the ability of BDNF to modulate mEPSC frequency was also similar between strains (Fig. 5).
Figure 5.

mEPSCs and their modulation by BDNF are not altered of Y365/7F mice. A, B, Representative traces of mESPCs in PFC pyramidal neurons from WT versus Y365/7F mice untreated (control) or treated with 100 ng/ml BDNF for 2 h at 32°C. Scale bars: 10 pA, 2 s. C, D, Bar graphs showing the mEPSC amplitude (C) or frequency (D) in control versus BDNF-treated PFC pyramidal neurons from either WT or Y365/7F mice (*p < 0.05).
These results suggest that BDNF exerts its actions on mIPSC amplitude by stimulating the phosphorylation of Y365/7 in the GABAAR γ2 subunit, thereby increasing the number of receptors at the cell surface. Its effect on frequency presumably reflects the ability of BDNF to potentiate the release of GABA from presynaptic terminals (Bardoni et al., 2007).
ACSF-induced increase in spontaneous action potentials of cortical circuits is diminished upon Y365/7F mutation
To determine whether altered GABAAR γ2 Y365/7 phosphorylation affects the overall excitability of cortical circuits, the frequency of sAPs was compared in PFC pyramidal neurons from WT and Y365/7F mice. Modified ACSF with reduced Mg2+ and slightly elevated K+, which is more similar to rodent CSF than standard ACSF (Maffei et al., 2004) was used. Spontaneous firing under the “active-slice” condition helps to reveal the circuit excitability changes induced by altered synaptic drive onto pyramidal neurons. As shown in Figure 6, the firing rate of sAP was significantly reduced (p < 0.05) in Y365/7F mice, compared with WT controls (WT: 2.0 ± 0.2 Hz, n = 8 neurons; Y365/7F: 0.7 ± 0.1 Hz, n = 8 neurons). The coefficient of variability (SD/mean) associated with sAP firing rates was modestly increased in Y365/7F mice (WT: 0.32, n = 8 neurons; Y365/7: 0.38, n = 8, neurons), consistent with the expectation from enhanced synaptic inhibition. We further compared the effect of BDNF treatment on sAP in PFC neurons from WT and Y365/7F mice. As shown in Figure 7, BDNF had little effect on firing rate of sAP in WT mice (control: 2.1 ± 0.3 Hz, n = 5 neurons; BDNF: 2.4 ± 0.3 Hz, n = 7 neurons), but significantly increased (p < 0.05) the sAP frequency in Y365/7F mutants (control: 0.8 ± 0.1 Hz, n = 5 neurons; BDNF: 1.9 ± 0.3 Hz, n = 7 neurons).
Figure 6.

Inhibitory drive is increased in the PFC of Y365/7F mice. A, Summarized input–output curves of GABAAR eIPSCs evoked by a series of stimulus intensities in PFC pyramidal neurons from WT versus Y365/7F mice. B, Representative traces of eIPSCs in PFC pyramidal neurons from WT versus Y365/7F mice. Scale bars: 100 pA, 100 ms. C, Representative spontaneous firing recordings of PFC pyramidal neurons from WT versus Y365/7F mice. Scale bars: 20 mV, 2 s. D, Bar graph showing sAP frequency in PFC pyramidal neurons from either WT or Y365/7F mice (*p < 0.05).
Figure 7.

ACSF-induced increase in sAPs of cortical circuits and their modulation by BDNF is modified in Y365/7F mice. A, B, Representative spontaneous firing recordings in PFC pyramidal neurons from WT versus Y365/7F mice untreated (control) or treated with 100 ng/ml BDNF for 2 h at 32°C. Scale bars: 20 mV, 2 s. C, D, Bar graph showing sAP frequency in control versus BDNF-treated PFC pyramidal neurons from either WT or Y365/7F mice (*p < 0.05).
These studies reveal that BDNF-dependent phosphorylation of the GABAR γ2 subunit at Y365/7 diminishes the ACSF-induced increase in sAPs of cortical networks by potentiating synaptic inhibition.
Depressive-like behaviors and the antidepressant efficacy of BDNF are dependent on γ2 Y365/7
Given the roles both the GABAAR γ2 subunit and BDNF are proposed to play in depressive disorders, we investigated the impact that the Y365/7F mutation has on validated models of depression. We did this by comparing the immobility time of Y365/F mice and WT littermate controls in the forced-swim task (FST) and the tail-suspension test (TST). In the FST, we found that, compared with WT controls, Y365/7F mice had an increased latency to the first immobile episode (Fig. 8A; WT: 74.71 ± 4.49s; Y365/7F: 110.57 ± 14.96s; p = 0.041, n = 10 mice per group) as well as decreased time spent immobile during the last 4 min of the test (Fig. 8A; WT: 166.20 ± 7.26s; Y365/7F: 142.00 ± 3.86s; p = 0.007, n = 10 mice per group). Similar to the FST, Y365/7F mice in the TST showed an increase in latency to immobility (Fig. 8B; WT: 58.41 ± 3.88s; Y365/7F: 76.67 ± 5.81s; p = 0.015, n = 18 mice per group) and a decrease in total immobility compared with WT controls (Fig. 8B; WT: 157.27 ± 8.04s; Y365/7F: 126.78 ± 12.31s; p = 0.046, n = 18 mice per group). In contrast to these differences in depressive-like behaviors, we found no difference in distance traveled (Fig. 8C) or average speed (Fig. 8D) between Y365/7F mice and WT littermate controls using the open-field test (OFT). In addition, both strains showed similar levels of habituation in response to the novel environment of the open field.
Figure 8.

Depression-related behaviors and the antidepressant efficacy of BDNF are reduced in Y365/7F mice. A, Latency to immobility and total immobility in the FST in WT and Y365/F mice. B, Latency to immobility and total immobility in the TST in WT and Y365/F mice. C, D, Distance and speed traveled of WT and Y365/F mice in the open field. E, Latency to immobility and total immobility in the TST in WT and Y365/F mice treated with either vehicle control or BDNF.
To assess whether Y365/7 contribute to the antidepressant efficacy of BDNF, we implanted WT and Y365/7F mice with intracerebroventricular cannulae to facilitate infusion of BDNF (or vehicle) directly into the brain. The effect of BDNF (0.1 μl/min; 10 min) on depressive-like behaviors using the TST was then measured 5 d later (Hoshaw et al., 2005). Similar to our observations comparing depressive-like behaviors in untreated WT and Y365/7F mice, in the TST we found that vehicle-treated Y365/7F mice showed an increased latency to immobility (Fig. 8E; WT vehicle: 25.54 ± 6.01s; Y365/7F vehicle: 58.02 ± 8.47s; p = 0.006, n = 8 mice per group) and reduced total time spent immobile (Fig. 8E; WT vehicle: 187.52 ± 17.67s; Y365/7F vehicle: 129.28 ± 12.04s; p = 0.017, n = 8 mice per group) compared with vehicle-treated WT controls. WT mice treated with BDNF showed a significant increase in latency to immobility (Fig. 8E; WT vehicle: 25.54 ± 6.01s; WT BDNF: 68.40 ± 7.05s; p < 0.001, n = 8 mice per group) and total immobility (Fig. 8E; WT vehicle: 187.52 ± 17.67s; WT BDNF: 102.02 ± 13.76s; p = 0.001, n = 8 mice per group) compared with vehicle-treated WT controls. In comparison, BDNF treatment did not significantly affect TST measures in Y365/7F mice compared with vehicle-treated Y365/7F controls (Fig. 8E).
These results suggest that Y365/7 in GABAAR γ2 subunit are important determinants of depressive-like behaviors and also contribute to the antidepressant efficacy of BDNF.
Basal hippocampal neurogenesis and BDNF-dependent neurogenesis are dependent on γ2 Y365/7
GABAAR γ2 subunit expression levels and BDNF are both determinants of neurogenesis. Therefore we compared the levels of basal and BDNF-induced neurogenesis in the dentate gyrus of WT and Y365/7F mice using Dcx staining. As illustrated in Figure 9A, there appeared to be a considerable increase in the number of Dcx-positive cells throughout the dentate gyrus of Y365/7F mice, compared with WT littermate controls. To quantify this, we costained hippocampal sections with Dcx and the nuclear marker DAPI to determine the percentage of Dcx-positive cells in hippocampal sections from WT and Y365/7F mice that had been treated with either vehicle or BDNF by infusion into the cerebral ventricles as above (Fig. 9B). We found a significant increase in the percentage of Dcx-positive cells in Y365/7F mice compared with WT controls treated with vehicle (Fig. 9C; WT vehicle: 8.82 ± 1.56; Y365/7F vehicle: 22.46 ± 2.31; p < 0.001, n = 8 mice). BDNF infusion increased the percentage of Dcx-positive cells in WT mice compared with vehicle (Fig. 9C; WT vehicle: 8.82 ± 1.56; WT BDNF: 20.02 ± 2.42; p < 0.001, n = 8 mice), but failed to produce a further increase in the percentage of Dcx-positive cells in the Y365/7F mutants (Fig. 9C). Similarly, stereological examination of the total number of Dcx-positive cells in the GCL revealed a significant increase in Y365/7F mice compared with WT controls treated with vehicle (Fig. 9C; WT vehicle: 3421.57 ± 642.49; Y365/7F vehicle: 7826.32 ± 1495.80; p = 0.016, n = 8 mice). BDNF infusion resulted in a significant increase in the total number of Dcx-positive cells in the GCL in WT mice (Fig. 9C; WT vehicle: 3421.57 ± 642.49; WT BDNF: 7011.06 ± 1375.17; p = 0.045, n = 8, mice), but failed to produce a further increase in the total Dcx-positive cells in the GCL of Y365/7F mutants (Fig. 9C).
Figure 9.
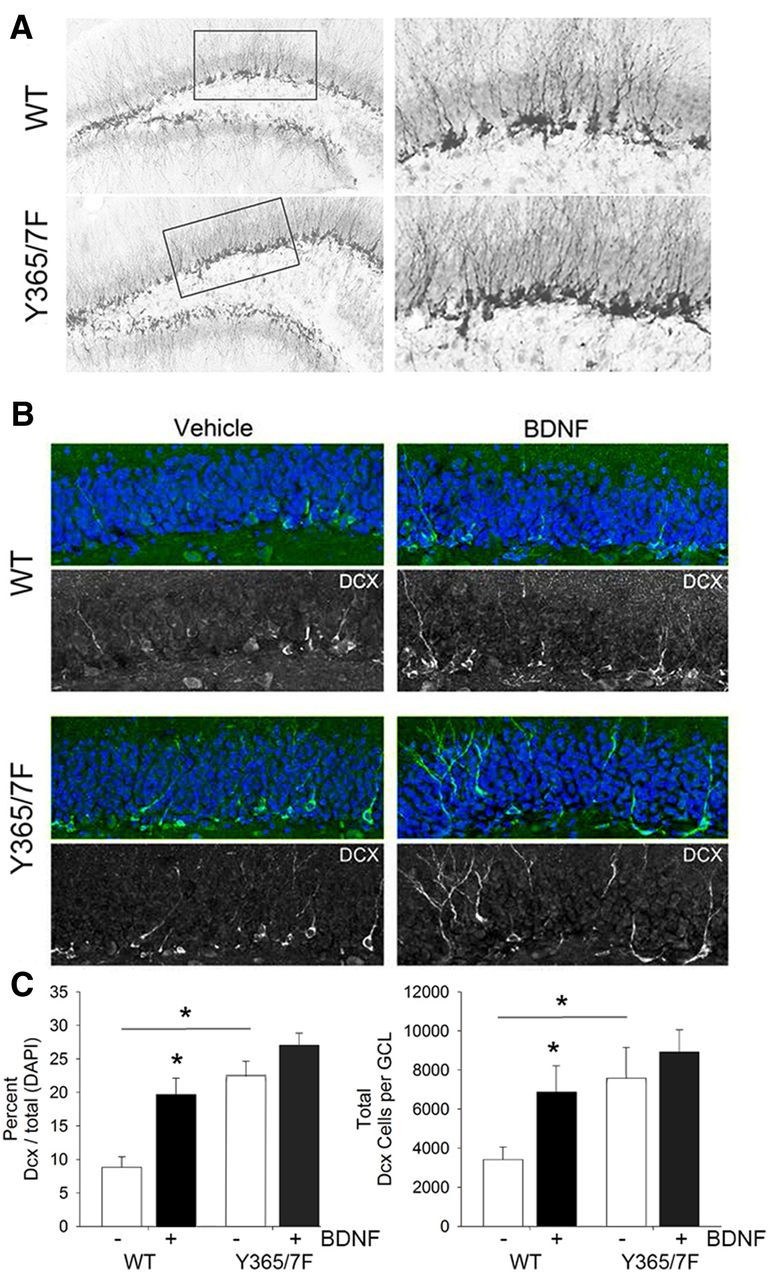
Basal hippocampal neurogenesis is elevated and insensitive to further modulation by BDNF in Y365/7F mice. A, Representative images of immunohistochemistry for Dcx in the dentate gyrus of the hippocampus in WT and Y365/7F mice. B, Representative images of immunofluorescent staining of Dcx in vehicle and BDNF-treated WT and Y365/F mice. C, Quantification of the percentage of Dcx-positive cells divided by the total DAPI-labeled nuclei (left) and the total number of Dcx-positive cells in the GCL (right).
This experiment reveals Y365/7F mutation leads to an enhancement in hippocampal neurogenesis that cannot be further modulated by BDNF. Therefore, in correlation with mediating the antidepressant efficacy of BDNF, Y365/7 are also key determinants of BDNF effects on neurogenesis.
Discussion
BDNF plays a central role in regulating neuronal activity and modifications in BDNF-dependent signaling are strongly implicated in a range of pathologies, including depression (Huang and Reichardt, 2001; Waterhouse and Xu, 2009). To determine how BDNF exerts its effects on neuronal activity, we examined its effects on synaptic inhibition mediated by γ2 subunit-containing GABAARs, deficits of which are also believed to contribute to depression. Our initial studies focused on the PFC, an area of the brain critical in regulating mood in humans and rodents.
Sustained treatment of PFC slices with BDNF increased the phosphorylation of Y365/7 in the γ2 subunit, previously identified substrates of Fyn and other Src-like kinases, which are intimately associated with GABAARs (Brandon et al., 2001; Jurd et al., 2010) and accepted downstream effectors of BDNF (Lu, 2003; Numakawa et al., 2010). In parallel, increased cell-surface expression of γ2 subunit-containing GABAARs was also evident following BDNF treatment. Next, using mice heterozygous for the Y365/7F mutation, we determined whether the effects of BDNF on cell-surface accumulation of the GABAAR γ2 subunit are directly dependent upon Y365/7. These animals exhibited enhanced cell-surface levels of the γ2 subunit in the PFC, an effect that was insensitive to further potentiation by BDNF. To independently corroborate these studies, we used mice with reduced levels of BDNF expression in the forebrain (Rios et al., 2006). These animals showed reduced cell-surface expression of the GABAAR γ2 subunit in the PFC that was accompanied by decreased Y365/7 phosphorylation. Therefore BDNF acts to enhance the cell-surface stability of GABAARs by stimulating the phosphorylation of Y365/7 in the receptor γ2 subunit to slow their endocytosis. This is consistent with our studies in cultured neurons, which have revealed that BDNF acts to increase the cell-surface accumulation of GABAARs by slowing their endocytosis (Jovanovic et al., 2004; Hewitt and Bains, 2006; Porcher et al., 2011).
BDNF-induced increase in GABAAR γ2 subunit phosphorylation and subsequent cell-surface accumulation was accompanied by an enhancement in the amplitude and frequency of mIPSCs in PFC neurons, events that principally arise from the activation of γ2 subunit-containing GABAARs (Essrich et al., 1998; Jacob et al., 2008; Luscher et al., 2011b). Thus BDNF induces an increase in the functional expression of γ2 subunit-containing GABAAR subtypes. Y365/7 within the γ2 subunit play a critical role in regulating GABAAR endocytosis as they mediate nanomolar affinity binding to AP2, which is ablated upon their phosphorylation and/or their mutation to phenylanine residues (Kittler et al., 2008). Significantly, the ability of BDNF to potentiate mIPSC amplitude was prevented by a peptide that abolishes the binding of the GABAAR γ2 subunit to AP2. To assess whether the effects of BDNF on mIPSC amplitude are directly dependent on Y365/7, we compared mIPSC properties in WT and Y365/F knock-in mice. Consistent with increased cell-surface accumulation of γ2 subunit-containing GABAARs, mIPSC amplitude, but not frequency, was increased in Y365/7F mice. To determine whether this increase in mIPSC amplitude affects the efficacy of synaptic inhibition, input–output relationships for eIPSCs were compared between genotypes. This revealed a significant increase in inhibitory drive in Y365/7F mice, which correlated with a reduction in the ACSF-induced increase in sAPs of the PFC. Interestingly, while BDNF had minimal efficacy on the ACSF-induced increase in sAPs of the PFC in WT mice, BDNF significantly increased this in the Y365/7F mutants. Presumably, this enhanced effect of BDNF reflects modified activity and/or synchronization of cortical circuits in the mutant mice due to enhanced synaptic inhibition.
Collectively our biochemical and electrophysiological studies suggest that BDNF acts to increase mIPSC amplitude by stimulating phosphorylation of Y365/7 in the GABAAR γ2 and thereby enhancing the cell-surface/synaptic accumulation of these receptor subtypes. This mechanism appears to be a key determinant for the excitability of the PFC.
In previous studies, BDNF has been shown to potentiate and/or reduce mIPSC amplitude (Brünig et al., 2001; Cheng and Yeh, 2003; Mizoguchi et al., 2003; Wardle and Poo, 2003; Jovanovic et al., 2004; Hewitt and Bains, 2006; Pattwell et al., 2012). In the majority of these papers, the effects of short-term application of BDNF (5–10 min, 50–100 ng/ml) on mIPSC properties were measured in cultured neurons (4–15 DIV cortical, cerebellar, or hippocampal), or in brain slices from immature mice/rats. In contrast, we assessed the effects of treating cortical slices from 2–4-month-old mice with BDNF for 2 h on mIPSC and IPSCs. Using this protocol, we establish that the ability of BDNF to increase mIPSC and IPSC amplitude, but not their frequency, is critically dependent on Y365/7 in the γ2 subunit. The reasons for the discrepancies between our results and those that show reductions in mIPSC amplitude may reside in the time of exposure to BDNF (5–10 min vs 2 h). Alternatively they may reflect differences in BDNF signaling and/or GABAAR structure between cultured neurons/immature brain slices compared with those seen in cortical slices from adult mice. Finally while PFC neurons in 2–3-month-old slices fired sAPs, their excitability was not enhanced by BDNF. In contrast, BDNF increases the excitability of cultured neurons (Desai et al., 1999). Presumably these differing effects of BDNF reflect distinctions in the intrinsic properties of neurons in culture and those in mature brain slices.
Modifications in the expression levels of γ2 subunit-containing GABAARs are correlated with a depression-like phenotype (Earnheart et al., 2007; Luscher et al., 2011a). Using the FST and TST, we assessed whether the enhanced functional expression of these receptors on the plasma membrane in the PFC of Y365/7F mice affects depressive-like behaviors. Y365/7F mice had significant reductions in immobility in both the FST and TST compared with WT controls, suggesting an antidepressant-like phenotype. In contrast, mutation of Y365/7 did not affect their behavior in the OFT, consistent with published studies revealing that male Y365/7F mice do not exhibit any gross behavioral deficits with regards to anxiolysis or motor coordination (Tretter et al., 2009). From this study, we can conclude that increasing the number of functional γ2 subunit-containing GABAAR subtypes on the cell surface in the PFC leads to a reduction in depressive-like behavior. Collectively, our results, together with those of Earnheart and colleagues (Earnheart et al., 2007), suggest that the expression levels of γ2 subunit-containing GABAARs and the efficacy of fast synaptic inhibition are critical determinants of depressive-like behaviors.
We also tested the role that BDNF-dependent phosphorylation of the GABAAR γ2 subunit at Y365/7 plays in mediating the antidepressant efficacy of this neurotrophin. BDNF induced a significant reduction in depressive-like behaviors in WT mice, but was without effect in Y365/7F mutants under the same conditions. The mechanism by which BDNF exerts its antidepressant activity is controversial but its ability to potentiate hippocampal neurogenesis is believed to be of significance (Scharfman et al., 2005). In addition, the expression levels of γ2 subunit-containing GABAARs have also shown to be of significance in determining hippocampal neurogenesis (Earnheart et al., 2007). Therefore, we examined basal and BDNF-dependent hippocampal neurogenesis in Y365/7F knock-in mice. Under basal conditions, the level of hippocampal neurogenesis was significantly increased in Y365/7F mice compared with WT controls. While BDNF produced a significant increase in hippocampal neurogenesis in WT mice, this effect was lost in the Y365/7F mutants. Thus Y365/7 are key determinants in mediating the antidepressant efficacy of BDNF and its ability to potentiate neurogenesis.
In summary, our correlated results reveal a critical role for BDNF-dependent phosphorylation of Y365/7 in the GABAAR γ2 subunit in determining the efficacy of synaptic inhibition, the activity of the PFC, and the expression of depressive-like behaviors. Moreover, Y365/7 are among the first molecular substrates linking the antidepressant efficacy of BDNF with its ability to potentiate hippocampal neurogenesis.
Footnotes
S.J.M. was supported by National Institutes of Health (NIH) Grants NS051195, NS056359, NS081735, and MH097446, and grants from Citizens United for Research in Epilepsy (PV0178) and the Simons Foundation (PV0138). Z.Y. was supported by NIH Grants MH084233 and MH085774. P.G.H. was supported by NIH Grants MH095385 and NS037585. M.R. was supported by NIH Grant DK073311. R.M.H. was supported by a Canadian Institutes of Health Research postdoctoral fellowship. M.T. received a National Scientist Development Award from the American Heart Association. D.J.H. was supported by a Heart and Stroke Foundation of Canada postdoctoral fellowship.
S.J.M. serves as a consultant for Sage Therapeutics and Astrazeneca, relationships that are regulated by Tufts University and do not impact on this study.
References
- Bardoni R, Ghirri A, Salio C, Prandini M, Merighi A. BDNF-mediated modulation of GABA and glycine release in dorsal horn lamina II from postnatal rats. Dev Neurobiol. 2007;67:960–975. doi: 10.1002/dneu.20401. [DOI] [PubMed] [Google Scholar]
- Brandon NJ, Delmas P, Hill J, Smart TG, Moss SJ. Constitutive tyrosine phosphorylation of the GABA(A) receptor gamma 2 subunit in rat brain. Neuropharmacology. 2001;41:745–752. doi: 10.1016/S0028-3908(01)00121-6. [DOI] [PubMed] [Google Scholar]
- Brown SD, Chambon P, de Angelis MH, de Angelis MH. EMPReSS: standardized phenotype screens for functional annotation of the mouse genome. Nat Genet. 2005;37:1155. doi: 10.1038/ng1105-1155. [DOI] [PubMed] [Google Scholar]
- Brünig I, Penschuck S, Berninger B, Benson J, Fritschy JM. BDNF reduces miniature inhibitory postsynaptic currents by rapid downregulation of GABA(A) receptor surface expression. Eur J Neurosci. 2001;13:1320–1328. doi: 10.1046/j.0953-816x.2001.01506.x. [DOI] [PubMed] [Google Scholar]
- Chen G, Kittler JT, Moss SJ, Yan Z. Dopamine D3 receptors regulate GABAA receptor function through a phospho-dependent endocytosis mechanism in nucleus accumbens. J Neurosci. 2006;26:2513–2521. doi: 10.1523/JNEUROSCI.4712-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Yeh HH. Brain-derived neurotrophic factor attenuates mouse cerebellar granule cell GABAA receptor-mediated responses via postsynaptic mechanisms. J Physiol. 2003;548:711–721. doi: 10.1113/jphysiol.2002.037846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai NS, Rutherford LC, Turrigiano GG. BDNF regulates the intrinsic excitability of cortical neurons. Learn Mem. 1999;6:284–291. [PMC free article] [PubMed] [Google Scholar]
- Earnheart JC, Schweizer C, Crestani F, Iwasato T, Itohara S, Mohler H, Lüscher B. GABAergic control of adult hippocampal neurogenesis in relation to behavior indicative of trait anxiety and depression states. J Neurosci. 2007;27:3845–3854. doi: 10.1523/JNEUROSCI.3609-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Essrich C, Lorez M, Benson JA, Fritschy JM, Lüscher B. Postsynaptic clustering of major GABAA receptor subtypes requires the gamma 2 subunit and gephyrin. Nat Neurosci. 1998;1:563–571. doi: 10.1038/2798. [DOI] [PubMed] [Google Scholar]
- Feighner JP, Aden GC, Fabre LF, Rickels K, Smith WT. Comparison of alprazolam, imipramine, and placebo in the treatment of depression. JAMA. 1983;249:3057–3064. doi: 10.1001/jama.1983.03330460039029. [DOI] [PubMed] [Google Scholar]
- Hewitt SA, Bains JS. Brain-derived neurotrophic factor silences GABA synapses onto hypothalamic neuroendocrine cells through a postsynaptic dynamin-mediated mechanism. J Neurophysiol. 2006;95:2193–2198. doi: 10.1152/jn.01135.2005. [DOI] [PubMed] [Google Scholar]
- Hoshaw BA, Malberg JE, Lucki I. Central administration of IGF-I and BDNF leads to long-lasting antidepressant-like effects. Brain Res. 2005;1037:204–208. doi: 10.1016/j.brainres.2005.01.007. [DOI] [PubMed] [Google Scholar]
- Huang EJ, Reichardt LF. Neurotrophins: roles in neuronal development and function. Annu Rev Neurosci. 2001;24:677–736. doi: 10.1146/annurev.neuro.24.1.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob TC, Moss SJ, Jurd R. GABA(A) receptor trafficking and its role in the dynamic modulation of neuronal inhibition. Nat Rev Neurosci. 2008;9:331–343. doi: 10.1038/nrn2370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Thomas P, Kittler JT, Smart TG, Moss SJ. Brain-derived neurotrophic factor modulates fast synaptic inhibition by regulating GABAA receptor phosphorylation, activity, and cell-surface stability. J Neurosci. 2004;24:522–530. doi: 10.1523/JNEUROSCI.3606-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurd R, Tretter V, Walker J, Brandon NJ, Moss SJ. Fyn kinase contributes to tyrosine phosphorylation of the GABA(A) receptor gamma2 subunit. Mol Cell Neurosci. 2010;44:129–134. doi: 10.1016/j.mcn.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalueff AV, Nutt DJ. Role of GABA in anxiety and depression. Depress Anxiety. 2007;24:495–517. doi: 10.1002/da.20262. [DOI] [PubMed] [Google Scholar]
- Kittler JT, Chen G, Kukhtina V, Vahedi-Faridi A, Gu Z, Tretter V, Smith KR, McAinsh K, Arancibia-Carcamo IL, Saenger W, Haucke V, Yan Z, Moss SJ. Regulation of synaptic inhibition by phospho-dependent binding of the AP2 complex to a YECL motif in the GABAA receptor gamma2 subunit. Proc Natl Acad Sci U S A. 2008;105:3616–3621. doi: 10.1073/pnas.0707920105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klumpers UM, Veltman DJ, Drent ML, Boellaard R, Comans EF, Meynen G, Lammertsma AA, Hoogendijk WJ. Reduced parahippocampal and lateral temporal GABAA-[11C]flumazenil binding in major depression: preliminary results. Eur J Nucl Med Mol Imaging. 2010;37:565–574. doi: 10.1007/s00259-009-1292-9. [DOI] [PubMed] [Google Scholar]
- Lu B. BDNF and activity-dependent synaptic modulation. Learn Mem. 2003;10:86–98. doi: 10.1101/lm.54603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher B, Shen Q, Sahir N. The GABAergic deficit hypothesis of major depressive disorder. Mol Psychiatry. 2011a;16:383–406. doi: 10.1038/mp.2010.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luscher B, Fuchs T, Kilpatrick CL. GABAA receptor trafficking-mediated plasticity of inhibitory synapses. Neuron. 2011b;70:385–409. doi: 10.1016/j.neuron.2011.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Turrigiano GG. Multiple modes of network homeostasis in visual cortical layer 2/3. J Neurosci. 2008;28:4377–4384. doi: 10.1523/JNEUROSCI.5298-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffei A, Nelson SB, Turrigiano GG. Selective reconfiguration of layer 4 visual cortical circuitry by visual deprivation. Nat Neurosci. 2004;7:1353–1359. doi: 10.1038/nn1351. [DOI] [PubMed] [Google Scholar]
- Maguire J, Mody I. GABA(A)R plasticity during pregnancy: relevance to postpartum depression. Neuron. 2008;59:207–213. doi: 10.1016/j.neuron.2008.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizoguchi Y, Kanematsu T, Hirata M, Nabekura J. A rapid increase in the total number of cell surface functional GABAA receptors induced by brain-derived neurotrophic factor in rat visual cortex. J Biol Chem. 2003;278:44097–44102. doi: 10.1074/jbc.M305872200. [DOI] [PubMed] [Google Scholar]
- Numakawa T, Suzuki S, Kumamaru E, Adachi N, Richards M, Kunugi H. BDNF function and intracellular signaling in neurons. Histol Histopathol. 2010;25:237–258. doi: 10.14670/HH-25.237. [DOI] [PubMed] [Google Scholar]
- Pattwell SS, Bath KG, Perez-Castro R, Lee FS, Chao MV, Ninan I. The BDNF Val66Met polymorphism impairs synaptic transmission and plasticity in the infralimbic medial prefrontal cortex. J Neurosci. 2012;32:2410–2421. doi: 10.1523/JNEUROSCI.5205-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. San Diego: Academic; 2001. [Google Scholar]
- Petty F, Trivedi MH, Fulton M, Rush AJ. Benzodiazepines as antidepressants: does GABA play a role in depression? Biol Psychiatry. 1995;38:578–591. doi: 10.1016/0006-3223(95)00049-7. [DOI] [PubMed] [Google Scholar]
- Porcher C, Hatchett C, Longbottom RE, McAinch K, Sihra TS, Moss SJ, Thomson AM, Jovanovic JN. Positive feedback regulation between gamma-aminobutyric acid type A (GABA(A)) receptor signaling and brain-derived neurotrophic factor (BDNF) release in developing neurons. J Biol Chem. 2011;286:21667–21677. doi: 10.1074/jbc.M110.201582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rios M, Lambe EK, Liu R, Teillon S, Liu J, Akbarian S, Roffler-Tarlov S, Jaenisch R, Aghajanian GK. Severe deficits in 5-HT2A-mediated neurotransmission in BDNF conditional mutant mice. J Neurobiol. 2006;66:408–420. doi: 10.1002/neu.20233. [DOI] [PubMed] [Google Scholar]
- Sanchez-Vives MV, McCormick DA. Cellular and network mechanisms of rhythmic recurrent activity in neocortex. Nat Neurosci. 2000;3:1027–1034. doi: 10.1038/79848. [DOI] [PubMed] [Google Scholar]
- Scharfman H, Goodman J, Macleod A, Phani S, Antonelli C, Croll S. Increased neurogenesis and the ectopic granule cells after intrahippocampal BDNF infusion in adult rats. Exp Neurol. 2005;192:348–356. doi: 10.1016/j.expneurol.2004.11.016. [DOI] [PubMed] [Google Scholar]
- Sieghart W. Structure, pharmacology, and function of GABAA receptor subtypes. Adv Pharmacol. 2006;54:231–263. doi: 10.1016/S1054-3589(06)54010-4. [DOI] [PubMed] [Google Scholar]
- Song J, Zhong C, Bonaguidi MA, Sun GJ, Hsu D, Gu Y, Meletis K, Huang ZJ, Ge S, Enikolopov G, Deisseroth K, Luscher B, Christian KM, Ming GL, Song H. Neuronal circuitry mechanism regulating adult quiescent neural stem-cell fate decision. Nature. 2012;489:150–154. doi: 10.1038/nature11306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svenningsson P, Tzavara ET, Liu F, Fienberg AA, Nomikos GG, Greengard P. DARPP-32 mediates serotonergic neurotransmission in the forebrain. Proc Natl Acad Sci U S A. 2002;99:3188–3193. doi: 10.1073/pnas.052712699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terunuma M, Xu J, Vithlani M, Sieghart W, Kittler J, Pangalos M, Haydon PG, Coulter DA, Moss SJ. Deficits in phosphorylation of GABAA receptors by intimately associated protein kinase C activity underlie compromised synaptic inhibition during status epilepticus. J Neurosci. 2008;28:376–384. doi: 10.1523/JNEUROSCI.4346-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tretter V, Revilla-Sanchez R, Houston C, Terunuma M, Havekes R, Florian C, Jurd R, Vithlani M, Michels G, Couve A, Sieghart W, Brandon N, Abel T, Smart TG, Moss SJ. Deficits in spatial memory correlate with modified {gamma}-aminobutyric acid type A receptor tyrosine phosphorylation in the hippocampus. Proc Natl Acad Sci U S A. 2009;106:20039–20044. doi: 10.1073/pnas.0908840106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vithlani M, Terunuma M, Moss SJ. The dynamic modulation of GABA(A) receptor trafficking and its role in regulating the plasticity of inhibitory synapses. Physiol Rev. 2011;91:1009–1022. doi: 10.1152/physrev.00015.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle RA, Poo MM. Brain-derived neurotrophic factor modulation of GABAergic synapses by postsynaptic regulation of chloride transport. J Neurosci. 2003;23:8722–8732. doi: 10.1523/JNEUROSCI.23-25-08722.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterhouse EG, Xu B. New insights into the role of brain-derived neurotrophic factor in synaptic plasticity. Mol Cell Neurosci. 2009;42:81–89. doi: 10.1016/j.mcn.2009.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuen EY, Liu W, Karatsoreos IN, Ren Y, Feng J, McEwen BS, Yan Z. Mechanisms for acute stress-induced enhancement of glutamatergic transmission and working memory. Mol Psychiatry. 2011;16:156–170. doi: 10.1038/mp.2010.50. [DOI] [PMC free article] [PubMed] [Google Scholar]