

Abstract
A new generation of vaccines containing multiple protein components that aim to provide broad protection against serogroup B meningococci has been developed. One candidate, 4CMenB (4 Component MenB), has been approved by the European Medicines Agency, but is predicted to provide at most 70–80 % strain coverage; hence there is a need for second-generation vaccines that achieve higher levels of coverage. Prior knowledge of the diversity of potential protein vaccine components is a key step in vaccine design. A number of iron import systems have been targeted in meningococcal vaccine development, including the HmbR and HpuAB outer-membrane proteins, which mediate the utilization of haemoglobin or haemoglobin–haptoglobin complexes as iron sources. While the genetic diversity of HmbR has been described, little is known of the diversity of HpuAB. Using whole genome sequences deposited in a Bacterial Isolate Genome Sequence Database (BIGSDB), the prevalence and diversity of HpuAB among Neisseria were investigated. HpuAB was widely present in a range of Neisseria species whereas HmbR was mainly limited to the pathogenic species Neisseria meningitidis and Neisseria gonorrhoeae. Patterns of sequence variation in sequences from HpuAB proteins were suggestive of recombination and diversifying selection consistent with strong immune selection. HpuAB was subject to repeat-mediated phase variation in pathogenic Neisseria and the closely related non-pathogenic Neisseria species Neisseria lactamica and Neisseria polysaccharea but not in the majority of other commensal Neisseria species. These findings are consistent with HpuAB being subject to frequent genetic transfer potentially limiting the efficacy of this receptor as a vaccine candidate.
Introduction
Neisseria meningitidis, the meningococcus, continues to cause meningitis and septicaemia worldwide with a high mortality rate and frequent debilitating sequelae. Meningococci are, however, normally commensal residents of the oropharynx of a high percentage (10–30 %) of the human population and can persist asymptomatically in the carrier state for periods of months to years (Caugant & Maiden, 2009). The closely related gonococcus, Neisseria gonorrhoeae, on the other hand, causes a widespread sexually transmitted disease, involving colonization and invasion of the mucosal surfaces of the genital tract with occasional ascension that leads to serious sequelae such as ectopic pregnancy, sterility and disseminated infection (Criss & Seifert, 2012). The gonococcus can also persist in asymptomatic carriers. Other Neisseria species are also localized to the upper respiratory tracts of humans, and related organisms occur in other vertebrates. Some of these have occasionally been associated with disease but they are essentially non-pathogenic. For example, Neisseria lactamica is a commensal frequently isolated in the upper respiratory tracts of young children but is very rarely associated with pathology (Bennett et al., 2012).
The most effective N. meningitidis vaccines are composed of capsular polysaccharide antigens conjugated to a carrier protein and these have been successfully used to induce protective immunity against N. meningitidis serogroups A, C, W and Y (Pollard, 2004). No such vaccine is available against serogroup B N. meningitidis due to similarities between the serogroup B polysaccharide and human glycoprotein structures, and no vaccines targeting N. gonorrhoeae isolates are available. Protein-based vaccines have been developed to protect against N. meningitidis strains expressing serogroup B capsules and these include a range of formulations such as 4CMenB (4 Component MenB, Bexsero®; Novartis Vaccines and Diagnostics) (Serruto et al., 2012) and rLP2086 (Pfizer) (Richmond et al., 2012); however, due to incomplete vaccine coverage, there is a requirement for second-generation vaccines which achieve higher levels of coverage by the inclusion of more variants of existing components and/or the inclusion of additional components (Lucidarme et al., 2010; Vogel et al., 2013).
Surface receptors that mediate iron acquisition are attractive potential vaccine components as they are essential for growth in the iron-limited conditions prevailing in host tissues. Meningococci express five such receptors: (i) the transferrin binding proteins TbpA and TbpB; (ii) the lactoferrin binding proteins LbpA and LbpB; (iii) the FrpB/FetA transporter; (iv) the haemoglobin receptor HmbR; and (v) the haptoglobin–haemoglobin receptors HpuAB (Perkins-Balding et al., 2004; Saleem et al., 2013). Transferrin and lactoferrin receptors are outer-membrane proteins (OMPs) involved in acquisition of iron from transferrin and lactoferrin and are almost universally present in meningococci and gonococci. Infection of human volunteers with gonococcal mutants has indicated a requirement for these receptors for efficient colonization of the male urethra (Cornelissen et al., 1998) and the recent publication of the TbpA and TbpB protein structures is prompting some renewed interest in these proteins as vaccine candidates (Calmettes et al., 2012; Noinaj et al., 2012b). HmbR is a transmembrane protein with specificity for haemoglobin, whilst the bipartite receptor HpuAB can bind both haemoglobin and haptoglobin–haemoglobin complexes (Lewis et al., 1999; Stojiljkovic et al., 1995).
The acquisition of iron from haemoglobin, lactoferrin and transferrin in Neisseria is mediated by receptors composed of two distinct surface-exposed OMPs with very different properties and specificities (Cornelissen & Hollander, 2011; Noinaj et al., 2012a). The first protein is TonB-dependent and functions as a pore through which the iron or haem is directly transported. The second is a fully surface-exposed lipoprotein which is thought to act as an accessory protein enhancing the specificity and strength of ligand binding. In the HpuAB system, HpuB is the TonB-dependent OMP acting as a pore with HpuA being the fully surface-exposed lipoprotein.
HmbR and HpuAB receptors are variably present in Neisseria isolates with four combinations of the receptors observed: (i) both receptors; (ii) HmbR only; (iii) HpuAB only; and (iv) neither receptor present (Tauseef et al., 2011). Among meningococci associated with disease, there is a significant over-representation of the presence of HmbR and an under-representation of an HpuAB only phenotype. Further there is a high prevalence of both receptors in clonal complexes frequently associated with invasion relative to their carriage prevalence (Harrison et al., 2009). Both of these receptors are subject to phase variation due to alterations in poly G repeat tracts present within the reading frame, with a high frequency of expression observed in meningococci associated with disease (Tauseef et al., 2011). These findings suggest that Tbp and Lbp are required for colonization of mucosal surfaces but that the haemoglobin receptors may facilitate invasion and dissemination in the vascular system.
While the molecular evolution of the HmbR protein has been investigated in meningococci (Evans et al., 2010), little is known about the diversity and selection pressures acting on the HpuAB OMPs and thus the suitability of this receptor as a vaccine candidate has not been explored. This study showed that HpuAB was present in a range of Neisseria species and that HmbR was mainly found in the pathogenic species Neisseria meningitidis and Neisseria gonorrhoeae. Two HpuAB families were evident: one present in non-pathogenic Neisseria species and having a non-phase variable hpuA gene and the other only observed in N. meningitidis, N. gonorrhoeae, Neisseria lactamica and Neisseria polysaccharea. Analysis of selection and recombination acting on HpuAB sequences revealed a number of residues were subject to immune selection and putatively located in surface-exposed regions of the protein. These findings were consistent with HpuAB being part of the accessory genome and subject to frequent exchange of allelic sequences possibly reducing the utility of this receptor as a vaccine antigen. Furthermore, the prevalence of these genes in the closely related non-pathogenic Neisseria population indicates that a vaccine targeting these antigens might have an effect on the commensal microbiota with implications for natural immunity.
Methods
Neisseria isolates.
A total of 218 isolates were investigated. These included the 107 N. meningitidis isolates employed in the evaluation of the MLST method (Maiden et al., 1998) (Table S1, available in Microbiology Online), additional N. meningitidis carriage isolates with distinct serogroups (L, H, I and K) (Harrison et al., 2013) along with N. meningitidis isolates obtained during a well-characterized disease outbreak in 1997 in Southampton, UK (Jolley et al., 2012) and several serogroup B isolates for which whole genome sequences were available (Budroni et al., 2011) (Table S1).
A total of 16 N. gonorrhoeae isolates, the genomes of which had been sequenced by the Broad Institute, were included (Neisseria gonorrhoeae group Sequencing Project, Broad Institute of Harvard and MIT (http://www.broadinstitute.org)), along with 81 commensal Neisseria obtained from a number of sources including: the Culture Collection of the University of Goteborg (CCUG); the American type Culture Collection (ATCC); and a study of asymptomatic carriers in Oxfordshire (Bennett et al., 2005) (Table S1). Species definitions were those based on ribosomal multi locus sequence typing (rMLST) (Bennett et al., 2012). Whole genome sequence data are available on the PubMLST website (www.pubmlst.org/neisseria).
Next generation sequencing and annotation of sequence data.
Sequencing, assembly, uploading and annotation of genomic sequences were performed as described previously (Bennett et al., 2012). Briefly, genomic DNA was prepared following overnight growth on Columbia horse blood agar plates (Oxoid) using a Wizard Genomic DNA Purification kit (Promega). Pooled libraries of sheared genomic DNA were subject to paired end sequencing on an Illumina Genome Analyser II platform. Genome sequence data were assembled using velvet version 1.0.10 with optimal parameters determined by the velvetoptimizer.pl script within the software package (Zerbino, 2010) with the resultant contigs uploaded into a Bacterial Isolate Genome Sequence Database (BIGSDB) along with any available provenance data. Genome sequence data from other studies were uploaded directly to the database (Table S1).
Sequence definitions were generated for hpuA, hpuB, and hmbR genes in the sequence definition database and seeded with corresponding reference nucleotide sequences. Iterative searches of genome sequence data using progressively decreasing stringency settings by means of blastn or tblastx algorithms (Altschul et al., 1997) identified hpuA, hpuB and hmbR genes which were then selected in the genomes enabling them to be extracted and exported for further analysis. Arbitrary allele numbers were assigned to each unique sequence for a given locus. Gene sequences were exported as XMFA files containing aligned sequence blocks and then converted to a fasta format for import into mega version 5.0 and Splitstree (Huson & Bryant, 2006; Tamura et al., 2011). In addition, genetic diversity arising from the slipped-strand mispairing region encoded by the polyG tract was removed in hpuA sequences. All hpuA and hpuB sequences are available through the PubMLST database (www.pubmlst.org/neisseria). In addition to the common gene name, these loci are assigned a value-free nomenclature (NEIS1946 and NEIS1947) following on from the FAM18 genome annotation but using the prefix NEIS instead of NMC.
Analysis of gene sequences.
Isolates containing complete nucleotide sequences for both hpuA and hpuB were selected for phylogenetic analyses (n = 151). mega version 5.0 was used to calculate overall p-distances of hpuA and hpuB nucleotide sequences as well as p-distance values within Family A or B genes or within each specific region from the putative HpuB structural topological model. neighbournet trees were constructed using Splitstree version 4.10 (Huson & Bryant, 2006). The characterization of selection in the presence of recombination was carried out using the omegamap software package (Wilson & McVean, 2006), which employs a Bayesian approach to parameter estimation that is independent of phylogeny and was therefore less likely to falsely identify sites subject to diversifying selection in sequences displaying clear evidence of recombination. The signature of natural selection was detected using the dN/dS ratio and the signature of recombination was detected from the patterns of linkage disequilibrium. In the present study, three runs composed of 100 000 iterations and 100 000 burn-ins each were undertaken, compared to assess convergence and combined. Output from the omegamap runs was used to visualize possible selection acting on the sequence by means of a graph indicating the posterior probability of positive selection along the sequence and using the statistics package R version 2.15.1 (http://www.r-project.org).
Generation of a structural topological model for HpuB.
Amino acid sequence alignments of HpuA and HpuB with TbpB or TbpA respectively, for which crystal structures have been published, did not reveal significant sequence identities. A structural model for HpuB was generated using swiss-model (Arnold et al., 2006) and the three-dimensional structure of the Shigella dysenteriae ShuA protein as a guide (Protein Data Bank PDB accession number 3FHH) (Cobessi et al., 2010). Even using this model, the overall sequence identity was 16 %; several loop regions were excised from the final model as they had low homology with the equivalent sequences in ShuA and were too long to be modelled effectively.
Results
Distribution of haemoglobin receptor genes in pathogenic and non-pathogenic Neisseria
The majority (88 %) of the N. meningitidis isolates examined contained hmbR genes with 37 % having hmbR alone whilst only 12 % had hpu alone (Table 1). The hmbR only genotype was particularly noticeable among serogroup B meningococci belonging to clonal complexes ST-18, ST-32 and ST-41/44 (Tauseef et al., 2011). All of the N. gonorrhoeae isolates contained hpuAB and hmbR genes, but the latter were all pseudogenes.
Table 1. Distribution of haemoglobin receptors in Neisseria species.
| Species* | Both | HpuAB only | HmbR only | Neither |
| N. meningitidis (n = 120) | 62 | 14 | 44 | 0 |
| N. gonorrhoeae (n = 16) | 16‡ | 0 | 0 | 0 |
| N. polysaccharea (n = 5) | 2‡ | 3 | 0 | 0 |
| N. ‘bergeri’ (n = 1) | 1 | 0 | 0 | 0 |
| N. lactamica (n = 22) | 0 | 22 | 0 | 0 |
| N. cinerea (n = 7) | 0 | 7 | 0 | 0 |
| N. subflava (n = 16) | 0 | 15 | 1 | 0 |
| N. mucosa (n = 12) | 0 | 10 | 2 | 0 |
| N. mucosa var. heidelbergensis† (n = 4) | 0 | 1 | 3 | 0 |
| N. (unknown) (n = 2) | 0 | 1 | 0 | 1 |
| N. bacilliformis (n = 6) | 0 | 0 | 0 | 6 |
| N. elongata variants (n = 4) | 0 | 0 | 1 | 3 |
| N. animalis (n = 1) | 0 | 0 | 0 | 1 |
| N. canis (n = 1) | 0 | 0 | 0 | 1 |
| N. dentiae (n = 1) | 0 | 0 | 0 | 1 |
| N. weaveri (n = 1) | 0 | 0 | 0 | 1 |
| Total | 81 | 73 | 51 | 14 |
Species were defined based on rMLST designations (Bennett et al., 2012).
Also known as N. oralis.
hmbR pseudogene.
Non-pathogenic Neisseria isolates exhibited a high prevalence of a hpu only genotype (71 %) with the majority (>80 %) of N. lactamica, Neisseria cinerea, N. polysaccharea and Neisseria subflava isolates containing this genotype (Table 1 and Table S1). In contrast, only 3 % (3/93) and 8 % (7/93) of the non-pathogenic isolates had genes encoding either receptors or a hmbR only genotype, respectively. The majority (n = 5) of hmbR only isolates among non-pathogenic Neisseria were present in Neisseria mucosa, which exhibited a split between a hmbR only genotype (31 %) (three of which in Neisseria mucosa var. heidelbergensis, which is also known as Neisseria oralis (Berger, 1971; Wolfgang et al., 2013)) and a hpuAB only genotype (59 %) (Table S1). Many (14/93; 15 %) of the non-pathogenic species Neisseria bacilliformis and Neisseria elongata lacked both receptors (Table 1).
Patterns of Hpu diversity
The nucleotide sequences for hpuA genes ranged in size from 909 to 1017 bp (303–339 amino acids) while hpuB sequences were substantially larger ranging in size from 2418 to 2439 bp (806–813 amino acid residues). Comparison of the hpuAB sequences and phylogenetic reconstructions indicated the presence of two divergent families of HpuAB receptor: Family A occurred in most of the non-pathogenic Neisseria species; while Family B was found in the pathogenic meningococcus and gonococcus and the closely related organisms N. polysaccharea, Neisseria bergeri and N. lactamica (Bennett et al., 2012) (Figs 1 and 2, Table 2). However, hpuA and hpuB sequences belonging to N. subflava isolates CCUG 24841 and CCUG 24844 and N. cinerea CCUG 5746 also contained Family B sequences (Table S1).
Fig. 1.

NEIGHBOURNET phylogeny created from hpuA nucleotide sequences from 151 Neisseria isolates. Families A and B are depicted. Circles denote N. meningitidis isolates and are colour coded by clonal complex. N. gonorrhoeae isolates are represented by black squares. Squares also depict the non-pathogenic Neisseria species which are colour coded. Scale bar represents 1% nucleotide substitutions per site with a fit index of 96%.
Fig. 2.

NEIGHBOURNET phylogeny created from hpuB nucleotide sequences from 151 Neisseria isolates. Families A and B are depicted. Circles denote N. meningitidis isolates and are colour coded by clonal complex. N. gonorrhoeae isolates are represented by black squares. Squares also depict the non-pathogenic Neisseria species which are colour coded. Scale bar represents 1% nucleotide substitutions per site with a fit index of 97%.
Table 2. Sequence diversity of hpuA and hpuB in 151 Neisseria isolates.
| Parameter | hpuA (906–1017 bp) | hpuB (2415–2436 bp) | ||||
| All | Family A | Family B | All | Family A | Family B | |
| Nucleotide sequences | 151 | 31 | 120 | 151 | 31 | 120 |
| Nucleotide alleles | 68 | 26 | 42 | 73 | 26 | 59 |
| Variable nucleotide sites | 622 | 527 | 356 | 1001 | 923 | 426 |
| p-distance | 0.195 | 0.147 | 0.100 | 0.100 | 0.102 | 0.038 |
| Amino acid alleles | 61 | 22 | 40 | 130 | 26 | 51 |
| Variable amino acid sites | 224 | 184 | 149 | 290 | 257 | 135 |
Within Family A, sequences derived from different Neisseria species were present in multiple, divergent lineages and did not form distinct clusters. In Family B, N. gonorrhoeae hpuA and hpuB sequences formed distinct clusters whilst hpuA and hpuB sequences from N. lactamica, N. polysaccharea and N. subflava were more diverse (Figs 1 and 2).
The hpuA genes of the Family A isolates were similar in length with only four short indels among the sequences examined – two of which were found in only one isolate. The hpuA sequences of the Family B isolates were variable in length with a ~60 nt insertion present in some isolates and numerous indels of 3–6 nt. The hpuB sequences of both families exhibited high levels of sequence similarity to each other with few indels. Extension of the sequence analysis to flanking regions showed that Family A isolates diverged immediately after the termination codon of hpuB, comprising variations in the types and numbers of repetitive sequences, whereas the Family B isolates displayed high levels of similarity even in this intergenic region.
Family A sequences did not contain the poly G repeat tract in hpuA. Translation of HpuA was predicted to start from an identical sequence in all of these isolates (5′ATGAAAATCA). Analysis of the flanking sequences detected variation immediately upstream of this initiation codon. In addition, further upstream there was a gene encoding a hypothetical protein that was conserved among some of the non-pathogenic species and exhibited a low level of amino acid homology to NMB1971 of N. meningitidis strain MC58 (data not shown). By contrast, all of the Family B sequences contained a poly G repeat tract starting at nucleotide 57 after the ATG start codon with the exception of those from N. subflava isolates CCUG 24841 and CCUG 24844 and N. cinerea CCUG 5746. A second feature of the Family A isolates is that the hpuA coding sequences terminate 53–56 nt upstream of the initiation codon of hpuB whereas the gap is only ~33 nt in the Family B sequences. Thus, the intergenic sequence of Family A sequences is large enough to contain a promoter and indeed there is a conserved putative −10 promoter sequence (5′-ATAATCA) and a conserved, putative Shine–Dalgarno sequence (5′-AGGC) (Figs S1 and S2) indicating a potential for a differential pattern of expression for HpuB between these two families of isolates.
Some association of sequence variants with particular N. meningitidis clonal complexes was evident in the phylogenetic constructions generated from the sequences of hpuA and hpuB (Figs 1 and 2). For example, distinct sequence clusters were associated with the ST-11 and ST-8 isolates with discrete clusters separating the two ST-11 electrophoretic types ET-37 and ET-15 consistent with previous studies describing this clonal complex (Jolley et al., 2012). However, extensive heterogeneity was observed with evidence of lateral gene transfer among lineages.
Detection of immune selection in the HpuAB proteins
There was no evidence of immune or diversifying selection acting on Family A HpuAB sequences when analysed with the OMEGAMAP algorithm; however, posterior probability of positive selection (dN/dS) values >1 were detected in 74 and 51 residues in Family B HpuA and HpuB respectively (Fig. 3). Variation in the amino acid sequences encoded by these genes was localized to distinct regions of the proteins. Comparisons of the HpuA amino acid sequences from both non-pathogenic and pathogenic Neisseria, which ranged in length from 302 to 338 (excluding the repeat tract), detected a number of conserved residues throughout the protein, but no extensive regions of sequence identity. The majority of variation in the Family A isolates was located to two regions between residues 40–61 and 187–201 of HpuA. The variation was much more extensive in the Family B isolates with the initial region of variation extending from 16 to 85 (including an indel of 17–18 amino acids in some strains), from 177 to 195 and multiple regions in the final 125 amino acids of the protein. Comparisons of Family B HpuB amino acid sequences revealed that most of the diversity occurred between sites 217 to 267 and 599 to 629 (Table 3). These sites also contained residues with dN/dS values >1 and corresponded with the location of putative surface-exposed loops (Fig. 3).
Fig. 3.

Detection of the posterior probability of positive selection acting upon Family A and B HpuAB proteins using the OMEGAMAP algorithm. Codon positions are on the x-axis while the y-axis denotes the posterior probability of positive selection. A value close to 1 is indicative of positive selection. Putative surface-exposed loops belonging to HpuB Family B proteins are denoted by black lines; the numbers indicating the loop number.
Table 3. Diversity of HpuB regions.
| Region* | All | Family A† | Family B† |
| plug {177 aa} | 0.078 (51) | 0.072 (32) | 0.018 (25) |
| t1 {10} | 0.080 (2) | 0.070 (2) | 0.000 (0) |
| Loop 1 {4} | 0.096 (1) | 0.051 (0) | 0.001 (0) |
| t2 {11} | 0.080 (4) | 0.060 (0) | 0.030 (4) |
| i1 {3} | 0.180 (2) | 0.126 (0) | 0.018 (2) |
| t3 {12} | 0.060 (2) | 0.040 (0) | 0.010 (2) |
| Loop 2 {50} | 0.192 (33) | 0.084 (25) | 0.099 (23) |
| t4 {16} | 0.050 (3) | 0.100 (3) | 0.020 (0) |
| i2 {2} | 0.276 (1) | 0.120 (1) | 0.252 (2) |
| t5 {15} | 0.080 (2) | 0.060 (2) | 0.020 (0) |
| Loop 3 {23} | 0.173 (13) | 0.085 (10) | 0.058 (7) |
| t6 {15} | 0.100 (4) | 0.070 (4) | 0.020 (1) |
| i3 {8} | 0.098 (4) | 0.063 (4) | 0.004 (1) |
| t7 {14} | 0.106 (7) | 0.069 (6) | 0.033 (2) |
| Loop 4 {27} | 0.131 (11) | 0.104 (7) | 0.061 (7) |
| t8 {8} | 0.117 (6) | 0.192 (5) | 0.002 (1) |
| i4 {4} | 0.075 (2) | 0.077 (2) | 0.000 (0) |
| t9 {22} | 0.099 (8) | 0.057 (7) | 0.015 (1) |
| Loop 5 {31} | 0.149 (21) | 0.042 (8) | 0.043 (14) |
| t10 {16} | 0.098 (7) | 0.022 (2) | 0.011 (2) |
| i5 {4} | 0.150 (4) | 0.069 (4) | 0.000 (0) |
| t11 {14} | 0.079 (7) | 0.095 (6) | 0.007 (0) |
| Loop 6 {21} | 0.110 (11) | 0.134 (8) | 0.013 (5) |
| t12 {14} | 0.110 (5) | 0.063 (5) | 0.021 (1) |
| i6 {5} | 0.156 (3) | 0.105 (3) | 0.000 (0) |
| t13 {12} | 0.069 (2) | 0.101 (0) | 0.000 (1) |
| Loop 7 {26} | 0.104 (4) | 0.055 (1) | 0.080 (3) |
| t14 {8} | 0.104 (2) | 0.049 (1) | 0.064 (2) |
| i7 {4} | 0.131 (1) | 0.063 (1) | 0.096 (0) |
| t15 {23} | 0.110 (5) | 0.065 (5) | 0.078 (5) |
| Loop 8 {30} | 0.132 (9) | 0.090 (9) | 0.109 (15) |
| t16 {21} | 0.080 (5) | 0.030 (1) | 0.071 (4) |
| i8 {10} | 0.139 (2) | 0.044 (0) | 0.130 (2) |
| t17 {13} | 0.107 (4) | 0.074 (2) | 0.085 (3) |
| Loop 9 {16} | 0.129 (5) | 0.071 (3) | 0.104 (3) |
| t18 {9} | 0.119 (3) | 0.055 (1) | 0.082 (3) |
| i9 {5} | 0.071 (2) | 0.047 (1) | 0.066 (1) |
| t19 {10} | 0.101 (4) | 0.099 (2) | 0.026 (4) |
| Loop 10 {27} | 0.057 (4) | 0.045 (2) | 0.013 (2) |
| t20 {11} | 0.058 (3) | 0.043 (3) | 0.002 (1) |
| i10 {4} | 0.096 (2) | 0.065 (2) | 0.000 (0) |
| t21 {8} | 0.096 (4) | 0.093 (3) | 0.021 (1) |
| Loop 11 {41} | 0.067 (8) | 0.094 (6) | 0.012 (3) |
| t22 {11} | 0.064 (2) | 0.124 (1) | 0.007 (1) |
Regions are numbered and designated as a periplasmic loop (i), a surface-exposed loop (Loop) or a transmembrane region (t); braces show the lengths of each region in aa.
Numbers in parentheses are non-synonymous sites. Values denote p-distances.
Structural model of HpuB and location of amino acid variation
There were no proteins with sufficiently high homology to HpuA present within the RCSB Protein database (PDB) (www.rcsb.org) for structural predictions for this protein. HpuB is a transmembrane protein and a member of the TonB-dependent transporter (TBDT) family (Noinaj et al., 2010). The closest orthologue for HpuB for which a crystal structure was available was ShuA, which is a 73 kDa OMP of Shigella dysenteriae required for extraction of haem from haemoglobin (Cobessi et al., 2010). The ShuA structure was therefore used as a template to generate a structural model for an N. meningitidis HpuB protein with an automated three-dimensional protein structure tool (swiss-model). The resulting structure comprised a 22-stranded β-barrel with an N-terminal ‘plug’ domain, characteristic of the TBDT family (Fig. 4a). Similarly to HmbR, it also contained 10 putative periplasmic loops and 11 surface-exposed loops (Evans et al., 2010). The overall sequence homology between the HpuB and ShuA was low (16 %) and, consequently, several longer exterior loops from HpuB could not be built reliably, and were omitted from the final three-dimensional structure. The HpuB structure preserved known features common to TonB-dependent haem transporters (Fig. S3): for example, two arginine residues found in the ShuA plug domain (R64 and R104), part of the lock region (Cobessi et al., 2010), were aligned with R91 and K145 in HpuB. Similarly, the ShuA FRAP/NPNL sequence, again characteristic of haem transporters (Perkins-Balding et al., 2004), was aligned with a well-conserved FRAP/NPEL motif found in all HpuB sequences.
Fig. 4.
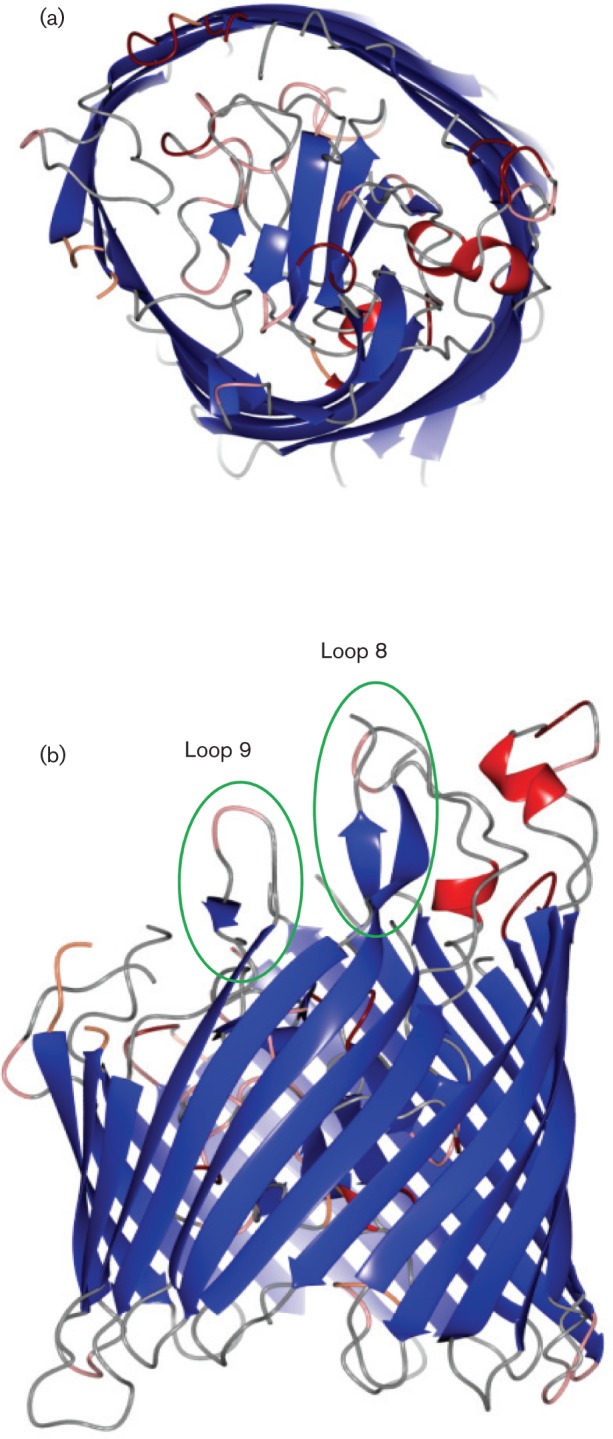
Structural model of HpuB. The structure is presented as a ribbon plot, with α-helices marked in red and β-strands in blue. (a) View from above, showing the location of the plug domain in the lumen of the β-barrel. (b) View from the side; the locations of two of the more variable sequence loops, 8 and 9, are circled and labelled.
The HpuB model assisted in defining the beginnings and ends of the 22 transmembrane beta strands (Table S2). As anticipated, the predicted extra-membrane regions on the periplasmic side of the membrane (numbered 2i, 4i, etc.), were shorter than those predicted to lie on the external surface. The putative surface-exposed regions that exhibited the highest sequence variation in both families were loops 2, 8 and 9: most variability occurred in loop 2, including differences in length by up to five residues (Table 3 & Fig. S4). Loop 2 was too long to be modelled effectively, but a complete model for loop 9 was constructed and part of loop 8 (Fig. 4b). Interestingly, loop 9 was not one of the longer external loop regions although it was subject to high sequence variation (Table 3 and Table S2). This could be because sequences within the longer loop regions are constrained in their variation for functional reasons or because they fold inwards and are less exposed at the surface. In addition, loop 9 does not appear to be subject to positive selection (Fig. 3). Compared to loop 8 which has among Family B isolates 15 variable sites, loop 9 only has three which were either Lys-Thr-Gln or Thr-Asn-Lys starting at residue 674 indicating that these may be compensatory mutations.
The HpuB proteins ranged from 805 to 812 amino acids in length and exhibited higher levels of conservation between the two families compared to HpuA (Table 2). The variability of all HpuB regions was examined in Family A and Family B isolates (Table 3). The ‘plug’ domain was highly conserved in Family B isolates but showed some variation in Family A (p-distances 0.018 and 0.072 respectively), indicating differences in selection pressures acting on these HpuB sequences among the non-pathogenic Neisseria species, possibly indicative of differences in function. High levels of variation were detected in external loops 2, 3, 4, 5 and 8 in the overall analysis but loop 2 and loop 8 had the highest level of variation in Family B strains. These results were consistent with loops 2 and 8 being subject to positive selection imposed by host immune responses.
Discussion
OMPs are considered to be among the best available candidates for a comprehensive vaccine targeting meningococci; however, these proteins are highly diverse, complicating vaccine design (Urwin et al., 2004). Analysis of the diversity and selection pressure acting on OMPs is thus an important part in assessing the suitability of a given protein as a vaccine component. OMPs involved in iron acquisition are attractive vaccine candidates as iron is essential for growth in vivo and systemic spread of meningococci. Iron-acquisition receptors have the added benefit of potentially providing protection against both meningococci and gonococci: the recent emergence of antibiotic-resistant gonococci (Lewis, 2010), combined with an increase in gonococcal infections, indicating a need for alternative treatment and prevention strategies targeting this organism (Chisholm et al., 2013; Noinaj et al., 2012a). Here, an analysis of the diversity of the hapto-haemoglobin receptor HpuAB in a collection of Neisseria isolates was used to assess the potential of the receptor as a vaccine candidate. In so doing, information on the dynamics of iron acquisition via haemoglobin or haemoglobin–haptoglobin by Neisseria was generated.
While there was some clustering of hpuAB sequences by species, the overall lack of distinct groups was consistent with frequent movement by lateral gene transfer among members of different species. This was similar to that seen with the iron-regulated TonB-dependent enterobactin receptor FetA, also known as FrpB, which is found among both pathogenic and non-pathogenic Neisseria and for which a common gene pool enabling frequent genetic exchange has been described (Bennett et al., 2009). Two major families, A and B of HpuAB, were identified (Figs 1 and 2). Family A hpuAB genes were not subject to mononucleotide repeat-mediated phase variation, which was prevalent in Family B. This suggests that additional phenotypes in haemoglobin acquisition, not based solely on the presence/absence of hpuAB genes, occur, including one in which hpuAB genes are constitutively expressed (in iron-deplete environments) and the other where hpuAB, specifically hpuA, translation can be switched on or off, the latter found in Neisseria associated with pathogenicity. The lack of positive selection evident in Family A HpuAB sequences was consistent with these proteins being less subject to selection pressures imposed by the immune system, which would be consistent with the absence of a need for phase variation of these variants (Fig. 3). In contrast, Family B HpuAB proteins exhibited evidence for strong diversifying selection that was consistent with immune selection acting on these variants. This was particularly apparent in HpuA sequences where 74 out of 344 amino acid residues were under positive selection as opposed to 51 out of 813 in HpuB. HpuA encodes a putative lipoprotein which is fully surface exposed when expressed, similar to the transferrin receptor, TbpB, which has been found to be particularly diverse (Rokbi et al., 1997). The surface-exposed nature of these proteins has possibly generated the hyper-variability seen and, for hpuA, resulted in the evolution of phase variation (tbpB sequences containing a homopolymeric repeat have not been found to date).
HpuB is a transmembrane protein, with the positive selection signal corresponding to the putative surface-exposed loops consistent with these eliciting a bactericidal immune response, while the transmembrane, periplasmic and plug regions were more conserved (Table 3). HpuB shares approximately 28 % sequence similarity with HmbR (Fig. S3) (Lewis et al., 1997) and, similarly to HmbR, the putative extracellular loops 6 and 7 identified in HpuB were conserved and had few sites under strong diversifying selection, particularly among Family B sequences (Evans et al., 2010). In addition, putative loop 7 contained the conserved NPEL haem transport motif also identified in loop 7 from HmbR (Perkins-Balding et al., 2004). These loops had been implicated in haemoglobin utilization in HmbR with deletion mutation resulting in reduced fitness of the organism consistent with a functional constraint limiting the diversity of these loops (Perkins-Balding et al., 2003). These functional studies of HmbR also indicated that loops 2 and 3 were necessary for haemoglobin binding with mutation resulting in reduced haemoglobin-binding activity. Although these loops contained regions implicated in haemoglobin acquisition in HmbR, the association of given variable region sequences within loops 2 and 3 with particular clonal complexes indicated conservation of the haemoglobin-binding site within lineages and, combined with the detection of positively selected residues, this suggested that these surface-exposed regions contained epitopes (Evans et al., 2010). Putative loop 2 from HpuB is in an equivalent position to loop 2 of HmbR and also displayed strong diversifying selection (Table 3 and Fig. 3). Some association of variable regions sequences in loop 2 with particular N. meningitidis clonal complexes was evident in Family B HpuB sequences (Fig. S4); however, evidence for lateral gene transfer was also apparent in this region both among meningococci and between this species and the closely related non-pathogenic species N. polysaccharea and N. lactamica, indicating that a common gene pool is associated with this receptor. In contrast, the amino acid sequence for this loop contained residues specific to N. gonorrhoeae, consistent with the absence of recombination in hpuAB between this species and other Neisseria.
In the related TonB-dependent receptors for transferrin and lactoferrin, the lipoproteins tbpB and lbpB are not essential and have been found to serve an accessory role by enhancing iron acquisition (Anderson et al., 1994; Biswas et al., 1999). In contrast, both HpuAB components are required in both gonococci and meningococci with RNA analysis demonstrating that hpuA and hpuB were co-transcribed on a single mRNA transcript (Chen et al., 1998; Lewis et al., 1997, 1999). Conflicting reports have, however, indicated that a 2.5 kb mRNA transcript consistent with the size of an independent mono-cistronic hpuB was sometimes found along with a separate report observing that point mutations in HpuB allowed gonococci to grow using haemoglobin without the expression of HpuA (Chen et al., 2002; Lewis et al., 1997). Eight non-polymorphisms were found in the N. gonorrhoeae HpuB proteins analysed in this study; however, these polymorphisms did not correlate with the previously described point mutations or with the on/off status of hpuA indicating that this mechanism for generating HpuA independent haemoglobin uptake is not widespread.
The distribution of the HpuAB and HmbR receptors among Neisseria species indicated the importance of iron acquisition from a haemoglobin source in this genus. Excluding N. bacilliformis, N. weaveri, N. animalis, N. canis, N. dentiae and N. elongata, which may inhabit diverse niches and have different nutritional requirements, all Neisseria species contained HpuAB alone, HmbR alone or both receptors for acquiring iron from haemoglobin. The non-pathogenic Neisseria species, in particular, were characterized by a high prevalence of a HpuAB only phenotype which was consistent with the observation that the haemoglobin receptor, HmbR, is a significant virulence factor in meningococcal pathogenesis (Harrison et al., 2009). The prevalence of hpuAB genes among the closely related non-pathogenic Neisseria population generates a global gene pool from which extensive allelic transfer is possible, and which ultimately will limit the efficacy of these receptors as vaccine candidates (Linz et al., 2000; Maiden et al., 1996). Carriage of non-pathogenic Neisseria may contribute to the development of natural immunity to meningococcal disease, and expression of OMPs such as HpuAB and FetA/FrpB may be implicated in this immunity. The use of such cross-reactive antigens in vaccine formulations could impede the acquisition of natural immunity to meningococcal disease by preventing colonization by non-pathogenic Neisseria and indicates caution when designing vaccines containing cross-reactive antigens. On the other hand, the prevalence of the HmbR receptor among N. meningitidis isolates associated with disease combined with the absence of this receptor among non-pathogenic Neisseria indicates that a vaccine would be unlikely to affect the host microbiota. Furthermore, the absence of this receptor in non-pathogenic Neisseria narrows the gene pool available for allelic exchange thereby probably generating a more promising vaccine candidate. The presence of an HmbR pseudogene among N. gonorrhoeae isolates, however, indicates that this would not be a suitable vaccine candidate for this species, although it would be important to determine whether all N. gonorrhoeae isolates exhibit the same phenotype as the 16 isolates analysed herein.
Acknowledgements
This work was funded by the Wellcome Trust, the University of Leicester, the University of Manchester and the University of Oxford. M. C. J. M. is a Wellcome Trust Senior Research Fellow. This work was instigated as part of Meningitis Research Foundation, grant number 1002.0 entitled ‘Examination of meningococcal haemoglobin receptors as potential vaccine targets’ following helpful discussions with Ian Feavers, Hannah Chan and Ed Kaczmarski.
Abbreviations:
- 4CMenB
4 Component MenB
- BIGSDB
Bacterial Isolate Genome Sequence Database
- CCUG
Culture Collection of the University of Goteborg
- OMP
outer-membrane protein
- TBDT
TonB-dependent transporter
Footnotes
Two supplementary tables and four supplementary figures are available with the online version of this paper.
References
- Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997). Gapped blastand psi-blast: a new generation of protein database search programs. Nucleic Acids Res 25, 3389–3402. 10.1093/nar/25.17.3389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson J. E., Sparling P. F., Cornelissen C. N. (1994). Gonococcal transferrin-binding protein 2 facilitates but is not essential for transferrin utilization. J Bacteriol 176, 3162–3170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnold K., Bordoli L., Kopp J., Schwede T. (2006). The swiss-model workspace: a web-based environment for protein structure homology modelling. Bioinformatics 22, 195–201. 10.1093/bioinformatics/bti770 [DOI] [PubMed] [Google Scholar]
- Bennett J. S., Griffiths D. T., McCarthy N. D., Sleeman K. L., Jolley K. A., Crook D. W., Maiden M. C. (2005). Genetic diversity and carriage dynamics of Neisseria lactamica in infants. Infect Immun 73, 2424–2432. 10.1128/IAI.73.4.2424-2432.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J. S., Thompson E. A., Kriz P., Jolley K. A., Maiden M. C. (2009). A common gene pool for the Neisseria FetA antigen. Int J Med Microbiol 299, 133–139. 10.1016/j.ijmm.2008.06.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J. S., Jolley K. A., Earle S. G., Corton C., Bentley S. D., Parkhill J., Maiden M. C. (2012). A genomic approach to bacterial taxonomy: an examination and proposed reclassification of species within the genus Neisseria. Microbiology 158, 1570–1580. 10.1099/mic.0.056077-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger U. (1971). [Neisseria mucosa var. heidelbergensis]. Z Med Mikrobiol Immunol 156, 154–158. 10.1007/BF02124646 [DOI] [PubMed] [Google Scholar]
- Biswas G. D., Anderson J. E., Chen C. J., Cornelissen C. N., Sparling P. F. (1999). Identification and functional characterization of the Neisseria gonorrhoeae lbpB gene product. Infect Immun 67, 455–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bracken C. S., Baer M. T., Abdur-Rashid A., Helms W., Stojiljkovic I. (1999). Use of heme-protein complexes by the Yersinia enterocolitica HemR receptor: histidine residues are essential for receptor function. J Bacteriol 181, 6063–6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budroni S., Siena E., Dunning Hotopp J. C., Seib K. L., Serruto D., Nofroni C., Comanducci M., Riley D. R., Daugherty S. C. & other authors (2011). Neisseria meningitidis is structured in clades associated with restriction modification systems that modulate homologous recombination. Proc Natl Acad Sci U S A 108, 4494–4499. 10.1073/pnas.1019751108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calmettes C., Alcantara J., Yu R. H., Schryvers A. B., Moraes T. F. (2012). The structural basis of transferrin sequestration by transferrin-binding protein B. Nat Struct Mol Biol 19, 358–360. 10.1038/nsmb.2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caugant D. A., Maiden M. C. (2009). Meningococcal carriage and disease – population biology and evolution. Vaccine 27 (Suppl 2), B64–B70. 10.1016/j.vaccine.2009.04.061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. J., Elkins C., Sparling P. F. (1998). Phase variation of hemoglobin utilization in Neisseria gonorrhoeae. Infect Immun 66, 987–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. J., Mclean D., Thomas C. E., Anderson J. E., Sparling P. F. (2002). Point mutations in HpuB enable gonococcal HpuA deletion mutants to grow on hemoglobin. J Bacteriol 184, 420–426. 10.1128/JB.184.2.420-426.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chisholm S. A., Unemo M., Quaye N., Johansson E., Cole M. J., Ison C. A., Van de Laar M. J. (2013). Molecular epidemiological typing within the European Gonococcal Antimicrobial Resistance Surveillance Programme reveals predominance of a multidrug-resistant clone. Euro surveillance: bulletin europeen sur les maladies transmissibles = European communicable disease bulletin 18. [PubMed] [Google Scholar]
- Cobessi D., Meksem A., Brillet K. (2010). Structure of the heme/hemoglobin outer membrane receptor ShuA from Shigella dysenteriae: heme binding by an induced fit mechanism. Proteins 78, 286–294. 10.1002/prot.22539 [DOI] [PubMed] [Google Scholar]
- Cornelissen C. N., Kelley M., Hobbs M. M., Anderson J. E., Cannon J. G., Cohen M. S., Sparling P. F. (1998). The transferrin receptor expressed by gonococcal strain FA1090 is required for the experimental infection of human male volunteers. Mol Microbiol 27, 611–616. 10.1046/j.1365-2958.1998.00710.x [DOI] [PubMed] [Google Scholar]
- Cornelissen C. N., Hollander A. (2011). TonB-dependent transporters expressed by Neisseria gonorrhoeae. Front Microbiol 2, 1–13. 10.1038/nrmicro2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Criss A. K., Seifert H. S. (2012). A bacterial siren song: intimate interactions between Neisseria and neutrophils. Nat Rev Microbiol 10, 178–190. 10.1038/nrmicro2713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans N. J., Harrison O. B., Clow K., Derrick J. P., Feavers I. M., Maiden M. C. J. (2010). Variation and molecular evolution of HmbR, the Neisseria meningitidis haemoglobin receptor. Microbiology 156, 1384–1393. 10.1099/mic.0.036475-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison O. B., Evans N. J., Blair J. M., Grimes H. S., Tinsley C. R., Nassif X., Kriz P., Ure R., Gray S. J. & other authors (2009). Epidemiological evidence for the role of the hemoglobin receptor, hmbR, in meningococcal virulence. J Infect Dis 200, 94–98. 10.1086/599377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison O. B., Claus H., Jiang Y., Bennett J. S., Bratcher H. B., Jolley K. A., Corton C., Care R., Poolman J. T. & other authors (2013). Description and nomenclature of Neisseria meningitidis capsule locus. Emerg Infect Dis 19, 566–573. 10.3201/eid1904.111799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huson D. H., Bryant D. (2006). Application of phylogenetic networks in evolutionary studies. Mol Biol Evol 23, 254–267. 10.1093/molbev/msj030 [DOI] [PubMed] [Google Scholar]
- Jolley K. A., Hill D. M., Bratcher H. B., Harrison O. B., Feavers I. M., Parkhill J., Maiden M. C. (2012). Resolution of a meningococcal disease outbreak from whole-genome sequence data with rapid Web-based analysis methods. J Clin Microbiol 50, 3046–3053. 10.1128/JCM.01312-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis D. A. (2010). The Gonococcus fights back: is this time a knock out? Sex Transm Infect 86, 415–421. 10.1136/sti.2010.042648 [DOI] [PubMed] [Google Scholar]
- Lewis L. A., Gray E., Wang Y.-P., Roe B. A., Dyer D. W. (1997). Molecular characterization of hpuAB, the haemoglobin–haptoglobin–utilization operon of Neisseria meningitidis. Mol Microbiol 23, 737–749. 10.1046/j.1365-2958.1997.2501619.x [DOI] [PubMed] [Google Scholar]
- Lewis L. A., Gipson M., Hartman K., Ownbey T., Vaughn J., Dyer D. W. (1999). Phase variation of HpuAB and HmbR, two distinct haemoglobin receptors of Neisseria meningitidis DNM2. Mol Microbiol 32, 977–989. 10.1046/j.1365-2958.1999.01409.x [DOI] [PubMed] [Google Scholar]
- Linz B., Schenker M., Zhu P., Achtman M. (2000). Frequent interspecific genetic exchange between commensal Neisseriae and Neisseria meningitidis. Mol Microbiol 36, 1049–1058. 10.1046/j.1365-2958.2000.01932.x [DOI] [PubMed] [Google Scholar]
- Lucidarme J., Comanducci M., Findlow J., Gray S. J., Kaczmarski E. B., Guiver M., Vallely P. J., Oster P., Pizza M. & other authors (2010). Characterization of fHbp, nhba (gna2132), nadA, porA, and sequence type in group B meningococcal case isolates collected in England and Wales during January 2008 and potential coverage of an investigational group B meningococcal vaccine. Clin Vaccine Immunol 17, 919–929. 10.1128/CVI.00027-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiden M. C. J., Malorny B., Achtman M. (1996). A global gene pool in the Neisseriae. Mol Microbiol 21, 1297–1298. 10.1046/j.1365-2958.1996.981457.x [DOI] [PubMed] [Google Scholar]
- Maiden M. C., Bygraves J. A., Feil E., Morelli G., Russell J. E., Urwin R., Zhang Q., Zhou J., Zurth K. & other authors (1998). Multilocus sequence typing: a portable approach to the identification of clones within populations of pathogenic microorganisms. Proc Natl Acad Sci U S A 95, 3140–3145. 10.1073/pnas.95.6.3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N., Guillier M., Barnard T. J., Buchanan S. K. (2010). TonB-dependent transporters: regulation, structure, and function. Annu Rev Microbiol 64, 43–60. 10.1146/annurev.micro.112408.134247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N., Buchanan S. K., Cornelissen C. N. (2012a). The transferrin–iron import system from pathogenic Neisseria species. Mol Microbiol 86, 246–257. 10.1111/mmi.12002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noinaj N., Easley N. C., Oke M., Mizuno N., Gumbart J., Boura E., Steere A. N., Zak O., Aisen P. & other authors (2012b). Structural basis for iron piracy by pathogenic Neisseria. Nature 483, 53–58. 10.1038/nature10823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkins-Balding D., Baer M. T., Stojiljkovic I. (2003). Identification of functionally important regions of a haemoglobin receptor from Neisseria meningitidis. Microbiology 149, 3423–3435. 10.1099/mic.0.26448-0 [DOI] [PubMed] [Google Scholar]
- Perkins-Balding D., Ratliff-Griffin M., Stojiljkovic I. (2004). Iron transport systems in Neisseria meningitidis. Microbiol Mol Biol Rev 68, 154–171. 10.1128/MMBR.68.1.154-171.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard A. J. (2004). Global epidemiology of meningococcal disease and vaccine efficacy. Pediatr Infect Dis J 23 (Suppl), S274–S279. [PubMed] [Google Scholar]
- Richmond P. C., Marshall H. S., Nissen M. D., Jiang Q., Jansen K. U., Garcés-Sánchez M., Martinón-Torres F., Beeslaar J., Szenborn L. & other authors (2012). Safety, immunogenicity, and tolerability of meningococcal serogroup B bivalent recombinant lipoprotein 2086 vaccine in healthy adolescents: a randomised, single-blind, placebo-controlled, phase 2 trial. Lancet Infect Dis 12, 597–607. 10.1016/S1473-3099(12)70087-7 [DOI] [PubMed] [Google Scholar]
- Rokbi B., Mignon M., Caugant D. A., Quentin-Millet M. J. (1997). Heterogeneity of tbpB, the transferrin-binding protein B gene, among serogroup B Neisseria meningitidis strains of the ET-5 complex. Clin Diagn Lab Immunol 4, 522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleem M., Prince S. M., Rigby S. E., Imran M., Patel H., Chan H., Sanders H., Maiden M. C., Feavers I. M., Derrick J. P. (2013). Use of a molecular decoy to segregate transport from antigenicity in the FrpB iron transporter from Neisseria meningitidis. PLoS ONE 8, e56746. 10.1371/journal.pone.0056746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serruto D., Bottomley M. J., Ram S., Giuliani M. M., Rappuoli R. (2012). The new multicomponent vaccine against meningococcal serogroup B, 4CMenB: immunological, functional and structural characterization of the antigens. Vaccine 30 (Suppl 2), B87–B97. 10.1016/j.vaccine.2012.01.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stojiljkovic I., Hwa V., de Saint Martin L., O’Gaora P., Nassif X., Heffron F., So M. (1995). The Neisseria meningitidis haemoglobin receptor: its role in iron utilization and virulence. Mol Microbiol 15, 531–541. 10.1111/j.1365-2958.1995.tb02266.x [DOI] [PubMed] [Google Scholar]
- Tamura K., Peterson D., Peterson N., Stecher G., Nei M., Kumar S. (2011). mega5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28, 2731–2739. 10.1093/molbev/msr121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tauseef I., Harrison O. B., Wooldridge K. G., Feavers I. M., Neal K. R., Gray S. J., Kriz P., Turner D. P., Ala’Aldeen D. A. & other authors (2011). Influence of the combination and phase variation status of the haemoglobin receptors HmbR and HpuAB on meningococcal virulence. Microbiology 157, 1446–1456. 10.1099/mic.0.046946-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urwin R., Russell J. E., Thompson E. A., Holmes E. C., Feavers I. M., Maiden M. C. (2004). Distribution of surface protein variants among hyperinvasive meningococci: implications for vaccine design. Infect Immun 72, 5955–5962. 10.1128/IAI.72.10.5955-5962.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogel U., Taha M. K., Vazquez J. A., Findlow J., Claus H., Stefanelli P., Caugant D. A., Kriz P., Abad R. & other authors (2013). Predicted strain coverage of a meningococcal multicomponent vaccine (4CMenB) in Europe: a qualitative and quantitative assessment. Lancet Infect Dis 13, 416–425. 10.1016/S1473-3099(13)70006-9 [DOI] [PubMed] [Google Scholar]
- Wilson D. J., McVean G. (2006). Estimating diversifying selection and functional constraint in the presence of recombination. Genetics 172, 1411–1425. 10.1534/genetics.105.044917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolfgang W. J., Passaretti T. V., Jose R., Cole J., Coorevits A., Carpenter A. N., Jose S., Van Landschoot A., Izard J. & other authors (2013). Neisseria oralis sp. nov., isolated from healthy gingival plaque and clinical samples. Int J Syst Evol Microbiol 63, 1323–1328. 10.1099/ijs.0.041731-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerbino D. R. (2010). Using the Velvet de novo assembler for short-read sequencing technologies. Curr Protoc Bioinformatics Chapter 11, 5.1–5.12. [DOI] [PMC free article] [PubMed] [Google Scholar]