

Abstract
Huntington disease is an autosomal-dominant neurodegenerative disorder caused by a polyglutamine (polyQ) expansion (> 35Q) in the first exon (EX1) of huntingtin protein (Htt). mHtt protein is thought to adopt one or more toxic conformation(s) that are involved in pathogenic interactions in cells . However, the structure of mHtt is not known. Here, we present a near atomic resolution structure of mHtt36Q-EX1. To facilitate crystallization, three histidine residues (3H) were introduced within the Htt36Q stretch resulting in the sequence of Q7HQHQHQ27. The Htt36Q3H region adopts α-helix, loop, β-hairpin conformations. Furthermore, we observed interactions between the backbone of the Htt36Q3H β-strand with the aromatic residues mimicking putative-toxic interactions with other proteins. Our findings support previous predictions that the expanded mHtt-polyQ region adopts a β-sheet structure. Detailed structural information about mHtt improves our understanding of the pathogenic mechanisms in HD and other polyQ expansion disorders and may form the basis for rational design of small molecules that target toxic conformations of disease-causing proteins.
Keywords: Huntingtin, aggregation, amyloid, ataxia, crystallography, hairpin, polyglutamine, prion, β-strand, β-turn
Introduction
Huntington disease (HD) is an autosomal-dominant neurodegenerative disorder. Neuropathological analysis of HD patients reveals selective and progressive neuronal loss in the striatum,1 particularly affecting the GABAergic medium spiny striatal neurons (MSN). At the molecular level, the cause of HD is a polyglutamine (polyQ) expansion (≥35Q) near the N-terminus of Huntingtin (Htt), a 350 kDa ubiquitously expressed cytoplasmic protein.2 The cellular mechanisms that link Htt polyQ expansion (mHtt) with the disease are under intense investigation. PolyQ-expanded mHtt is thought to adopt one or more toxic conformation(s) that are involved in pathogenic protein-protein interactions in cells.3,4 A number of toxic functions have been assigned to mHtt, including formation of toxic aggregates, effect on gene transcription, induction of apoptosis and disruption of key neuronal functions such as proteosomal function, ubiquitination, axonal transport, endocytosis, synaptic transmission and Ca2+ signaling.3-10 Many of the proposed mechanisms suggest that the mHtt is involved in pathological interactions with other signaling proteins in cells, leading to neuronal dysfunction and death.
Information about the structure of Htt polyQ region is critical for understanding mHtt toxicity and may aid in the development of potential HD therapies. However, the structure determination of the polyQ region of Htt has proven to be an extremely difficult problem.10,11 In the aggregated form polyQ sequence most likely adopts β-sheet structure.12-21 The biophysical studies of soluble monomeric form of polyQ fragments of various length provided evidence for random coil conformation,15,22-24 for α-helical conformation,25,26 for β-sheet19,26 and for extended helix.27 We previously solved the crystal structure of the first exon (EX1) of Htt-17Q28 and the other group reported crystal structure of GQ10G peptide complexed with the polyQ-specific antibody.29 However, the near atomic structure of mHtt with a disease-associated polyQ expansion is not known. Here, we present a near atomic resolution structure of exon 1 of Htt-36Q with insertion of 3 histidine residues within 36Q stretch resulting in the sequence Q7HQHQHQ27. We discovered that Htt-36Q3H-EX1 protein fragment adopts α-helix, loop and β-hairpin conformations. Our findings support previous predictions that the expanded polyQ region adopts a β-sheet structure and provide first near atomic resolution structure of pathogenic form of mHtt or any other polyQ-expanded protein.
Results
Crystallization of Htt36Q exon 1 fragment
In the previous study we used protein crystallography approach to determine near atomic resolution structure of exon 1 of Htt17Q (Htt17Q-EX1).28 In this study Htt17Q-EX1 protein fragment was expressed, purified and crystallized as N-terminal MBP-fusion protein with addition of 19 amino-acids C-terminal tag28 (Fig. 1A). In the present study we aimed to use the same approach to solve the structure of mHtt exon 1 containing expanded glutamine repeat region. By following procedures described in the previous study28 we generated, expressed, purified and crystallized MBP-Htt36Q-EX1 protein with C-terminal tag. However, resulting crystals diffracted at resolution less than 6 Å, preventing structure determination of Htt36Q-EX1 protein (data not shown). We reasoned that poor diffraction quality of MBP-Htt36Q-EX1 crystals is due to a random helix-to-loop transition within 36Q region, as observed in our previous structural studies of Htt17Q-EX1.28 To reduce the randomness within polyQ structure, we inserted XQXQX interrupter sequence within the Htt-polyQ region in order to either break or stabilize the α-helical conformation of Htt-polyQ. As an X amino acid we selected potential helix breaker Gly and potential helix stabilizer Ala. In addition, we also tested turn-promoting His. We selected His because insertion of His within a polyQ stretch in Ataxin-1 exerts strong disease-modifying effect in SCA1 patients,30-32 and a number of detailed biophysical studies have been performed with polyQ peptides containing His insertions.33-35 The XQXQX interrupters (X = G, A, H) were introduced at several locations within Htt-polyQ-EX1 constructs. The polyQ constructs of various length (17Q, 23Q, 36Q and 45Q) were generated in our experiments. From all constructs that we tested, only MBP-Htt17Q3A-EX1 (first X at position 8) and MBP-Htt36Q3H-EX1 (first X at position 8) yielded diffraction quality crystals (Table S1). The Htt17Q3A protein adopted α-helical conformation in the crystals (data not shown), in agreement with our previous studies of Htt17Q-EX1.28 Thus, in order to explore the Htt-polyQ conformational space further we focused on Htt36Q3H-EX1 construct for our further studies.

Figure 1. The structure of MBP-Htt36Q3H-EX1. (A) Amino acid sequence of MBP-Htt17Q-EX128 and MBP-Htt36Q3H-EX1 expression constructs. MBP3A denotes the maltose binding protein followed by a three alanines linker. In both constructs position M371 corresponds to M1 in Htt sequence. The sequence of Htt-Ex1 is composed of N-terminal N17 region (green), polyQ region (orange), His (pink), PolyP region (blue) and mixed P/Q region (purple). Three His residues (pink) are inserted within poly36Q stretch in MBP-Htt36Q3H-EX1 construct. In both constructs identical 19 amino acids C-terminal tag was added to facilitate crystallization (black).(B) The trimer of MBP-Htt36Q3H-EX1. (C, D) Htt36Q3H-EX1 trimers in the crystal X1 (C) and X2 (D). On panels (B), (C) and (D) the MBP protein (gray), 3A linker (gray), Htt-N17 (green) and Htt-polyQ (orange) regions are shown for each of the three MBP-Htt36Q3H-EX1 molecules (A, B, C) in the asymmetric unit of the crystal.
The overall design of Htt36Q3H-Ex1 expression construct (Fig. 1a) was similar to the design of the Htt17Q-EX1 expression construct used in the previous study.28 The N-terminal MBP molecule was connected with Htt36Q3H-Ex1 via 3 alanines (3A) linker (Fig. 1A). The amino-acid (aa) numbering in MBP-Htt36Q3H-Ex1 construct starts with MBP, so that Met1 in the Htt corresponds to Met371 in the MBP-Htt36Q3H-Ex1. The N-terminal region of Htt36Q3H-Ex1 starting at Met371 (Met1 in Htt sequence) extends for 17aa until Phe387 (Fig. 1a). The next region Gln388 - Gln426 contains a stretch of 36Gln (poly36Q) with addition of 3 Histidines and immediately followed by a stretch of 11Pro from Pro427 until Pro437 (poly11P) (Fig. 1A). The proline stretch is followed by 15aa mixed Pro and Gln regions (polyP/Q) that spans from Gln438 until Gln452 (Fig. 1A). The native Htt sequence ends at Gln452 and followed by an exogenous C-terminal 19 aa tag added to facilitate crystallization (Fig. 1A).
MBP-Htt36Q3H-EX1 protein was expressed, purified and crystallized according to the procedures described in our previous paper28 (see Experimental Procedures for details). X-ray diffraction data were collected for several crystals of MBP-Htt36Q3H-EX1, with the best crystals diffracting up to 2.5 Å resolution. All crystals were monoclinic, space group C2. The asymmetric unit contained 3 molecules of MBP-Htt36Q-EX1 (designated as molecules A, B, C) in all crystals. The phasing was done for the two best crystals (X1 and X2) by molecular replacement (MR) using MBP structural models (see Experimental Procedures for details). MR gave solutions for both crystals with complete maps for MBP and partial maps for Htt36Q3H-EX1. The detailed procedures used to generate the maps are described in Methods section.
The crystallographic parameters for X1 and X2 crystals are listed in Table 1. In the process of refinement we discovered that molecule C adopts dual conformation in both crystals. The model for each of these conformations (C1 and C2) was built and refined independently. The final refinement statistics for MBP3A-Htt36Q3H-Ex1 structure determination in X1 and X2 crystals are summarized in Table 1. The packing of 3 MBP-Htt36Q3H-Ex1 molecules in the asymmetric unit (Fig. 1B) was nearly identical to the trimer formed by 3 MBP-Htt17Q-Ex1 molecules in the previous study.28
Table 1. Diffraction data collection and refinement statistics for two crystals used for MBP-Htt36Q3H-EX1 structure determination.
| Crystal | X1 | X2 | ||||
|---|---|---|---|---|---|---|
| Data collection | ||||||
| Wavelength (λ) |
0.98 |
0.98 |
||||
| Space group |
C2 |
C2 |
||||
| Unit Cell parameter (Å, o) | ||||||
| a |
155.052 |
153.182 |
||||
| b |
177.278 |
177.357 |
||||
| c |
78.868 |
78.346 |
||||
| β |
109.025 |
108.674 |
||||
| Resolution limit (Å) |
2.8–40.0/2.8–2.9 |
2.8–40.0/2.8–2.9 |
||||
| Rsym(%) |
11.0/53.2 |
6.6/65.3 |
||||
| I/σI (overall/outer shell) |
27.9/2.1 |
17.2/1.0 |
||||
| Redundancy (overall/outer shell) |
3.6/3.1 |
3.6/2.4 |
||||
| Overall completeness (%) |
99.2 |
99.2 |
||||
| Refinement |
X1-C1 |
X1-C2 |
X2-C1 |
X2-C2 |
||
| B-factor | ||||||
| Overall (Å2) |
54.3 |
55.8 |
56.2 |
56.1 |
||
| Solvent |
46.9 |
47.7 |
27.3 |
30.8 |
||
| No of data |
40324 |
39566 |
||||
| RWork/RFree |
0.23/0.27 |
0.23/0.27 |
0.23/0.27 |
0.23/0.27 |
||
| RMS_D (Å) |
0.006 |
0.006 |
0.005 |
0.005 |
||
| RMS_A (o) |
0.912 |
0.912 |
0.789 |
0.789 |
||
| Ramachandran map | ||||||
| Favored (%) |
97.2 |
97.4 |
97.1 |
96.9 |
||
| Allowed (%) |
2.6 |
2.5 |
2.7 |
2.7 |
||
| Outliner (%) |
0.2 |
0.1 |
0.2 |
0.4 |
||
| PDB accession number | 4FE8 | 4FEB | 4FEC | 4FED | ||
Secondary structure of the Htt36Q3H-EX1
The ribbon structure diagrams of Htt36Q3H-Ex1 trimers resolved in crystals X1 and X2 are shown on Figure 1C and Figure 1D. All 6 molecules composed of identical α-helical N17 N-terminal Htt region (green) and polyQ regions (orange) which adopted multiple conformations in the crystals. Similar to the previous structure of Htt17Q-Ex1,28 the trimer packing of Htt36Q3H-Ex1 molecules is stabilized by hydrophobic interactions between Leu and Phe residues from Htt-N17 regions of molecules A, B and C. The polyQ region initially adopts α-helical structure, which extends for 7 Gln until Gln394 residue in all 6 molecules (Fig. 1C and 1D). The Htt36Q3H structures starting from His395 adopt diverse conformations as summarized in Table 2. The A and B molecules in both crystals adopt β-hairpin conformations starting from Gln396 (Table 2). The C molecules in both crystals adopt dual conformations (C1 and C2), which have been modeled independently. The C1 conformations in crystal X1 and X2 are an α-helix and a loop respectively, while C2 conformation in both crystals is the hairpin (Table 2).
Table 2. Conformations adopted by the Htt36Q3H region starting at position Q396 in MBP-Htt36Q3H-EX1 for crystals X1 and X2.
| Crystal | Molecule | Residues | Conformation | ||
|---|---|---|---|---|---|
|
X1 |
A |
396Q-401Q |
Hairpin |
||
| B |
396Q-401Q |
Hairpin |
|||
| C |
C1 |
396Q-410Q |
α-helix |
||
| C2 |
396Q-401Q |
Hairpin |
|||
| X2 | A |
396Q-401Q |
Hairpin |
||
| B |
396Q-401Q |
Hairpin |
|||
| C | C1 |
396Q-407Q |
Loop |
||
| C2 | 396Q-401Q | Hairpin | |||
Multiple conformations of polyglutamine region in Htt36Q3H-EX1
The ribbon-stick structures of C molecules in α-helical and loop conformations are shown on Figure 2A and Figure 2B together with the supporting electron density difference maps (2Fo-Fc). The α-helical conformation of polyQ region of C1 molecule in crystal X1 extends until Gln410 according to the electron density map (Fig. 2A, Table 2). In contrast, for C1 molecule in crystal X2 the α-helix breaks from H395 and becomes a loop starting at Q396 (Fig. 2B, Table 2). The loop can be resolved until Q407 according to the electron density map (Fig. 2B). The polyQ regions in α-helix and loop conformations have been observed in our previous analysis of Htt17Q-Ex1 structure.28 Both α-helical and loop conformations of Htt36Q3H-Ex1 (Fig. 2A and 2B) and Htt17Q-Ex128 are located in the solvent area and lack any obvious protein interactions.

Figure 2. The Htt36Q3H region in α-helical, loop and β-hairpin conformations. (A) The α-helical structure of Htt36Q3H (Q388-Q410) (C1 molecule from crystal X1). (B) The α-helical structure of Htt36Q3H (Q388-H395) followed by a loop (Q396-Q407) (C1 molecule from crystal X2). (C) The α-helical structure of Htt36Q3H (Q388-Q394) followed by a β-hairpin (H395-Q401) (C2 molecule from crystal X1). On (A), (B) and (C) Htt-N17 (green), Htt-polyQ (orange) and His residues (pink) are labeled and numbered on the ribbon diagram. The supportive electron density maps (2Fo-Fc) at 1σ are shown by a green mesh.
In addition to α-helix and loop conformations, we discovered a β-hairpin conformation of Htt36Q3H region. The example of such conformation with the supporting electron density map (2Fo-Fc) is shown on Figure 2C. These structural elements have not been observed in our previous analysis of Htt17Q-Ex1,28 but they have been discovered for Q396–Q401 region of most Htt36Q3H-Ex1 molecules (Table 2). The example of β-hairpin structure formed by Q396, H397, Q398, H399, Q400, Q401 is shown in details on Figure 3. The β-hairpin consists of β-strand (Q396 and H397), turn (Q398 and H399) and β-strand (Q400 and Q401) (Fig. 3, diagram). This hairpin is stabilized by intramolecular double hydrogen bonds between H397 and Q400 (Fig. 3). We propose that the transition of Htt-polyQ conformation from α-helix (Fig. 2A) to loop (Fig. 2B) to β-hairpin (Fig. 2C) captured in our crystals provides a plausible sequential conformational transition from α-helical to β−sheet conformation in mHtt. These transitions have not been previously observed for Htt or for any other protein using near near atomic resolution techniques.
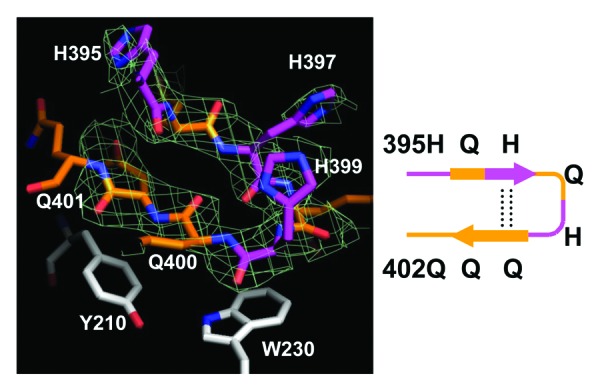
Figure 3. The Htt36Q3H region in β-hairpin conformation. The hairpin structure of Htt36Q3H (H395-Q401) (C1 molecule from crystal X1). The Gln (orange) and His (pink) residues are labeled and numbered on the stick diagram. The hydrogen bonds are shown by a dotted line. The Y210 and W230 proximal residues from MBP are also shown. The supportive electron density maps (2Fo-Fc) at 1σ are shown by green mesh for Htt36Q3H. The diagram of structure is depicted in a schematic form on the right panel.
Intermolecular protein interactions of polyglutamine region in Htt36Q3H-EX1
In contrast to “inert” α-helical and loop conformations of Htt36Q3H-Ex1 located in the solvent region (Fig. 2A and 2B), the β-hairpin of Htt36Q3H-Ex1 was resolved fastened to the aromatic residues in the MBP protein (Fig. 2C and 3). The hairpin in these crystals was stabilized by intermolecular interactions between the amine of amide backbone (HNb) of Q398, H399, Q400 and Q401 in Htt and aromatic rings of Y155, Y210 and W230 in MBP (Fig. 4). The amide backbone (HNb) of each residue interacts with aromatic rings in a parallel configuration (Fig. 4). In this geometry HNb is capable to form two simultaneous hydrogen bonds, intramolecular with the carboxyl on the other side of the Htt hairpin and intermolecular with the aromatic ring provided by the MBP amino acids. This type of interactions between aromatic rings and amide backbone (Ar-HNb) has been observed in many protein structures previously.36-39 The interaction distances between HNb and aromatic rings are in the range 4.3–4.5Ǻ (Fig. 4), which is a typical distance for Ar-HNb interactions.36-39 The Ar-HNb interaction is a weak polar interaction with an interaction energy of approximately 1.5 Kcal/mol, and usually multiple Ar-HNb interactions are needed to stabilize the ternary local structure of a protein.39 Consistent with these general features, there are four parallel Ar-HNb interactions between β-hairpin of Htt36Q3H-Ex1 and aromatic residues of MBP (Fig. 4).
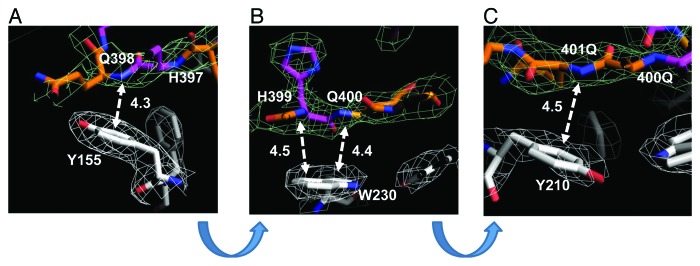
Figure 4. Intermolecular interactions between β−hairpin of Htt36Q3H and aromatic residues in MBP. (A) The interaction between the amide backbone of Q398 in Htt36Q3H and Y155 in MBP. (B) The interactions between the amide backbones of H399 and Q400 in Htt36Q3H and W230 in MBP. (C) The interactions between the amide backbone of Q401 in Htt36Q3H and Y210 in MBP. On panels (A), (B) and (C) the interactions at different angles are shown for C1 molecule in crystal X1. The Gln (orange) and His (pink) residues are labeled on the stick diagram The interacting residues are numbered and the interactions are shown with a dotted arrow line with interaction distances indicated in angstroms (Å). The interaction distances were measured from the backbone of the residues in the Htt36Q3H β-strand to the center of the aromatic ring residues in MBP. The supportive electron density maps (2Fo-Fc) at 1σ are shown by a green mesh for Htt38Q3H and a white mesh for MBP.
Discussion
There are a number of conclusions that can be reached from the analysis of the Htt17Q-EX128 and Htt36Q3H-EX1 (present study) structures. Both structures indicate that most N-terminal fragment of Htt protein (N17 region) exists as α-helix. This conclusion is also in agreement with previous computational and biochemical analysis of Htt-N17 region,40,41 but not with the solution NMR studies.42 However, recent magic-angle-spinning solid-state NMR studies indicated that Htt-N17 domain is in α-helical conformation, in agreement with our results.20 Both crystal structures further indicate that polyQ region can also exist in α-helical configuration, in agreement with CD spectra analysis25,26,43 and modeling results.44-46 However, in both structures the polyQ region was “conformationally unstable,” indicating that polyQ α-helix could easily unfold into a loop. A very shallow equilibrium between α-helical and loop conformations of Htt-polyQ is in agreement with various conformations reported for the same region by molecular dynamics stimulations, by biophysical measurements and by epitope mapping.15,20,22-27,43,46-49 In the Htt17Q-Ex1 structure helix-to-loop transition occurred at random positions within poly17Q region.28 In Htt36Q3H-EX1 structure helix-to-loop transition was facilitated by insertion of three histidine residues after Q394 within polyQ stretch. Thus we conjectured that the “synchronized” transition from α-helix to loop at H395 in most Htt36Q3H-Ex1 molecules increased diffraction quality of the Htt36Q3H-EX1 crystals. Consistent with this hypothesis, we could not obtain well diffracted crystals for Htt36Q-EX1 protein without His insertion.
In some Htt36Q3H-Ex1 molecules the polyQ region maintained an α-helical structure after His395 (Fig. 2A) and in other molecules remaining portion of polyQ region adopted a loop conformation (Fig. 2B). Both α-helical and loop conformations of Htt-polyQ were also observed in the Htt17Q-Ex1 structure.28 The α-helical and loop conformations of Htt-polyQ were located in the solvent region and were not involved in any protein-protein interactions in both Htt36Q3H-Ex1 and Htt17Q-Ex1 structures. Thus, we confirmed our previous conclusions28 that the α-helical and loop conformations of polyQ region are likely to be “inert” and non-pathogenic.
When compared with the previous analysis of Htt17Q-EX1 structure,28 the main new discovery in the present study is the ability of expanded polyQ region in Htt36Q3H-EX1 to adopt β-hairpin conformations. The β-strand structures observed in our crystals for Htt36Q3H were stabilized by an intramolecular hydrogen double bond (Fig. 3). It is likely that the hairpin structures constitute a nucleation site for the anti-parallel β-sheet formed by expanded polyQ tracks. Such β-sheet structure was predicted for expanded polyQ based on biophysical and computational analysis.12-21,26,46 We propose that the transition of Htt36Q3H from α-helix (Fig. 2a) to loop (Fig. 2B) to β-hairpin (Fig. 2C) captured in our crystals provides a plausible sequential conformational transition from α-helical to β-sheet conformation of expanded polyQ region in mHtt. These transitions have not been previously observed for Htt or for any other protein using near near atomic resolution techniques.
In contrast to “inert” α-helical and loop conformations of polyQ, we discovered that all hairpin structures of Htt-polyQ were stabilized by additional interactions with aromatic residues in the MBP protein (Fig. 4). It is well established that the presence of aromatic residues in a target protein facilitates interactions with expanded polyQ proteins, such as in the case of QBP1 peptide50 or HQP09 peptoid.51 Our results indicate that these interactions most likely mediated by Ar-HNb interactions between a backbone of Htt-polyQ exposed in the β-strand structure and the aromatic residues in the mHtt ligands (Fig. 4). We uncovered similar Ar-HNb interactions in the recently reported crystal structure of extended conformation of GQ10G peptide complexed with the polyQ-specific antibody.29 The potential interactions in this structure (pdb structure file 2OTW) are between backbones of Q4 and Q7 in the GQ10G peptide and Y102 and Y31 residues in the antibody, with the measured distance of 4.0 Å for both interaction pairs (data not shown). Similar interactions are likely to be involved in association of mutant Htt with Hsp70 and Hsp40 chaperons.52 We suggest that the presence of the MBP aromatic residues at positions optimal for formation Ar-HNb contacts with HQHQH β-hairpin residues may have facilitated crystallization of MBP-Htt36Q3H-EX1. This may explain why insertion of HQHQH in other positions within MBP-Htt36Q-EX1 has not resulted in the diffracting crystals (Table S1). We reasoned that association between HNb backbone groups in polyQ β-strands and aromatic residues in biologically unrelated proteins (such as MBP in our crystals) is a mimic for pathogenic interactions of mHtt in cells. We further propose that the observed β-hairpin structure constitutes a seed for β-sheet conformation of expanded polyQ in a mHtt. This conclusion is in agreement with the recent biochemical studies,19 solid-state NMR analysis20 and computational modeling studies.46
One potential limitation of the approach used in our studies is that HQHQH sequence was inserted into Htt36Q region. Interruption of polyQ track in Ataxin-1 by His insertion is known to affect pathogenesis of SCA1.30-32 Moreover, it was directly demonstrated that centrally inserted HQH sequence slows down the rates of Q30 aggregation in vitro.33 However, biophysical and modeling studies suggest that insertion of His residues within polyQ track does not result in major alterations in the polyQ structure. Indeed, it has been demonstrated that K2Q15HQHQ15K2 peptide is able to seed formation of K2Q30K2 aggregates as well as K2Q30K2 peptide itself.33 These results suggest that insertion of HQH sequence slowed down aggregation of K2Q15HQHQ15K2 peptides but did not altered properties of resulting aggregates. The β-hairpin structure observed in our crystals (Fig. 2C and 3) is in complete agreement with intramolecular β-sheet structure proposed for HQH peptides based on spectroscopic studies.34,35 As discussed by these authors, the pure polyQ peptides have tendency to form large intermolecular β-sheet structures through extended hydrogen bonding and thus tend to aggregate quickly.34,35 In contrast, polyQ peptides with His interruptions demonstrate a greater propensity for intramolecular hydrogen bonding and greater solubility.34,35 Most likely these physical properties of His-interrupted polyQ peptides facilitated formation of well diffracted MBP-Htt36Q3H-EX1 crystals and enabled us to solve the structure of β-hairpin at high resolution (Fig. 2C and 3).
Conclusions
It is likely that the conformational transitions of Htt-polyQ region from α-helix to loop to β-hairpin that we captured in our crystals of Htt36Q3H-EX1 (Fig. 2) may reflect potential behavior of many amyloidogenic proteins, such as proteins involved in AD, PD and Prion Disease (PrP).11,53-56 Similar to Htt-polyQ, the sequences of these amyloidogenic proteins have also been predicted to have an ambiguous secondary structure and similar to Htt these proteins are prone to aggregation. Also similar to Htt-polyQ many of these proteins share propensity for association with aromatic residues and aromatic molecules, such as for example with Congo Red.57 Thus, the structural information obtained in our study may have implications not only for understanding the toxicity of mHtt protein, but also for understanding the toxicity of other amyloidogenic proteins as well.
Methods
Cloning, protein expression, purification, crystallization and X-ray diffraction data collection
The first exon (EX1) fragment of human Htt containing polyQ tracks of various length (17Q, 23Q, 36Q and 45Q) were synthesized by GenScript. XQXQX interrupters (X = G, A, H) were introduced after 7th, 9th, 10th, 12th and 15th glutamine residue within a polyQ track (Table S1). The 19 aa tag (QSYQITAGKLGTGRRFTTS) was added at C-terminal (C-terminal) of each construct as previously described.28 Generated constructs were cloned into NotI and PstI sites of the pMAL vector with modified 3Ala-linker.58 The amino-acid (aa) numbering in MBP-HttPolyQ-EX1 construct starts with MBP, so that Met1 in the Htt corresponds to Met371 in the MBP-HttPolyQ-EX1. The MBP-HttPolyQ-EX1 proteins were expressed and purified according to the procedures described in our previous paper.28 Briefly, the MBP-HttPolyQ-EX1 protein was expressed in BL21 bacteria and purified on Maltose-binding affinity column, followed by molecular exclusion chromatography (SuperdexTM200 from Pharmacia Biotech) using FPLC. The purified MBP-HttPolyQ-EX1 proteins were crystallized in a hanging drop without removing MBP or C-terminal tag in order to improve protein solubility and to facilitate crystallization. Among all constructs tested the best diffracting crystals were obtained for MBP-Htt36Q3H-EX1 protein with HQHQH insertion after 7th glutamine residue (Table S1). For MBP-Htt36Q3H-EX1 proteins single crystals in size of 50/50/200 µm grew in the mother drop containing 15% polyethylene glycol MW12K, 100 mM Zn Acetate, 200 mM Sodium Floride, 100 mM Sodium Cacodylate pH = 6.5 to 7.4 at 4 to 16°C in 6 to 12 weeks. To check protein integrity, MBP-Htt36Q3H-EX1 crystals have been harvested, washed three times in the crystal-mother-solution buffer and analyzed by SDS-PAGE gel electrophoresis. The crystals with mother liquid in a loop were directly frozen at liquid-nitrogen temperature. X-ray diffraction data from the frozen crystals were collected at APS 19ID and ALS Bl8.2.2. The diffraction data collected from several MBP-Htt36Q3H-Ex1 crystals were indexed and processed by HKL2000 and two of best diffracted data sets (crystals X1 and X2) were selected to solve the MBP-Htt36Q3H-Ex1 structures.
Phasing by molecular replacement
All crystals of MBP-Htt36Q3H-EX1 had monoclinic symmetry, space group C2, with three molecules of MBP-Htt36Q3H-EX1 in the asymmetric unit (ASU). For each crystal, phasing was done by molecular replacement (MR) using the Phaser program. 40 MBP pdb files were downloaded from the protein databank and different permutations of 3 MBP models were used as possible models for three proteins in the ASU. The best solutions for both data sets (X1 and X2 crystals) from MR consisted of a trimer of 1LLS models for MBP. The two crystals were nearly isomorphous but insufficient for averaging them. Thus, the structure determination was performed separately for each crystal. The electron density maps generated by MR consisted of MBP map connected to the electron density representing Htt36Q3H-EX1. The part of the electron density corresponding to MBP protein and N17 region of Huntingtin were resolved at 2.8Å. The part of the map corresponding to polyQ region of Htt36Q3H was less clear, indicating that this part of the protein adopts multiple conformation in the crystals. This part of the map was improved in the process of iterative model building with Coot and refinement with Refmac. However, only backbone structure could be resolved for this region, forcing us to build the models initially just for the backbone of the protein. The side-chains were incorporated into the backbone model by using Ramanchadran plot and side-chain polarity information. In order to validate the resulting model we calculated Omit map using CCP4–6.2 (Fig. S1) and determined map-model correlation coefficient (Table S2). Table 1 summaries the properties of the two crystals and corresponding diffraction data sets used in solving Htt36Q3H-EX1 structure.
Model building and refinement
When the two maps generated by MR for X1 and X2 crystals were compared, it became apparent that the Htt36Q3H-EX1 fragments adopt slightly different configuration in the two crystals. To account for this fact, the models of Htt36Q3H-EX1 fragments in the two crystals were built separately but simultaneously. As these models were built, it became apparent that the molecule C in each trimer existed in a double conformation (C1 and C2). To account for a double conformation of the C molecule, the C1 and C2 models were built separately and refined with the occupancy in the range between 0.70–0.60 for each model to yield the best RWork and RFree factors during refinement using refmac5. In the process of model building and refinement the RWork and RFree were improved and the electron density map of each Htt36Q3H-EX1 became more clear. Table 1 summarizes the final refinement statistics for the two crystals with C1 and C2 conformations used for Htt36Q3H-EX1 structure determination. The ribbon diagrams for the final Htt36Q3H models were prepared using PyMOL.
Supplementary Material
Acknowledgments
I thank the personnel of ID-19 beamline at the Advanced Photon Source (APS) of Argonne National Laboratory and the bl8.2.2 bealine at ALS of Berkely Laboratories for help with data collection and processing. I thank Drs Tomchick and Chad Brautigam for organizing APS trip and providing Crystallography Core Facility, Leah Taylor for administrative assistance and Dr Ilya Bezprozvanny for comments on the paper. MWK is a Young Investigator of the National Ataxia Foundation. Funding: This work was supported by the HHMI (Johann Deisenhofer), NIH R01 NS056224 and R01NS074376 (Ilya Bezprozvanny), Hereditary Disease Foundation (MWK) and CHDI foundation (MWK, Nick Grishin and Ilya Bezprozvanny).
Glossary
Abbreviations:
- Huntington disease
HD
- Huntingtin
Htt
- polyglutamine
polyQ
- Htt polyQ expansion
mHtt
- three histidine residues
3H, the GABAergic medium spiny striatal neurons, MSN
- the first exon
EX1
- maltose binding protein
MBP
- spinal cerebral ataxia
SCA1
- polyQ binding protein1
QBP1
- Huntingtin polyQ binding peptoid 09
HQP09
- Alzheimer disease
AD
- Parkinson disease
PD
- prion disease
PrP
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/prion/article/23807
References
- 1.Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–84. doi: 10.1097/00005072-199805000-00001. [DOI] [PubMed] [Google Scholar]
- 2.The Huntington's Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington disease chromosomes. Cell. 1993;72:971–83. doi: 10.1016/0092-8674(93)90585-E. [DOI] [PubMed] [Google Scholar]
- 3.Tobin AJ, Signer ER. Huntington’s disease: the challenge for cell biologists. Trends Cell Biol. 2000;10:531–6. doi: 10.1016/S0962-8924(00)01853-5. [DOI] [PubMed] [Google Scholar]
- 4.Ross CA. Polyglutamine pathogenesis: emergence of unifying mechanisms for Huntington’s disease and related disorders. Neuron. 2002;35:819–22. doi: 10.1016/S0896-6273(02)00872-3. [DOI] [PubMed] [Google Scholar]
- 5.Rubinsztein DC. Lessons from animal models of Huntington’s disease. Trends Genet. 2002;18:202–9. doi: 10.1016/S0168-9525(01)02625-7. [DOI] [PubMed] [Google Scholar]
- 6.Li SH, Li XJ. Huntingtin-protein interactions and the pathogenesis of Huntington’s disease. Trends Genet. 2004;20:146–54. doi: 10.1016/j.tig.2004.01.008. [DOI] [PubMed] [Google Scholar]
- 7.Bezprozvanny I. Calcium signaling and neurodegenerative diseases. Trends Mol Med. 2009;15:89–100. doi: 10.1016/j.molmed.2009.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cha JH. Transcriptional signatures in Huntington’s disease. Prog Neurobiol. 2007;83:228–48. doi: 10.1016/j.pneurobio.2007.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Truant R, Atwal RS, Desmond C, Munsie L, Tran T. Huntington’s disease: revisiting the aggregation hypothesis in polyglutamine neurodegenerative diseases. FEBS J. 2008;275:4252–62. doi: 10.1111/j.1742-4658.2008.06561.x. [DOI] [PubMed] [Google Scholar]
- 10.Takahashi T, Katada S, Onodera O. Polyglutamine diseases: where does toxicity come from? what is toxicity? where are we going? J Mol Cell Biol. 2010;2:180–91. doi: 10.1093/jmcb/mjq005. [DOI] [PubMed] [Google Scholar]
- 11.Temussi PA, Masino L, Pastore A. From Alzheimer to Huntington: why is a structural understanding so difficult? EMBO J. 2003;22:355–61. doi: 10.1093/emboj/cdg044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Perutz MF. Glutamine repeats and inherited neurodegenerative diseases: molecular aspects. Curr Opin Struct Biol. 1996;6:848–58. doi: 10.1016/S0959-440X(96)80016-9. [DOI] [PubMed] [Google Scholar]
- 13.Perutz MF, Finch JT, Berriman J, Lesk A. Amyloid fibers are water-filled nanotubes. Proc Natl Acad Sci U S A. 2002;99:5591–5. doi: 10.1073/pnas.042681399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Singer SJ, Dewji NN. Evidence that Perutz’s double-beta-stranded subunit structure for beta-amyloids also applies to their channel-forming structures in membranes. Proc Natl Acad Sci U S A. 2006;103:1546–50. doi: 10.1073/pnas.0509892103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen S, Berthelier V, Yang W, Wetzel R. Polyglutamine aggregation behavior in vitro supports a recruitment mechanism of cytotoxicity. J Mol Biol. 2001;311:173–82. doi: 10.1006/jmbi.2001.4850. [DOI] [PubMed] [Google Scholar]
- 16.Tanaka M, Morishima I, Akagi T, Hashikawa T, Nukina N. Intra- and intermolecular beta-pleated sheet formation in glutamine-repeat inserted myoglobin as a model for polyglutamine diseases. J Biol Chem. 2001;276:45470–5. doi: 10.1074/jbc.M107502200. [DOI] [PubMed] [Google Scholar]
- 17.Bevivino AE, Loll PJ. An expanded glutamine repeat destabilizes native ataxin-3 structure and mediates formation of parallel beta -fibrils. Proc Natl Acad Sci U S A. 2001;98:11955–60. doi: 10.1073/pnas.211305198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Takahashi T, Kikuchi S, Katada S, Nagai Y, Nishizawa M, Onodera O. Soluble polyglutamine oligomers formed prior to inclusion body formation are cytotoxic. Hum Mol Genet. 2008;17:345–56. doi: 10.1093/hmg/ddm311. [DOI] [PubMed] [Google Scholar]
- 19.Zhang QC, Yeh TL, Leyva A, Frank LG, Miller J, Kim YE, et al. A compact beta model of huntingtin toxicity. J Biol Chem. 2011;286:8188–96. doi: 10.1074/jbc.M110.192013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sivanandam VN, Jayaraman M, Hoop CL, Kodali R, Wetzel R, van der Wel PC. The aggregation-enhancing huntingtin N-terminus is helical in amyloid fibrils. J Am Chem Soc. 2011;133:4558–66. doi: 10.1021/ja110715f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fiumara F, Fioriti L, Kandel ER, Hendrickson WA. Essential role of coiled coils for aggregation and activity of Q/N-rich prions and PolyQ proteins. Cell. 2010;143:1121–35. doi: 10.1016/j.cell.2010.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Altschuler EL, Hud NV, Mazrimas JA, Rupp B. Random coil conformation for extended polyglutamine stretches in aqueous soluble monomeric peptides. J Pept Res. 1997;50:73–5. doi: 10.1111/j.1399-3011.1997.tb00622.x. [DOI] [PubMed] [Google Scholar]
- 23.Masino L, Kelly G, Leonard K, Trottier Y, Pastore A. Solution structure of polyglutamine tracts in GST-polyglutamine fusion proteins. FEBS Lett. 2002;513:267–72. doi: 10.1016/S0014-5793(02)02335-9. [DOI] [PubMed] [Google Scholar]
- 24.Bennett MJ, Huey-Tubman KE, Herr AB, West AP, Jr., Ross SA, Bjorkman PJ. A linear lattice model for polyglutamine in CAG-expansion diseases. Proc Natl Acad Sci U S A. 2002;99:11634–9. doi: 10.1073/pnas.182393899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhattacharyya A, Thakur AK, Chellgren VM, Thiagarajan G, Williams AD, Chellgren BW, et al. Oligoproline effects on polyglutamine conformation and aggregation. J Mol Biol. 2006;355:524–35. doi: 10.1016/j.jmb.2005.10.053. [DOI] [PubMed] [Google Scholar]
- 26.Nagai Y, Inui T, Popiel HA, Fujikake N, Hasegawa K, Urade Y, et al. A toxic monomeric conformer of the polyglutamine protein. Nat Struct Mol Biol. 2007;14:332–40. doi: 10.1038/nsmb1215. [DOI] [PubMed] [Google Scholar]
- 27.Chellgren BW, Miller AF, Creamer TP. Evidence for polyproline II helical structure in short polyglutamine tracts. J Mol Biol. 2006;361:362–71. doi: 10.1016/j.jmb.2006.06.044. [DOI] [PubMed] [Google Scholar]
- 28.Kim MW, Chelliah Y, Kim SW, Otwinowski Z, Bezprozvanny I. Secondary structure of Huntingtin amino-terminal region. Structure. 2009;17:1205–12. doi: 10.1016/j.str.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Li P, Huey-Tubman KE, Gao T, Li X, West AP, Jr., Bennett MJ, et al. The structure of a polyQ-anti-polyQ complex reveals binding according to a linear lattice model. Nat Struct Mol Biol. 2007;14:381–7. doi: 10.1038/nsmb1234. [DOI] [PubMed] [Google Scholar]
- 30.Zoghbi HY, Orr HT. Spinocerebellar ataxia type 1. Semin Cell Biol. 1995;6:29–35. doi: 10.1016/1043-4682(95)90012-8. [DOI] [PubMed] [Google Scholar]
- 31.Quan F, Janas J, Popovich BW. A novel CAG repeat configuration in the SCA1 gene: implications for the molecular diagnostics of spinocerebellar ataxia type 1. Hum Mol Genet. 1995;4:2411–3. doi: 10.1093/hmg/4.12.2411. [DOI] [PubMed] [Google Scholar]
- 32.Klement IA, Skinner PJ, Kaytor MD, Yi H, Hersch SM, Clark HB, et al. Ataxin-1 nuclear localization and aggregation: role in polyglutamine-induced disease in SCA1 transgenic mice. Cell. 1998;95:41–53. doi: 10.1016/S0092-8674(00)81781-X. [DOI] [PubMed] [Google Scholar]
- 33.Jayaraman M, Kodali R, Wetzel R. The impact of ataxin-1-like histidine insertions on polyglutamine aggregation. Protein Eng Des Sel. 2009;22:469–78. doi: 10.1093/protein/gzp023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma D, Sharma S, Pasha S, Brahmachari SK. Peptide models for inherited neurodegenerative disorders: conformation and aggregation properties of long polyglutamine peptides with and without interruptions. FEBS Lett. 1999;456:181–5. doi: 10.1016/S0014-5793(99)00933-3. [DOI] [PubMed] [Google Scholar]
- 35.Sen S, Dash D, Pasha S, Brahmachari SK. Role of histidine interruption in mitigating the pathological effects of long polyglutamine stretches in SCA1: A molecular approach. Protein Sci. 2003;12:953–62. doi: 10.1110/ps.0224403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levitt M, Perutz MF. Aromatic rings act as hydrogen bond acceptors. J Mol Biol. 1988;201:751–4. doi: 10.1016/0022-2836(88)90471-8. [DOI] [PubMed] [Google Scholar]
- 37.Babu MM. NCI: A server to identify non-canonical interactions in protein structures. Nucleic Acids Res. 2003;31:3345–8. doi: 10.1093/nar/gkg528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Palermo NY, Csontos J, Murphy RF, Lovas S. The Role of Aromatic Residues in Stabilizing the Secondary and Tertiary Structure of Avian Pancreatic Polypeptide. Int J Quantum Chem. 2008;108:814–9. doi: 10.1002/qua.21521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tóth G, Watts CR, Murphy RF, Lovas S. Significance of aromatic-backbone amide interactions in protein structure. Proteins. 2001;43:373–81. doi: 10.1002/prot.1050. [DOI] [PubMed] [Google Scholar]
- 40.Kelley NW, Huang X, Tam S, Spiess C, Frydman J, Pande VS. The predicted structure of the headpiece of the Huntingtin protein and its implications on Huntingtin aggregation. J Mol Biol. 2009;388:919–27. doi: 10.1016/j.jmb.2009.01.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Atwal RS, Xia J, Pinchev D, Taylor J, Epand RM, Truant R. Huntingtin has a membrane association signal that can modulate huntingtin aggregation, nuclear entry and toxicity. Hum Mol Genet. 2007;16:2600–15. doi: 10.1093/hmg/ddm217. [DOI] [PubMed] [Google Scholar]
- 42.Thakur AK, Jayaraman M, Mishra R, Thakur M, Chellgren VM, Byeon IJ, et al. Polyglutamine disruption of the huntingtin exon 1 N terminus triggers a complex aggregation mechanism. Nat Struct Mol Biol. 2009;16:380–9. doi: 10.1038/nsmb.1570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singer D, Zauner T, Genz M, Hoffmann R, Zuchner T. Synthesis of pathological and nonpathological human exon 1 huntingtin. J Pept Sci. 2010;16:358–63. doi: 10.1002/psc.1252. [DOI] [PubMed] [Google Scholar]
- 44.Lathrop RH, Casale M, Tobias DJ, Marsh JL, Thompson LM. Modeling protein homopolymeric repeats: possible polyglutamine structural motifs for Huntington’s disease. Proc Int Conf Intell Syst Mol Biol. 1998;6:105–14. [PubMed] [Google Scholar]
- 45.Wang X, Vitalis A, Wyczalkowski MA, Pappu RV. Characterizing the conformational ensemble of monomeric polyglutamine. Proteins. 2006;63:297–311. doi: 10.1002/prot.20761. [DOI] [PubMed] [Google Scholar]
- 46.Lakhani VV, Ding F, Dokholyan NV. Polyglutamine induced misfolding of huntingtin exon1 is modulated by the flanking sequences. PLoS Comput Biol. 2010;6:e1000772. doi: 10.1371/journal.pcbi.1000772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Darnell G, Orgel JP, Pahl R, Meredith SC. Flanking polyproline sequences inhibit beta-sheet structure in polyglutamine segments by inducing PPII-like helix structure. J Mol Biol. 2007;374:688–704. doi: 10.1016/j.jmb.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 48.Legleiter J, Lotz GP, Miller J, Ko J, Ng C, Williams GL, et al. Monoclonal antibodies recognize distinct conformational epitopes formed by polyglutamine in a mutant huntingtin fragment. J Biol Chem. 2009;284:21647–58. doi: 10.1074/jbc.M109.016923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kar K, Jayaraman M, Sahoo B, Kodali R, Wetzel R. Critical nucleus size for disease-related polyglutamine aggregation is repeat-length dependent. Nat Struct Mol Biol. 2011;18:328–36. doi: 10.1038/nsmb.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nagai Y, Tucker T, Ren H, Kenan DJ, Henderson BS, Keene JD, et al. Inhibition of polyglutamine protein aggregation and cell death by novel peptides identified by phage display screening. J Biol Chem. 2000;275:10437–42. doi: 10.1074/jbc.275.14.10437. [DOI] [PubMed] [Google Scholar]
- 51.Chen X, Wu J, Luo Y, Liang X, Supnet C, Kim MW, et al. Expanded polyglutamine-binding peptoid as a novel therapeutic agent for treatment of Huntington’s disease. Chem Biol. 2011;18:1113–25. doi: 10.1016/j.chembiol.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lotz GP, Legleiter J, Aron R, Mitchell EJ, Huang SY, Ng C, et al. Hsp70 and Hsp40 functionally interact with soluble mutant huntingtin oligomers in a classic ATP-dependent reaction cycle. J Biol Chem. 2010;285:38183–93. doi: 10.1074/jbc.M110.160218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Murphy RM. Peptide aggregation in neurodegenerative disease. Annu Rev Biomed Eng. 2002;4:155–74. doi: 10.1146/annurev.bioeng.4.092801.094202. [DOI] [PubMed] [Google Scholar]
- 54.Blondelle SE, Forood B, Houghten RA, Pérez-Payá E. Polyalanine-based peptides as models for self-associated beta-pleated-sheet complexes. Biochemistry. 1997;36:8393–400. doi: 10.1021/bi963015b. [DOI] [PubMed] [Google Scholar]
- 55.Fraser PE, Nguyen JT, Surewicz WK, Kirschner DA. pH-dependent structural transitions of Alzheimer amyloid peptides. Biophys J. 1991;60:1190–201. doi: 10.1016/S0006-3495(91)82154-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nguyen J, Baldwin MA, Cohen FE, Prusiner SB. Prion protein peptides induce alpha-helix to beta-sheet conformational transitions. Biochemistry. 1995;34:4186–92. doi: 10.1021/bi00013a006. [DOI] [PubMed] [Google Scholar]
- 57.Sánchez I, Mahlke C, Yuan J. Pivotal role of oligomerization in expanded polyglutamine neurodegenerative disorders. Nature. 2003;421:373–9. doi: 10.1038/nature01301. [DOI] [PubMed] [Google Scholar]
- 58.Center RJ, Kobe B, Wilson KA, Teh T, Howlett GJ, Kemp BE, et al. Crystallization of a trimeric human T cell leukemia virus type 1 gp21 ectodomain fragment as a chimera with maltose-binding protein. Protein Sci. 1998;7:1612–9. doi: 10.1002/pro.5560070715. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.