

Abstract
The ductal epithelium plays a key role in physiological secretion of pancreatic enzymes into the digestive system. Loss of barrier properties of the pancreatic duct may contribute to the development of pancreatitis and metastatic dissemination of pancreatic tumors. Proinflammatory cytokines are essential mediators of pancreatic inflammation and tumor progression; however, their effects on the integrity and barrier properties of the ductal epithelium have not been previously addressed. In the present study, we investigate mechanisms of cytokine-induced disassembly of tight junctions (TJs) and adherens junctions (AJs) in a model pancreatic epithelium. Exposure of HPAF-II human pancreatic epithelial cell monolayers to interferon (IFN)γ disrupted integrity and function of apical junctions as manifested by increased epithelial permeability and cytosolic translocation of AJ and TJ proteins. Tumor necrosis factor (TNF)α potentiated the effects of IFNγ on pancreatic epithelial junctions. The cytokine-induced increase in epithelial permeability and AJ/TJ disassembly was attenuated by pharmacological inhibition of Janus kinase (JAK) and protein kinase D (PKD). Loss of apical junctions in IFNγ/TNFα-treated HPAF-II cells was accompanied by JAK and PKD dependent decrease in expression of AJ (E-cadherin, p120 catenin) and TJ (occludin, ZO-1) proteins. Depletion of E-cadherin or p120 catenin recapitulated the effects of cytokines on HPAF-II cell permeability and junctions. Our data suggests that proinflammatory cytokines disrupt pancreatic epithelial barrier via expressional downregulation of key structural components of AJs and TJs. This mechanism is likely to be important for pancreatic inflammatory injury and tumorigenesis.
Keywords: tight junctions, interferon, tumor necrosis factor, adherens junctions, pancreatitis
Introduction
Epithelial layers create protective barriers between the body interior and the surrounding environment and preserve unique biochemical composition of different tissues and organs.1-3 Disruption of epithelial barriers is a hallmark of tissue inflammation that leads to body exposure to environmental pathogens and alterations of tissue homeostasis.2-4 Integrity and barrier properties of various epithelia are determined by multiprotein adhesive complexes known as epithelial junctions that are formed between adjacent cells.5 It is well-established that disruption of intercellular junctions in the intestinal epithelium, airway epithelium, and skin epidermis can contribute to the pathogenesis of different inflammatory disorders such as inflammatory bowel disease, asthma, and atopic dermatitis.4,6-8
However, there are other types of epithelia in the human body, which normal physiological functions and roles in organ pathology remain poorly understood. A vivid example is the epithelium that forms the pancreatic duct, an elaborate tubular complex interconnecting digestive enzyme-producing acinar cells of the pancreas and collecting secreted zymogens for their delivery into the duodenum.9 Evidence suggests that disruption of this ductal barrier is a common manifestation of pancreatic pathology such as pancreatitis and pancreatic ductal adenocarcinoma. For example, clinical studies observed appearance of serum proteins in the pancreatic juice of patients with chronic pancreatitis, which is indicative of increased ductal permeability.10,11 Furthermore, disruption of paracellular sealing in ductal epithelium was found to be an early feature of animal models of acute pancreatitis.12-15 Finally, progression of pancreatic intraepithelial neoplasm in mice was recently associated with abnormal organization of epithelial cell-cell contacts.16
Similar to other types of vertebrate epithelia, pancreatic ductal epithelial cells are linked by several types of junctional complexes. Among these complexes, the most apically-located tight junctions (TJs) and adherens junctions (AJs) play key roles in epithelial morphogenesis and establishment of the paracellular barrier.17 TJs and AJs mediate cell-cell adhesions through homotypical interactions of their transmembrane proteins such as occludin, claudins, junctional adhesion molecule-A (JAM-A) and E-cadherin that are assisted by a number of cytoplasmic scaffolds.18-22 Different scaffolding proteins participate in the organization of distinct junctional complexes. Thus, members of the so called “zonula occludens” (ZO) protein family interact with occludin, claudins and JAM-A at TJs,18,19 whereas β- and p120 catenins bind to E-cadherin creating a core complex of epithelial AJs.20-22 Abnormal localization of TJ proteins, ZO-1, occludin and claudins was observed in ductal pancreatic epithelium during experimental pancreatitis15,23 and neoplastic transformation,16 although biological roles and molecular mechanisms of such abnormalities remain unknown.
Disruption of epithelial barriers in inflamed mucosa is known to be mediated by proinflamatory cytokines, most notably, interferon-γ (IFNγ) and tumor necrosis factor-α (TNFα).3,24,25 Interestingly, blood or tissue levels of these cytokines are increased in patients with chronic or acute pancreatitis and pancreatic adenocarcinoma.26-29 Furthermore, IFNγ administration aggravates autoimmune pancreatitis in mice30 and case reports describe induction of pancreatitis by anti-viral interferon-based therapy in human patients.26 Nevertheless, the involvement of proinflammatory cytokines in disruption of the pancreatic ductal barrier has not been previously addressed. In this study, we examined the effects of IFNγ and TNFα on permeability and structure of AJs and TJs in model pancreatic epithelial cell monolayers. We describe a profound dissociation of epithelial junctions in IFNγ/TNFα-treated pancreatic epithelium and dissect key molecular events that mediate such cytokine-induced junctional disassembly.
Results
Cytokines disrupt the paracellular barrier and induce AJ/TJ disassembly in model pancreatic epithelial cell monolayers
Finding an adequate in vitro model is crucial for examining signaling mechanisms that mediate establishment and disassembly of the pancreatic ductal barrier. HPAF-II human pancreatic adenocarcinoma cells have a striking resemblance to well-polarized columnar epithelium found in normal pancreatic duct.31,32 These cells readily differentiate to form robust apical junctions, whose architecture can be modulated by external stimuli.31,33 In order to investigate the effects of proinflammatory cytokines on barrier properties and integrity of pancreatic epithelial junctions, polarized HPAF-II cells were treated with either vehicle, TNFα (10 ng/ml), IFNγ (50 ng/ml) or both cytokines for 48 h. These experimental conditions were chosen based on previous studies conducted in other types of cultured epithelial cells.34-38 After 48h incubation, TNFα and IFNγ caused ~35% and 90% drop in transepithelial electrical resistance (TEER) respectively, whereas combination of these cytokines appeared to be a more potent barrier disruptor decreasing TEER by up to 98% (Fig. 1A). Additionally, TNFα alone did not affect transepithelial fluxes of fluoresceinated dextrans (Fig. 1B), while IFNγ caused approximately 4 fold and 2 fold increase in permeability of 4,000 Da and 40,000 Da dextrans respectively (Fig. 1B). Furthermore, co-treatment with TNFα and IFNγ more potently (up to 22 fold) enhanced transepithelial flux of both fluorescent markers (Fig. 1B). To investigate the effects of proinflammatory cytokines on structure of pancreatic epithelial junctions, localization of different AJ/TJ proteins was analyzed using immunofluorescence labeling and confocal microscopy. Incubation of HPAF-II cell monolayers with TNFα did not affect the integrity of AJs and TJs as was made evident by intact “chicken wire” labeling patterns of E-cadherin, ZO-1and claudin-4 at the cell apex (Fig. 2, arrows). By contrast, IFNγ alone disrupted the linear junctional labeling of ZO-1 and caused accumulation of intracellular vesicles containing E-cadherin, claudin-4 (Fig. 2, arrowheads), p-120-catenin and β-catenin (data not shown). The combination of TNFα and IFNγ induced more profound disassembly of pancreatic epithelial junctions comparing to IFNγ-only treatment (Fig. 2). Together, these results suggest that IFNγ is a potent disruptor of pancreatic epithelial AJs and TJs and that TNFα enhances the effects of interferon on the pancreatic ductal barrier.
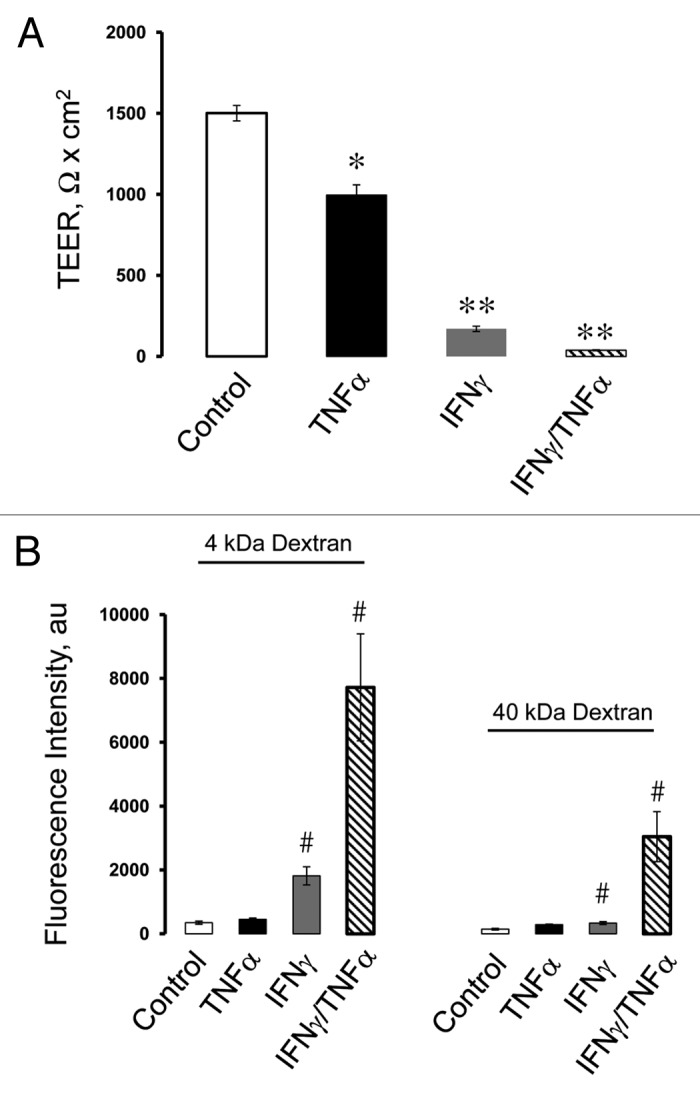
Figure 1. Proinflammatory cytokines increase permeability of human pancreatic epithelial cell monolayers. Confluent HPAF-II cell monolayers were exposed for 48 h to either vehicle, TNFα (10 ng/ml), IFNγ (50 ng/ml) or a combination of these two cytokines. Integrity of the epithelial barrier was evaluated by measuring transepithelial electrical resistance (TEER) and fluorescent dextran fluxes. Permeability assays show significant decrease in TEER in all cytokine-treated groups (A) and enhancement of dextran fluxes in IFNγ and IFNγ/TNFα exposed cells (B). Data are presented as mean ± SE (n = 3); #p < 0.05 *p < 0.01; **p < 0.001compared to control cells.

Figure 2. IFNγ induces disassembly of pancreatic epithelial tight junctions and adherens junctions. Confluent HPAF-II cell monolayers were treated for 48 h with either vehicle, TNFα, IFNγ or a combination of these two cytokines. Localization of AJ and TJ proteins, E-cadherin, ZO-1 and claudin-4, was determined by fluorescence labeling and confocal microscopy. IFNγ induces translocation of AJ and TJ proteins from the areas of cell-cell contact (arrows) into cytosol (arrowheads), while TNFα potentiates IFNγ-driven junctional disassembly. Bar, 20 µm.
Cytokine-dependent disruption of the pancreatic ductal barrier is accompanied by downregulation of AJ/TJ proteins
Several different mechanisms have been implicated in disassembly of epithelial junctions during experimental or clinical inflammation. These mechanisms include decreased expression of junctional proteins, endocytosis of TJ components and reorganization of the perijunctional actin cytoskeleton.4,24,39,40 Since IFNγ is known to be a potent modulator of protein expression in different cell types,24,41,42 we investigated whether decreased expression of molecular constituents of AJs or TJs can be involved in cytokine-induced disassembly of pancreatic epithelial junctions. Immunoblotting analysis revealed that exposure of HPAF-II cells to IFNγ and TNFα caused a marked (50–85%) decrease in the levels of several AJ (E-cadherin, β-catenin and p120 catenin) and TJ (occludin, ZO-1and JAM-A) proteins (Fig. 3). Such expressional downregulation was evident already after 24 h of cytokine exposure, thus preceding morphological signs of AJ/TJ disassembly. IFNγ alone also decreased expression of junctional proteins although not as fast and efficiently as in combination with TNFα (Fig. 3).

Figure 3. Cytokines decrease expression of different AJ and TJ proteins. Representative immunoblots (A) and densitometric quantification (B) show that exposure of HPAF-II cells to either IFNγ or IFNγ plus TNFα markedly decreases the levels of different AJ/TJ proteins. Data are presented as mean ± SE (n = 3); #p < 0.05, *p < 0.01, ** p < 0.001 compared with appropriate controls.
To gain insight into molecular mechanisms of cytokine-induced loss of AJ/TJ proteins, we analyzed their mRNA expression. Interestingly, quantitative RT-PCR analysis revealed differential effects of cytokines on mRNA transcription of AJ and TJ proteins. Indeed, transcription of occludin and ZO-1 was significantly decreased in HPAF-II cells after 24 h incubation with IFNγ/TNFα as compared with control cells (Fig. 4A). By contrast, no such decrease was observed for E-cadherin and p120-catenin mRNA levels (Fig. 4A). This data suggests that cytokines diminished expression of AJ proteins via post-transcriptional mechanisms. One likely mechanism involves accelerated degradation of junctional components by either lysosomes or proteasomes. To test this possibility, we exposed IFNγ/TNFα-treated cells to either vehicle, lysosomal (concanamycin A (100 nM) and chloroquine (100 µM)) or proteasomal (MG-132 (25 µM) and epoxomicin, (1 µM)) inhibitors. However, blockage of protein degradation failed to prevent cytokine-induced decrease in the expression of E-cadherin, p120 catenin, occludin and ZO-1 in HPAF-II cells (Fig. 4B). These results indicate that accelerated degradation cannot be responsible for the decreased expression of AJ/TJ proteins caused by IFNγ and TNFα.

Figure 4. Cytokines inhibit expression of AJ/TJ proteins by mRNA transcription-dependent and independent mechanisms. (A) Real-time quantitative RT-PCR data shows a significant decrease in mRNA levels for major TJ but not AJ proteins in IFNγ/TNFα-treated HPAF-II cells, compared with the control. Data are presented as mean ± SE (n = 3); *p < 0.01compared to control cells. (B) Immunoblotting analysis shows that 48 h blockage of lysosomal proteolysis with chloroquine (100 µM) or concanamycin A (100 nM), or inhibition of proteosomal protein degradation with MG-132 (25 µM) or epoxomicin (1 µM) does not restore normal expression of AJ/TJ proteins in cytokine-treated HPAF-II cells. (C) Immunoblotting with anti-puromycin antibody shows that cytokines inhibit puromycin incorporation in different proteins extracted from HPAF-II cells thereby indicating inhibition of global protein translation.
Proinflammatory cytokines inhibit protein translation in pancreatic epithelial cells
An alternative mechanism of posttranscriptional regulation of protein expression involves translation of the polypeptide chain. Therefore, we investigated if proinflammatory cytokines affect protein translation in HPAF-II cells. Global protein translation was examined using a recently developed technique involving labeling of newly-synthesized polypeptides with low concentrations of puromycin with subsequent detection of these proteins with anti-puromycin antibody.43 Immunoblotting analysis demonstrated that IFNγ alone or in combination with TNFα markedly decreased incorporation of puromycin into polypeptide chains (Fig. 4C) thereby indicating inhibition of protein translation. A combination of IFNγ and TNFα appeared to be a more efficient inhibitor compared with IFNγ alone which is consistent with the synergistic effects of these cytokines on the expression of AJ and TJ proteins. Suppression of puromycin immunoreactivity after exposure of control HPAF-II cells to a classical inhibitor of protein translation, cycloheximide, served as a positive control for this assay (Fig. 4C).
Janus kinase mediates cytokine-induced junctional disassembly
Next we sought to elucidate which intracellular signals mediate cytokine-dependent disruption of pancreatic epithelial junctions. A canonical signaling pathway initiated by IFNγ binding to its receptor on the plasma membrane involves activation of Janus kinases (JAK) 1 and 2 with subsequent phosphorylation and activation of members of the signal-transducing activators of transcription (STAT) protein family.42,44 Since the roles of JAK-STAT activation in IFNγ-induced disruption of epithelial barriers remains controversial,24 we investigated if this signaling is essential for cytokine-dependent disassembly of pancreatic epithelial junctions. Incubation of control HPAF-II monolayers with either pharmacological pan-JAK inhibitor (10 µM) or a specific JAK2 inhibitor (50 µM) increased a steady-state TEER comparing to vesicle-treated cells (Fig. 5A). More importantly, total JAK inhibitor efficiently prevented the decrease in TEER caused by 48 h exposure of HPAF-II cells to IFNγ and TNFα (Fig. 5A). The selective JAK-2 inhibitor partially ameliorated the effects of cytokines of the TEER but not as effectively as the inhibitor of all JAK isoforms (Fig. 5A). Furthermore, immunocytochemical analysis demonstrated that total JAK and JAK2 inhibitors prevented cytokine induced disruption of AJs and TJs in HPAF-II cell monolayers (Fig. 5B). Together, this data highlights JAK as a crucial component of the IFNγ/TNFα-induced signaling cascade that leads to disruption of the pancreatic epithelial barrier.

Figure 5. Janus kinase mediates cytokine-induced disruption of pancreatic epithelial junctions. HPAF-II cells were treated for 48 h with either medium or proinflammatory cytokines with and without pharmacological inhibitors of Janus kinase (JAK). Barrier properties of epithelial cell monolayers were determined by TEER measurement (A), and the integrity of AJs and TJs was determined by immunofluorescence labeling for E-cadherin and ZO-1 respectively (B). A total JAK inhibitor (10 µM) and a selective JAK2 inhibitor (50 µM) significantly attenuate IFNγ/TNFα-driven decrease in TEER and TJ/AJ disassembly (arrows). Data are presented as mean ± SE (n = 3); *p < 0.01; **p < 0.001 compared with cytokine/vehicle-treated cells. Bar, 20 µm.
Activation of protein kinase D mediates the effects of IFNγ on pancreatic epithelial junctions
We next sought to determine downstream signaling events that mediate barrier-disrupting effects of proinflammatory cytokines in pancreatic epithelium. We focused on the role of protein kinase D (PKD), a family of serine/threonine protein kinases consisting of three closely-related isoforms (PKD 1–3).45,46 There are several reasons for investigating possible roles of PKD in regulation of pancreatic epithelial junctions. First, PKD activation was shown to contribute to pathogenesis of experimental pancreatitis47,48 and pancreatic cancer.49 Second, PKD is known to be involved in disassembly of the airway epithelial barrier caused by double-stranded RNA50 and in disruption of endothelial junctions triggered by phorbol ester and diacylglycerol.51,52 Finally, unlike other major epithelial kinases, PKD has not been previously implicated in cytokine-induced disassembly of epithelial barriers.
To determine if cytokines can activate PKD in model pancreatic duct epithelium, expression of either total PKD of its autophosphorylated Ser744 and Ser912 forms53 was determined by immunoblotting. Exposure of HPAF-II cell monolayers to either IFNγ or IFNγ/TNFα resulted in the appearance of both phosphorylated forms of PKD, indicating activation of this enzyme (Fig. 6A). The effect was already evident at 8 h and lasted for at least 24 h of cytokine treatment. Interestingly, cytokines also stimulated expression of total PKD in HPAF-II cells (Fig. 6A). As a positive control, we used two known chemical PKD activators, phorbol 12 myristate -13-acetate (PMA) or octylindolactam-V (OI-V), that rapidly induced PKD phosphorylation at Ser744 and Ser912 residues (Fig. S1) that paralleled AJ/TJ disassembly (ref. 31 and data not shown). Since PKD activation is known to trigger translocation of this enzyme from the cytoplasm to the cellular membranes,50 we investigated the effects of cytokines on intracellular localization of these enzymes. Immunofluorescence analysis demonstrated diffuse distribution of PKD isoforms 2 (Fig. 6B) and 3 (data not shown) in control HPAF-II cells. By contrast, cell exposure to IFNγ/TNFα lead to the appearance of prominent PKD labeling at the lateral plasma membrane (Fig. 6B, arrows), which indicates translocation of PKD to areas of disassembling cell-cell contacts. To gain insight into functional effects of PKD activation on epithelial junctions, we performed cytokine treatment of HPAF-II cells with and without two structurally unrelated PKD inhibitors, Gö 697654 and CID755673.55 Both inhibitors significantly attenuated the decrease in TEER (Fig. 7A) and the increase in transepithelial dextran fluxes (Fig. 7B) caused by 48 h exposure of HPAF-II cells to IFNγ/TNFα. Furthermore, PKD inhibition prevented cytokine-induced disruption of AJs and TJs (Fig. 8, arrows). Given the fact that PKD has been initially described as a member of the protein kinase C family (PKCµ isoform), it is possible that pharmacological inhibitors of PKD can also block activity of certain PKC isoforms. To rule out this possibility, the effects of several known PKC inhibitors on cytokine-induced disruption of the pancreatic ductal barrier and junctional disassembly were tested. They include GF-109203X, which blocks both classical (PKCα, βI, βII, and γ) and novel (PKCδ, θ, ε and η) PKC isoforms,56 a selective inhibitor of classical PKC, Gö 6983,54 and an inhibitor of novel PKC δ and θ, rottlerin.57 Incubation of control HPAF-II cells with either GF-109203X or Gö 6983 (10 µM for 48h) did not affect the paracellular barrier or integrity of normal junctions, whereas exposure to rottlerin (25 µM) caused a dramatic drop in TEER accompanied by disruption of AJs and TJ (data not shown). Interestingly, neither PKC inhibitor was able to ameliorate decreased TEER and disassembled junctions in cytokine-challenged HPAF-II cells (Fig. S2). These results highlight a specific role of PKD in cytokine-induced disassembly of the pancreatic ductal barrier and suggest that members of the PKC family are not involved in this process. We next sought to investigate if the signaling events activated by IFNγ are essential for the observed downregulation of junctional protein expression. Incubation of HPAF-II cells with total JAK inhibitor abrogated cytokine-dependent decrease in expression of E-cadherin, p120 catenin, occludin and ZO-1 (Fig. 9). Furthermore, PKD inhibitor, Gö 6976, also significantly attenuated loss of AJ/TJ protein expression in cytokine-treated pancreatic epithelial cells (Fig. 9).

Figure 6. Cytokines trigger upregulation and membrane translocation of protein kinase D. (A) Representative immunoblots demonstrate an increase in the amounts of phosphorylated (p) protein kinase D (PKD) species in HPAF-II lysates after 24 h exposure to either IFNγ alone or IFNγ plus TNFα. (B) Dual fluorescence immunolabeling of PKD2 (green) and β-Catenin (red) shows a diffuse apical staining of PKD2 in control HPAF-II cells and accumulation of this enzyme at the areas of E-cadherin-based cell-cell contact after 24 h of the IFNγ/TNFα treatment (arrows). Bar, 20 μm.

Figure 7. PKD activity is involved in cytokine-induced disruption of the pancreatic epithelial barrier. HPAF-II cells were treated for 48 h with IFNγ plus TNFα in the presence of either vehicle or two different pharmacological inhibitors of PKD, Gö 6976 (10 µM) and CID755673 (50 µM). Barrier properties of the epithelial monolayers were determined by TEER (A) and fluorescent dextran flux (B) measurements. Both PKD inhibitors significantly attenuate cytokine-induced drop of TEER and increase in transepithelial dextran fluxes. Data are presented as mean ± SE (n = 3); *p < 0.01; **p < 0.001; #p < 0.05 compared with cytokine/vehicle-treated cells.

Figure 8. PKD mediates cytokine-induced disassembly of pancreatic epithelial junctions. HPAF-II cells were treated for 48 h with IFNγ plus TNFα in the presence of either vehicle or two different pharmacological inhibitors of PKD, Gö 6976 (10 µM) and CID755673 (50 µM). Localization of AJ and TJ proteins E-cadherin, β-Catenin, ZO-1 and claudin-4 was determined by fluorescence labeling and confocal microscopy. Both PKD inhibitors prevent IFNγ/ TNFα induced AJ and TJ disassembly (arrows). Bar, 20 µm.

Figure 9. Cytokines decrease expression of different AJ and TJ proteins via JAK and PKD-dependent mechanisms. Representative immunoblots (A) and densitometric quantification (B) show that pharmacological inhibitors of JAK or PKD significantly attenuates cytokine-induced decrease in junctional protein expression. Data are presented as mean ± SE (n = 3); *p < 0.01; **p < 0.001; #p < 0.05 compared with cytokine/vehicle-treated cells.
To further clarify whether PKD plays a unique role in cytokine-dependent disruption of pancreatic epithelial junctions, we next tested the involvement of alternative signaling pathways previously implicated in disassembly of cell-cell adhesions in different types of epithelia.35,58-61 We used pharmacological inhibitors, LY294002 (20 µM), Compound C (100 µM), PP2 (20 µM) and U0126 (10 µM), which block the activity of phosphatidylinositide (PI)-3 kinase, AMP-activated protein kinase (AMPK), Src kinase and extracellular signal regulated kinases (ERK) 1/2 respectively. Remarkably, none of the aforementioned inhibitors reversed cytokine-induced drop in TEER nor ameliorated AJ/TJ disassembly in HPAF-II cells (Fig. S3). Likewise, inhibition of caspases by z-VAD-fmk did not affect cytokine dependent disruption of epithelial junctions (data not shown), thereby ruling out the involvement of apoptotic cell death. This data highlights the unique role of PKD in the molecular cascade, initiated by IFNγ signaling and leading to junctional breakdown.
Knockdown of individual AJ proteins disrupts TJ integrity in model pancreatic epithelium
It is logical to assume that decreased expression of different AJ and/or TJ proteins plays a causal role in observed junctional disassembly and barrier defects of cytokine-treated pancreatic epithelial cells. However, previous studies suggest that loss of individual proteins such as occludin, ZO-1 or E-cadherin does not always compromise normal architecture of epithelial junctions.62-64 Therefore, we investigated if decreased expression of major AJ constituents, E-cadherin and p120 catenin, is sufficient to trigger AJ and TJ disassembly in HPAF-II cells. Cells were transfected with control, E-cadherin, or p120 catenin specific siRNA SmartPools and were examined on day 4 post-transfection. Immunoblotting analysis indicated that E-cadherin-specific siRNAs selectively decreased expression of the targeted protein, whereas p120-specific siRNAs decreased expression of both p120 catenin and E-cadherin (Fig. S4A). Knockdown of either E-cadherin or p120 catenin attenuated establishment of the paracellular barrier as indicated by significantly lower TEER values (Fig. S4B). Furthermore, immunolabeling and confocal microscopy indicated that control siRNA-transfected HPAF-II cells developed normal junctional staining of ZO-1 (Fig. 10, arrows), occludin and β-catenin (data not shown). By contrast, cells depleted of either E-cadherin or p120 catenin showed disorganized and fragmented ZO-1 labeling at the areas of cell-cell contacts (Fig. 10, arrowheads) and diffuse intracellular localization of β-catenin and occludin (data not shown). Together, these results demonstrate that decreased expression of AJ protein is sufficient to trigger AJ and TJ disassembly in model pancreatic epithelium and strongly suggest that loss of AJ/TJ proteins can mediate the cytokine-induced disruption of the pancreatic ductal barrier.

Figure 10. siRNA-mediated depletion of E-cadherin or p120 catenin prevents TJ assembly in pancreatic epithelium. Immunolabeling and confocal microscopy show formation of normal ZO-1-based TJs in control siRNA-treated HPAF-II cell monolayers on day 4 post-transfection (arrows). In contrast, E-cadherin or p120 catenin-depleted pancreatic epithelial cells demonstrate poorly-developed and fragmented TJ strands (arrowheads). Bar, 20 µm.
Discussion
Ductal epithelium is essential for normal pancreatic function and the impairment of its integrity plays key roles in the pathogenesis of pancreatitis and pancreatic cancer. In this study, we investigate molecular mechanisms that mediate disruption of the pancreatic epithelial barrier by proinflammatory cytokines. We report several major novel observations: (1) IFNγ alone or in combination with TNFα impairs the model ductal pancreatic barrier by causing orchestrated AJ and TJ disassembly; (2) proinflammatory cytokines decrease expression of AJ/TJ proteins by mRNA transcription dependent and independent mechanisms; (3) JAK and PKD are major players in the signaling cascade triggered by IFNγ and resulting in disassembly of pancreatic epithelial junctions. A hypothetical sequence of events, likely to be involved in cytokine-induced disruption of AJs and TJs, is presented in Figure 11.
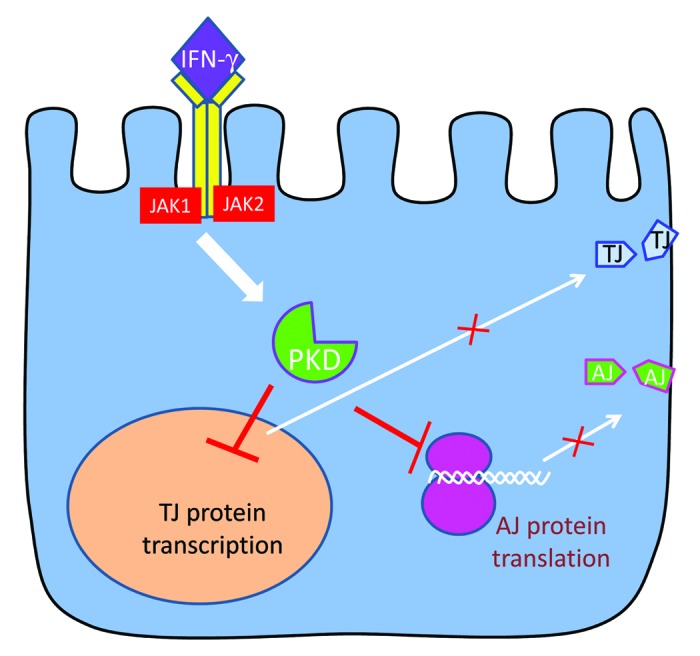
Figure 11. A summary of proposed mechanisms that mediate cytokine-induced junctional disassembly in model pancreatic epithelium. . Binding of IFNγ to its receptor elicits an intracellular signaling cascade that involves sequential activation of JAK and PKD. This signaling results in inhibition of TJ protein transcription in the nucleus and attenuation of AJ protein translation. Decreased expression of major TJ and AJ proteins interrupts the supply of new junctional components to the plasma membrane leading to disassembly and internalization of epithelial apical junctions.
Little is known about the effects of proinflammatory cytokines on the integrity of the pancreatic epithelial barrier. The only published study on the subject documented a selective increase in tricellulin level with no reported change in the expression of other junctional proteins or TJ integrity in interleukin 1β and TNFα treated HPAC pancreatic epithelial cells.65 In the present study, we observed that IFNγ acts as a potent disruptor of AJs and TJs in HPAF-II pancreatic epithelial cells. TNFα alone did not alter structure of pancreatic junctions but this cytokine potentiated the effects of IFNγ (Figs. 1 and 2). Similar synergistic effects of IFNγ and TNFα on epithelial permeability and tight junctions were previously reported in colonic and airway epithelial cells.36,66,67 The combination of IFNγ and TNFα exerted profound effects on model pancreatic epithelium by increasing permeability to small ions and large uncharged molecules and inducing disassembly of both AJs and TJs (Figs. 1 and 2). While disassembly of TJs is a common consequence of cytokine exposure in many types of human epithelia,34,36,38,66 disruption of AJs is a rather unusual effect of IFNγ 68. For example, previous studies of cultured intestinal epithelial cells observed largely unaltered localization and detergent solubility of E-cadherin and β-catenin even after prolonged incubation with IFNγ alone or in combination with TNFα.36,38 By contrast in HPAF-II cells, IFNγ/TNFα treatment caused a profound AJ disassembly accompanied by translocation of major AJ proteins into intracellular vehicles enriched along the lateral plasma membrane (Figs. 2 and 8). These vesicles resemble AJ protein-containing vacuoles that accumulate during a total loss of epithelial cell-cell contacts by extracellular calcium depletion or chemical tumor promoters.69 Hence in pancreatic epithelium, proinflammatory cytokines are likely to trigger large-scale rearrangements of the plasma membrane that lead to dissociation of different junctional complexes.
In a search for mechanisms mediating disruption of the pancreatic epithelial barrier, we found that cytokines downregulate expression of major AJ/TJ proteins. Previously, the effects of IFNγ and TNFα on TJ protein expression have been investigated in a number of studies, with conflicting results. For example, one group of studies demonstrated a significant decrease in the levels of occludin and ZO-1 in intestinal and airway epithelial cells exposed to IFNγ and TNFα,37,38,60,66,70,71 whereas other studies described unaltered expression of these TJ proteins in intestinal epithelial cells treated with the same cytokines.34,36,72 In HPAF-II cells, IFNγ markedly decreased expression not only of TJ proteins, occludin, ZO-1 and JAM-A, but also of major AJ constituents such as E-cadherin, p120 catenin and β-catenin (Fig. 3). To the best of our knowledge, this is the first report of cytokine-dependent decrease of junctional protein expression in the pancreatic ductal epithelium. Interestingly, a marked decrease of occludin and E-cadherin levels was observed in mouse pancreas during experimentally-induced pancreatitis,15,73 suggesting that loss of junctional proteins can contribute to pancreatic pathology in vivo. Similar to the reported effects of IFNγ and TNFα in intestinal and airway epithelial cells,37,38,66 the decrease of TJ protein expression in HPAF-II cells was at least partially mediated by inhibition of their mRNA transcription (Fig. 4A). On the contrary, no effect of the cytokines on mRNA expression of AJ protein was observed (Fig. 4A). Furthermore, we did not find evidence of accelerated degradation of major junctional constituents in IFNγ/TNFα-treated HPAF-II cells (Fig. 4B). Taken together, these results strongly indicate inhibited translation as the most likely mechanism of decreased expression of AJ proteins in cytokine-exposed pancreatic epithelium. IFNγ and TNFα are known inhibitors of protein translation in different cell types including human epithelia.74,75 We observed that IFNγ globally blocked protein translation in HPAF-II cells and that such interference was exaggerated by TNFα. These observations correlate well with the effects of these cytokines on epithelial junctions (Fig. 4C). Although we did not directly analyze translation of individual AJ/TJ proteins, the described data strongly suggests that inhibition of translation can be a major mechanism underlying IFNγ/TNFα-dependent loss of E-cadherin, p120 catenin, and other AJ components. How can proinflammatory cytokines affect translation of junctional proteins? The definitive answer to this question is lacking, however, the most likely stage of IFNγ actions is initiation of protein translation. Translational initiation represents the rate-limiting step for the entire process controlled by multiprotein complexes composed of different translation initiation factors.76 Previous studies have shown that IFNγ signaling can inhibit activity of translational initiating factors.74,75 It would be interesting to investigate which of these factors plays a role in cytokine-induced loss of AJ/TJ protein expression.
Another important observation of this study is that loss of individual AJ proteins such as E-cadherin and p120 catenin was sufficient to impair formation of junctions (Fig. 10) and establishment of the paracellular barrier (Fig. S4B) in pancreatic epithelium. Similar effects have been previously reported in cultured intestinal epithelial cells,77,78 as well as in the gut mucosa of E-cadherin or p120 knockout mice in vivo.78,79 Given the dramatic downregulation of AJ/TJ proteins by IFNγ and TNFα in HPAF-II cells, it is reasonable to suggest that this mechanism is essential for the disruption of the pancreatic epithelial barrier during experimental inflammation.
The majority of biological effects of IFNγ are mediated by a canonical signaling cascade initiated by the heterodimerization of IFNγ receptor resulting in the activation of JAK and STAT-1.42,44 Recent studies suggest the existence of alternative signaling events that either substitute for or act in parallel with JAK-STAT signaling.80,81 It is of note that such JAK-STAT-independent mechanisms have been previously implicated in IFNγ-induced impairment of the paracellular barrier in intestinal epithelial cells.82 However, cytokine-induced disruption of the pancreatic epithelial barrier must involve the established JAK signaling pathway since all effects of IFNγ and TNFα on structure and permeability of AJs and TJs in HPAF-II cells were abrogated by JAK inhibitors (Fig. 5). Based on this data, one can suggest that IFNγ elicits distinct signaling pathways in different types of epithelia. JAK-STAT inhibition was recently shown to decrease severity of experimental pancreatitis in rats.83 It is tempting to speculate that activation of the JAK-STAT signaling can compromise the integrity of the ductal barrier thereby contributing to inflammatory pancreatic injury in vivo.
Another key step in the signaling cascade triggered by IFNγ and leading to disassembly of pancreatic epithelial junctions involves activation of PKD. PKD is a family of closely related calcium and calmodulin-dependent serine/threonine kinases that were previously included in the PKC superfamily.45,46 PKD controls multiple cellular processes by phosphorylating a number of transcriptional regulators, cytoplasmic scaffolds, cytoskeletal proteins and membrane receptors.45,46 While the involvement of PKC isoenzymes in regulation of epithelial barriers has been intensively studied,84-86 little is known about the role(s) of PKD at epithelial cell-cell contacts. We found that PKD is markedly upregulated by IFNγ in model pancreatic epithelium (Fig. 6) and demonstrated that PKD activity is essential for cytokine-dependent increase in epithelial permeability, AJ and TJ disassembly and downregulation of junctional proteins (Figs. 7–9). This data concurs with previous reports of negative regulation of cell-cell adhesions by PKD. For example, PKD activation was shown to accompany dissociation of cell-cell contacts during extracellular calcium depletion in keratinocytes87 and different epithelial cells.88 Furthermore, pharmacological inhibition of PKD stimulated intercellular adhesions in invasive bladder carcinoma cells.89 Finally, PKD inactivation suppressed AJ/TJ disassembly triggered by either synthetic double-stranded RNA50 or autoantibodies targeting desmosomal proteins.90 While some data suggests that PKD can affect epithelial junctions via direct phosphorylation of E-cadherin,91 this kinase mediates cytokine-dependent disruption of pancreatic epithelial barrier by diminishing expression of AJ/TJ proteins (Fig. 9). Cytokine-dependent activation of PKD likely decreased mRNA levels of TJ proteins but inhibited expression of AJ components in transcription-independent fashion. The former effect is consistent with the reported ability of PKD to modulate protein expression by phosphorylating transcription factors and chromatin regulators.45,46 One possible target is a transcriptional repressor, Snail1, that is phosphorylated and activated by PKD92 and can suppress transcription of TJ proteins.93 Yet, mechanisms of transcription-independent effects of PKD on AJ proteins remain elusive, since the current list of PKD targets does not include any regulators of protein translation.45,46 However, PKD activation can inhibit translation of AJ proteins indirectly, by affecting the structure and function of the Golgi complex. Indeed, a significant pool of PKD is located in the Golgi where it participates in formation of transport vesicles.94 Active PKD phosphorylates a large number of Golgi resident proteins95 leading to disruption of Golgi integrity under different experimental conditions.96,97 Interestingly, IFNγ/TNFα treatment caused a noticeable fragmentation of the Golgi in HPAF-II cells that parallel PKD activation and junctional disassembly (data not shown). The Golgi and the endoplasmic reticulum (ER) control each other’s structure and function via bi-directional vesicle trafficking between the two organelles. As a result, molecular constituents of the Golgi may regulate protein synthesis at the ER. In fact, our recent studies demonstrate that Golgi fragmentation suppresses synthesis of different proteins in a transcription-independent fashion.77,98 It is possible, therefore, that proinflammatory cytokines may impair AJ/TJ protein synthesis at the ER via PKD-dependent fragmentation of the Golgi and interruption of normal vesicle cycling between these respective organelles. It should be noted that inhibition of PKD did not completely prevent disruption of the paracellular barrier in IFNγ/TNFα-treated HPAF-II cells (Fig. 7). This indicates that PKD is not the only effector molecule in the signaling cascade activated in inflamed pancreatic epithelium. Surprisingly, neither of the major alternative signals previously implicated in cytokine-mediated disruption of the intestinal epithelial barrier appear to be involved (Fig. S3) in this specific signaling cascade. This highlights the existence of unique signaling pathways that target pancreatic epithelial junctions. The molecular nature of these additional pathways awaits future investigation.
In conclusion, this study demonstrates that combination of IFNγ and TNFα readily disrupts integrity of the pancreatic epithelial barrier by triggering profound AJ and TJ disassembly. The cytokines prompt a unique signaling cascade involving activation of JAK and PKD and eventuating in the expressional downregulation of major AJ and TJ proteins. The newly-discovered mechanism may play a role in inflammatory pancreatic injury during chronic pancreatitis and also contribute to invasive behavior and dissemination of pancreatic cancer.
Methods
Antibodies and other reagents
The following primary polyclonal (pAb) and monoclonal (mAb) antibodies were used to detect junctional and signaling proteins in epithelial cells: anti-occludin, ZO-1, and claudin-4 mAbs and anti-claudin-1 pAb (Invitrogen); anti-E-cadherin, p120 catenin, and β-catenin mAbs (BD Biosciences) anti-total PKD and phosphorylated S744 PKD and S916 PKD pAbs (Cell Signaling Technology Inc.); anti PKD2 and anti PKD3 pAbs (Bethyl Laboratories); anti-puromycin mAb (KeraFAST); anti-gamma actin mAb (Sigma-Aldrich). Anti-JAM-A mAb was provided by Dr. C. A. Parkos (Emory University). Alexa-488 or Alexa-568 dye conjugated donkey anti-rabbit and goat anti-mouse secondary antibodies were obtained from Invitrogen; horseradish peroxidase-conjugated goat anti-rabbit and anti-mouse secondary antibodies were obtained from BioRad Laboratories. Human recombinant IFNγ and TNFα were purchased from R and D Systems. PMA, concanamycine A and chloroquine were obtained from Sigma. JAK inhibitor I, JAK2 inhibitor V, Gö 6976, Gö 6983, GF-109203X, rottlerin, Compound C, PP2, LY294002, MGM-132, epoximycin, Y-27632 and U0126 were purchased from EMD Biosciences. CID755673 was obtained from Tocris Bioscience. OI-V was purchased from Biomol International. All other reagents were of the highest analytical grade and obtained from Sigma.
Cell culture and cytokine-induced junctional disassembly
HPAF-II human pancreatic epithelial cells (American Type Culture Collection) were grown in RPMI medium supplemented with 10% fetal bovine serum, 10 mM HEPES, 1 mM sodium pyruvate, 2 mM L-glutamine, 100 IU/ml penicillin and 100 μg/ml streptomycin, pH 7.4. For all experiments, cells were grown for 6–10 d on collagen-coated permeable polycarbonate Transwell filters with 3 μm pore size (Costar). Membrane filters with diameter 6.5 and 24 mm were used for immunolabeling and biochemical experiments respectively. For cytokine treatment, IFNγ and TNFα were added to both the apical and the basolateral Transwell chambers for 48 h. Pharmacological inhibitors were added at the onset of cytokine treatment and were readministered along with cytokines after the first 24 h incubation. The appropriate vehicle (DMSO) was added to control samples.
Immunofluorescence labeling and image analysis
For immunolabeling of AJ and TJ proteins, HPAF II cell monolayers were fixed/permeabilized in 100% methanol for 20 min at -20°C. For PKD immunolabeling, the cells were fixed in 3.7% paraformaldehyde for 15 min and permeabilized with 0.5% Triton X-100 for 10 min at room temperature. Fixed cells were blocked in HEPES-buffered Hanks balanced salt solution (HBSS+) containing 1% bovine serum albumin (blocking buffer) for 60 min at room temperature and incubated for another 60 min with primary antibodies diluted in blocking buffer. Washed cells were subsequently incubated for 60 min with Alexa dye-conjugated secondary antibodies, rinsed with blocking buffer and mounted with ProLong Antifade medium (Invitrogen). Immunofluorescently-labeled cell monolayers were examined using an Olympus FluoView 1000 confocal microscope (Olympus America). The Alexa Fluor 488 and 555 signals were imaged sequentially in frame-interlace mode to eliminate cross talk between channels. The images were processed using the Olympus FV10-ASW 2.0 Viewer software and Adobe Photoshop. Images shown are representative of at least 3 experiments, with multiple images taken per slide.
Immunoblotting
Cells were homogenized in RIPA lysis buffer (20 mM Tris, 50 mM NaCl, 2 mM EDTA, 2 mM EGTA, 1% sodium deoxycholate, 1% Triton X-100 (TX-100), and 0.1% SDS, pH 7.4), containing a protease inhibitor cocktail (1:100, Sigma) and phosphatase inhibitor cocktails 1 and 3 (both at 1:200, Sigma). Lysates were cleared by centrifugation (20 min at 14,000 × g), diluted with 2 x SDS sample buffer and boiled. SDS-PAGE and immunoblotting were conducted by standard protocols with an equal amount of total protein (10 or 20 μg) per lane. Protein expression was quantified by densitometry of three immunoblot images, each representing an independent experiment, with Image J (NIH). Data are presented as normalized values assuming the expression levels in vehicle-treated groups were at 100%. Statistical analysis was performed with row densitometric data using Microsoft Excel.
RNA interference
Small interfering (si) RNA-mediated knockdown of E-cadherin and p120 catenin in HPAF-II cells was performed as previously described69,77 using siRNA SmartPools (Dharmacon). A non-coding siRNA duplex-2 was used as a control. Cells were transfected using the DharmaFect 1 reagent (Dharmacon) in Opti-MEM I medium (Invitrogen) according to the manufacturer’s protocol with a final siRNA concentration of 50 nM. Cells were utilized in experiments on day 4, post-transfection.
Quantitative REAL-TIME RT-PCR
Total RNA was isolated from confluent HPAF-II cells using RNeasy mini Kit (Qiagen) according to manufacturer’s protocol followed by DNase I digestion (Promega). Equal amounts of total RNA were reverse transcribed using an iScript cDNA synthesis kit (Bio-Rad). Specificity of amplification was verified by running agarose electrophoresis of each amplicon and obtaining a single band of expected size. The following primers were used, E-cadherin (forward primer TTACTGCCCCCAGAGGATGA and reverse TGCAACGTCGTTACGAGTCA), p 120 catenin (forward primer CAGAGAGGAGTGAAGCTCGC and reverse TTCCCTCTGGAGAACCACTCA), occludin (forward primer ACAGACTACACAACTGGCGG and reverse TCATCAGCAGCAGCCATGTA), β-actin (forward primer CGAGGCCCAGAGCAAGAGAG and reverse CGGTTGGCCTTAGGGTTCAG), while primers for amplifying ZO-1 mRNA were obtained and utilized according to.99 Real time reaction was performed using iTaq universal SYBR green Supermix (Bio-Rad) and Applied Biosystems 7900HT real time PCR system. The relative expression of each gene was determined with the comparative cycle threshold method using β-actin mRNA levels for normalization.
Protein translation assay
Effect of cytokines on global protein translation was examined as described elsewhere.43 Briefly, control and cytokine treated HPAF II cells were exposed for 1 h to either vehicle or puromycin (10 µg/µl), with subsequent cell lysis in RIPA buffer and immunoblotting using anti-puromycin antibody. Preincubation of HPAF-II cells with known protein synthesis inhibitor, cycloheximide (20 µM for 1 h), served as a control for the specificity of puromycin labeling of newly-synthesized polypeptides.
Epithelial barrier permeability measurements
Transepithelial electrical resistance (TEER) was measured with an EVOMX voltohmmeter (World Precision Instruments). The resistance of cell-free collagen-coated filters was subtracted from each experimental point. Dextran flux assay was performed as described elsewhere.100 Briefly, on day 4 post-siRNA transfection, HPAF II cell monolayers growing on Transwell filters were exposed to 1 mg/ml of either FITC dextran 4,000 Da or 40,000 Da in HBSS added to the upper chamber, whereas only HBSS was added the lower chamber. After 60 min incubation, HBSS samples were collected from the lower chamber and FITC fluorescence intensity was measured using a Victor V plate reader (Applied Biosystems) with excitation and emission wavelengths 485 and 544 nm respectively. After subtraction of fluorescence of the dextran-free HBSS, relative intensity was calculated by using Prism 5 software.
Statistics
Numerical values from individual experiments were pooled and expressed as mean ± standard error of the mean (S.E.) throughout. Obtained numbers were compared by two-tailed Student’s t-test, with statistical significance assumed at p < 0.05.
Supplementary Material
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Charles A. Parkos for providing JAM-A antibody and Mr. Gianni Harris for excellent technical assistance. This work was supported in part by the National Institute of Health grants DK084953, DK083968 and Virginia Commonwealth University Trauma Fund grant to A.I.I.
Supplemental Materials
Supplemental materials may be found here:
http://www.landesbioscience.com/journals/tissuebarriers/article/25231/
Footnotes
Previously published online: www.landesbioscience.com/journals/tissuebarriers/article/25231
References
- 1.Cheng CY, Mruk DD. The blood-testis barrier and its implications for male contraception. Pharmacol Rev. 2012;64:16–64. doi: 10.1124/pr.110.002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Duerr CU, Hornef MW. The mammalian intestinal epithelium as integral player in the establishment and maintenance of host-microbial homeostasis. Semin Immunol. 2012;24:25–35. doi: 10.1016/j.smim.2011.11.002. [DOI] [PubMed] [Google Scholar]
- 3.Marchiando AM, Graham WV, Turner JR. Epithelial barriers in homeostasis and disease. Annu Rev Pathol. 2010;5:119–44. doi: 10.1146/annurev.pathol.4.110807.092135. [DOI] [PubMed] [Google Scholar]
- 4.Turner JR. Intestinal mucosal barrier function in health and disease. Nat Rev Immunol. 2009;9:799–809. doi: 10.1038/nri2653. [DOI] [PubMed] [Google Scholar]
- 5.Franke WW. Discovering the molecular components of intercellular junctions--a historical view. Cold Spring Harb Perspect Biol. 2009;1:a003061. doi: 10.1101/cshperspect.a003061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kubo A, Nagao K, Amagai M. Epidermal barrier dysfunction and cutaneous sensitization in atopic diseases. J Clin Invest. 2012;122:440–7. doi: 10.1172/JCI57416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roda G, Sartini A, Zambon E, Calafiore A, Marocchi M, Caponi A, et al. Intestinal epithelial cells in inflammatory bowel diseases. World J Gastroenterol. 2010;16:4264–71. doi: 10.3748/wjg.v16.i34.4264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Swindle EJ, Collins JE, Davies DE. Breakdown in epithelial barrier function in patients with asthma: identification of novel therapeutic approaches. J Allergy Clin Immunol. 2009;124:23–34, quiz 35-6. doi: 10.1016/j.jaci.2009.05.037. [DOI] [PubMed] [Google Scholar]
- 9.Grapin-Botton A. Ductal cells of the pancreas. Int J Biochem Cell Biol. 2005;37:504–10. doi: 10.1016/j.biocel.2004.07.010. [DOI] [PubMed] [Google Scholar]
- 10.Clemente F, Ribeiro T, Colomb E, Figarella C, Sarles H. [Comparison of the proteins in pancreatic juice in normal and diseased humans. Determination of serum proteins and demonstration of a particular protein in chronic calcifying pancreatitis] Biochim Biophys Acta. 1971;251:456–66. doi: 10.1016/0005-2795(71)90135-8. [DOI] [PubMed] [Google Scholar]
- 11.Kennedy RH, Bockman DE, Uscanga L, Choux R, Grimaud JA, Sarles H. Pancreatic extracellular matrix alterations in chronic pancreatitis. Pancreas. 1987;2:61–72. doi: 10.1097/00006676-198701000-00010. [DOI] [PubMed] [Google Scholar]
- 12.Coskun T, Bozoklu S, Ozenç A, Ozdemir A. Effect of hydrogen peroxide on permeability of the main pancreatic duct and morphology of the pancreas. Am J Surg. 1998;176:53–8. doi: 10.1016/S0002-9610(98)00096-8. [DOI] [PubMed] [Google Scholar]
- 13.Harvey MH, Wedgwood KR, Austin JA, Reber HA. Pancreatic duct pressure, duct permeability and acute pancreatitis. Br J Surg. 1989;76:859–62. doi: 10.1002/bjs.1800760832. [DOI] [PubMed] [Google Scholar]
- 14.Lüthen RE, Niederau C, Grendell JH. Effects of bile and pancreatic digestive enzymes on permeability of the pancreatic duct system in rabbits. Pancreas. 1993;8:671–81. doi: 10.1097/00006676-199311000-00002. [DOI] [PubMed] [Google Scholar]
- 15.Schmitt M, Klonowski-Stumpe H, Eckert M, Lüthen R, Häussinger D. Disruption of paracellular sealing is an early event in acute caerulein-pancreatitis. Pancreas. 2004;28:181–90. doi: 10.1097/00006676-200403000-00010. [DOI] [PubMed] [Google Scholar]
- 16.Westmoreland JJ, Drosos Y, Kelly J, Ye J, Means AL, Washington MK, et al. Dynamic distribution of claudin proteins in pancreatic epithelia undergoing morphogenesis or neoplastic transformation. Dev Dyn. 2012;241:583–94. doi: 10.1002/dvdy.23740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hartsock A, Nelson WJ. Adherens and tight junctions: structure, function and connections to the actin cytoskeleton. Biochim Biophys Acta. 2008;1778:660–9. doi: 10.1016/j.bbamem.2007.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Anderson JM, Van Itallie CM. Physiology and function of the tight junction. Cold Spring Harb Perspect Biol. 2009;1:a002584. doi: 10.1101/cshperspect.a002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Furuse M. Molecular basis of the core structure of tight junctions. Cold Spring Harb Perspect Biol. 2010;2:a002907. doi: 10.1101/cshperspect.a002907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ishiyama N, Ikura M. The three-dimensional structure of the cadherin-catenin complex. Subcell Biochem. 2012;60:39–62. doi: 10.1007/978-94-007-4186-7_3. [DOI] [PubMed] [Google Scholar]
- 21.Shapiro L, Weis WI. Structure and biochemistry of cadherins and catenins. Cold Spring Harb Perspect Biol. 2009;1:a003053. doi: 10.1101/cshperspect.a003053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Meng W, Takeichi M. Adherens junction: molecular architecture and regulation. Cold Spring Harb Perspect Biol. 2009;1:a002899. doi: 10.1101/cshperspect.a002899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fallon MB, Gorelick FS, Anderson JM, Mennone A, Saluja A, Steer ML. Effect of cerulein hyperstimulation on the paracellular barrier of rat exocrine pancreas. Gastroenterology. 1995;108:1863–72. doi: 10.1016/0016-5085(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 24.Beaurepaire C, Smyth D, McKay DM. Interferon-γ regulation of intestinal epithelial permeability. J Interferon Cytokine Res. 2009;29:133–44. doi: 10.1089/jir.2008.0057. [DOI] [PubMed] [Google Scholar]
- 25.Capaldo CT, Nusrat A. Cytokine regulation of tight junctions. Biochim Biophys Acta. 2009;1788:864–71. doi: 10.1016/j.bbamem.2008.08.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.da Silva J, Giroldi SB, de Oliveira Basso F, Antunes GN, Borba LA, de Lima CR. Acute pancreatitis during interferon-α and ribavirin treatment for hepatitis C. BMJ Case Rep. 2009;2009:bcr09.2008.0998. doi: 10.1136/bcr.09.2008.0998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pavan Kumar P, Radhika G, Rao GV, Pradeep R, Subramanyam C, Talukdar R, et al. Interferon γ and glycemic status in diabetes associated with chronic pancreatitis. Pancreatology. 2012;12:65–70. doi: 10.1016/j.pan.2011.12.005. [DOI] [PubMed] [Google Scholar]
- 28.Pietruczuk M, Dabrowska MI, Wereszczynska-Siemiatkowska U, Dabrowski A. Alteration of peripheral blood lymphocyte subsets in acute pancreatitis. World J Gastroenterol. 2006;12:5344–51. doi: 10.3748/wjg.v12.i33.5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Talar-Wojnarowska R, Gasiorowska A, Smolarz B, Romanowicz-Makowska H, Kulig A, Malecka-Panas E. Tumor necrosis factor alpha and interferon gamma genes polymorphisms and serum levels in pancreatic adenocarcinoma. Neoplasma. 2009;56:56–62. doi: 10.4149/neo_2009_01_56. [DOI] [PubMed] [Google Scholar]
- 30.Fitzner B, Holzhueter SA, Ibrahim S, Nizze H, Jaster R. Interferon-gamma treatment accelerates and aggravates autoimmune pancreatitis in the MRL/Mp-mouse. Pancreatology. 2009;9:233–9. doi: 10.1159/000199434. [DOI] [PubMed] [Google Scholar]
- 31.Ivanov AI, Samarin SN, Bachar M, Parkos CA, Nusrat A. Protein kinase C activation disrupts epithelial apical junctions via ROCK-II dependent stimulation of actomyosin contractility. BMC Cell Biol. 2009;10:36. doi: 10.1186/1471-2121-10-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rajasekaran SA, Gopal J, Espineda C, Ryazantsev S, Schneeberger EE, Rajasekaran AK. HPAF-II, a cell culture model to study pancreatic epithelial cell structure and function. Pancreas. 2004;29:e77–83. doi: 10.1097/00006676-200410000-00016. [DOI] [PubMed] [Google Scholar]
- 33.Orlichenko L, Weller SG, Cao H, Krueger EW, Awoniyi M, Beznoussenko G, et al. Caveolae mediate growth factor-induced disassembly of adherens junctions to support tumor cell dissociation. Mol Biol Cell. 2009;20:4140–52. doi: 10.1091/mbc.E08-10-1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baker OJ, Camden JM, Redman RS, Jones JE, Seye CI, Erb L, et al. Proinflammatory cytokines tumor necrosis factor-alpha and interferon-gamma alter tight junction structure and function in the rat parotid gland Par-C10 cell line. Am J Physiol Cell Physiol. 2008;295:C1191–201. doi: 10.1152/ajpcell.00144.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Boivin MA, Roy PK, Bradley A, Kennedy JC, Rihani T, Ma TY. Mechanism of interferon-gamma-induced increase in T84 intestinal epithelial tight junction. J Interferon Cytokine Res. 2009;29:45–54. doi: 10.1089/jir.2008.0128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, et al. Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol. 2003;171:6164–72. doi: 10.4049/jimmunol.171.11.6164. [DOI] [PubMed] [Google Scholar]
- 37.Mankertz J, Tavalali S, Schmitz H, Mankertz A, Riecken EO, Fromm M, et al. Expression from the human occludin promoter is affected by tumor necrosis factor alpha and interferon gamma. J Cell Sci. 2000;113:2085–90. doi: 10.1242/jcs.113.11.2085. [DOI] [PubMed] [Google Scholar]
- 38.Youakim A, Ahdieh M. Interferon-gamma decreases barrier function in T84 cells by reducing ZO-1 levels and disrupting apical actin. Am J Physiol. 1999;276:G1279–88. doi: 10.1152/ajpgi.1999.276.5.G1279. [DOI] [PubMed] [Google Scholar]
- 39.Ivanov AI, Nusrat A, Parkos CA. Endocytosis of the apical junctional complex: mechanisms and possible roles in regulation of epithelial barriers. Bioessays. 2005;27:356–65. doi: 10.1002/bies.20203. [DOI] [PubMed] [Google Scholar]
- 40.Ivanov AI, Parkos CA, Nusrat A. Cytoskeletal regulation of epithelial barrier function during inflammation. Am J Pathol. 2010;177:512–24. doi: 10.2353/ajpath.2010.100168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Noon-Song EN, Ahmed CM, Dabelic R, Canton J, Johnson HM. Controlling nuclear JAKs and STATs for specific gene activation by IFNγ. Biochem Biophys Res Commun. 2011;410:648–53. doi: 10.1016/j.bbrc.2011.06.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schroder K, Hertzog PJ, Ravasi T, Hume DA. Interferon-gamma: an overview of signals, mechanisms and functions. J Leukoc Biol. 2004;75:163–89. doi: 10.1189/jlb.0603252. [DOI] [PubMed] [Google Scholar]
- 43.Schmidt EK, Clavarino G, Ceppi M, Pierre P. SUnSET, a nonradioactive method to monitor protein synthesis. Nat Methods. 2009;6:275–7. doi: 10.1038/nmeth.1314. [DOI] [PubMed] [Google Scholar]
- 44.O’Shea JJ, Plenge R. JAK and STAT signaling molecules in immunoregulation and immune-mediated disease. Immunity. 2012;36:542–50. doi: 10.1016/j.immuni.2012.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fu Y, Rubin CS. Protein kinase D: coupling extracellular stimuli to the regulation of cell physiology. EMBO Rep. 2011;12:785–96. doi: 10.1038/embor.2011.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rozengurt E. Protein kinase D signaling: multiple biological functions in health and disease. Physiology (Bethesda) 2011;26:23–33. doi: 10.1152/physiol.00037.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thrower EC, Yuan J, Usmani A, Liu Y, Jones C, Minervini SN, et al. A novel protein kinase D inhibitor attenuates early events of experimental pancreatitis in isolated rat acini. Am J Physiol Gastrointest Liver Physiol. 2011;300:G120–9. doi: 10.1152/ajpgi.00300.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuan J, Liu Y, Tan T, Guha S, Gukovsky I, Gukovskaya A, et al. Protein kinase d regulates cell death pathways in experimental pancreatitis. Front Physiol. 2012;3:60. doi: 10.3389/fphys.2012.00060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guha S, Tanasanvimon S, Sinnett-Smith J, Rozengurt E. Role of protein kinase D signaling in pancreatic cancer. Biochem Pharmacol. 2010;80:1946–54. doi: 10.1016/j.bcp.2010.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rezaee F, Meednu N, Emo JA, Saatian B, Chapman TJ, Naydenov NG, et al. Polyinosinic:polycytidylic acid induces protein kinase D-dependent disassembly of apical junctions and barrier dysfunction in airway epithelial cells. J Allergy Clin Immunol. 2011;128:1216–, e11. doi: 10.1016/j.jaci.2011.08.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tinsley JH, Teasdale NR, Yuan SY. Involvement of PKCdelta and PKD in pulmonary microvascular endothelial cell hyperpermeability. Am J Physiol Cell Physiol. 2004;286:C105–11. doi: 10.1152/ajpcell.00340.2003. [DOI] [PubMed] [Google Scholar]
- 52.Konstantoulaki M, Kouklis P, Malik AB. Protein kinase C modifications of VE-cadherin, p120, and β-catenin contribute to endothelial barrier dysregulation induced by thrombin. Am J Physiol Lung Cell Mol Physiol. 2003;285:L434–42. doi: 10.1152/ajplung.00075.2003. [DOI] [PubMed] [Google Scholar]
- 53.Rozengurt E, Rey O, Waldron RT. Protein kinase D signaling. J Biol Chem. 2005;280:13205–8. doi: 10.1074/jbc.R500002200. [DOI] [PubMed] [Google Scholar]
- 54.Gschwendt M, Dieterich S, Rennecke J, Kittstein W, Mueller HJ, Johannes FJ. Inhibition of protein kinase C μ by various inhibitors. Differentiation from protein kinase c isoenzymes. FEBS Lett. 1996;392:77–80. doi: 10.1016/0014-5793(96)00785-5. [DOI] [PubMed] [Google Scholar]
- 55.Sharlow ER, Giridhar KV, LaValle CR, Chen J, Leimgruber S, Barrett R, et al. Potent and selective disruption of protein kinase D functionality by a benzoxoloazepinolone. J Biol Chem. 2008;283:33516–26. doi: 10.1074/jbc.M805358200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, et al. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. J Biol Chem. 1991;266:15771–81. [PubMed] [Google Scholar]
- 57.Gschwendt M, Müller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, et al. Rottlerin, a novel protein kinase inhibitor. Biochem Biophys Res Commun. 1994;199:93–8. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 58.Diraimondo TR, Klöck C, Khosla C. Interferon-γ activates transglutaminase 2 via a phosphatidylinositol-3-kinase-dependent pathway: implications for celiac sprue therapy. J Pharmacol Exp Ther. 2012;341:104–14. doi: 10.1124/jpet.111.187385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Petecchia L, Sabatini F, Usai C, Caci E, Varesio L, Rossi GA. Cytokines induce tight junction disassembly in airway cells via an EGFR-dependent MAPK/ERK1/2-pathway. Lab Invest. 2012;92:1140–8. doi: 10.1038/labinvest.2012.67. [DOI] [PubMed] [Google Scholar]
- 60.Scharl M, Paul G, Barrett KE, McCole DF. AMP-activated protein kinase mediates the interferon-gamma-induced decrease in intestinal epithelial barrier function. J Biol Chem. 2009;284:27952–63. doi: 10.1074/jbc.M109.046292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Smyth D, Phan V, Wang A, McKay DM. Interferon-γ-induced increases in intestinal epithelial macromolecular permeability requires the Src kinase Fyn. Lab Invest. 2011;91:764–77. doi: 10.1038/labinvest.2010.208. [DOI] [PubMed] [Google Scholar]
- 62.Capaldo CT, Macara IG. Depletion of E-cadherin disrupts establishment but not maintenance of cell junctions in Madin-Darby canine kidney epithelial cells. Mol Biol Cell. 2007;18:189–200. doi: 10.1091/mbc.E06-05-0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McNeil E, Capaldo CT, Macara IG. Zonula occludens-1 function in the assembly of tight junctions in Madin-Darby canine kidney epithelial cells. Mol Biol Cell. 2006;17:1922–32. doi: 10.1091/mbc.E05-07-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Van Itallie CM, Fanning AS, Holmes J, Anderson JM. Occludin is required for cytokine-induced regulation of tight junction barriers. J Cell Sci. 2010;123:2844–52. doi: 10.1242/jcs.065581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kojima T, Fuchimoto J, Yamaguchi H, Ito T, Takasawa A, Ninomiya T, et al. c-Jun N-terminal kinase is largely involved in the regulation of tricellular tight junctions via tricellulin in human pancreatic duct epithelial cells. J Cell Physiol. 2010;225:720–33. doi: 10.1002/jcp.22273. [DOI] [PubMed] [Google Scholar]
- 66.Coyne CB, Vanhook MK, Gambling TM, Carson JL, Boucher RC, Johnson LG. Regulation of airway tight junctions by proinflammatory cytokines. Mol Biol Cell. 2002;13:3218–34. doi: 10.1091/mbc.E02-03-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Fish SM, Proujansky R, Reenstra WW. Synergistic effects of interferon gamma and tumour necrosis factor alpha on T84 cell function. Gut. 1999;45:191–8. doi: 10.1136/gut.45.2.191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smyth D, Leung G, Fernando M, McKay DM. Reduced surface expression of epithelial E-cadherin evoked by interferon-gamma is Fyn kinase-dependent. PLoS One. 2012;7:e38441. doi: 10.1371/journal.pone.0038441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Naydenov NG, Ivanov AI. Adducins regulate remodeling of apical junctions in human epithelial cells. Mol Biol Cell. 2010;21:3506–17. doi: 10.1091/mbc.E10-03-0259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Scharl M, Paul G, Weber A, Jung BC, Docherty MJ, Hausmann M, et al. Protection of epithelial barrier function by the Crohn’s disease associated gene protein tyrosine phosphatase n2. Gastroenterology. 2009;137:2030–40, e5. doi: 10.1053/j.gastro.2009.07.078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sugi K, Musch MW, Field M, Chang EB. Inhibition of Na+,K+-ATPase by interferon gamma down-regulates intestinal epithelial transport and barrier function. Gastroenterology. 2001;120:1393–403. doi: 10.1053/gast.2001.24045. [DOI] [PubMed] [Google Scholar]
- 72.Wang F, Graham WV, Wang Y, Witkowski ED, Schwarz BT, Turner JR. Interferon-gamma and tumor necrosis factor-alpha synergize to induce intestinal epithelial barrier dysfunction by up-regulating myosin light chain kinase expression. Am J Pathol. 2005;166:409–19. doi: 10.1016/S0002-9440(10)62264-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lerch MM, Lutz MP, Weidenbach H, Müller-Pillasch F, Gress TM, Leser J, et al. Dissociation and reassembly of adherens junctions during experimental acute pancreatitis. Gastroenterology. 1997;113:1355–66. doi: 10.1053/gast.1997.v113.pm9322531. [DOI] [PubMed] [Google Scholar]
- 74.Hu S, Ciancio MJ, Lahav M, Fujiya M, Lichtenstein L, Anant S, et al. Translational inhibition of colonic epithelial heat shock proteins by IFN-gamma and TNF-alpha in intestinal inflammation. Gastroenterology. 2007;133:1893–904. doi: 10.1053/j.gastro.2007.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu S, Claud EC, Musch MW, Chang EB. Stress granule formation mediates the inhibition of colonic Hsp70 translation by interferon-gamma and tumor necrosis factor-alpha. Am J Physiol Gastrointest Liver Physiol. 2010;298:G481–92. doi: 10.1152/ajpgi.00234.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–45. doi: 10.1016/j.cell.2009.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Naydenov NG, Brown B, Harris G, Dohn MR, Morales VM, Baranwal S, et al. A membrane fusion protein αSNAP is a novel regulator of epithelial apical junctions. PLoS One. 2012;7:e34320. doi: 10.1371/journal.pone.0034320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Smalley-Freed WG, Efimov A, Burnett PE, Short SP, Davis MA, Gumucio DL, et al. p120-catenin is essential for maintenance of barrier function and intestinal homeostasis in mice. J Clin Invest. 2010;120:1824–35. doi: 10.1172/JCI41414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Schneider MR, Dahlhoff M, Horst D, Hirschi B, Trülzsch K, Müller-Höcker J, et al. A key role for E-cadherin in intestinal homeostasis and Paneth cell maturation. PLoS One. 2010;5:e14325. doi: 10.1371/journal.pone.0014325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gough DJ, Levy DE, Johnstone RW, Clarke CJ. IFNgamma signaling-does it mean JAK-STAT? Cytokine Growth Factor Rev. 2008;19:383–94. doi: 10.1016/j.cytogfr.2008.08.004. [DOI] [PubMed] [Google Scholar]
- 81.Hill T, 3rd, Xu C, Harper RW. IFNgamma mediates DUOX2 expression via a STAT-independent signaling pathway. Biochem Biophys Res Commun. 2010;395:270–4. doi: 10.1016/j.bbrc.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Watson JL, Ansari S, Cameron H, Wang A, Akhtar M, McKay DM. Green tea polyphenol (-)-epigallocatechin gallate blocks epithelial barrier dysfunction provoked by IFN-gamma but not by IL-4. Am J Physiol Gastrointest Liver Physiol. 2004;287:G954–61. doi: 10.1152/ajpgi.00302.2003. [DOI] [PubMed] [Google Scholar]
- 83.Chen P, Huang L, Zhang Y, Qiao M, Yao W, Yuan Y. The antagonist of the JAK-1/STAT-1 signaling pathway improves the severity of cerulein-stimulated pancreatic injury via inhibition of NF-κB activity. Int J Mol Med. 2011;27:731–8. doi: 10.3892/ijmm.2011.632. [DOI] [PubMed] [Google Scholar]
- 84.Clarke H, Marano CW, Peralta Soler A, Mullin JM. Modification of tight junction function by protein kinase C isoforms. Adv Drug Deliv Rev. 2000;41:283–301. doi: 10.1016/S0169-409X(00)00047-8. [DOI] [PubMed] [Google Scholar]
- 85.Farhadi A, Keshavarzian A, Ranjbaran Z, Fields JZ, Banan A. The role of protein kinase C isoforms in modulating injury and repair of the intestinal barrier. J Pharmacol Exp Ther. 2006;316:1–7. doi: 10.1124/jpet.105.085449. [DOI] [PubMed] [Google Scholar]
- 86.Kojima T, Sawada N. Regulation of tight junctions in human normal pancreatic duct epithelial cells and cancer cells. Ann N Y Acad Sci. 2012;1257:85–92. doi: 10.1111/j.1749-6632.2012.06579.x. [DOI] [PubMed] [Google Scholar]
- 87.Jadali A, Ghazizadeh S. Protein kinase D is implicated in the reversible commitment to differentiation in primary cultures of mouse keratinocytes. J Biol Chem. 2010;285:23387–97. doi: 10.1074/jbc.M110.105619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cowell CF, Yan IK, Eiseler T, Leightner AC, Döppler H, Storz P. Loss of cell-cell contacts induces NF-kappaB via RhoA-mediated activation of protein kinase D1. J Cell Biochem. 2009;106:714–28. doi: 10.1002/jcb.22067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Koivunen J, Aaltonen V, Koskela S, Lehenkari P, Laato M, Peltonen J. Protein kinase C alpha/beta inhibitor Go6976 promotes formation of cell junctions and inhibits invasion of urinary bladder carcinoma cells. Cancer Res. 2004;64:5693–701. doi: 10.1158/0008-5472.CAN-03-3511. [DOI] [PubMed] [Google Scholar]
- 90.Spindler V, Endlich A, Hartlieb E, Vielmuth F, Schmidt E, Waschke J. The extent of desmoglein 3 depletion in pemphigus vulgaris is dependent on Ca(2+)-induced differentiation: a role in suprabasal epidermal skin splitting? Am J Pathol. 2011;179:1905–16. doi: 10.1016/j.ajpath.2011.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jaggi M, Rao PS, Smith DJ, Wheelock MJ, Johnson KR, Hemstreet GP, et al. E-cadherin phosphorylation by protein kinase D1/protein kinase Cmu is associated with altered cellular aggregation and motility in prostate cancer. Cancer Res. 2005;65:483–92. [PubMed] [Google Scholar]
- 92.Eiseler T, Köhler C, Nimmagadda SC, Jamali A, Funk N, Joodi G, et al. Protein kinase D1 mediates anchorage-dependent and -independent growth of tumor cells via the zinc finger transcription factor Snail1. J Biol Chem. 2012;287:32367–80. doi: 10.1074/jbc.M112.370999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ikenouchi J, Matsuda M, Furuse M, Tsukita S. Regulation of tight junctions during the epithelium-mesenchyme transition: direct repression of the gene expression of claudins/occludin by Snail. J Cell Sci. 2003;116:1959–67. doi: 10.1242/jcs.00389. [DOI] [PubMed] [Google Scholar]
- 94.Malhotra V, Campelo F. PKD regulates membrane fission to generate TGN to cell surface transport carriers. Cold Spring Harb Perspect Biol. 2011;3:3. doi: 10.1101/cshperspect.a005280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Franz-Wachtel M, Eisler SA, Krug K, Wahl S, Carpy A, Nordheim A, et al. Global detection of protein kinase D-dependent phosphorylation events in nocodazole-treated human cells. Mol Cell Proteomics. 2012;11:160–70. doi: 10.1074/mcp.M111.016014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Díaz Añel AM. Phospholipase C beta3 is a key component in the Gbetagamma/PKCeta/PKD-mediated regulation of trans-Golgi network to plasma membrane transport. Biochem J. 2007;406:157–65. doi: 10.1042/BJ20070359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fuchs YF, Eisler SA, Link G, Schlicker O, Bunt G, Pfizenmaier K, et al. A Golgi PKD activity reporter reveals a crucial role of PKD in nocodazole-induced Golgi dispersal. Traffic. 2009;10:858–67. doi: 10.1111/j.1600-0854.2009.00918.x. [DOI] [PubMed] [Google Scholar]
- 98.Naydenov NG, Harris G, Brown B, Schaefer KL, Das SK, Fisher PB, et al. Loss of soluble N-ethylmaleimide-sensitive factor attachment protein α (αSNAP) induces epithelial cell apoptosis via down-regulation of Bcl-2 expression and disruption of the Golgi. J Biol Chem. 2012;287:5928–41. doi: 10.1074/jbc.M111.278358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Go M, Kojima T, Takano K, Murata M, Koizumi J, Kurose M, et al. Connexin 26 expression prevents down-regulation of barrier and fence functions of tight junctions by Na+/K+-ATPase inhibitor ouabain in human airway epithelial cell line Calu-3. Exp Cell Res. 2006;312:3847–56. doi: 10.1016/j.yexcr.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 100.Baranwal S, Naydenov NG, Harris G, Dugina V, Morgan KG, Chaponnier C, et al. Nonredundant roles of cytoplasmic β- and γ-actin isoforms in regulation of epithelial apical junctions. Mol Biol Cell. 2012;23:3542–53. doi: 10.1091/mbc.E12-02-0162. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.