


Abstract
Selective inhibitory crosstalk has been known to occur within the signaling pathways of the dioxin (AhR) and estrogen (ERα) receptors. More specifically, ERα represses a cytochrome P450-encoding gene (CYP1A1) that converts cellular estradiol into a metabolite that inhibits the cell cycle, while it has no effect on a P450-encoding gene (CYP1B1) that converts estrodiol into a genotoxic product. Here we show that ERα represses CYP1A1 by targeting the Dnmt3B DNA methyltransferase and concomitant DNA methylation of the promoter. We also find that histone H2A.Z can positively contribute to CYP1A1 gene expression, and its presence at that gene is inversely correlated with DNA methylation. Taken together, our results provide a framework for how ERα can repress transcription, and how that impinges on the production of an enzyme that generates genotoxic estradiol metabolites, and potential breast cancer progression. Finally, our results reveal a new mechanism for how H2A.Z can positively influence gene expression, which is by potentially competing with DNA methylation events in breast cancer cells.
INTRODUCTION
Breast cancer is the major type of cancer that affects women worldwide (http://globocan.iarc.fr/). One well known factor involved in the development of mammary tumors is estrogen. The carcinogenic effect of this hormone has several documented modes of action, one of those is through the estrogen receptor α (ERα). ERα is a transcriptional regulator that belongs to the nuclear receptor family, which regulates the expression of genes involved in cellular proliferation in response to estrogen (1,2). As a transcriptional activator, ERα is able to recruit many cofactors, such as general transcription factors, histone-modifying enzymes and ATP-dependent chromatin remodeling complexes (3). ERα has also been shown to negatively regulate gene expression but not much is currently know on how it can achieve this (4). A second mode of action by which ERα can promote breast carconogenesis is through the formation of metabolites that possess mutagenic properties. Estrogen metabolism is mediated in part by Phase I metabolizing enzymes such as CYP1A1 and CYP1B1, which can convert 17β-estradiol into 2-hydroxy-estradiol (2-OHE2) and 4-hydroxy-estradiol (4-OHE2), respectively (5,6). Numerous studies have shown that 4-OHE2 possesses genotoxic properties whereas 2-OHE2 can actually inhibit the cell cycle (7–10). Others have suggested a critical role of the CYP1B1/CYP1A1 enzyme ratio in mammary carcinogenesis (11). The major transcription factor involved in CYP1 gene expression is the Aryl hydrocarbon Receptor (AhR), also known as dioxin receptor, a ligand-activated molecule that belongs to the basic helix–loop–helix/Per–Arnt–Sim (bHLH/PAS) family of proteins (12). Pollutants such as halogenated aromatic hydrocarbons (HAHs) and polycyclic aromatic hydrocarbons (PAHs) are well characterized AhR ligands (13). AhR is sequestered into the cytoplasm; after ligand binding, it is translocated into the nucleus where it heterodimerizes with Arnt and binds to Xenobotic Response Elements (XRE’s). Importantly, there are reports showing that ERα is involved in a two-way inhibitory crosstalk with AhR. Interestingly, ERα selectively represses CYP1A1 but not CYP1B1 (14–16). Numerous mechanisms have been proposed to explain how AhR represses transcription of ERα regulated genes (17–21), but little is known about how ERα inhibits CYP1A1.
Our laboratory has previously investigated the role of histone variant H2A.Z and the p400/Tip60 complex in ERα-mediated target gene expression (4). H2A.Z is a very well conserved histone variant involved in the regulation of gene expression in many organisms from yeast to human cells (4,22,23). In mammalian cells, H2A.Z is predominantly localized in a region that surrounds the transcriptional start site (TSS) of genes, as well as distal regulatory elements (24). H2A.Z binding to these regulatory regions positively correlates with the presence of RNA polymerase II—whether active or inactive—in human cells (25). A hint for how H2A.Z may modulate gene expression emerged from studies in our laboratory (4,26). Our observations suggest that incorporation of H2A.Z within specific chromatin loci may allow ‘regulatory’ nucleosomes to adopt a stable preferred position along the translational axis of DNA, which could either favor or disfavor the recruitment of components of the transcriptional machinery to nucleosome-embedded DNA [see (27), for a more elaborate discussion of the subject]. Another hint as to how H2A.Z contributes to regulate gene expression comes from recent studies in plants and in mammals that have shown that within regulatory regions, the presence of H2A.Z and DNA methylation are mutually antagonistic (28–30). However, how H2A.Z is able to exclude DNA methylation—or vice versa—remains to be elucidated.
In this study, we investigate the mechanism of repression of CYP1A1 by ERα and the role of H2A.Z in that process. We observe that H2A.Z depletion, or ERα recruitment to the CYP1A1 proximal promoter region, impairs AhR binding. We also find that inhibition of DNA methylation with 5-azacytidine, or by cellular depletion of Dnmt3B, restores CYP1A1 expression levels in the presence of ERα. Furthermore, we show that ERα is able to interact directly with Dnmt3B. Importantly, depletion of H2A.Z leads to an increase in DNA methylation at the CYP1A1 promoter region. Taken together, our results propose a novel unexpected mechanism of repression of CYP1A1 by ERα, and a link between H2A.Z and DNA methylation in human cells.
MATERIALS AND METHODS
Chemicals and reagents
2,3,7,8-Tetrachlorodibenzo-p-dioxin (TCDD) was obtained from Cerilliant. 17β-Estradiol (E2), 4-hydroxytamoxifen (TAM), 5-azacytosine, cycloheximide and ICI 182,780 were purchased from Sigma-Aldrich. shRNA directed against different Dnmts and cloned in pLKO.1-puro lentiviral vector were bought from Sigma. The same Dnmt-targeting sequences were also cloned in the pLVTHM lentiviral vector (Trono lab). All the other shRNAs were cloned in either pLKO.1-puro lentiviral vector or in pLVTHM lentiviral vector. Their targeting sequences were listed in Supplementary Table S1.
Cell culture, lentiviral infection and treatments
All the cell lines (MCF7, MDA-MB-231, T47D and HepG2) were maintained in DMEM medium (Wisent) containing 10% fetal bovine serum (FBS, VWR) and antibiotics (Invitrogen). The cells were transduced with lentiviruses in the presence of polybrene (10 µg/ml) for 24 h immediately following cell passage. On the fifth day following infections, the cells were treated with 10 nM TCDD for 90 min (ChIP experiments) or 24 h (RT-qPCR experiments). For estrogen-induction assays, cells were grown in phenol red-free DMEM medium (Wisent) containing 5% dextran-coated charcoal-treated fetal bovin serum and antibiotics for 3 days and then treated for 90 min or 24 h with 10 nM TCDD and/or 100 nM E2.
RT-qPCR
Human CYP1A1 and CYP1B1 mRNAs were quantified by RT-qPCR with 36B4 as an internal control. Total RNA was extracted from cultured cells using GenElute (Sigma) and reverse transcribed using the M-MLV reverse transcriptase enzyme (Promega). The RT-qPCR primer sequences were listed in Supplementary Table S2.
ChIP assays
ChIP assays were performed essentially as described previously (23) using the antibodies listed in Supplementary Table S3. The recovered DNA was analyzed by qPCR using sets of primers relevant to the promoter regions of the CYP1A1 and CYP1B1 genes. The qPCR primers were listed in Supplementary Table S4. Results were shown as percent of maximum signal except for H2A.Z where results are normalized to H3 to account for nucleosome density.
MeDIP
Methylated DNA immunoprecipitation (MeDIP) experiments were performed as described previously (31). An amount of 5 µg of DNA was immunoprecipitated with 10 µg of monoclonal antibody against 5-methylcytidine (A-1014) from Eurogentec.
Bisulfite sequencing
Genomic DNA was extracted as for the MeDIP experiment. For the bisulfite conversion, we used EZ DNA Methylation-Gold Kit (Zymo Research) on 2 µg of DNA. Two rounds of PCR were performed with specific primers (see in Supplementary Table S4). PCR products were cloned in pGEM-T-easy vector (Promega). After transformation, 10 clones for each different experiment were sequenced. The sequences were analyzed with QUMA (32).
Immunoprecipitation and western blot
For whole cell extract for western blotting experiments, cells were washed with PBS, harvested, resuspended in lysis buffer (50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% Na-deoxycholate, 0.2% SDS) and disrupted by passing cells through a 23G1 needle. Lysis was performed at 4°C for 1 h with continuous agitation, and the lysate was cleared by centrifugation at 14 000 rpm.
For immunoprecipitation experiments, cells were washed with PBS, harvested, resuspended in 50 mM Tris–HCl, pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% Na-deoxycholate, 0.1% SDS, 1 mM EDTA and disrupted by passing cells through a 23G1 needle. Lysis was performed at 4°C for 1 h with continuous agitation, and the lysate was cleared by centrifugation at 14 000 rpm. Dnmt3b was immunoprecipitated with 4 µg of H-230 (Santa Cruz Biotechnology). The antibodies used in the western blot experiments are listed in Supplementary Table S5.
RESULTS
ERα specifically represses CYP1A1 but not CYP1B1
As ERα was previously shown to contribute to CYP1A1 and CYP1B1 gene expression, we decided to investigate how ERα affects CYP1A1 and CYP1B1 induction by TCDD in the MCF7 breast cancer cell line. To test this, we measured the expression of both CYP1 genes by RT-qPCR after treatment with TCDD alone or in combination with estradiol (E2) for 24 h in MCF7 cells pre-grown in estrogen-free media during three days. We observed that ERα specifically represses CYP1A1 in the presence of E2 (Figure 1A) while it has no effect on CYP1B1 induction (Figure 1B). Next, to assess whether an ERα antagonist induces more repression of CYP1A1 expression than E2, we made use of tamoxifen (TAM). TAM is an E2 competitor that prevents ERα from recruiting coactivators, and it is used in breast cancer hormone therapy (33). MCF7 cells grown in estrogen-free media were treated with a combination of TCDD and TAM for 24 h. We observed that unlike E2 treatment, TAM has no effect on CYP1A1 induction (Figure 1A). To ensure that the repression of CYP1A1 was due to ERα and not E2 itself, we carried out the same experiments in the presence of ICI 182.720—a specific pharmalogical inhibitor of ERα that promotes its degradation—for 24 h prior to TCDD and TCDD+E2 treatements (Supplementary Figure S1A and B). We observe that in the absence of ERα addition of E2 has no significant effect on CYP1A1 induction by TCDD. Moreover, ICI 182.720 treatment appears to globally increase CYP1A1 and CYP1B1 basal and induced levels of gene transcription. Altogether, our results show that ERα specifically represses CYP1A1 in an E2-dependant manner in MCF7 cells.
Figure 1.
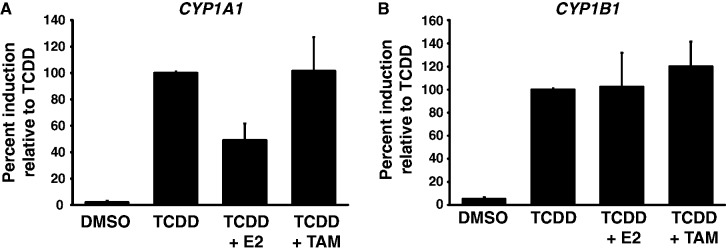
Estrogen specifically represses CYP1A1 expression in MCF7 cells. CYP1A1 (A) and CYP1B1 (B) mRNA levels were quantified in MCF7 cells grown in estrogen-free media for 3 days, and then treated with DMSO, 10 nM TCDD, 10 nM TCDD + 100 nM E2 or 10 nM TCDD + 500 nM TAM for 24 h. The results are expressed as a percentage of induction in the TCDD-treated sample.
H2A.Z depletion impairs AhR-mediated activation in ERα-positive cell lines
Since we have previously demonstrated that H2A.Z is an important positive regulator of ERα signaling, we wanted to investigate how depletion of H2A.Z would impact on TCDD-induced CYP gene expression in ERα-positive cells compared with ERα-negative cells. To test this, we depleted H2A.Z in MCF7 and T47D cells (ERα positive), and in MDA MB-231 and HepG2 cells (ERα negative) using a lentiviral shRNA construct directed to H2A.Z (4). Figure 2A shows that the H2A.Z shRNA construct is efficient at specifically depleting H2A.Z protein levels. TCDD treatment, as expected, strongly induces both CYP genes in all four cell lines (Figure 2B and C, black bars). Interestingly, knockdown of H2A.Z significantly impairs TCDD-mediated induction of both CYP1A1 and CYP1B1 in ERα-positive cell lines (Figure 2B, white bars). In both ERα-negative cell lines, knockdown of H2A.Z does not impair the induction potential of CYP1A1, while it still appears to affect induction of CYP1B1 (Figure 2C). Taken together, these result show that H2A.Z is involved in CYP1A1 and CYP1B1 gene expression, but its apparent contribution in regulation differs between the two genes.
Figure 2.

H2A.Z depletion impairs AhR-mediated activation in ERα-positive cell lines. (A) MCF7 cells were infected with shCT or shH2A.Z constructs for 5 days, then protein extraction and western blots were performed. Analysis of CYP1A1 and CYP1B1 mRNA expression was performed in ERα-positive cell lines (B) or in ERα-negative cell lines (C). The different cell lines were infected with shCT or shH2A.Z constructs for 5 days and then treated with 10 nM TCDD for 24 h.
ERα-mediated repression of CYP1A1 as well as depletion of H2A.Z reduce AhR binding to the promoter region
The effect of H2A.Z depletion on CYP1A1 expression lead us to ask whether the presence of H2A.Z at its promoter is required to allow full expression and recruitment of AhR and RNA polII. ChIP experiments in MCF7 cells show that H2A.Z is enriched at the CYP1A1 XRE’s under uninduced conditions, whereas its binding is significantly reduced upon induction of the gene by TCDD (Figure 3B). Next, we depleted H2A.Z in untreated and TCDD-treated cells to score for AhR and RNA polII binding at the CYP1A1 promoter. The results of Figure 3C and D show that recruitment of both AhR and RNA polII are significantly impaired upon knockdown of H2A.Z. Surprisingly, depletion of H2A.Z has no effect on AhR binding at the CYP1B1 promoter (Supplementary Figure S2B). This result supports our previous observation that the role of H2A.Z in the regulation of these two genes is different.
Figure 3.

AhR binding at the CYP1A1 promoter is impaired in H2A.Z-depleted cells or in the presence of E2. (A) Schematic representation of the CYP1A1 promoter. The position of the amplicons A, B, C and D used in the qPCR analyses are illustrated. (B) ChIP of H2A.Z were performed in MCF7 cells treated or not with 10 nM TCDD during 90 min. ChIPs of AhR (C) and RNA polymerase II (D) were performed in MCF7 cells infected with shCT or shH2A.Z constructs for 5 days and then treated or not with 10 nM TCDD during 90 min. ChIPs of AhR (E), ERα (F) and RNA polymerase II (G) were performed in MCF7 cells grown in estrogen-free medium for 3 days, then treated with DMSO, 10 nM TCDD or 10 nM TCDD + 100 nM E2 for 90 min.
To gain insight into how ERα mediates repression of CYP1A1, we carried out several ChIP experiments in the promoter region using antibodies raised against AhR, ERα and RNA polII. In a first set of experiments, we monitored AhR binding upon ERα-mediated repressive conditions. As expected, AhR efficiently binds the CYP1A1 XRE’s when cells are treated with TCDD in estrogen-depleted culture medium (Figure 3E). However, upon addition of estradiol, we observe a significant decrease in AhR binding, which is concomitant with a significant increase in ERα recruitment (Figure 3E and F). Predictively, RNA polII levels also significantly decrease upon addition of estradiol (Figure 3G). Likewise, AhR binding at the CYP1A1 promoter is increased in the presence of the ERα inhibitor, ICI 182.720, in TCDD-treated MCF7 cells (Supplementary Figure S1C and D).
Next, we wanted to verify whether the recruitment of ERα to CYP1B1 would also affect AhR and RNA polII binding. The results show that while ERα is efficiently recruited to the CYP1B1 XRE’s upon treatment of MCF7 cells with both TCDD and E2 (Supplementary Figure S2C), it does not appear to influence the ability of AhR and RNA polII to be recruited (Supplementary Figure S2D and E). Consequently, these results support the expression data we have obtained in Figure 1 and confirm that, contrary to what is observed for CYP1A1, ERα has no effect on CYP1B1 induction.
Taken together, our results show that whatever the mechanism by which ERα represses CYP1A1, it actually results in reduced binding of the AhR activator. Moreover, H2A.Z is required for efficient binding of AhR and RNA polII at the CYP1A1 promoter.
Inhibition of de novo DNA methylation reverses the repressive effect of ERα on dioxin-induced CYP1A1 gene expression
Previous reports have shown that AhR binding to its cognate XRE sequences was significantly reduced when these binding sites were methylated in vitro (34), and in vivo (35). Because we observe that AhR binding is affected upon ERα-mediated repression of CYP1A1, we wished to verify whether inhibiting de novo DNA methylation could alleviate repression by ERα. To achieve this, we made use of 5-azacytidine (5-azaC), a cytosine analogue that prevents de novo DNA methylation (36). 5-azaC was added to cultured MCF7 cells grown in the absence of estradiol, and CYP1A1 expression was monitored by qRT-PCR after treatment with TCDD alone or in combination with E2 for 24 h. Our results show that E2 significantly inhibits CYP1A1 expression, while addition of 5-azaC reverses ERα-mediated repression (Figure 4A). Interestingly, 5-azaC actually increases TCDD-dependent expression of both CYP1A1 and CYP1B1(Figure 4A and B). Next, we wanted to verify how 5-azaC would influence AhR binding after treating cells with both TCDD and E2. As expected, E2 treatment reduces AhR binding to the CYP1A1 XRE’s upon TCDD-mediated activation of the gene (Figure 4C). However, in the presence of 5-azaC, AhR levels remain unaffected when cells are treated with both E2 and TCDD (Figure 4C). We also monitored the presence of ERα under the same conditions in the presence or absence of 5-azaC. Strikingly, the levels of ERα are not diminished in the presence of 5-azaC and E2 (Figure 4D). Taken together, these results suggest that ERα mediates CYP1A1 repression by virtue of DNA methylation.
Figure 4.

Inhibition of DNA methylation restores full induction of CYP1A1 and AhR binding at the CYP1A1 promoter in presence of E2. CYP1A1 (A) and CYP1B1 (B) mRNA levels were quantified in MCF7 cells treated with 10 µM 5-azacytidine for 5 days and grown in estrogen-free medium for 3 days, then treated with 10 nM TCDD or 10 nM TCDD + 100 nM E2 during 24 h. ChIPs of AhR (C) and ERα (D) were performed in MCF7 cells treated with 10 µM 5-azacytidine for 5 days and grown in estrogen-free medium for 3 days, then treated with 10 nM TCDD or 10 nM TCDD + 100 nM E2 for 90 min. Primer B was used for the qPCR analysis.
Dnmt3B is involved in ERα-mediated repression of CYP1A1
We next sought to identify potential DNA methyltransferases that could be involved in repression of CYP1A1. To do this, we engineered lentivirus-expressed shRNA constructs directed against three DNA methyltransferases Dnmt1, Dnmt3A and Dnmt3B (Supplementary Figure S3). To monitor the effect of Dnmt’s on CYP expression, selected shRNA constructs were expressed in MCF7 cells prior to treatment with or without TCDD and E2. Knockdown of either Dnmt1 or Dnmt3B alleviates the ERα-mediated repression of TCDD-induced CYP1A1, while knockdown of Dnmt3A has no effect (Figure 5A). Knockdown of Dnmt1 or Dnmt3a has no significant effect at CYP1B1, but knockdown of Dnmt3B appears to increase its expression independently of ERα (Figure 5B). As a control, we wished to investigate whether cellular depletion of the Dnmt1 and Dnmt3B DNA methyltransferases would have any effect on ERα expression itself, a result that could account for the derepression observed at CYP1A1. Immunoblotting experiments show that knockdown of Dnmt3B has no effect on ERα expression, while knockdown of Dnmt1 significantly reduces ERα expression (Figure 5C). While this result does not completely rule out a potential role for Dnmt1 in mediating repression at CYP1A1, it certainly complicates further investigations. We have thus pursued our investigations only with Dnmt3B for our studies. An important prediction of the aforementioned results is that Dnmt3B should be associated to the CYP1A1 locus upon treatment of cells with E2. ChIP experiments using an anti-Dnmt3B antibody show a significant enrichment of Dmnt3B at the CYP1A1 XRE’s after treatment with TCDD + E2, but not with TCDD alone (Figure 5D). These results suggest that ERα directly recruits Dnmt3B to CYP1A1 to repress its expression. In line with this possibility, we have been able to detect direct protein–protein interactions between the ERα and Dnmt3B (Supplementary Figure S4). Taken together, our results suggest that Dnmt3B functions downstream of ERα to mediate repression of TCDD-induced CYP1A1 but not CYP1B1, and that ERα might directly recruit Dnmt3B in the process.
Figure 5.

ERα can not repress CYP1A1 induction in Dnmt3B-depleted cells. CYP1A1 (A) and CYP1B1 (B) expression was measured in MCF7 cells infected with shCT, shDnmt1, shDnmt3A or shDnmt3B constructs for 5 days and then treated with DMSO (D), 10 nM TCDD (T) or 10 nM TCDD + 100 nM E2 (T+E2) for 24 h. (C) MCF7 cells were infected with shCT, shDnmt1 and shDnmt3b constructs for 5 days, and then proteins were extracted and western blot performed to verify ERα protein levels. Actin is used as loading control. ChIP of Dnmt3B (D) was performed in MCF7 cells grown in estrogen-free media for 3 days, and then treated with DMSO, TCDD or TCDD + E2 for 90 min.
ERα-Dnmt3B direct a specific methylation pattern at the CYP1A1 promoter
Because Dnmt3B is essential to mediate ERα-directed repression of CYP1A1, we wanted to monitor how its presence at CYP1A1 could influence the methylation pattern of the proximal promoter region (Figure 6A). We chose that particular region of the gene because it is where we observed a decrease in AhR binding upon ERα-mediated repression (Figure 5C). We performed bisulfite sequencing on genomic DNA extracts from MCF7 cells grown either in the presence or absence of E2 and TCDD, and with or without prior treatment with an shRNA directed to Dnmt3B. Figure 6A shows the raw bisulfite sequencing data and Figure 6B shows a the percentage of methylation at the CYP1A1 XRE3 obtained from Figure 6A. Two significant observations can be made: (i) addition of E2 to MCF7 cells increases DNA methylation at XRE3; (ii) knockdown of Dnmt3B greatly decreases E2-mediated methylation of XRE3. Taken together these results suggest that ERα/Dnmt3B appear to direct a specific methylation pattern at XRE3, which combined with expression results showed in Figures 4A and 5A seems important to mediate repression of CYP1A1 in presence of E2.
Figure 6.

ERα induces DNA methylation at the XRE-3 of the CYP1A1 promoter. (A) Schematic representation of the CYP1A1 promoter and XRE positions. Bisulfite sequencing was performed in MCF7 cells infected with shCT or shDnmt3B constructs, grown in estrogen-free media for 3 days and treated with DMSO, TCDD or TCDD + E2 for 24 h. XRE’s are numbered relatively to their positions from the TSS of CYP1A1, and each red rectangles represent one XRE. (B) Graphical representation of the percentage of unmethylated and methylated CpGs in XRE-3 (*P < 0.05).
H2A.Z antagonizes DNA methylation at the CYP1A1 proximal promoter
Because H2A.Z is important for the ability of AhR to bind its cognate XRE’s under repressive conditions (Figure 2), we wanted to verify whether the histone variant could directly regulate DNA methylation levels at the CYP1A1 promoter. This notion is supported by the fact that the presence of H2A.Z at regulatory regions genome wide has been found to be mutually exclusive with DNA methylation (28–30). As a first approch, we investigated DNA methylation levels at the entire CYP1A1 promoter by bisulfite sequencing (Supplementary Figure S5). We observed that most of the promoter is unmethylated except for two regions (E and F) that are localized upstream the XRE4. Next, we performed MeDIP experiments on MCF7 cells which express either control (CT) or H2A.Z-directed shRNA. The MeDIP experiments make use of an antibody that specifically recognizes methylated DNA. Two amplicons were used for the MeDIP qPCR analysis (Figure 7A): amplicon A is located in the methylated region that is devoid in H2A.Z whereas amplicon B is in an unmethylated region that is strongly enriched in H2A.Z (Figure 3B). The results shown in Figure 7B represent a ratio of immunoprecipitated methylated DNA from cells depleted for H2A.Z over control cells (i.e. using a scrambled shRNA construct); thus, it is representative of de novo methylation at these loci in the absence of the histone variant. Strikingly, we observe that the knockdown of H2A.Z significantly increases DNA methylation levels at amplicon B by about 3.5-fold, whereas no significant increase is observed at amplicon A (Figure 7B). We next wanted to substantiate this finding by using bisulfite sequencing of the regions encompassing XRE’s 2, 3 and 4 in control versus H2A.Z-depleted cells. Consistent with our MeDIP results, we find that absence of H2A.Z globally increases DNA methylation in that area, particularly around XRE 2 and 3 (Figure 7C). Taken together, our results suggest that the presence of H2A.Z can exclude DNA methylation at the CYP1A1 proximal XRE’s, and as such, favors AhR recruitment upon induction.
Figure 7.

H2A.Z depletion promotes DNA methylation at the CYP1A1 promoter. (A) Schematic representation of the CYP1A1 promoter. The position of the amplicons A and B used in the qPCR analyses are illustrated. (B) MeDIP was performed in MCF7 cells infected with shCT or shH2A.Z constructs for 5 days. (C) Bisulfite sequencing were performed in MCF7 cells infected with shCT or shH2A.Z constructs and grown in DMEM 10% FBS for 5 days.
DISCUSSION
Carcinogenesis is a multistep process, and in breast cancer, estrogen and ERα are critical players in the initiation and progression stages. Most mechanistic studies on ERα have focussed on its positive role in gene transcription, but less is known about how it represses transcription, as well as which cofactors are involved in this repression process. In breast tissues, maintenance of a high CYP1A1/CYP1B1 enzyme ratio ensures that intracellular levels of 2-OHE2 are high and levels of 4-OHE2 are low. However, in cancer cells and tumors, elevated concentrations of 4-OHE2 are predominant as compared with normal tissus, and these observations are correlated with a higher expression of CYP1B1. In this study, we observe that ERα represses CYP1A1 specifically without affecting CYP1B1 expression, which is consistent with previous findings (14,15). Our results suggest a mechanism for how ERα represses CYP1A1: in the absence of estradiol and following TCDD treatment, ERα is absent from the promoter and maximal AhR binding is achieved, thus allowing CYP1A1 transcription (Figure 8A). In the presence of estradiol and TCDD, ERα, by virtue of its interaction with AhR-Arnt, is recruited to the CYP1A1 promoter, which in turn directs the recruitment of Dnmt3B, an outcome that promotes DNA methylation of the AhR response elements. Methylation of specific CYP1A1 XRE’s impairs AhR binding and consequently decreases CYP1A1 expression (Figure 8B). We suggest that increasing DNA methylation levels at CYP1A1 XRE’s can impair H2A.Z incorporation at the end of the activation process. Taken together, we describe a novel mechanism by which ERα can repress transcription of an AhR target gene. Nevertheless, our study reveals a mechanism for how ERα could promote breast tumorigenesis by differentially regulating the expression of enzymes involved in estrogen metabolism. Indeed, other laboratories have previously observed specific repression of CYP1A1 by ERα without affecting CYP1B1 expression (14,15). In our model, the presence of E2 and TCDD are both necessary for ERα recrutment to the CYP1A1 promoter. It remains to be determined, however, whether Dnmt3B is also involved in the inhibition of other genes that are repressed by ERα.
Figure 8.

Proposed model for CYP1A1 gene regulation by AhR and ERα. (A) In the absence of estradiol, when TCDD is added in the media, AhR /Arnt binds the XRE’s located in the CYP1A1 promoter. At the same time, the histone variant H2A.Z is removed from the XRE’s. (B) In the presence of estradiol, ERα displaces AhR /Arnt by promoting DNA methylation on the XRE’s of the CYP1A1 promoter, thus resulting in less AhR activating surfaces available to stimulate CYP1A1 expression.
Changes in DNA methylation patterns are frequently observed in cancer cells when compared with normal cells (37). Despite hypermethylation of tumor suppressor gene promoters (38,39), global 5-methylcytosine content is decreased in tumor cells (40). This global hypomethylation observed in cancer cells can be explained by a drastic decrease of DNA methylation in repeated sequences such as LINEs and SINEs, which would then result in an increase in genome instability caused by recombination or displacement of these sequences. Our results show that Dmnt3B is specifically required for ERα-dependent gene repression without altering ERα expression itself. This result suggests that Dnmt3B may play a greater role in cancer progression than other Dnmt’s. This is supported by Girault et al. (41) who analyzed the expression of each DNMT gene (DNMT1, DNMT3A and DNMT3B) in breast carcinomas isolated from 130 patients. Dnmt3B was shown to be overexpressed in 30% of the tumors, and the authors proposed that Dnmt3B may play a predominant role over Dnmt3A and Dnmt1 in breast carcinogenesis. Interestingly, Dnmt3B possesses numerous splice variants that are differentialy expressed in normal and cancer cell lines (42). One of these variants, Dnmt3B7, is able to significantly change DNA methylation patterns when expressed to high levels (42). Whether each of the Dnmt3B isoforms is capable of interacting with ERα and mediate repression of CYP1A1 expression (and potentially other genes repressed by ERα) is a matter of further investigations. Variation in the expression level of Dnmt3B isoforms between breast cancer cell lines might explain, at least in part, why there are some discrepancies in the literature regarding the positive or negative role of ERα in CYP1A1 expression.
Genome-wide studies have shown an enrichment of H2A.Z in promoters, enhancers and insulators in numerous species (24–26). However, little is known about the function of H2A.Z at these regions. In 2009 and 2010, the Henikoff and Zilberman laboratories have elegantly demonstrated an antagonistic relationship between H2A.Z and DNA methylation, first in Arabidopsis thaliana and fungi and animals (28,30). It has been proposed that methylation is the default state of nucleosomal DNA and that unmethylated regions are protected from DNA methylation by histone modifications such as H3K4me, or deposition of histone variant H2A.Z (43). From these observations, it has been suggested that the presence of H2A.Z could prevent DNA methylation at CpG islands located within regulatory regions, and thus protect those regions from silencing. A previous study also showed that removing DNA methylation by 5-azacytidine treatment quickly induces H2A.Z incorporation in a subset of genes in colon cancer cell lines (44). However, incorporation of H2A.Z within DNA was not sufficient to restore gene expression in that context. In our study, we demonstrate that depletion of H2A.Z leads to a 3.5-fold increase in DNA methylation of the CYP1A1 promoter after only 5 days. We also show that the increase in DNA methylation induced by H2A.Z depletion impairs CYP1A1 induction following TCDD treatement. However, we hypothesize that extended loss of H2A.Z could lead to a more important increase in DNA methylation than what we currently observe. Another open question is whether the regulated methylation events that we observe at CYP1A1 are actively reversible. Interestingly, a study by Metivier et al. (45) has demonstrated that both Dnmt3A and Dnmt3B are involved in cyclical methylation and demethylation (by deamination) of the ERα-target gene, TFF1. In fact these authors have shown that this dual event of methylation and demethylation by the same enzymes was necessary for the activation process. It remains to be determined whether similar mechanisms of action are involved in repression of CYP1A1 by the ERα and whether other enzymes are also involved. Finally, it will be interesting to investigate whether H2A.Z deposition is also dependent or influenced by such potential demethylation cycles.
Taken together, our study unravels two new keys players (H2A.Z and Dnmt3B) in the regulation of CYP1A1 expression. These two factors play a crucial role in de novo DNA methylation establishment, which is thought to be a major early event in the initiation of tumor formation. Methylation of the CYP1A1 promoter is already associated with prostate and lung cancers (35,46). It will be interesting to test whether this observation is also true in mammary tumors and more generaly to all hormone responding tissues.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Cancer Research Society of Canada (to L.G.) [018924001]. LG holds a Canada Research Chair on Mechanisms of Gene Transcription [014077001]. Funding for open access charge: Cancer Research Society of Canada.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr. Benoit Leblanc for artwork presented in Figure 8 and Dr. Benoit Guillemette for critical reading of the manuscript. We also thank Dr. Matthew Lorincz for help in primer design, and analysis of bisulfite sequencing experiments.
REFERENCES
- 1.Dubik D, Dembinski TC, Shiu RP. Stimulation of c-myc oncogene expression associated with estrogen-induced proliferation of human breast cancer cells. Cancer Res. 1987;47:6517–6521. [PubMed] [Google Scholar]
- 2.Altucci L, Addeo R, Cicatiello L, Dauvois S, Parker MG, Truss M, Beato M, Sica V, Bresciani F, Weisz A. 17beta-Estradiol induces cyclin D1 gene transcription, p36D1-p34cdk4 complex activation and p105Rb phosphorylation during mitogenic stimulation of G(1)-arrested human breast cancer cells. Oncogene. 1996;12:2315–2324. [PubMed] [Google Scholar]
- 3.Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–763. doi: 10.1016/s0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 4.Gevry N, Hardy S, Jacques PE, Laflamme L, Svotelis A, Robert F, Gaudreau L. Histone H2A.Z is essential for estrogen receptor signaling. Genes Dev. 2009;23:1522–1533. doi: 10.1101/gad.1787109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Martucci CP, Fishman J. P450 enzymes of estrogen metabolism. Pharmacol. Ther. 1993;57:237–257. doi: 10.1016/0163-7258(93)90057-k. [DOI] [PubMed] [Google Scholar]
- 6.Tsuchiya Y, Nakajima M, Yokoi T. Cytochrome P450-mediated metabolism of estrogens and its regulation in human. Cancer Lett. 2005;227:115–124. doi: 10.1016/j.canlet.2004.10.007. [DOI] [PubMed] [Google Scholar]
- 7.Liehr JG, Ricci MJ. 4-Hydroxylation of estrogens as marker of human mammary tumors. Proc. Natl Acad. Sci. USA. 1996;93:3294–3296. doi: 10.1073/pnas.93.8.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liehr JG, Fang WF, Sirbasku DA, Ari-Ulubelen A. Carcinogenicity of catechol estrogens in Syrian hamsters. J. Steroid. Biochem. 1986;24:353–356. doi: 10.1016/0022-4731(86)90080-4. [DOI] [PubMed] [Google Scholar]
- 9.Yager JD, Liehr JG. Molecular mechanisms of estrogen carcinogenesis. Annu. Rev. Pharmacol. Toxicol. 1996;36:203–232. doi: 10.1146/annurev.pa.36.040196.001223. [DOI] [PubMed] [Google Scholar]
- 10.Newbold RR, Liehr JG. Induction of uterine adenocarcinoma in CD-1 mice by catechol estrogens. Cancer Res. 2000;60:235–237. [PubMed] [Google Scholar]
- 11.Coumoul X, Diry M, Robillot C, Barouki R. Differential regulation of cytochrome P450 1A1 and 1B1 by a combination of dioxin and pesticides in the breast tumor cell line MCF-7. Cancer Res. 2001;61:3942–3948. [PubMed] [Google Scholar]
- 12.Hankinson O. The aryl hydrocarbon receptor complex. Annu. Rev. Pharmacol. Toxicol. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 13.Poland A, Knutson JC. 2,3,7,8-tetrachlorodibenzo-p-dioxin and related halogenated aromatic hydrocarbons: examination of the mechanism of toxicity. Annu. Rev. Pharmacol Toxicol. 1982;22:517–554. doi: 10.1146/annurev.pa.22.040182.002505. [DOI] [PubMed] [Google Scholar]
- 14.Kietz S, Feng S, Agoulnik A, Hombach-Klonisch S. Estrogen and TCDD influence RLN2 gene activity in estrogen receptor-positive human breast cancer cells. Ann. NY Acad. Sci. 2009;1160:367–373. doi: 10.1111/j.1749-6632.2009.03836.x. [DOI] [PubMed] [Google Scholar]
- 15.Kharat I, Saatcioglu F. Antiestrogenic effects of 2,3,7,8-tetrachlorodibenzo-p-dioxin are mediated by direct transcriptional interference with the liganded estrogen receptor. Cross-talk between aryl hydrocarbon- and estrogen-mediated signaling. J. Biol. Chem. 1996;271:10533–10537. doi: 10.1074/jbc.271.18.10533. [DOI] [PubMed] [Google Scholar]
- 16.Bjeldanes LF, Kim JY, Grose KR, Bartholomew JC, Bradfield CA. Aromatic hydrocarbon responsiveness-receptor agonists generated from indole-3-carbinol in vitro and in vivo: comparisons with 2,3,7,8-tetrachlorodibenzo-p-dioxin. Proc. Natl Acad. Sci. USA. 1991;88:9543–9547. doi: 10.1073/pnas.88.21.9543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nguyen TA, Hoivik D, Lee JE, Safe S. Interactions of nuclear receptor coactivator/corepressor proteins with the aryl hydrocarbon receptor complex. Arch. Biochem. Biophys. 1999;367:250–257. doi: 10.1006/abbi.1999.1282. [DOI] [PubMed] [Google Scholar]
- 18.Kumar MB, Tarpey RW, Perdew GH. Differential recruitment of coactivator RIP140 by Ah and estrogen receptors. Absence of a role for LXXLL motifs. J. Biol. Chem. 1999;274:22155–22164. doi: 10.1074/jbc.274.32.22155. [DOI] [PubMed] [Google Scholar]
- 19.Kumar MB, Perdew GH. Nuclear receptor coactivator SRC-1 interacts with the Q-rich subdomain of the AhR and modulates its transactivation potential. Gene Expr. 1999;8:273–286. [PMC free article] [PubMed] [Google Scholar]
- 20.Krishnan V, Porter W, Santostefano M, Wang X, Safe S. Molecular mechanism of inhibition of estrogen-induced cathepsin D gene expression by 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) in MCF-7 cells. Mol. Cell. Biol. 1995;15:6710–6719. doi: 10.1128/mcb.15.12.6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohtake F, Baba A, Takada I, Okada M, Iwasaki K, Miki H, Takahashi S, Kouzmenko A, Nohara K, Chiba T, et al. Dioxin receptor is a ligand-dependent E3 ubiquitin ligase. Nature. 2007;446:562–566. doi: 10.1038/nature05683. [DOI] [PubMed] [Google Scholar]
- 22.Meneghini MD, Wu M, Madhani HD. Conserved histone variant H2A.Z protects euchromatin from the ectopic spread of silent heterochromatin. Cell. 2003;112:725–736. doi: 10.1016/s0092-8674(03)00123-5. [DOI] [PubMed] [Google Scholar]
- 23.Gevry N, Chan HM, Laflamme L, Livingston DM, Gaudreau L. p21 transcription is regulated by differential localization of histone H2A.Z. Genes Dev. 2007;21:1869–1881. doi: 10.1101/gad.1545707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. doi: 10.1016/j.cell.2007.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Hardy S, Jacques PE, Grévry N, Forest A, Fortin ME, Laflamme L, Gaudreau L, Robert F. The euchromatic and heterochromatic landscapes are shaped by antagonizing effects of transcription on H2A.Z deposition. PLoS Genet. 2009;5:e1000687. doi: 10.1371/journal.pgen.1000687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guillemette B, Bataille AR, Gevry N, Adam M, Blanchette M, Robert F, Gaudreau L. Variant histone H2A.Z is globally localized to the promoters of inactive yeast genes and regulates nucleosome positioning. PLoS Biol. 2005;3:e384. doi: 10.1371/journal.pbio.0030384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marques M, Laflamme L, Gervais AL, Gaudreau L. Reconciling the positive and negative roles of histone H2A.Z in gene transcription. Epigenetics. 2010;5:267–272. doi: 10.4161/epi.5.4.11520. [DOI] [PubMed] [Google Scholar]
- 28.Zilberman D, Coleman-Derr D, Ballinger T, Henikoff S. Histone H2A.Z and DNA methylation are mutually antagonistic chromatin marks. Nature. 2008;456:125–129. doi: 10.1038/nature07324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Conerly ML, Teves SS, Diolaiti D, Ulrich M, Eisenman RN, Henikoff S. Changes in H2A.Z occupancy and DNA methylation during B-cell lymphomagenesis. Genome Res. 2010;20:1383–1390. doi: 10.1101/gr.106542.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zemach A, McDaniel IE, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. doi: 10.1126/science.1186366. [DOI] [PubMed] [Google Scholar]
- 31.Sorensen AL, Collas P. Immunoprecipitation of methylated DNA. Methods Mol. Biol. 2009;567:249–262. doi: 10.1007/978-1-60327-414-2_16. [DOI] [PubMed] [Google Scholar]
- 32.Kumaki Y, Oda M, Okano M. QUMA: quantification tool for methylation analysis. Nucleic Acids Res. 2008;36:W170–W175. doi: 10.1093/nar/gkn294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bernstein L, Deapen D, Cerhan JR, Schwartz SM, Liff J, McGann-Maloney E, Perlman JA, Ford L. Tamoxifen therapy for breast cancer and endometrial cancer risk. J. Natl Cancer Inst. 1999;91:1654–1662. doi: 10.1093/jnci/91.19.1654. [DOI] [PubMed] [Google Scholar]
- 34.Shen ES, Whitlock JP., Jr The potential role of DNA methylation in the response to 2,3,7,8-tetrachlorodibenzo-p-dioxin. J. Biol. Chem. 1989;264:17754–17758. [PubMed] [Google Scholar]
- 35.Okino ST, Pookot D, Li LC, Zhao H, Urakami S, Shiina H, Igawa M, Dahiya R. Epigenetic inactivation of the dioxin-responsive cytochrome P4501A1 gene in human prostate cancer. Cancer Res. 2006;66:7420–7428. doi: 10.1158/0008-5472.CAN-06-0504. [DOI] [PubMed] [Google Scholar]
- 36.Santi DV, Norment A, Garrett CE. Covalent bond formation between a DNA-cytosine methyltransferase and DNA containing 5-azacytosine. Proc. Natl Acad. Sci. USA. 1984;81:6993–6997. doi: 10.1073/pnas.81.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat. Genet. 2009;41:178–186. doi: 10.1038/ng.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC, Baylin SB, Sidransky D. 5′ CpG island methylation is associated with transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat. Med. 1995;1:686–692. doi: 10.1038/nm0795-686. [DOI] [PubMed] [Google Scholar]
- 39.Esteller M, Silva JM, Dominguez G, Bonilla F, Matias-Guiu X, Lerma E, Bussaglia E, Prat J, Harkes IC, Repasky EA, et al. Promoter hypermethylation and BRCA1 inactivation in sporadic breast and ovarian tumors. J. Natl Cancer Inst. 2000;92:564–569. doi: 10.1093/jnci/92.7.564. [DOI] [PubMed] [Google Scholar]
- 40.Wilson AS, Power BE, Molloy PL. DNA hypomethylation and human diseases. Biochim. Biophys. Acta. 2007;1775:138–162. doi: 10.1016/j.bbcan.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 41.Girault I, Tozlu S, Lidereau R, Bieche I. Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin. Cancer Res. 2003;9:4415–4422. [PubMed] [Google Scholar]
- 42.Ostler KR, Davis EM, Payne SL, Gosalia BB, Exposito-Cespedes J, Le Beau MM, Godley LA. Cancer cells express aberrant DNMT3B transcripts encoding truncated proteins. Oncogene. 2007;26:5553–5563. doi: 10.1038/sj.onc.1210351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Edwards JR, O'Donnell AH, Rollins RA, Peckham HE, Lee C, Milekic MH, Chanrion B, Fu Y, Su T, Hibshoosh H, et al. Chromatin and sequence features that define the fine and gross structure of genomic methylation patterns. Genome Res. 2010;20:972–980. doi: 10.1101/gr.101535.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang X, Noushmehr H, Han H, Andreu-Vieyra C, Liang G, Jones PA. Gene reactivation by 5-aza-2′-deoxycytidine-induced demethylation requires SRCAP-mediated H2A.Z insertion to establish nucleosome depleted regions. PLoS Genet. 2012;8:e1002604. doi: 10.1371/journal.pgen.1002604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Metivier R, Gallais R, Tiffoche C, Le Peron C, Jurkowska RZ, Carmouche RP, Ibberson D, Barath P, Demay F, Reid G, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature. 2008;452:45–50. doi: 10.1038/nature06544. [DOI] [PubMed] [Google Scholar]
- 46.Tekpli X, Zienolddiny S, Skaug V, Stangeland L, Haugen A, Mollerup S. DNA methylation of the CYP1A1 enhancer is associated with smoking-induced genetic alterations in human lung. Int. J. Cancer. 2012;131:1509–1516. doi: 10.1002/ijc.27421. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.