

Abstract
Cancer stem cells (CSCs) are cells that exist within a tumor with a capacity of self-renewal and an ability to differentiate, giving rise to heterogeneous populations of cancer cells. These cells are increasingly being implicated in resistance to conventional therapeutics and have also been implicated in tumor recurrence. Several cellular signaling pathways including Notch, Wnt, phosphoinositide-3-kinase–Akt–mammalian target of rapamycin pathways, and known markers such as CD44, CD133, CD166, ALDH, etc. have been associated with CSCs. Here, we have reviewed our current understanding of self-renewal pathways and factors that help in the survival of CSCs with special emphasis on those that have been documented to be modulated by well characterized natural agents such as curcumin, sulforaphane, resveratrol, genistein, and epigallocatechin gallate. With the inclusion of a novel derivative of curcumin, CDF, we showcase how natural agents can be effectively modified to increase their efficacy, particularly against CSCs. We hope that this article will generate interest among researchers for further mechanistic and clinical studies exploiting the cancer preventive and therapeutic role of nutraceuticals by targeted elimination of CSCs.
Keywords: Cancer stem cells, Phytochemicals, CDF, Notch, EMT
Introduction
Cancer is a major disease that is often incurable; hence, efforts are constantly being made for designing effective drug(s) for its treatment. In spite of exhaustive research on therapeutic target identification, establishing cancer cell pathways, and anticancer drug development, cancer still remains the second largest cause of deaths in USA. In 2011, a total of 15,96,670 new cancer cases were recorded with 5,71,950 deaths resulting from it [1]. It has been well-established that multiple pathways are involved in cancer growth, development, and metastasis [2]. Anticancer drug resistance and tumor recurrence after surgery remains a major problem to be tackled in the field of cancer research [3, 4]. Recent reports have suggested the existence of cancer stem cells (CSCs) in several cancers which have the capacity of self-renewal and differentiation in the heterogeneous lineages of cancer cells which comprise of the whole tumor [5]. These tumor-initiating cells provide a reservoir of cells that can cause tumor recurrence after therapy [6]. Self-renewal pathways have the ability to regulate CSCs and promote drug resistance [6, 7]. Consequently there has been a surge of research amongst cancer research groups to identify signaling pathways involved in the generation of CSCs and designing their possible natural or synthetic inhibitors for eliminating them. Translation research thus offers an effective tool to enhance the effectiveness and speed of cancer drug discovery and a way to avoid failures in clinical setting.
Current literature suggests that nature is a rich source of multi-targeting phytochemicals (nutraceuticals) (Fig. 1) that have the ability to eliminate CSCs. However, many of these phytochemicals like curcumin or resveratrol have not done well in translational studies due to their limited water solubility, consequent poor bioavailability, and metabolic instability [8, 9]. In our group, we have overcome these drawbacks to some extent by the synthesis of novel analog approach and employing different polymeric materials as drug carriers. We believe that formulation and development has a definitive role in translation research to evolve a drug from laboratory to ultimately into clinical practice.
Fig. 1.

Structures of phytochemicals that target CSCs
Our group has been exploring the origin, survival, and proliferative pathways of cancer cells and CSCs [10–18] and has been reporting on the synthetic analogs of some phytochemicals which show a great promise for the elimination of CSCs in different cancers [19–23]. We have recently summarized the biology of CSCs [24] and important roles of micro-RNA (miRNAs) in drug resistance [22]. In the present review, we have summarized role of EMT and miRNAs in CSC maintenance and also the potential role of nutraceuticals and their synthetic analogs for the elimination of CSCs.
Cancer stem cells
Many tumor cells are known to show several histological characteristics with variable morphology, differentiation status, and molecular features exhibiting tumor initiating stem cell-like properties which can self-renew and further differentiate to produce the tumor cell population within a tumor mass [25–27]. Due to these histological differences, these stem-like cells behave in a different way and undergo adaptation in response to anticancer drugs which might be a major reason for their drug resistance characteristics [28]. These adaptations in tumors involve epithelial-to-mesenchymal transition (EMT) in epithelial cancers which may trigger conversion of EMT phenotypic cells to CSCs [29, 30]. Studies have clearly suggested a molecular link between EMT, CSCs, and drug resistance in cancer [17, 31, 32].
Importance of EMT
Several progressing cancers have been found to exhibit EMT phenotypic cells that are characterized by increased cell motility and invasion, which have an ability to infiltrate into surrounding tissues leading to cancer metastasis in different parts of the body [33–37]. Some of the recent reports have suggested that EMT is not only involved in cancer metastasis but also in cancer progression as well [38–42]. The process by which epithelial cells undergo major morphological changes from an epithelial cobblestone phenotype to an elongated fibroblastic phenotype has been termed as “epithelial-to-mesenchymal transition” [43]. This process also involves disturbance of cell–cell junctions [44], reorganization of actin cytoskeleton [45], enhanced cell motility, and invasion [34, 43]. Other cellular features of EMT include down-regulation and relocation of E-cadherin and zonula occludens-1 [10, 46], translocation of β-catenin from the cell membrane to nucleus, and up-regulation of mesenchymal molecular markers such as vimentin [10, 45], fibronectin, and N-cadherin [38, 43], respectively. The enhanced cell motility and invasion of cells [33] in tumor development is linked with accumulation of mesenchymal phenotype along with the loss of epithelial marker expression and up-regulation of mesenchymal molecular markers which leads to cancer metastasis [37] and drug resistance [17]. These processes are accompanied with the acquisition of a “cancer stem-like cell (CSLC)” phenotype which is also known as “stemness” or CSC characteristics [35]. The development and recurrence of tumors is believed to be strongly linked with the biology of CSCs or cancer-initiating cells [47–49]. Emerging literature reports have shown that the cells with an EMT phenotype induced by different factors are rich sources for cancer stem-like cells [29, 37, 50, 51], suggesting the biological similarities between CSCs, CSLCs, or cancer-initiating cells and EMT-phenotypic cells [24].
Armstrong et al. [52] have examined the existence of EMT in circulating tumor cells (CTC) from patients with progressive metastatic solid tumors, with a focus on men with castration-resistant prostate cancer (CRPC) and women with metastatic breast cancer. They found that the majority (>80 %) of these CTCs in patients with metastatic CRPC co-express epithelial proteins such as epithelial cell adhesion molecule (EpCAM), cytokeratins, and E-cadherin, with mesenchymal proteins including vimentin, N-cadherin, and O-cadherin, along with the stem cell marker CD133. It has been shown that cells with EMT phenotypic characteristics are the major source of CSLCs indicating the link between CSCs, CSLCs, EMT phenotypic cells, and cancer-initiating cells [29, 50, 51].
CSCs are the cells present within a tumor that possess the capacity of self-renewal and differentiation capacity, suggesting that these are the tumor-initiating cells which provide a reservoir of cells that may be responsible for tumor recurrence after therapy. The presence of CSCs was first shown in leukemic cells [53]. It was found that only a minor subset of leukemic cells with cell surface markers CD34 +CD38− and when transplanted into severely combined immune deficient (SCID) mice resulted in a pattern of dissemination and leukemic cell morphology similar to that seen in the original patient. Recently, CSCs have also been identified in solid tumors of breast, colon, brain, prostate [54–57], pancreatic [26, 58], liver [59, 60], ovarian [61, 62], bladder [63, 64], and lung cancers [65, 66]. In the primary human pancreatic cancer tissues and some cancer cell lines, CD133 was found to be expressed which showed increased cell proliferation under anchorage-independent conditions and enhanced migration and invasion capacity, particularly when co-cultured with primary pancreatic stromal cells [67].
Importance of microRNA in EMT and CSC’s regulation
Several recent reports, including our own, have suggested that miRNAs regulate EMT through the regulation of E-cadherin and other molecules such as ZEB and vimentin [68–72]. For example, while over-expression of miR-200 was found to have dramatic effects on the expression of ZEB, a negative correlation was found between ZEB and miR-200 expression in many human cancer cell lines [70, 72–74]. Gregory et al. [46] have found that all five members of the miR-200 family (miR-200a, miR-200b, miR-200c, miR-141, and miR-429) and miR-205 were markedly down-regulated in cells that had undergone EMT in response to transforming growth factor β (TGF-β) or to ectopic expression of the protein tyrosine phosphatase, viz. Pez. Enforced expression of the miR-200 family alone was sufficient to prevent TGF-β-induced EMT. Together, these miRNAs regulate the expression of E-cadherin transcriptional repressors like ZEB1 (also known as deltaEF1) and ZEB2 (also known as SIP1), which were previously implicated in EMT and tumor metastasis. Inhibition of the miRNAs was sufficient to induce EMT in a process requiring up-regulation of ZEB1 and/or SIP1. Recently, miR-661 was found in Snail-induced EMT-type cells and required for efficient invasion of breast cancer cells by destabilizing two of its predicted mRNA targets, viz. cell–cell adhesion protein Nectin-1 and lipid transferase StarD10, respectively, resulting in the down-regulation of epithelial markers [75]. In our group, we have found that miR-200a, miR-200b, and miR-200c were down-regulated in gemcitabine-resistant pancreatic cancer cells, which showed the acquisition of EMT phenotype [23]. Re-expression of the miR-200 family members resulted in the up-regulation of epithelial marker E-cadherin and down-regulation of the mesenchymal markers, including ZEB1 and vimentin, respectively. Members of the let-7 family are also down-regulated in EMT-type gemcitabine-resistant (GR) cells, suggesting that the loss of some critical miRNAs contributes to tumor aggressiveness via the acquisition of EMT phenotype or increasing the population of CSCs in the tumor. Thus, novel strategy by which these miRNAs could be up-regulated would certainly provide a newer approach for the elimination of EMT or CSCs, which will improve the treatment outcome of cancer patients.
Stinson et al. have identified miR-221 and miR-222 (miR-221/222) as basal-like subtype-specific miRNAs whose presence has been shown to decrease the expression of epithelial-specific genes and increase the expression of mesenchymal-specific genes, leading to increased cell migration and invasion in a manner consistent with the characteristic of the EMT phenotype [76]. Li et al. have reported that miR-448 is the most down-regulated miRNA following chemotherapy with cyclophosphamide (CP), epirubicin+taxotere/CP, epirubicin+5-fluorouracil chemotherapy which correlates with EMT induction in breast cancer in vitro and in vivo [77]. Peng et al. have indicated that miRNA-143 and miRNA-145 are associated with bone metastasis of cancer which may play important role in the bone metastasis and could be involved in the regulation of EMT [78]. Recent studies have shown the involvement of several miRNAs in the regulation of CSCs. For example, Lulla et al. have found 22 differentially expressed miRNAs, four of which (miR-135b, miR-150, miR-542-5p, and miR-652) were confirmed and validated in a different group of tumors that are associated with CSCs. Both miR-135b and miR-150 have previously shown to be important in cancer [79]. Shimono and co-workers [80] observed that miR-200c strongly suppressed the ability of normal mammary stem cells to form mammary ducts and tumor formation driven by human breast CSCs in vivo. The miR-200 family miRNAs were found to be strongly suppressed in CD44+/CD24−lineage human breast cancer cells. Jung and co-workers [81] have found differentially expressed miRNAs including miR-99a, miR-100, miR-125b, miR-192, and miR-429 in pancreatic CSCs. Hao et al. [82] have reported that the expression of miR-483-3p is strongly enhanced in pancreatic cancer tissues compared to normal tissues. Ectopic expression of miR-483-3p significantly represses DPC4/Smad4 protein levels in pancreatic cancer cell lines and simultaneously promotes cell proliferation and colony formation in vitro. Liu et al. [83] have shown that miR-34a could function as a key negative regulator of CD44 (+) prostate cancer cells. Recently, our group has shown loss of expression of miR-34a in prostate cancer, which is in part due to epigenetic silencing and that the expression of miR-34a could be up-regulated by a natural agent, 3,3′-diindolyl-methane both in cell culture model as well as in clinical trial [84].
In summary, EMT induction in cells not only leads to the generation of CSCs and its maintenance but also leads to cancer cell invasion, metastasis, and drug resistance [22, 32, 38], suggesting that future targeted therapies need to be directed toward these cells for complete eradication of tumors, which certainly would improve the overall survival of cancer patients.
Self-renewal pathways of cancer stem cells
Recent studies on CSC identification and pathways involved in their growth have suggested that CSCs can follow a number of self renewal pathways for their survival. Also, a number of markers have been associated with CSC populations in different human cancers (Fig. 2). In this section, we have summarized some major and important self-renewal pathways and have provided an account of the phytochemicals (nutraceuticals) that are capable of influencing some of these pathways (Fig. 3).
Fig. 2.
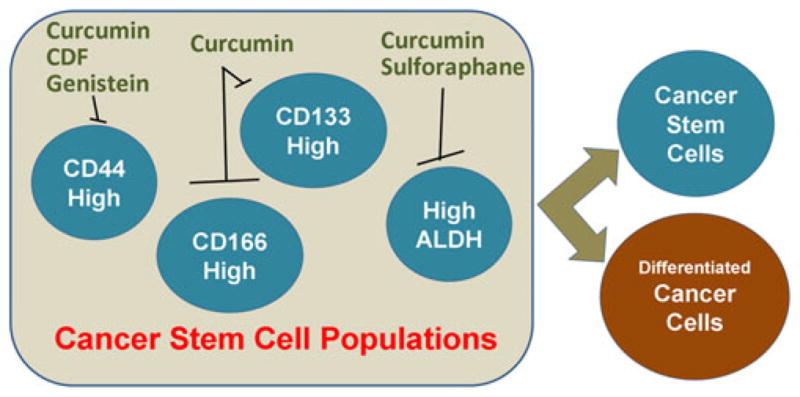
Markers of cancer stem cell populations that have been shown to be modulated by phytochemicals. A number of cell surface as well as cytosolic markers have been associated with CSC phenotype. Accumulating evidence suggests that the individual populations enriched for the proposed CSC markers are sensitive to treatment with various phytochemicals
Fig. 3.
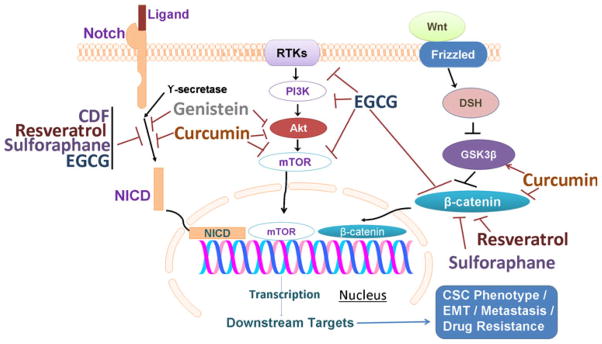
Several key factors/signaling pathways in CSCs that are targets of phytochemicals. Notch signaling pathway activation results in cleavage of Notch by γ-secretase into Notch intracellular domain (NICD) that translocates to nucleus, a process that is known to be inhibited by multiple phytochemicals. Receptor tyrosine kinases (RTKs), such as PDGFR, IGFR, EGFR etc., activate PI3K–Akt–mTOR signaling pathway, and this pathway is also known to be regulated by phytochemicals at different steps. Binding of wnt ligand to its receptor Frizzled activates Dishevelled (DSH) leading to attenuation of GSK-3β-mediated repression of β-catenin. β-Catenin also translocates to nucleus resulting in induction of downstream signaling molecules, and this pathway is modulated by phytochemicals as well. Inhibition of these multiple signaling pathways represents a mechanism by which phytochemicals influence the phenotype of CSCs
Notch signaling pathway
The Notch signaling pathway is a conserved ligand–receptor signaling pathway which plays important role in maintaining the balance of cell proliferation, survival, apoptosis, and differentiation which affects development and function of many organs [85]. Hence, alteration in Notch signaling prevents differentiation thereby guiding undifferentiated cells toward malignant transformation leading to many human cancers [86–92]. Notch signaling also functions as anti-proliferative in a limited number of cancers like those of skin, medullary thyroid, cervical, small cell lung cancer, and human hepatocellular carcinoma [93–97]. The cross-talks between Notch and other oncogenic signaling pathways like NF-κB, Akt, Sonic hedgehog (Shh), mammalian target of rapamycin (mTOR), Ras, Wnt, estrogen receptor (ER), androgen receptor (AR), epidermal growth factor receptor (EGFR), and platelet-derived growth factor (PDGF) have also been thought to play important role in tumor development [98–103]. Several recent studies have suggested that Notch pathway is involved in the regulation of CSC and the acquisition of the EMT phenotype which are involved in drug resistance [15, 104]. Thus, targeting Notch pathway can be a useful therapeutic strategy for eliminating drug resistant CSCs or EMT cells in order for overcoming the drug-resistant cancer phenotypes.
Notch signaling pathway has been reported to be involved in the acquisition of EMT in drug-resistant cancer cells. We also found that Notch-2 and Jagged-1 are highly up-regulated in GR pancreatic cancer cells. Moreover, down-regulation of Notch signaling by siRNA approach led to a partial reversal of the EMT phenotype, resulting in the MET, which was associated with decreased expression of vimentin, ZEB1, Slug, Snail, and NF-κB [15]. These results provide molecular evidence indicating that the activation of Notch signaling is mechanistically linked with chemo-resistance phenotype, which is consistent with the acquisition of EMT phenotype by pancreatic cancer cells. Notch signaling has also been found to be activated via the activity of γ-secretase which has become a target for cancer therapy. Several forms of γ-secretase inhibitors (GSIs) have been tested for their anti-tumor effects. The GSI inhibits cell growth and could induce apoptosis in many human cancer cells, such as hepatoma, breast cancer, pancreatic cancer, and myeloma cells [85, 105]. Song et al. have evaluated the effects of Notch-1 silencing on cisplatin-induced cytotoxicity in CaSki cervical cancer cells. These authors found that Notch-1 knock-down by siRNA significantly potentiated cisplatin-induced cytotoxicity, lowering the IC50 value of cisplatin in CaSki cells by almost two orders of magnitude [106]. Hence, compounds inhibiting Notch pathways can be implicated in the elimination of EMT phenotypic cells as well as CSCs, which would likely improve treatment outcome.
Wnt and TCF/β-catenin signaling pathway
A mutation in Drosophila initiating absence of wing and haltere led to the discovery of Wnt gene and due to its mutant phenotype it was named as wingless [107]. Subsequently, the homologous gene with 15 chromosome sequence was identified in mouse where proviral addition at 5′ or 3′ end induced cancerous transformation in mammary cells [108]. This signaling pathway is a complex canonical pathway in which β-catenin act as the major regulator, whereas Wnt signal cascade may also proceed without β-catenin, accomplishing its effects where calcium signaling is the major mediator [109]. Further studies have shown that several molecules in the Wnt signaling pathways are involved in the embryonic development processes [110, 111].
Wnt ligands interact with both secreted and membrane-associated proteins including, at least, ten seven-pass trans-membrane Frizzled (Fzd) receptors, two low-density lipoprotein receptor-related proteins (LRP), and a number of extracellular Wnt-modulating proteins such as Kremlin, Dickkopf, Wnt-inhibitory factor, secreted Fzds, and Norrin to initiate signaling processes [112, 113]. The Wnt signals are transduced through membrane frizzled (FZD) receptors and low-density LRP to the β-catenin signaling cascade. In the absence of active Wnt ligands, β-catenin is complexed with scaffold proteins axis inhibition protein (axin) and adenomatosis polyposis coli (APC) and phosphorylated by glycogen synthase kinase (GSK)-3β and casein kinase Iα at N-terminal residues. Phosphorylated β-catenin is then ubiquitinated and undergoes proteasome-mediated degradation, while in the presence of active Wnt signaling, Wnt ligands bind to FZD and LRP, resulting in the phosphorylation of LRP6 by GSK-3β in its cytoplasmic region, leading to the recruitment of the cytosolic proteins dishevelled and axin. Because of the formation of FZD–DVL complex and LRP–Axin–FRAT complex, β-catenin is released from phosphorylation by GSK-3β and degradation by proteosome. Then, the accumulated β-catenin translocates to the nucleus and regulates the expression of target genes. In the absence of nuclear β-catenin, T-cell factor (TCF) and groucho form a constitutive repressor complex. β-catenin translocated into the nucleus disrupts this interaction between TCF and groucho. The nuclear β-catenin is then complexed with T cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors and Legless family docking proteins (BCL9 and BCL9L) associated with PYGO family co-activators. The TCF/LEF–β-catenin–legless–PYGO nuclear complex activates the transcription of Wnt signaling target genes such as FGF20, DKK1, Myc, cyclin D1, etc. Several proteins that are secreted and present intracellularly play critical roles in regulating the activation of Wnt signaling, while secreted frizzled receptor proteins and dickkopf proteins lead to reduce the activation of Wnt signaling by inhibiting active Wnt ligands binding to FZD and LRP, respectively.
Apart from well-studied canonical Wnt pathway, there are non-canonical Wnt/Ca2+ signaling pathways such as Wnt planer cell polarity pathway, Wnt-JNK signaling pathway, Wnt/Ror receptor pathway, Wnt-GSK3MT pathway, Wnt-aPKC pathway, Wnt-RYK pathway, and Wnt-mTOR pathway [114]. In non-canonical Wnt pathway [114], active signals are transduced through FZD receptors and co-receptors including ROR2 and RYK. DVL, c-jun NH2-terminal kinase, and small G-proteins such as RHOA, RHOU, RAC, and CDC42 are also involved in the non-canonical Wnt pathway. De has recently reviewed the participation of Wnt5a in the non-canonical Wnt signaling pathway [114]. Wnt5a has been reported as a tumor suppressor gene in breast cancer [115], thyroid carcinoma [116], and colon carcinoma [117] while it is also reported to be a proto-oncogene in prostate cancer [118, 119], melanoma [120], breast cancer [121], and pancreatic cancer [122, 123]. A tumor suppressor effect of Wnt5a in thyroid cancer occurred via down-regulation of c-myc, a well-known proto-oncogene activated via the Wnt/b-catenin pathway [116]. As oncogene, Wnt5a was found to induce invasiveness in breast cancer cell lines, while the expression of TNF-α and matrix metalloproteases-7 were found to regulate the invasiveness [121]. Wnt5a mediated signaling has also found to be expressed in melanoma cells which lead to enhanced cell motility and invasiveness and found to be mediated by protein kinase C involving calcium signaling [124, 125]. Upon hypomethylation of Wnt5a promoter region, wnt5a signaling pathway has been found to be up-regulated in prostate cancer cells [126] causing high motility, invasiveness, and displayed different morphological characteristics than wild type. These effects were reported to be regulated via Ca2+/CaMKII.
It is known that small G-proteins are implicated in the cytoskeletal reorganization during invasion and metastasis, suggesting the importance of non-canonical wnt signaling in cancer invasion and metastasis. The aberrant activation of the canonical Wnt/β-catenin signaling is one of the most frequent signaling abnormalities known in human cancers. During the development of human cancer, activated Wnt signaling promotes β-catenin accumulation in the nucleus, resulting in the consequent transcriptional activation of specific target genes [127, 128].
The deregulated expression of Wnt-ligand and Wnt-binding proteins and inappropriate activation of the Wnt signaling have been found in a variety of human cancers [129–132]. Recently, it was shown that Wnt activity could define colon CSCs and could be regulated by the microen-vironment [133]. The β-catenin was found to enhance the function of AR, and nuclear translocation of β-catenin was observed in prostate cancer tissues, suggesting important role of Wnt signaling in the progression of prostate cancer [134]. Thus, loss or decrease in E-cadherin with abnormal expression of Wnt ligands, receptors, inhibitors, and other co-regulators could also contribute to the activation of Wnt signaling in cancers with the acquisition of EMT phenotype of cancer cells [129, 135]. Therefore, targeting Wnt activity in cancer cells may provide future treatment options, which in part would be due to the elimination of CSCs [130, 135, 136].
Hedgehog signaling pathway
Hedgehog signaling pathway is another important pathway involved in cell development and proliferation which is a major regulator of cell differentiation, tissue polarity, and cell proliferation [137, 138]. Hedgehog signaling pathway is known to secrete three proteins viz. Shh, Desert Hedgehog, and Indian Hedgehog which undergoes different processing to become activated. They stimulate Gli transcription factors which constitute the final effectors of the Hedgehog signaling pathway. It has been found that germline mutations that subtly affect Hedgehog pathway activity are associated with developmental disorders, while various somatic mutations which could activate Hedgehog pathway are thought to be linked with numerous human cancers [139]. Recent reports suggest that activation of the Hedgehog signaling has been found in various human cancers including basal cell carcinomas, medulloblastomas, leukemia, gastrointestinal, lung, ovarian, breast, and prostate cancers [138]. It was found that Hedgehog pathway is involved in the control of proliferation and differentiation of both embryonic stem cells and adult stem cells, while activation of Hedgehog signaling leads to generation of CSCs and the development of cancer [140]. The epithelial expression of hedgehog ligand during prostate development exerted autocrine and paracrine signaling activities that regulates the growth and differentiation of the prostate gland. Enhanced Hedgehog signaling was found to be associated with progression and acceleration of prostate cancer. Recent experiments have shown important role of Hedgehog signaling in prostate cancer, and autocrine Hedgehog signaling by tumor cells is required for proliferation, viability, and invasive behavior of prostate cancer [141]. It has been shown that activation of Hedgehog signaling is involved in CSCs and EMT features in several types of cancers including colon, gastric, esophagus, hepatic, and other cancers [142–146]. Hence, the development of Hedgehog inhibitors could be the future promising strategy of targeting CSC’s and EMT cells for inhibiting recurrence of cancer.
PI3K/mTOR pathway
Several lines of evidence suggest that phosphoinositide-3-kinase (PI3K)/Akt/mTOR signaling system plays a key role in CSC biology. CSCs are more sensitive to pathway inhibition with small molecules when compared to healthy stem cells. This observation provided evidence suggesting that there are functional differences in signaling transduction pathways between CSCs and healthy stem cells [147]. Aberrant signaling via the lipid kinase PI3K, and its main effector kinase Akt, is believed to operate in many tumor tissues [148]. Eyler and group [149] have demonstrated that populations enriched for CSCs are preferentially sensitive to an inhibitor of Akt, a prominent cell survival and invasion signaling node. Treatment with an Akt inhibitor more potently reduced the numbers of viable brain CSCs relative to matched non-stem cancer cells associated with a preferential induction of apoptosis and a suppression of neurosphere formation. Akt inhibition also reduced the motility and invasiveness of all tumor cells but had a greater impact on CSC characteristics. It has been found that the inhibition of Akt activity in CSCs increased the survival of immune-compromised mice bearing human glioma xenografts in vivo. These results have suggested that Akt inhibitors may function as an effective anticancer stem cell therapy [149]. Dubrovska et al. [150] have suggested that the PTEN/PI3K/AKT pathway is critical for the in vitro maintenance of CD133 (+)/CD44 (+) prostate cancer progenitors and consequently suggested that targeting PI3K signaling may be beneficial for the treatment of prostate cancer. Furthermore, it has been shown that the inhibition of PI3K activity by the dual PI3K/mTOR inhibitor NVP-BEZ235 leads to a decrease in the population of CD133 (+)/CD44 (+) prostate cancer progenitor cells in vivo. Moreover, the combination of the PI3K/mTOR modulator NVP-BEZ235, which eliminates prostate cancer progenitor populations, and the chemotherapeutic drug taxotere, which targets the bulk of the differentiated tumor cells, was significantly more effective in eradicating tumors in a prostate, and could be useful for the elimination of CSCs.
Zhou et al. [151] have characterized side population (SP) cells from the human breast cancer cell line MCF-7 as a model for CSLCs. Compared with non-SP cells, MCF-7 SP cells had higher colony formation ability in vitro and greater tumorigenicity in vivo, suggesting that MCF-7 SP cells enriched in CSLCs or CSCs. They identified signal pathways important for CSLCs that were altered in the SP cells, by using cDNA microarray data. Notably, signaling of PI3K/mTOR, signal transduction and activator of transcription (STAT3), and phosphatase and tensin homolog (PTEN) was confirmed to be very critical for MCF-7 SP cell survival and proliferation as tested by pathway specific inhibitors, selected gene knockdown, and in vivo tumorigenicity assays. The STAT3 pathway was found to be positively regulated by mTOR signaling whereas PTEN served as a negative regulator of both STAT3 and mTOR signaling. This study suggested the existence of pro-survival signaling pathways that are critical for the maintenance of CSLCs, which could be selectively targeted in order for improving treatment outcome [151]. Yasuda et al. [152] have examined the role of c-kit receptor (KIT) signal transduction on the proliferation and invasion of colorectal cancer cells and found that c-kit was expressed in two colorectal cancer cell lines. In KIT-positive lines, KIT was activated by stem cell factor (SCF). SCF enhanced cellular proliferation of positive lines as demonstrated by the WST-1 proliferation assay. Furthermore, SCF enhanced the invasive ability of KIT-positive cell lines. SCF stimulation was up-regulated by p44/42 mitogen-activated protein kinase (MAPK) and Akt. PI3K/Akt activity was strongly correlated with proliferation and invasion and p44/42 MAPK associated with invasive capacity. In conclusion, the SCF-enhanced proliferation and invasion of KIT-positive colorectal cancer cells is partly mediated through PI3K/Akt pathway. Thus, PI3K/mTOR pathway appears to play a crucial role in cell proliferation and invasion of CSCs or CSLCs, and thus, targeting this pathway could provide a useful treatment option for eliminating drug resistant cancer cells.
Phytochemicals (nutraceuticals) targeting cancer stem cells
It has been well documented that several cellular and sub-cellular pathways are significantly involved in tumor development, metastasis, and drug resistance. The discovery of CSCs, CSLC, and EMT phenotypic cells and their role in cancer recurrence, metastasis, and drug resistance has been reported for many cancers. Several pathways like Notch, Wnt, PI3K/Akt, and others have been shown to be critically involved in the development of CSCs as discussed above. It is evident that compounds (Fig. 1) that can target these multiple pathways could be useful not only for treatment of cancers but also for elimination of CSCs as well. It has been well-established that many of the naturally occurring phytochemicals can target multiple pathways involved in cancer cells and are considered as promising candidates for anticancer drug development. In this part, we will summarize the potential of few well-known phytochemicals (nutraceuticals) in eliminating CSCs including the new cur-cumin analog developed in our laboratory.
Curcumin
Curcumin (Fig. 1) is a natural polyphenolic compound derived from the plant Curcuma longa on which extensive research has been carried out over the past 50 years. The compound has been implicated as a multi-targeting (pleiotropic) molecule capable of targeting several key oncogenic proteins. Literature reports continue to grow on curcumin with reference to new structural modifications which indicate selective targeting of tissues and abilities to eliminate CSCs.
We have recently examined the effectiveness of the combination therapy involving dasatinib (Src inhibitor) and curcumin in inhibiting the growth and other properties of chemo-resistant colon cancer cells that are enriched in CSCs sub-population. The residual tumors from APCMin+/− mice treated with dasatinib and/or curcumin showed 80–90 % decrease in the expression of the CSC markers ALDH, CD44, CD133, and CD166. The colon cancer cells resistant to therapeutic agents (CR cells) showed higher expression of CSCs markers, cell invasion potential, and ability to form colonospheres, compared to the corresponding parental cells. The combination therapy of dasatinib and curcumin demonstrated synergistic interactions in CR HCT-116 and CR HT-29 cells. The combinatorial therapy inhibited cellular growth, invasion, and colonosphere formation and also showed reduced CSC population as evidenced by decreased expression of CSC-specific markers: CD133, CD44, CD166, and ALDH [153]. Remnants of spontaneous adenomas from APCMin+/− mice treated with dasatinib and/or curcumin were analyzed for several CSC markers such as ALDH, CD44, CD133, and CD166, respectively. Moreover, human colon cancer cells HCT-116 (p53 wild type; K-ras mutant) and HT-29 (p53 mutant; K-ras wild type) were used to generate 5-FU + oxaliplatin resistant (CR) cells for additional experiments. In these studies, the residual tumors from APCMin+/− mice treated with dasatinib and/or curcumin showed 80–90 % decrease in the expression of above CSC markers. The results suggest that such combination therapies may emerge as useful therapeutic strategy for treating chemo-resistant colon cancers. Recently, Lin and coworkers [154] have shown that ALDH(+)/CD133(+) colon cancer cells expressed higher levels of phosphorylated STAT3 than ALDH-negative/CD133-negative colon cancer cells, suggesting that STAT3 is activated in colon CSCs. They found that curcumin and its analog GO-Y030 could inhibit STAT3 phosphorylation, cell viability, colonosphere formation, and induced apoptosis in colon CSCs. GO-Y030 was also found to reduce growth of CSCs in SW480 and HCT-116 colon cancer cell lines transplanted in the mouse model. Ryu et al. [155] have shown that demethoxycurcumin and bisdemethoxycurcumin suppressed β-catenin responsive transcription (CRT) that was activated by Wnt-3a conditioned-medium without altering the level of intracellular β-catenin, with comparable potency to curcumin. Additionally, these compounds have down-regulated p300 protein which is a positive regulator of the Wnt/β-catenin pathway. Another analog, viz. tetrahydrocurcumin, also inhibited CRT and cell proliferation, but to a much lesser degree than curcumin, demethoxycurcumin, or bisdemethoxycurcumin, indicating that the conjugated bonds in the central seven-carbon chain of curcuminoids are essential for the inhibition of Wnt/β-catenin pathway. These findings suggest that curcumin derivatives could inhibit Wnt/β-catenin pathway by decreasing the amount of the transcriptional co-activator p300.
Another report [156] showed that curcumin treatment could lead to p53- and p21-independent G2/M phase arrest and apoptosis in HCT-116 (p53(+/+)), HCT-116(p53(−/−)), and HCT-116(p21(−/−)) cell lines. These activities were found to be mediated by caspase-3-mediated cleavage of β-catenin, decreased trans-activation of β-catenin/Tcf-Lef, decreased promoter DNA binding activity of the β-catenin/Tcf-Lef complex, and decreased levels of c-Myc protein. These activities were linked with decreased Cdc2/cyclin B1 kinase activity, a function of the G2/M phase arrest. Curcumin treatment also induced caspase-3-mediated degradation of cell–cell adhesion proteins β-catenin, E-cadherin, and APC, which were linked with apoptosis. This degradation could be prevented with caspase-3 inhibitors. Thus, it was concluded that curcumin treatment impairs both Wnt signaling and cell–cell adhesion pathways, resulting in G(2)/M phase arrest and apoptosis in HCT-116 cells.
The anticancer effects of curcumin were also found to be mediated through up- or down-regulation of number of miRNAs. Alterations in miRNA expression were seen in curcumin-treated A549 cells, including significant down-regulation of miRNA-186 expression. Additionally, caspase-10 was identified as a target of miRNA-186, suggesting that curcumin induces A549 cell apoptosis through a miRNA pathway involving miRNA-186 as potential target [157]. Mudduluru et al. [158] have examined the potential of curcumin to regulate miR-21, tumor growth, invasion, and in vivo metastasis in colorectal cancer. Curcumin treatment reduced miR-21 promoter activity and expression in a dose-dependent manner by inhibiting AP-1 binding to the promoter and induced the expression of the tumor suppressor programmed cell death protein 4, which is a target of miR-21. Curcumin-treated Rko and HCT116 cells were arrested in the G2/M phase with increasing concentrations. Furthermore, the compound inhibited tumor growth, invasion, and in vivo metastasis in the chicken-embryo-metastasis assay. Additionally, curcumin significantly inhibited miR-21 expression in primary tumors generated in vivo in the CAM assay by Rko and HCT116 cells.
In another study, combination of curcumin and piperine treatment was found to inhibit mammosphere formation, serial passaging, percent of ALDH+ cells, and Wnt signaling by 50 % in normal and malignant breast cells. Both curcumin and piperine inhibited mammosphere formation, serial passaging, and percent of ALDH+ cells by 50 % at 5 μM and completely at 10 μM concentration in normal and malignant breast cells. Wnt signaling was inhibited by both curcumin and piperine by 50 % at 5 μM and completely at 10 μM. The compounds separately or in combination inhibited breast stem cell self-renewal without causing toxicity to differentiated cells [159]. Curcumin has been found to exhibit cytotoxic effect on MCF-7 and MDA-MB-231 cells with IC50 values between 30 and 35 μM and inhibition of several Wnt/β-catenin pathway components. The expression levels of two integral proteins of Wnt signaling, GSK-3β, and E-cadherin were also altered by curcumin treatment [160]. Curcumin also inhibited proliferation (IC50 value 1–5 μM) in tumorigenic MDA-MB-468 human breast cell lines with more pronounced apoptosis. Curcumin inhibited basal phosphorylation of Akt/protein kinase-B (PKB) in both cell lines, but more consistently and to a greater extent in the MDA-MB-468 cells. These results suggest that cur-cumin has several different molecular targets within the MAPK and PI3K/PKB signaling pathways that could contribute to the inhibition of cell proliferation and induction of apoptosis, inhibition of basal activity of Akt/PKB, but not ERK and may facilitate apoptosis in the tumor cell line [161].
Curcumin was found to affect cell proliferation of androgen-dependent prostate cancer through the induction of cell cycle arrest in G2 and modulation of Wnt signaling [162]. It decreased the level of Tcf-4, CBP, and p300 proteins implicated in the Wnt transcriptional complex that leads to the decrease of β-catenin/Tcf-4 transcriptional activity and the expression of β-catenin target genes (cyclin D1 and c-myc). Subsequent cell death induction was linked to autophagy but, interestingly, curcumin did not affect Wnt/β-catenin transcriptional activity in androgen-independent prostate cancer cells. It has also been shown that curcumin targets downstream β-catenin activity and effectively represses HBx-mediated regulation of c-myc and E-cadherin in tumor [163]. In another study, curcumin was shown to induce dose-dependent inhibition of AR expression and β-catenin in the nuclear and cytoplasmic extracts as well as whole cell lysates. The target gene of the β-catenin/T-cell factor transcriptional complex, viz. cyclin D1 and c-myc, were also decreased. These findings suggest that curcumin modulates the Wnt/β-catenin signaling pathway and might have a significant role in mediating inhibitory effects on LNCaP prostate cancer cells [164].
Curcumin treatment has been found to induce cell growth inhibition and apoptosis in pancreatic cancer cell lines which is found to be mediated by down-regulation of Notch-1, Hes-1, and Bcl-XL expression levels. These results were correlated with the inactivation of NF-κB activity and increased apoptosis induced by curcumin. The down-regulation of Notch-1 by small-interfering RNA prior to curcumin treatment resulted in enhanced cell growth inhibition and apoptosis [165]. These and many other in vitro studies showed multi-targeting effects of curcumin; however, curcumin has significant limitation in its activity in vivo [166].
Curcumin analog
Among the multi-targeting natural compounds, curcumin is unique in having high potency yet negligible toxicity. However, its therapeutic utility has remained limited due to inadequate water solubility and lesser bioavailability [166]. In order to overcome these limitations, our group has developed a series of curcumin analogs with different pharmacophores embedded in its structural motif and have evaluated their anticancer effects through in vitro and in vivo studies. Out of these efforts, one has emerged as very effective such as difluorinated curcumin analog, referred as CDF (Fig. 1), with superior biological activity which exhibited inhibitory effects on purified rabbit 26S proteasome and growth inhibition and induction of apoptosis in colon and pancreatic cancer cell lines [167]. Molecular docking of CDF into COX-2 protein cavity revealed that the compound did not introduce any major steric changes in the parent curcumin molecule except promoting additional hydrogen bonding interactions involving residues GLU 346, PHE 580, ASN101, and GLN 350 resulting in higher binding energy than curcumin. Curcumin and CDF both down-regulated NF-κB expression in MIAPaCa-2 cells, but the effect was more pronounced at much lower concentration in case of CDF [168]. Both lowered PGE2 levels in MIAPaCa-2 cells although in case of BxPC-3 cells lowering was found only in CDF-treated cells and not with curcumin treatment. These results suggest that CDF targets COX-2 in a more pronounced manner than curcumin [168].
Pharmacokinetic studies [168] on curcumin and CDF at a single oral dose of 250 mg/kg to female ICR-SCID mice revealed that CDF achieved Cmax (0.21 μg/mL) with a relatively slow oral absorption with Tmax of 8 h, although it achieved 2.7-fold higher systemic drug level than curcumin (AUClast, 1.22 vs. 0.44 μg/mL h). Interestingly, CDF accumulated preferentially in pancreas (10.6-fold higher then curcumin) with Cmax 44.5-fold higher than in serum, suggesting that CDF has a better bioavailability profile, especially in pancreatic tissue. This property of CDF makes it a promising candidate for either inclusion in the prevention strategy of pancreatic cancer or for the treatment of pancreatic cancer in conjunction with cytotoxic agents similar to those reported for curcumin. The combination of CDF with gemcitabine exhibited a significant reduction in cell viability as compared to CDF alone in BxPC-3 cell lines [169]. The combination caused a significant induction of apoptosis in BxPC-3, MIAPaCa-E, and MIAPaCa-M cell lines when compared with single agent alone which was attributed to inactivation of NF-κB in both gemcitabine-resistant and gemcitabine-sensitive cell lines.
CDF in combination with gemcitabine brings about a remarkable reduction of cell survival and completely eliminates pancreatospheres formation after 4 weeks of treatment, suggesting that CDF could be a useful therapeutic agent for eliminating CSCs [19]. CDF also decreased CD44 and EpCAM expressions in pancreatospheres, suggesting its inhibitory effect on pancreatospheres formation may be associated with the inhibition of CD44 and EpCAM expression. Using in vivo subcutaneous xenograft tumor model induced by MIAPaCa-2 cells in CB17-SCID mice, we were able to show that CDF treatment in combination with gemcitabine significantly inhibited tumor growth of MIAPaCa-2 tumors. The mice did not show any weight loss during the treatment period (30 days), suggesting that these treatment had no major adverse effects on animals. CDF treatment with or without gemcitabine combination showed increased expression of miR-200b and miR-200c. Similar results were also found in case of orthotropic xenograft model of human pancreatic cancers where administration of CDF inhibited tumor growth associated with reduced expression of EZH2, Notch-1, CD44, EpCAM, Nanog, and increased expression of let-7, miR-26a, and miR-101 [170]. These results indicate that CDF inhibits PC tumor growth and aggressiveness by targeting an EZH2-miRNA regulatory circuit through epigenetic control of specific gene expression.
We have extended our studies to test the effects of CDF in combination with 5-fluorouracil and oxaliplatin (5-FU+ Ox) against colon cancer [21]. The chemo-resistant HCT-116 and HT-29 cells were analyzed for cellular growth following incubation with 5-FU+Ox alone or in combination with increasing concentrations (up to 8 μM) of either CDF or curcumin. A significant inhibition of cellular growth of chemo-resistant cells was observed when the chemo-resistant HCT-116 or HT-29 cells were incubated for 48 h with the combination of 4 or 8 μM of CDF and 5-FU+Ox. The expression of ABC transporter protein ABCG2 in chemo-resistant HCT-116 cells was inhibited by more than 50 % in response to above combination. These results suggest that CDF together with the conventional chemotherapeutics could be an effective treatment strategy for preventing the emergence of chemo-resistant colon cancer cells by eliminating CSCs.
Sulforaphane
Sulforaphanes are isothiocyanate compounds which are formed by hydrolysis of their precursor parent glucosinolates compounds found in cruciferous vegetables such as broccoli, cauliflower, kale, and cabbage. As an inducer of phase II enzymes [171], the compounds enhance detoxification of carcinogens based on a mechanism contributing to anti-carcinogenic activity [172, 173]. A large number of studies have shown that sulforaphane (Fig. 1) can prevent, delay, or reverse pre-neoplastic lesions and has anti-proliferative effects on several cancers including breast, hepatic, bladder, osteosarcoma, pancreatic, and melanoma [174–178].
The effects of R-sulforaphane on the biology on human mesenchymal stem cells (MSCs), along with their ability to differentiate into mesenchymal tissues contribute to the homeostatic maintenance of many organs and tissues, have been evaluated [179]. It was found that low doses of R-sulforaphane promote MSCs proliferation and protect them from apoptosis and senescence, while higher doses have a cytotoxic effect, leading to the induction of cell cycle arrest, programmed cell death and senescence. The beneficial effects of R-sulforaphane may be ascribed to its antioxidant properties, which were observed when MSC cultures were incubated with low doses of R-sulforaphane. Its cytotoxic effects observed after treating MSCs with high doses of R-sulforaphane could be attributed to its HDAC inhibitory activity.
Srivastava and coworkers [180] have evaluated the effects of sulforaphane alone and in combination with quercetin on self-renewal capacity of pancreatic CSCs. Compounds inhibited self-renewal capacity of pancreatic CSCs, while enhanced effects were observed with inhibition of Nanog by lentiviral-mediated short hairpin RNA (shRNA) expression. Sulforaphane induced apoptosis by inhibiting the expression of Bcl-2 and XIAP, phosphorylation of FKHR, and activating caspase-3 enzyme. It inhibited the expression of proteins involved in the EMT (β-catenin, vimentin, twist-1, and ZEB1), suggesting blockade of signaling involved in early metastasis. Furthermore, the combination of quercetin with sulforaphane had synergistic effects on self-renewal capacity of pancreatic CSCs. These data suggest that sulforaphane either alone or in combination with quercetin can eliminate CSC characteristics. In another study, quercetin was found to reduce self-renewal as measured by spheroid and colony formation pancreatic CSCs without causing pronounced toxicity on normal cells or mice [181].
The effect of sulforaphane in combination with existing anticancer compounds was evaluated in pancreatic CSCs. Cisplatin, gemcitabine, doxorubicin, and 5-flurouracil effectively induced apoptosis and prevented cell viability, while the combination of these drugs with sulforaphane increased toxicity. Sulforaphane potentiated the drug effect in established prostate CSCs revealing that the compound enhances drug cytotoxicity in other tumor entities. Most importantly, combined treatment enhanced the inhibition of clonogenicity, spheroid formation, and aldehyde dehydrogenase 1 (ALDH1) activity along with Notch-1 and c-Rel expressions. In vivo studies indicated that the combination treatment was most effective and totally abolished growth of CSC xenografts and tumor-initiating potential without affecting normal cells or mouse health [182].
The co-administration of sulforaphane with sorafenib led to complete eradication of sorafenib-induced NF-κB binding, associated with abrogated clonogenicity, spheroid formation, ALDH1 activity, migratory capacity, and induction of apoptosis [183]. Combination therapy under in vivo studies reduced the tumor size in a synergistic manner due to the induction of apoptosis, inhibition of proliferation as well as angiogenesis, and down-regulation of sorafenib-induced expression of proteins involved in EMT. Sulforaphane (1–5 μM/L) has been found to decrease aldehyde dehydrogenase-positive cell population by 65 to 80 % in human breast cancer cells and reduced the size and number of primary mammospheres [184]. Sulforaphane also eliminated breast CSCs in vivo, abrogating tumor growth after the re-implantation of primary tumor cells into the secondary mice. Western blot analysis and β-catenin reporter assay showed that the compound down-regulated the Wnt/β-catenin self-renewal pathway.
Resveratrol
Resveratrol (3, 5, 4′-trihydroxy-trans-stilbene) (Fig. 1) was first isolated in 1940 as an ingredient of the roots of white hellebore (Veratrum grandiflorum O. Loes) and has since been found in a wide variety of plant species, including grapes, mulberries, and peanuts [185]. Identified as the active constituent of the dried roots of Polygonum cuspidatum and grapevines (Vitis vinifera), it is well-known for its biological activities like antioxidant, anti-inflammatory, antibacterial, and anticancer activity against breast, prostate, colon, thyroid, skin, and pancreatic cancers [185, 186].
Shankar et al. [187] have studied the molecular mechanisms by which resveratrol inhibits stem cell characteristics of pancreatic CSCs derived from human primary tumors and K-Ras (G12D) transgenic mice. The compound was found to induce apoptosis by activating capase-3 and caspase-7 enzymes and inhibiting the expression of Bcl-2 and XIAP in human CSCs. It also inhibited pluripotency maintaining factors (Nanog, Sox-2, c-Myc, and Oct-4) and drug resistance gene ABCG2 in CSCs. The effect of resveratrol on lipid synthesis in CSLCs isolated from both ER+ and ER-breast cancer cell lines has been examined. The compound was found to reduce cell viability and mammosphere formation followed by inducing apoptosis. It was accompanied by the reduction in lipid synthesis due to down-regulation of the fatty acid synthase gene and up-regulation of pro-apoptotic genes, DAPK2 and BNIP3. Resveratrol was able to significantly suppress the growth of CSLCs in an animal model without showing apparent toxicity [188].
Resveratrol was also found to inhibit Wnt signaling and repressed colorectal cell proliferation, but it was ineffective at concentrations less than 10 μM. In order to understand the structure–activity relationship of stilbene derivatives and for further development of more efficacious Wnt inhibitors than these natural products, Zhang et al. [189] have synthesized and evaluated a panel of fluorinated N, N-dialkyl-aminostilbenes. Among this panel, (E)-4-(2, 6-difluorostyryl)-N, N-dimethylaniline inhibited Wnt signaling at nanomolar levels associated with inhibition of human colorectal tumor in a xenograft nude mice model at a dosage of 20 mg/kg. These fluorinated N, N-dialkylaminostilbenes appear to inhibit Wnt signaling downstream of β-catenin at the transcriptional level.
Genistein
Research from last few decades has provided convincing evidence that isoflavones in soy rich foods contribute to relatively lower rates of prostate and breast cancers in Asian countries such as China and Japan than in the Western population. Genistein (4,5,7-trihydroxyisoflavone) (Fig. 1) has been identified as the pre-dominant isoflavone in soybean enriched foods which comprises a significant portion of the Asian diet and provides 10 % of the total per capita protein intake in Japan and China [190]. Asian men who consume a soy rich diet were found to have high levels of soy isoflavone in the serum, urine, and prostatic fluid which has been suggested to contribute to lowering of the incidence of prostate cancer [191, 192]. Similar results have also been found with high consumption of soymilk [193]. Isoflavone was found to sensitize the effect of radiotherapy and cytotoxic chemotherapeutic drugs used in a variety of cancers including pancreatic, breast, prostate, head and neck, lung, and bladder cancer [194].
We have established that FoxM1 is over-expressed in pancreatic cancer cells AsPC-1, which corresponds to increased cell growth, clonogenicity, and cell migration [13]. Moreover, over-expression of FoxM1 led to the acquisition of EMT phenotype by activation of mesenchymal cell markers, ZEB1, ZEB2, Snail2, E-cadherin, and vimentin, which is consistent with increased sphere-forming (pancreatospheres) capacity and expression of CSC surface markers (CD44 and EpCAM). Over-expression of FoxM1 also led to decreased expression of miRNAs (let-7a, let-7b, let-7c, miR-200b, and miR-200c). Genistein was found to inhibit cell growth, clonogenicity, cell migration and invasion, EMT phenotype, and formation of pancreatospheres consistent with reduced expression of CD44 and EpCAM. These results suggest that FoxM1 over-expression is responsible for the acquisition of EMT and CSC phenotype, which is in part mediated through regulation of miR-200b, and these processes could be attenuated by genistein. It was also found that genistein up-regulated ARHI by down-regulating miR-221 and mir-222 in PC-3 prostate cancer cells further demonstrating that prostate cancer cells have decreased level of ARHI which could be caused by direct targeting of 3′UTR of ARHI by miR221/222 [195]. Genistein restored the expression of ARHI which may be an important dietary therapeutic agent for treating prostate cancer. We have investigated the cellular consequences of Notch-1 down-regulation as well as the molecular consequences of Notch-1-mediated alterations of its downstream targets on cell viability and apoptosis in prostate cancer (PCa) cells [20]. Genistein could cause reduction in cell viability and induced apoptosis of PCa cells, which was consistent with down-regulation of Notch-1, Akt, and FoxM1.
Tea polyphenols
The leaves of Camellia sinensis plant contain an assortment of compounds, most significant being catechins or polyphenols. The catechins account for 30–42 % of the dry weight of the solids in brewed green tea. The most abundant catechin present is epigallocatechin gallate (EGCG) (Fig. 1) which has been the subject of intense research in recent years and has been shown to exert anticancer effects against several cancers including pancreatic, breast, colon, lung, prostate cancer, and many others [196].
The major signaling pathway affected by green tea extract and EGCG are EGFR and Notch pathways, which in turn affect cell cycle-related networks resulting in the collective inhibition of cancer cell growth [197]. Wnt signaling was also found to be inhibited by EGCG in a dose-dependent manner in breast cancer cells. EGCG treatment-induced HBP1 transcriptional repressor levels through an increase in HBP1 mRNA stability, but not transcriptional initiation. To test functionality, shRNA was used to knock down the endogenous HBP1 gene. The knockdown reduced sensitivity to EGCG in the suppression of Wnt signaling and c-myc gene. These studies seem to suggest that EGCG blocks Wnt signaling by inducing the HBP1 transcriptional repressor and inhibiting invasive breast cancer [198]. Van Aller and group [199] have shown that EGCG is an ATP-competitive inhibitor of both PI3K and mTOR with K(i) values of 380 and 320 nM, respectively, that are within physiologically relevant concentrations. In addition, EGCG inhibits cell proliferation and Akt phosphorylation at Ser473 in MDA-MB-231 and A549 cells. Molecular docking studies have shown that EGCG binds well to the PI3K kinase domain active site, suggesting that EGCG competes for ATP binding.
Fix et al. [200] have reported, for the first time, the effect of green tea on the miRNA expression profile of human breast cancer MCF-7 cells. Low concentration treatment with the green tea extract (Polyphenon-60) significantly altered the miRNA expression profile in MCF-7 cells which included down-regulation of miR-21 and miR-27, respectively. These two miRNAs have previously been identified as being over-expressed in MCF-7 breast cancer cells, with miR-21 specifically implicated in down-regulating the tumor suppressor gene, tropomyosin-1. These data support the hypothesis that green tea extract treatment of the breast cancer alters miRNA expression profile, which in part contributes to the efficacy of green tea-derived compounds. EGCG was found to be a direct antagonist of androgen action [201]. In silico modeling and FRET-based competition assay showed that EGCG physically interacts with the ligand-binding domain of AR by replacing a high-affinity labeled ligand (IC50 value 0.4 μM). The functional consequence of this interaction was a decrease in AR-mediated transcriptional activation, which was due to EGCG mediated inhibition of inter-domain N-C terminal interaction of AR. In a xenograft model, EGCG was found to inhibit AR nuclear translocation and protein expression. In further studies, these workers observed a significant down-regulation of androgen-regulated miR-21 and up-regulation of a tumor suppressor miRNA, miR-330, in tumors of mice treated with EGCG. These results indicate that EGCG could functionally antagonize androgen action at multiple levels, resulting in the inhibition of prostate cancer growth.
Summary and conclusions
It has been known for quite some time that cancer cells grow relentlessly and metastasize through deregulation of multiple pathways. It is only recently that researchers have confirmed that cancer cells have stem cell populations which are responsible for drug resistance and tumor recurrence. Consequently, for combating cancer, potent multi-targeting compounds that can block the self-renewal capacity and survival pathways of CSCs would be required. Nature is a rich source of such multi-targeting compounds, and attempts have been made in recent years to explore their potential for eliminating CSCs and overcoming therapeutic resistance. Current literature review suggests that some of the well-studied phytochemicals (nutraceuticals) and their novel derivatives/analogs (particularly curcumin-derivative, CDF) can effectively modulate multiple self-renewal pathways/factors associated with CSCs (Figs. 2 and 3). With the issue of bioavailability hampering the progress of phytochemicals in clinical settings, it is pertinent to design novel analogs/derivatives of these natural anticancer agents with special emphasis on targeting CSCs. It is expected that some of these phytochemicals, their analogs, and their carrier-mediated delivery may finally provide the solution for eliminating CSCs, which would strengthen our efforts to find a cure for human malignancies.
Acknowledgments
Part of the work cited in this article was funded by National Cancer Institute, NIH grant 1R01CA154321 (F.H. Sarkar).
Contributor Information
Prasad Dandawate, ISTRA, Department of Chemistry, Abeda Inamdar Senior College, University of Pune, Pune 411001, India.
Subhash Padhye, ISTRA, Department of Chemistry, Abeda Inamdar Senior College, University of Pune, Pune 411001, India. Department of Pathology, Barbara Ann Karmanos Cancer Institute, Wayne State University School of Medicine, 740 HWCRC Bldg, 4100 John R. Street, Detroit, MI 48201, USA.
Aamir Ahmad, Department of Pathology, Barbara Ann Karmanos Cancer Institute, Wayne State University School of Medicine, 740 HWCRC Bldg, 4100 John R. Street, Detroit, MI 48201, USA.
Fazlul H. Sarkar, Email: fsarkar@med.wayne.edu, Department of Pathology, Barbara Ann Karmanos Cancer Institute, Wayne State University School of Medicine, 740 HWCRC Bldg, 4100 John R. Street, Detroit, MI 48201, USA. Department of Oncology, Barbara Ann Karmanos Cancer Institute, Wayne State University School of Medicine, Detroit, MI 48201, USA
References
- 1.Siegel R, Ward E, Brawley O, Jemal A. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J Clin. 2011;61:212–36. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat Med. 2004;10:789–99. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 3.Gomez-Veiga F, Marino A, Alvarez L, Rodriguez I, Fernandez C, Pertega S, Candal A. Brachytherapy for the treatment of recurrent prostate cancer after radiotherapy or radical prostatectomy. BJU Int. 2012;109 (Suppl 1):17–21. doi: 10.1111/j.1464-410X.2011.10826.x. [DOI] [PubMed] [Google Scholar]
- 4.Meguid RA, Hooker CM, Taylor JT, Kleinberg LR, Cattaneo SM, Sussman MS, Yang SC, Heitmiller RF, Forastiere AA, Brock MV. Recurrence after neoadjuvant chemoradiation and surgery for esophageal cancer: does the pattern of recurrence differ for patients with complete response and those with partial or no response? J Thorac Cardiovasc Surg. 2009;138:1309–17. doi: 10.1016/j.jtcvs.2009.07.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li Y, Laterra J. Cancer stem cells: distinct entities or dynamically regulated phenotypes? Cancer Res. 2012;72:576–80. doi: 10.1158/0008-5472.CAN-11-3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer. 2005;5:275–84. doi: 10.1038/nrc1590. [DOI] [PubMed] [Google Scholar]
- 7.Subramaniam D, Ramalingam S, Houchen CW, Anant S. Cancer stem cells: a novel paradigm for cancer prevention and treatment. Mini Rev Med Chem. 2010;10:359–71. doi: 10.2174/138955710791330954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Padhye S, Chavan D, Pandey S, Deshpande J, Swamy KV, Sarkar FH. Perspectives on chemopreventive and therapeutic potential of curcumin analogs in medicinal chemistry. Mini Rev Med Chem. 2010;10:372–87. doi: 10.2174/138955710791330891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neves AR, Lucio M, Lima JL, Reis S. Resveratrol in medicinal chemistry: a critical review of its pharmacokinetics, drug-delivery, and membrane interactions. Curr Med Chem. 2012;19:1663–81. doi: 10.2174/092986712799945085. [DOI] [PubMed] [Google Scholar]
- 10.Kong D, Wang Z, Sarkar SH, Li Y, Banerjee S, Saliganan A, Kim HR, Cher ML, Sarkar FH. Platelet-derived growth factor-D over-expression contributes to epithelial–mesenchymal transition of PC3 prostate cancer cells. Stem Cells. 2008;26:1425–35. doi: 10.1634/stemcells.2007-1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kong D, Banerjee S, Ahmad A, Li Y, Wang Z, Sethi S, Sarkar FH. Epithelial to mesenchymal transition is mechanistically linked with stem cell signatures in prostate cancer cells. PLoS One. 2010;5:e12445. doi: 10.1371/journal.pone.0012445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sethi S, Sarkar FH, Ahmed Q, Bandyopadhyay S, Nahleh ZA, Semaan A, Sakr W, Munkarah A, Li-Fehmi R. Molecular markers of epithelial-to-mesenchymal transition are associated with tumor aggressiveness in breast carcinoma. Transl Oncol. 2011;4:222–6. doi: 10.1593/tlo.10244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bao B, Wang Z, Ali S, Kong D, Banerjee S, Ahmad A, Li Y, Azmi AS, Miele L, Sarkar FH. Over-expression of FoxM1 leads to epithelial–mesenchymal transition and cancer stem cell phenotype in pancreatic cancer cells. J Cell Biochem. 2011;112:2296–306. doi: 10.1002/jcb.23150. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 14.Bao B, Wang Z, Ali S, Kong D, Li Y, Ahmad A, Banerjee S, Azmi AS, Miele L, Sarkar FH. Notch-1 induces epithelial–mesenchymal transition consistent with cancer stem cell phenotype in pancreatic cancer cells. Cancer Lett. 2011;307:26–36. doi: 10.1016/j.canlet.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Wang Z, Li Y, Kong D, Banerjee S, Ahmad A, Azmi AS, Ali S, Abbruzzese JL, Gallick GE, Sarkar FH. Acquisition of epithelial–mesenchymal transition phenotype of gemcitabine-resistant pancreatic cancer cells is linked with activation of the notch signaling pathway. Cancer Res. 2009;69:2400–7. doi: 10.1158/0008-5472.CAN-08-4312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang Z, Li Y, Banerjee S, Sarkar FH. Emerging role of Notch in stem cells and cancer. Cancer Lett. 2009;279:8–12. doi: 10.1016/j.canlet.2008.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang Z, Li Y, Ahmad A, Azmi AS, Kong D, Banerjee S, Sarkar FH. Targeting miRNAs involved in cancer stem cell and EMT regulation: an emerging concept in overcoming drug resistance. Drug Resist Updat. 2010;13:109–18. doi: 10.1016/j.drup.2010.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang Z, Li Y, Sarkar FH. Signaling mechanism(s) of reactive oxygen species in epithelial–mesenchymal transition reminiscent of cancer stem cells in tumor progression. Curr Stem Cell Res Ther. 2010;5:74–80. doi: 10.2174/157488810790442813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bao B, Ali S, Kong D, Sarkar SH, Wang Z, Banerjee S, Aboukameel A, Padhye S, Philip PA, Sarkar FH. Anti-tumor activity of a novel compound-CDF is mediated by regulating miR-21, miR-200, and PTEN in pancreatic cancer. PLoS One. 2011;6:e17850. doi: 10.1371/journal.pone.0017850. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D, Wojewoda C, Miele L, Sarkar FH. Down-regulation of Notch-1 is associated with Akt and FoxM1 in inducing cell growth inhibition and apoptosis in prostate cancer cells. J Cell Biochem. 2011;112:78–88. doi: 10.1002/jcb.22770. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 21.Kanwar SS, Yu Y, Nautiyal J, Patel BB, Padhye S, Sarkar FH, Majumdar AP. Difluorinated-curcumin (CDF): a novel curcumin analog is a potent inhibitor of colon cancer stem-like cells. Pharm Res. 2011;28:827–38. doi: 10.1007/s11095-010-0336-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sarkar FH, Li Y, Wang Z, Kong D, Ali S. Implication of micro-RNAs in drug resistance for designing novel cancer therapy. Drug Resist Updat. 2010;13:57–66. doi: 10.1016/j.drup.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y, Vandenboom TG, Kong D, Wang Z, Ali S, Philip PA, Sarkar FH. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69:6704–12. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kong D, Li Y, Wang Z, Sarkar FH. Cancer stem cells and epithelial-to-mesenchymal transition (EMT)-phenotypic cells: are they cousins or twins? Cancers (Basel) 2011;3:716–29. doi: 10.3390/cancers30100716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pardal R, Clarke MF, Morrison SJ. Applying the principles of stem-cell biology to cancer. Nat Rev Cancer. 2003;3:895–902. doi: 10.1038/nrc1232. [DOI] [PubMed] [Google Scholar]
- 26.Hermann PC, Huber SL, Herrler T, Aicher A, Ellwart JW, Guba M, Bruns CJ, Heeschen C. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1:313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 27.O’Brien CA, Pollett A, Gallinger S, Dick JE. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature. 2007;445:106–10. doi: 10.1038/nature05372. [DOI] [PubMed] [Google Scholar]
- 28.Zhou BB, Zhang H, Damelin M, Geles KG, Grindley JC, Dirks PB. Tumour-initiating cells: challenges and opportunities for anticancer drug discovery. Nat Rev Drug Discov. 2009;8:806–23. doi: 10.1038/nrd2137. [DOI] [PubMed] [Google Scholar]
- 29.Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M, Campbell LL, Polyak K, Brisken C, Yang J, Weinberg RA. The epithelial–mesenchymal transition generates cells with properties of stem cells. Cell. 2008;133:704–15. doi: 10.1016/j.cell.2008.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Polyak K, Weinberg RA. Transitions between epithelial and mesenchymal states: acquisition of malignant and stem cell traits. Nat Rev Cancer. 2009;9:265–73. doi: 10.1038/nrc2620. [DOI] [PubMed] [Google Scholar]
- 31.Wang Z, Li Y, Ahmad A, Banerjee S, Azmi AS, Kong D, Sarkar FH. Pancreatic cancer: understanding and overcoming chemo-resistance. Nat Rev Gastroenterol Hepatol. 2011;8:27–33. doi: 10.1038/nrgastro.2010.188. [DOI] [PubMed] [Google Scholar]
- 32.Singh A, Settleman J. EMT, cancer stem cells and drug resistance: an emerging axis of evil in the war on cancer. Oncogene. 2010;29:4741–51. doi: 10.1038/onc.2010.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thiery JP. Epithelial–mesenchymal transitions in tumour progression. Nat Rev Cancer. 2002;2:442–54. doi: 10.1038/nrc822. [DOI] [PubMed] [Google Scholar]
- 34.Brabletz T, Jung A, Reu S, Porzner M, Hlubek F, Kunz-Schughart LA, Knuechel R, Kirchner T. Variable beta-catenin expression in colorectal cancers indicates tumor progression driven by the tumor environment. Proc Natl Acad Sci U S A. 2001;98:10356–61. doi: 10.1073/pnas.171610498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brabletz T, Jung A, Spaderna S, Hlubek F, Kirchner T. Opinion: migrating cancer stem cells—an integrated concept of malignant tumour progression. Nat Rev Cancer. 2005;5:744–9. doi: 10.1038/nrc1694. [DOI] [PubMed] [Google Scholar]
- 36.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial–mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172:973–81. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hollier BG, Evans K, Mani SA. The epithelial-to-mesenchymal transition and cancer stem cells: a coalition against cancer therapies. J Mammary Gland Biol Neoplasia. 2009;14:29–43. doi: 10.1007/s10911-009-9110-3. [DOI] [PubMed] [Google Scholar]
- 38.Hugo H, Ackland ML, Blick T, Lawrence MG, Clements JA, Williams ED, Thompson EW. Epithelial–mesenchymal and mesenchymal–epithelial transitions in carcinoma progression. J Cell Physiol. 2007;213:374–83. doi: 10.1002/jcp.21223. [DOI] [PubMed] [Google Scholar]
- 39.Christiansen JJ, Rajasekaran AK. Reassessing epithelial to mesenchymal transition as a prerequisite for carcinoma invasion and metastasis. Cancer Res. 2006;66:8319–26. doi: 10.1158/0008-5472.CAN-06-0410. [DOI] [PubMed] [Google Scholar]
- 40.Chaffer CL, Brennan JP, Slavin JL, Blick T, Thompson EW, Williams ED. Mesenchymal-to-epithelial transition facilitates bladder cancer metastasis: role of fibroblast growth factor receptor-2. Cancer Res. 2006;66:11271–8. doi: 10.1158/0008-5472.CAN-06-2044. [DOI] [PubMed] [Google Scholar]
- 41.Irie HY, Pearline RV, Grueneberg D, Hsia M, Ravichandran P, Kothari N, Natesan S, Brugge JS. Distinct roles of Akt1 and Akt2 in regulating cell migration and epithelial–mesenchymal transition. J Cell Biol. 2005;171:1023–34. doi: 10.1083/jcb.200505087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moustakas A, Heldin CH. Signaling networks guiding epithelial–mesenchymal transitions during embryogenesis and cancer progression. Cancer Sci. 2007;98:1512–20. doi: 10.1111/j.1349-7006.2007.00550.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Thiery JP, Sleeman JP. Complex networks orchestrate epithelial–mesenchymal transitions. Nat Rev Mol Cell Biol. 2006;7:131–42. doi: 10.1038/nrm1835. [DOI] [PubMed] [Google Scholar]
- 44.Schmalhofer O, Brabletz S, Brabletz T. E-cadherin, beta-catenin, and ZEB1 in malignant progression of cancer. Cancer Metastasis Rev. 2009;28:151–66. doi: 10.1007/s10555-008-9179-y. [DOI] [PubMed] [Google Scholar]
- 45.Berx G, Raspe E, Christofori G, Thiery JP, Sleeman JP. Pre-EMTing metastasis? Recapitulation of morphogenetic processes in cancer. Clin Exp Metastasis. 2007;24:587–97. doi: 10.1007/s10585-007-9114-6. [DOI] [PubMed] [Google Scholar]
- 46.Gregory PA, Bert AG, Paterson EL, Barry SC, Tsykin A, Farshid G, Vadas MA, Khew-Goodall Y, Goodall GJ. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol. 2008;10:593–601. doi: 10.1038/ncb1722. [DOI] [PubMed] [Google Scholar]
- 47.Kasper S. Stem cells: the root of prostate cancer? J Cell Physiol. 2008;216:332–6. doi: 10.1002/jcp.21489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kasper S. Identification, characterization, and biological relevance of prostate cancer stem cells from clinical specimens. Urol Oncol. 2009;27:301–3. doi: 10.1016/j.urolonc.2008.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Marian CO, Shay JW. Prostate tumor-initiating cells: a new target for telomerase inhibition therapy? Biochim Biophys Acta. 2009;1792:289–96. doi: 10.1016/j.bbadis.2009.02.012. [DOI] [PubMed] [Google Scholar]
- 50.Peter ME. Let-7 and miR-200 microRNAs: guardians against pluripotency and cancer progression. Cell Cycle. 2009;8:843–52. doi: 10.4161/cc.8.6.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Santisteban M, Reiman JM, Asiedu MK, Behrens MD, Nassar A, Kalli KR, Haluska P, Ingle JN, Hartmann LC, Manjili MH, Radisky DC, Ferrone S, Knutson KL. Immune-induced epithelial to mesenchymal transition in vivo generates breast cancer stem cells. Cancer Res. 2009;69:2887–95. doi: 10.1158/0008-5472.CAN-08-3343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Armstrong AJ, Marengo MS, Oltean S, Kemeny G, Bitting RL, Turnbull JD, Herold CI, Marcom PK, George DJ, Garcia-Blanco MA. Circulating tumor cells from patients with advanced prostate and breast cancer display both epithelial and mesenchymal markers. Mol Cancer Res. 2011;9:997–1007. doi: 10.1158/1541-7786.MCR-10-0490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lapidot T, Sirard C, Vormoor J, Murdoch B, Hoang T, Caceres-Cortes J, Minden M, Paterson B, Caligiuri MA, Dick JE. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367:645–8. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 54.Al-Hajj M, Wicha MS, Ito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci U S A. 2003;100:3983–8. doi: 10.1073/pnas.0530291100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ricci-Vitiani L, Lombardi DG, Pilozzi E, Biffoni M, Todaro M, Peschle C, De MR. Identification and expansion of human colon-cancer-initiating cells. Nature. 2007;445:111–5. doi: 10.1038/nature05384. [DOI] [PubMed] [Google Scholar]
- 56.Singh SK, Clarke ID, Terasaki M, Bonn VE, Hawkins C, Squire J, Dirks PB. Identification of a cancer stem cell in human brain tumors. Cancer Res. 2003;63:5821–8. [PubMed] [Google Scholar]
- 57.Singh SK, Hawkins C, Clarke ID, Squire JA, Bayani J, Hide T, Henkelman RM, Cusimano MD, Dirks PB. Identification of human brain tumour initiating cells. Nature. 2004;432:396–401. doi: 10.1038/nature03128. [DOI] [PubMed] [Google Scholar]
- 58.Li C, Heidt DG, Dalerba P, Burant CF, Zhang L, Adsay V, Wicha M, Clarke MF, Simeone DM. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67:1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 59.Eguchi S, Kanematsu T, Arii S, Omata M, Kudo M, Sakamoto M, Takayasu K, Makuuchi M, Matsuyama Y, Monden M. Recurrence-free survival more than 10 years after liver resection for hepatocellular carcinoma. Br J Surg. 2011;98:552–7. doi: 10.1002/bjs.7393. [DOI] [PubMed] [Google Scholar]
- 60.Zhu Z, Hao X, Yan M, Yao M, Ge C, Gu J, Li J. Cancer stem/progenitor cells are highly enriched in CD133+CD44+ population in hepatocellular carcinoma. Int J Cancer. 2010;126:2067–78. doi: 10.1002/ijc.24868. [DOI] [PubMed] [Google Scholar]
- 61.Curley MD, Therrien VA, Cummings CL, Sergent PA, Koulouris CR, Friel AM, Roberts DJ, Seiden MV, Scadden DT, Rueda BR, Foster R. CD133 expression defines a tumor initiating cell population in primary human ovarian cancer. Stem Cells. 2009;27:2875–83. doi: 10.1002/stem.236. [DOI] [PubMed] [Google Scholar]
- 62.Suzuki S, Terauchi M, Umezu T, Kajiyama H, Shibata K, Nawa A, Kikkawa F. Identification and characterization of cancer stem cells in ovarian yolk sac tumors. Cancer Sci. 2010;101:2179–85. doi: 10.1111/j.1349-7006.2010.01672.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ning ZF, Huang YJ, Lin TX, Zhou YX, Jiang C, Xu KW, Huang H, Yin XB, Huang J. Subpopulations of stem-like cells in side population cells from the human bladder transitional cell cancer cell line T24. J Int Med Res. 2009;37:621–30. doi: 10.1177/147323000903700304. [DOI] [PubMed] [Google Scholar]
- 64.She JJ, Zhang PG, Wang ZM, Gan WM, Che XM. Identification of side population cells from bladder cancer cells by DyeCycle Violet staining. Cancer Biol Ther. 2008;7:1663–8. doi: 10.4161/cbt.7.10.6637. [DOI] [PubMed] [Google Scholar]
- 65.Janikova M, Skarda J, Dziechciarkova M, Radova L, Chmelova J, Krejci V, Sedlakova E, Zapletalova J, Langova K, Klein J, Grygarkova I, Kolek V. Identification of CD133+/nestin+ putative cancer stem cells in non-small cell lung cancer. Biomed Pap Med Fac Univ Palacky Olomouc Czech Repub. 2010;154:321–6. doi: 10.5507/bp.2010.048. [DOI] [PubMed] [Google Scholar]
- 66.Leung EL, Fiscus RR, Tung JW, Tin VP, Cheng LC, Sihoe AD, Fink LM, Ma Y, Wong MP. Non-small cell lung cancer cells expressing CD44 are enriched for stem cell-like properties. PLoS One. 2010;5:e14062. doi: 10.1371/journal.pone.0014062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moriyama T, Ohuchida K, Mizumoto K, Cui L, Ikenaga N, Sato N, Tanaka M. Enhanced cell migration and invasion of CD133+ pancreatic cancer cells cocultured with pancreatic stromal cells. Cancer. 2010;116:3357–68. doi: 10.1002/cncr.25121. [DOI] [PubMed] [Google Scholar]
- 68.Bracken CP, Gregory PA, Khew-Goodall Y, Goodall GJ. The role of microRNAs in metastasis and epithelial–mesenchymal transition. Cell Mol Life Sci. 2009;66:1682–99. doi: 10.1007/s00018-009-8750-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gibbons DL, Lin W, Creighton CJ, Rizvi ZH, Gregory PA, Goodall GJ, Thilaganathan N, Du L, Zhang Y, Pertsemlidis A, Kurie JM. Contextual extracellular cues promote tumor cell EMT and metastasis by regulating miR-200 family expression. Genes Dev. 2009;23:2140–51. doi: 10.1101/gad.1820209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Gregory PA, Bracken CP, Bert AG, Goodall GJ. MicroRNAs as regulators of epithelial–mesenchymal transition. Cell Cycle. 2008;7:3112–8. doi: 10.4161/cc.7.20.6851. [DOI] [PubMed] [Google Scholar]
- 71.Wellner U, Schubert J, Burk UC, Schmalhofer O, Zhu F, Sonntag A, Waldvogel B, Vannier C, Darling D, zur Hausen A, Brunton VG, Morton J, Sansom O, Schuler J, Stemmler MP, Herzberger C, Hopt U, Keck T, Brabletz S, Brabletz T. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11:1487–95. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 72.Ahmad A, Aboukameel A, Kong D, Wang Z, Sethi S, Chen W, Sarkar FH, Raz A. Phosphoglucose isomerase/autocrine motility factor mediates epithelial–mesenchymal transition regulated by miR-200 in breast cancer cells. Cancer Res. 2011;71:3400–9. doi: 10.1158/0008-5472.CAN-10-0965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial–mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910–4. doi: 10.1074/jbc.C800074200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894–907. doi: 10.1101/gad.1640608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Vetter G, Saumet A, Moes M, Vallar L, Le BA, Laurini C, Sabbah M, Arar K, Theillet C, Lecellier CH, Friederich E. miR-661 expression in SNAI1-induced epithelial to mesenchymal transition contributes to breast cancer cell invasion by targeting Nectin-1 and StarD10 messengers. Oncogene. 2010;29:4436–48. doi: 10.1038/onc.2010.181. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 76.Stinson S, Lackner MR, Adai AT, Yu N, Kim HJ, O’Brien C, Spoerke J, Jhunjhunwala S, Boyd Z, Januario T, Newman RJ, Yue P, Bourgon R, Modrusan Z, Stern HM, Warming S, de Sauvage FJ, Amler L, Yeh RF, Dornan D. TRPS1 targeting by miR-221/222 promotes the epithelial-to-mesenchymal transition in breast cancer. Sci Signal. 2011;4:ra41. doi: 10.1126/scisignal.2001538. [DOI] [PubMed] [Google Scholar]
- 77.Li QQ, Chen ZQ, Cao XX, Xu JD, Xu JW, Chen YY, Wang WJ, Chen Q, Tang F, Liu XP, Xu ZD. Involvement of NF-kappaB/miR-448 regulatory feedback loop in chemotherapy-induced epithelial–mesenchymal transition of breast cancer cells. Cell Death Differ. 2011;18:16–25. doi: 10.1038/cdd.2010.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Peng X, Guo W, Liu T, Wang X, Tu X, Xiong D, Chen S, Lai Y, Du H, Chen G, Liu G, Tang Y, Huang S, Zou X. Identification of miRs-143 and -145 that is associated with bone metastasis of prostate cancer and involved in the regulation of EMT. PLoS One. 2011;6:e20341. doi: 10.1371/journal.pone.0020341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lulla RR, Costa FF, Bischof JM, Chou PM, de Bonaldo F, Vanin EF, Soares MB. Identification of differentially expressed Micro-RNAs in osteosarcoma. Sarcoma. 2011;2011:732690. doi: 10.1155/2011/732690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, Diehn M, Liu H, Panula SP, Chiao E, Dirbas FM, Somlo G, Pera RA, Lao K, Clarke MF. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell. 2009;138:592–603. doi: 10.1016/j.cell.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jung DE, Wen J, Oh T, Song SY. Differentially expressed micro-RNAs in pancreatic cancer stem cells. Pancreas. 2011;40:1180–7. doi: 10.1097/MPA.0b013e318221b33e. [DOI] [PubMed] [Google Scholar]
- 82.Hao J, Zhang S, Zhou Y, Hu X, Shao C. MicroRNA 483-3p suppresses the expression of DPC4/Smad4 in pancreatic cancer. FEBS Lett. 2011;585:207–13. doi: 10.1016/j.febslet.2010.11.039. [DOI] [PubMed] [Google Scholar]
- 83.Liu C, Kelnar K, Liu B, Chen X, Calhoun-Davis T, Li H, Patrawala L, Yan H, Jeter C, Honorio S, Wiggins JF, Bader AG, Fagin R, Brown D, Tang DG. The microRNA miR-34a inhibits prostate cancer stem cells and metastasis by directly repressing CD44. Nat Med. 2011;17:211–5. doi: 10.1038/nm.2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kong D, Heath E, Chen W, Cher M, Powell I, Heilbrun L, Li Y, Ali S, Sethi S, Hassan O, Hwang C, Gupta N, Chitale D, Sakr WA, Menon M, Sarkar FH. Epigenetic silencing of miR-34a in human prostate cancer cells and tumor tissue specimens can be reversed by BR-DIM treatment. Am J Transl Res. 2012;4:14–23. [PMC free article] [PubMed] [Google Scholar]
- 85.Miele L. Notch signaling. Clin Cancer Res. 2006;12:1074–9. doi: 10.1158/1078-0432.CCR-05-2570. [DOI] [PubMed] [Google Scholar]
- 86.Weng AP, Lau A. Notch signaling in T-cell acute lymphoblastic leukemia. Future Oncol. 2005;1:511–9. doi: 10.2217/14796694.1.4.511. [DOI] [PubMed] [Google Scholar]
- 87.Sjolund J, Manetopoulos C, Stockhausen MT, Axelson H. The Notch pathway in cancer: differentiation gone awry. Eur J Cancer. 2005;41:2620–9. doi: 10.1016/j.ejca.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 88.Li JL, Harris AL. Notch signaling from tumor cells: a new mechanism of angiogenesis. Cancer Cell. 2005;8:1–3. doi: 10.1016/j.ccr.2005.06.013. [DOI] [PubMed] [Google Scholar]
- 89.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–89. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 90.Real PJ, Ferrando AA. NOTCH inhibition and glucocorticoid therapy in T-cell acute lymphoblastic leukemia. Leukemia. 2009;23:1374–7. doi: 10.1038/leu.2009.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Qiao L, Wong BC. Role of Notch signaling in colorectal cancer. Carcinogenesis. 2009;30:1979–86. doi: 10.1093/carcin/bgp236. [DOI] [PubMed] [Google Scholar]
- 92.Sandy AR, Maillard I. Notch signaling in the hematopoietic system. Expert Opin Biol Ther. 2009;9:1383–98. doi: 10.1517/14712590903260777. [DOI] [PubMed] [Google Scholar]
- 93.Wang M, Xue L, Cao Q, Lin Y, Ding Y, Yang P, Che L. Expression of Notch1, Jagged1 and beta-catenin and their clinicopathological significance in hepatocellular carcinoma. Neoplasma. 2009;56:533–41. doi: 10.4149/neo_2009_06_533. [DOI] [PubMed] [Google Scholar]
- 94.Gao J, Chen Y, Wu KC, Liu J, Zhao YQ, Pan YL, Du R, Zheng GR, Xiong YM, Xu HL, Fan DM. RUNX3 directly interacts with intracellular domain of Notch1 and suppresses Notch signaling in hepatocellular carcinoma cells. Exp Cell Res. 2010;316:149–57. doi: 10.1016/j.yexcr.2009.09.025. [DOI] [PubMed] [Google Scholar]
- 95.Wang C, Qi R, Li N, Wang Z, An H, Zhang Q, Yu Y, Cao X. Notch1 signaling sensitizes tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis in human hepatocellular carcinoma cells by inhibiting Akt/Hdm2-mediated p53 degradation and up-regulating p53-dependent DR5 expression. J Biol Chem. 2009;284:16183–90. doi: 10.1074/jbc.M109.002105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 96.Dotto GP. Notch tumor suppressor function. Oncogene. 2008;27:5115–23. doi: 10.1038/onc.2008.225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Nicolas M, Wolfer A, Raj K, Kummer JA, Mill P, van Noort M, Hui CC, Clevers H, Dotto GP, Radtke F. Notch1 functions as a tumor suppressor in mouse skin. Nat Genet. 2003;33:416–21. doi: 10.1038/ng1099. [DOI] [PubMed] [Google Scholar]
- 98.De La OJ, Murtaugh LC. Notch and Kras in pancreatic cancer: at the crossroads of mutation, differentiation and signaling. Cell Cycle. 2009;8:1860–4. doi: 10.4161/cc.8.12.8744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Osipo C, Golde TE, Osborne BA, Miele LA. Off the beaten pathway: the complex cross talk between Notch and NF-kappaB. Lab Invest. 2008;88:11–7. doi: 10.1038/labinvest.3700700. [DOI] [PubMed] [Google Scholar]
- 100.Sundaram MV. The love-hate relationship between Ras and Notch. Genes Dev. 2005;19:1825–39. doi: 10.1101/gad.1330605. [DOI] [PubMed] [Google Scholar]
- 101.Wang Z, Sengupta R, Banerjee S, Li Y, Zhang Y, Rahman KM, Aboukameel A, Mohammad R, Majumdar AP, Abbruzzese JL, Sarkar FH. Epidermal growth factor receptor-related protein inhibits cell growth and invasion in pancreatic cancer. Cancer Res. 2006;66:7653–60. doi: 10.1158/0008-5472.CAN-06-1019. [DOI] [PubMed] [Google Scholar]
- 102.Wang Z, Kong D, Banerjee S, Li Y, Adsay NV, Abbruzzese J, Sarkar FH. Down-regulation of platelet-derived growth factor-D inhibits cell growth and angiogenesis through inactivation of Notch-1 and nuclear factor-kappaB signaling. Cancer Res. 2007;67:11377–85. doi: 10.1158/0008-5472.CAN-07-2803. [DOI] [PubMed] [Google Scholar]
- 103.Weijzen S, Rizzo P, Braid M, Vaishnav R, Jonkheer SM, Zlobin A, Osborne BA, Gottipati S, Aster JC, Hahn WC, Rudolf M, Siziopikou K, Kast WM, Miele L. Activation of Notch-1 signaling maintains the neoplastic phenotype in human Ras-transformed cells. Nat Med. 2002;8:979–86. doi: 10.1038/nm754. [DOI] [PubMed] [Google Scholar]
- 104.Wang Z, Li Y, Kong D, Ahmad A, Banerjee S, Sarkar FH. Crosstalk between miRNA and Notch signaling pathways in tumor development and progression. Cancer Lett. 2010;292:141–8. doi: 10.1016/j.canlet.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rizzo P, Osipo C, Foreman K, Golde T, Osborne B, Miele L. Rational targeting of Notch signaling in cancer. Oncogene. 2008;27:5124–31. doi: 10.1038/onc.2008.226. [DOI] [PubMed] [Google Scholar]
- 106.Song LL, Peng Y, Yun J, Rizzo P, Chaturvedi V, Weijzen S, Kast WM, Stone PJ, Santos L, Loredo A, Lendahl U, Sonenshein G, Osborne B, Qin JZ, Pannuti A, Nickoloff BJ, Miele L. Notch-1 associates with IKKalpha and regulates IKK activity in cervical cancer cells. Oncogene. 2008;27:5833–44. doi: 10.1038/onc.2008.190. [DOI] [PubMed] [Google Scholar]
- 107.Sharma RP, Chopra VL. Effect of the Wingless (wg1) mutation on wing and haltere development in Drosophila melanogaster. Dev Biol. 1976;48:461–5. doi: 10.1016/0012-1606(76)90108-1. [DOI] [PubMed] [Google Scholar]
- 108.Nusse R, van Ooyen A, Cox D, Fung YK, Varmus H. Mode of proviral activation of a putative mammary oncogene (int-1) on mouse chromosome 15. Nature. 1984;307:131–6. doi: 10.1038/307131a0. [DOI] [PubMed] [Google Scholar]
- 109.Kohn AD, Moon RT. Wnt and calcium signaling: beta-catenin-independent pathways. Cell Calcium. 2005;38:439–46. doi: 10.1016/j.ceca.2005.06.022. [DOI] [PubMed] [Google Scholar]
- 110.Woll PS, Morris JK, Painschab MS, Marcus RK, Kohn AD, Biechele TL, Moon RT, Kaufman DS. Wnt signaling promotes hematoendothelial cell development from human embryonic stem cells. Blood. 2008;111:122–31. doi: 10.1182/blood-2007-04-084186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Angers S, Moon RT. Proximal events in Wnt signal transduction. Nat Rev Mol Cell Biol. 2009;10:468–77. doi: 10.1038/nrm2717. [DOI] [PubMed] [Google Scholar]
- 112.Semenov MV, Habas R, Macdonald BT, He X. SnapShot: non-canonical Wnt signaling pathways. Cell. 2007;131:1378. doi: 10.1016/j.cell.2007.12.011. [DOI] [PubMed] [Google Scholar]
- 113.Macdonald BT, Semenov MV, He X. SnapShot: Wnt/beta-catenin signaling. Cell. 2007;131:1204. doi: 10.1016/j.cell.2007.11.036. [DOI] [PubMed] [Google Scholar]
- 114.De A. Wnt/Ca2+ signaling pathway: a brief overview. Acta Biochim Biophys Sin (Shanghai) 2011;43:745–56. doi: 10.1093/abbs/gmr079. [DOI] [PubMed] [Google Scholar]
- 115.Leris AC, Roberts TR, Jiang WG, Newbold RF, Mokbel K. WNT5A expression in human breast cancer. Anticancer Res. 2005;25:731–4. [PubMed] [Google Scholar]
- 116.Kremenevskaja N, von Wasielewski R, Rao AS, Schofl C, Andersson T, Brabant G. Wnt-5a has tumor suppressor activity in thyroid carcinoma. Oncogene. 2005;24:2144–54. doi: 10.1038/sj.onc.1208370. [DOI] [PubMed] [Google Scholar]
- 117.MacLeod RJ, Hayes M, Pacheco I. Wnt5a secretion stimulated by the extracellular calcium-sensing receptor inhibits defective Wnt signaling in colon cancer cells. Am J Physiol Gastrointest Liver Physiol. 2007;293:G403–11. doi: 10.1152/ajpgi.00119.2007. [DOI] [PubMed] [Google Scholar]
- 118.Wang Q, Symes AJ, Kane CA, Freeman A, Nariculam J, Munson P, Thrasivoulou C, Masters JR, Ahmed A. A novel role for Wnt/Ca2+ signaling in actin cytoskeleton remodeling and cell motility in prostate cancer. PLoS One. 2010;5:e10456. doi: 10.1371/journal.pone.0010456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Yamamoto H, Oue N, Sato A, Hasegawa Y, Yamamoto H, Matsubara A, Yasui W, Kikuchi A. Wnt5a signaling is involved in the aggressiveness of prostate cancer and expression of metalloproteinase. Oncogene. 2010;29:2036–46. doi: 10.1038/onc.2009.496. [DOI] [PubMed] [Google Scholar]
- 120.Weeraratna AT, Jiang Y, Hostetter G, Rosenblatt K, Duray P, Bittner M, Trent JM. Wnt5a signaling directly affects cell motility and invasion of metastatic melanoma. Cancer Cell. 2002;1:279–88. doi: 10.1016/s1535-6108(02)00045-4. [DOI] [PubMed] [Google Scholar]
- 121.Pukrop T, Klemm F, Hagemann T, Gradl D, Schulz M, Siemes S, Trumper L, Binder C. Wnt 5a signaling is critical for macrophage-induced invasion of breast cancer cell lines. Proc Natl Acad Sci U S A. 2006;103:5454–9. doi: 10.1073/pnas.0509703103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ripka S, Konig A, Buchholz M, Wagner M, Sipos B, Kloppel G, Downward J, Gress T, Michl P. WNT5A—target of CUTL1 and potent modulator of tumor cell migration and invasion in pancreatic cancer. Carcinogenesis. 2007;28:1178–87. doi: 10.1093/carcin/bgl255. [DOI] [PubMed] [Google Scholar]
- 123.Pilarsky C, Ammerpohl O, Sipos B, Dahl E, Hartmann A, Wellmann A, Braunschweig T, Lohr M, Jesenofsky R, Friess H, Wente MN, Kristiansen G, Jahnke B, Denz A, Ruckert F, Schackert HK, Kloppel G, Kalthoff H, Saeger HD, Grutzmann R. Activation of Wnt signalling in stroma from pancreatic cancer identified by gene expression profiling. J Cell Mol Med. 2008;12:2823–35. doi: 10.1111/j.1582-4934.2008.00289.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Dissanayake SK, Wade M, Johnson CE, O’Connell MP, Leotlela PD, French AD, Shah KV, Hewitt KJ, Rosenthal DT, Indig FE, Jiang Y, Nickoloff BJ, Taub DD, Trent JM, Moon RT, Bittner M, Weeraratna AT. The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J Biol Chem. 2007;282:17259–71. doi: 10.1074/jbc.M700075200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Dissanayake SK, Weeraratna AT. Detecting PKC phosphorylation as part of the Wnt/calcium pathway in cutaneous melanoma. Methods Mol Biol. 2008;468:157–72. doi: 10.1007/978-1-59745-249-6_12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang Q, Williamson M, Bott S, Brookman-Amissah N, Freeman A, Nariculam J, Hubank MJ, Ahmed A, Masters JR. Hypomethylation of WNT5A, CRIP1 and S100P in prostate cancer. Oncogene. 2007;26:6560–5. doi: 10.1038/sj.onc.1210472. [DOI] [PubMed] [Google Scholar]
- 127.Behrens J. Control of beta-catenin signaling in tumor development. Ann N Y Acad Sci. 2000;910:21–33. doi: 10.1111/j.1749-6632.2000.tb06698.x. [DOI] [PubMed] [Google Scholar]
- 128.Peifer M, Polakis P. Wnt signaling in oncogenesis and embryogenesis—a look outside the nucleus. Science. 2000;287:1606–9. doi: 10.1126/science.287.5458.1606. [DOI] [PubMed] [Google Scholar]
- 129.Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–54. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- 130.Verras M, Sun Z. Roles and regulation of Wnt signaling and beta-catenin in prostate cancer. Cancer Lett. 2006;237:22–32. doi: 10.1016/j.canlet.2005.06.004. [DOI] [PubMed] [Google Scholar]
- 131.Reya T, Clevers H. Wnt signalling in stem cells and cancer. Nature. 2005;434:843–50. doi: 10.1038/nature03319. [DOI] [PubMed] [Google Scholar]
- 132.Clevers H. Wnt breakers in colon cancer. Cancer Cell. 2004;5:5–6. doi: 10.1016/s1535-6108(03)00339-8. [DOI] [PubMed] [Google Scholar]
- 133.Vermeulen L, De Sousa E, van der Melo HM, Cameron K, de Jong JH, Borovski T, Tuynman JB, Todaro M, Merz C, Rodermond H, Sprick MR, Kemper K, Richel DJ, Stassi G, Medema JP. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010;12:468–76. doi: 10.1038/ncb2048. [DOI] [PubMed] [Google Scholar]
- 134.Chesire DR, Ewing CM, Gage WR, Isaacs WB. In vitro evidence for complex modes of nuclear beta-catenin signaling during prostate growth and tumorigenesis. Oncogene. 2002;21:2679–94. doi: 10.1038/sj.onc.1205352. [DOI] [PubMed] [Google Scholar]
- 135.Barker N, Clevers H. Mining the Wnt pathway for cancer therapeutics. Nat Rev Drug Discov. 2006;5:997–1014. doi: 10.1038/nrd2154. [DOI] [PubMed] [Google Scholar]
- 136.Dihlmann S, von Knebel DM. Wnt/beta-catenin-pathway as a molecular target for future anti-cancer therapeutics. Int J Cancer. 2005;113:515–24. doi: 10.1002/ijc.20609. [DOI] [PubMed] [Google Scholar]
- 137.Gritli-Linde A, Bei M, Maas R, Zhang XM, Linde A, McMahon AP. Shh signaling within the dental epithelium is necessary for cell proliferation, growth and polarization. Development. 2002;129:5323–37. doi: 10.1242/dev.00100. [DOI] [PubMed] [Google Scholar]
- 138.Yang L, Xie G, Fan Q, Xie J. Activation of the hedgehog-signaling pathway in human cancer and the clinical implications. Oncogene. 2010;29:469–81. doi: 10.1038/onc.2009.392. [DOI] [PubMed] [Google Scholar]
- 139.Varjosalo M, Taipale J. Hedgehog: functions and mechanisms. Genes Dev. 2008;22:2454–72. doi: 10.1101/gad.1693608. [DOI] [PubMed] [Google Scholar]
- 140.Medina V, Calvo MB, az-Prado S, Espada J. Hedgehog signalling as a target in cancer stem cells. Clin Transl Oncol. 2009;11:199–207. doi: 10.1007/s12094-009-0341-y. [DOI] [PubMed] [Google Scholar]
- 141.Anton Aparicio LM, Garcia CR, Cassinello EJ, Valladares AM, Reboredo LM, Diaz PS, Aparicio GG. Prostate cancer and Hedgehog signalling pathway. Clin Transl Oncol. 2007;9:420–8. doi: 10.1007/s12094-007-0080-x. [DOI] [PubMed] [Google Scholar]
- 142.Varnat F, Duquet A, Malerba M, Zbinden M, Mas C, Gervaz P, Altaba A. Human colon cancer epithelial cells harbour active HEDGEHOG-GLI signalling that is essential for tumour growth, recurrence, metastasis and stem cell survival and expansion. EMBO Mol Med. 2009;1:338–51. doi: 10.1002/emmm.200900039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Choi SS, Omenetti A, Witek RP, Moylan CA, Syn WK, Jung Y, Yang L, Sudan DL, Sicklick JK, Michelotti GA, Rojkind M, Diehl AM. Hedgehog pathway activation and epithelial-to-mesenchymal transitions during myofibroblastic transformation of rat hepatic cells in culture and cirrhosis. Am J Physiol Gastrointest Liver Physiol. 2009;297:G1093–106. doi: 10.1152/ajpgi.00292.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Isohata N, Aoyagi K, Mabuchi T, Daiko H, Fukaya M, Ohta H, Ogawa K, Yoshida T, Sasaki H. Hedgehog and epithelial–mesenchymal transition signaling in normal and malignant epithelial cells of the esophagus. Int J Cancer. 2009;125:1212–21. doi: 10.1002/ijc.24400. [DOI] [PubMed] [Google Scholar]
- 145.Ohta H, Aoyagi K, Fukaya M, Danjoh I, Ohta A, Isohata N, Saeki N, Taniguchi H, Sakamoto H, Shimoda T, Tani T, Yoshida T, Sasaki H. Cross talk between hedgehog and epithelial–mesenchymal transition pathways in gastric pit cells and in diffuse-type gastric cancers. Br J Cancer. 2009;100:389–98. doi: 10.1038/sj.bjc.6604846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Omenetti A, Porrello A, Jung Y, Yang L, Popov Y, Choi SS, Witek RP, Alpini G, Venter J, Vandongen HM, Syn WK, Baroni GS, Benedetti A, Schuppan D, Diehl AM. Hedgehog signaling regulates epithelial–mesenchymal transition during biliary fibrosis in rodents and humans. J Clin Invest. 2008;118:3331–42. doi: 10.1172/JCI35875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Martelli AM, Evangelisti C, Follo MY, Ramazzotti G, Fini M, Giardino R, Manzoli L, McCubrey JA, Cocco L. Targeting the phosphatidylinositol 3-kinase/Akt/mammalian target of rapamycin signaling network in cancer stem cells. Curr Med Chem. 2011;18:2715–26. doi: 10.2174/092986711796011201. [DOI] [PubMed] [Google Scholar]
- 148.Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–41. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- 149.Eyler CE, Foo WC, LaFiura KM, McLendon RE, Hjelmeland AB, Rich JN. Brain cancer stem cells display preferential sensitivity to Akt inhibition. Stem Cells. 2008;26:3027–36. doi: 10.1634/stemcells.2007-1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Dubrovska A, Elliott J, Salamone RJ, Kim S, Aimone LJ, Walker JR, Watson J, Sauveur-Michel M, Garcia-Echeverria C, Cho CY, Reddy VA, Schultz PG. Combination therapy targeting both tumor-initiating and differentiated cell populations in prostate carcinoma. Clin Cancer Res. 2010;16:5692–702. doi: 10.1158/1078-0432.CCR-10-1601. [DOI] [PubMed] [Google Scholar]
- 151.Zhou J, Wulfkuhle J, Zhang H, Gu P, Yang Y, Deng J, Margolick JB, Liotta LA, Petricoin E, III, Zhang Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc Natl Acad Sci U S A. 2007;104:16158–63. doi: 10.1073/pnas.0702596104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Yasuda A, Sawai H, Takahashi H, Ochi N, Matsuo Y, Funahashi H, Sato M, Okada Y, Takeyama H, Manabe T. Stem cell factor/c-kit receptor signaling enhances the proliferation and invasion of colorectal cancer cells through the PI3K/Akt pathway. Dig Dis Sci. 2007;52:2292–300. doi: 10.1007/s10620-007-9759-7. [DOI] [PubMed] [Google Scholar]
- 153.Nautiyal J, Banerjee S, Kanwar SS, Yu Y, Patel BB, Sarkar FH, Majumdar AP. Curcumin enhances dasatinib-induced inhibition of growth and transformation of colon cancer cells. Int J Cancer. 2011;128:951–61. doi: 10.1002/ijc.25410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lin L, Liu Y, Li H, Li PK, Fuchs J, Shibata H, Iwabuchi Y, Lin J. Targeting colon cancer stem cells using a new curcumin analogue, GO-Y030. Br J Cancer. 2011;105:212–20. doi: 10.1038/bjc.2011.200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ryu MJ, Cho M, Song JY, Yun YS, Choi IW, Kim DE, Park BS, Oh S. Natural derivatives of curcumin attenuate the Wnt/beta-catenin pathway through down-regulation of the transcriptional coactivator p300. Biochem Biophys Res Commun. 2008;377:1304–8. doi: 10.1016/j.bbrc.2008.10.171. [DOI] [PubMed] [Google Scholar]
- 156.Jaiswal AS, Marlow BP, Gupta N, Narayan S. beta-Catenin-mediated transactivation and cell-cell adhesion pathways are important in curcumin (diferuylmethane)-induced growth arrest and apoptosis in colon cancer cells. Oncogene. 2002;21:8414–27. doi: 10.1038/sj.onc.1205947. [DOI] [PubMed] [Google Scholar]
- 157.Zhang J, Du Y, Wu C, Ren X, Ti X, Shi J, Zhao F, Yin H. Curcumin promotes apoptosis in human lung adenocarcinoma cells through miR-186* signaling pathway. Oncol Rep. 2010;24:1217–23. doi: 10.3892/or_00000975. [DOI] [PubMed] [Google Scholar]
- 158.Mudduluru G, George-William JN, Muppala S, Asangani IA, Kumarswamy R, Nelson LD, Allgayer H. Curcumin regulates miR-21 expression and inhibits invasion and metastasis in colorectal cancer. Biosci Rep. 2011;31:185–97. doi: 10.1042/BSR20100065. [DOI] [PubMed] [Google Scholar]
- 159.Kakarala M, Brenner DE, Korkaya H, Cheng C, Tazi K, Ginestier C, Liu S, Dontu G, Wicha MS. Targeting breast stem cells with the cancer preventive compounds curcumin and piperine. Breast Cancer Res Treat. 2010;122:777–85. doi: 10.1007/s10549-009-0612-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Prasad CP, Rath G, Mathur S, Bhatnagar D, Ralhan R. Potent growth suppressive activity of curcumin in human breast cancer cells: modulation of Wnt/beta-catenin signaling. Chem Biol Interact. 2009;181:263–71. doi: 10.1016/j.cbi.2009.06.012. [DOI] [PubMed] [Google Scholar]
- 161.Squires MS, Hudson EA, Howells L, Sale S, Houghton CE, Jones JL, Fox LH, Dickens M, Prigent SA, Manson MM. Relevance of mitogen activated protein kinase (MAPK) and phosphotidylinositol-3-kinase/protein kinase B (PI3K/PKB) pathways to induction of apoptosis by curcumin in breast cells. Biochem Pharmacol. 2003;65:361–76. doi: 10.1016/s0006-2952(02)01517-4. [DOI] [PubMed] [Google Scholar]
- 162.Teiten MH, Gaascht F, Cronauer M, Henry E, Dicato M, Diederich M. Anti-proliferative potential of curcumin in androgen-dependent prostate cancer cells occurs through modulation of the Wingless signaling pathway. Int J Oncol. 2011;38:603–11. doi: 10.3892/ijo.2011.905. [DOI] [PubMed] [Google Scholar]
- 163.Hsieh A, Kim HS, Lim SO, Yu DY, Jung G. Hepatitis B viral X protein interacts with tumor suppressor adenomatous polyposis coli to activate Wnt/beta-catenin signaling. Cancer Lett. 2011;300:162–72. doi: 10.1016/j.canlet.2010.09.018. [DOI] [PubMed] [Google Scholar]
- 164.Choi HY, Lim JE, Hong JH. Curcumin interrupts the interaction between the androgen receptor and Wnt/beta-catenin signaling pathway in LNCaP prostate cancer cells. Prostate Cancer Prostatic Dis. 2010;13:343–9. doi: 10.1038/pcan.2010.26. [DOI] [PubMed] [Google Scholar]
- 165.Wang Z, Zhang Y, Banerjee S, Li Y, Sarkar FH. Notch-1 down-regulation by curcumin is associated with the inhibition of cell growth and the induction of apoptosis in pancreatic cancer cells. Cancer. 2006;106:2503–13. doi: 10.1002/cncr.21904. [DOI] [PubMed] [Google Scholar]
- 166.Sarkar FH, Li Y, Wang Z, Padhye S. Lesson learned from nature for the development of novel anti-cancer agents: implication of isoflavone, curcumin, and their synthetic analogs. Curr Pharm Des. 2010;16:1801–12. doi: 10.2174/138161210791208956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Padhye S, Yang H, Jamadar A, Cui QC, Chavan D, Dominiak K, McKinney J, Banerjee S, Dou QP, Sarkar FH. New difluoro Knoevenagel condensates of curcumin, their Schiff bases and copper complexes as proteasome inhibitors and apoptosis inducers in cancer cells. Pharm Res. 2009;26:1874–80. doi: 10.1007/s11095-009-9900-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Padhye S, Banerjee S, Chavan D, Pandye S, Swamy KV, Ali S, Li J, Dou QP, Sarkar FH. Fluorocurcumins as cyclooxygenase-2 inhibitor: molecular docking, pharmacokinetics and tissue distribution in mice. Pharm Res. 2009;26:2438–45. doi: 10.1007/s11095-009-9955-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Ali S, Ahmad A, Banerjee S, Padhye S, Dominiak K, Schaffert JM, Wang Z, Philip PA, Sarkar FH. Gemcitabine sensitivity can be induced in pancreatic cancer cells through modulation of miR-200 and miR-21 expression by curcumin or its analogue CDF. Cancer Res. 2010;70:3606–17. doi: 10.1158/0008-5472.CAN-09-4598. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 170.Bao B, Ali S, Banerjee S, Wang Z, Logna F, Azmi AS, Kong D, Ahmad A, Li Y, Padhye S, Sarkar FH. Curcumin analogue CDF inhibits pancreatic tumor growth by switching on suppressor microRNAs and attenuating EZH2 expression. Cancer Res. 2012;72:335–45. doi: 10.1158/0008-5472.CAN-11-2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang Y, Talalay P, Cho CG, Posner GH. A major inducer of anticarcinogenic protective enzymes from broccoli: isolation and elucidation of structure. Proc Natl Acad Sci U S A. 1992;89:2399–403. doi: 10.1073/pnas.89.6.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Verhoeven DT, Verhagen H, Goldbohm RA, van den Brandt PA, van Poppel G. A review of mechanisms underlying anticarcinogenicity by brassica vegetables. Chem Biol Interact. 1997;103:79–129. doi: 10.1016/s0009-2797(96)03745-3. [DOI] [PubMed] [Google Scholar]
- 173.Zhang Y, Tang L, Gonzalez V. Selected isothiocyanates rapidly induce growth inhibition of cancer cells. Mol Cancer Ther. 2003;2:1045–52. [PubMed] [Google Scholar]
- 174.Cho SD, Li G, Hu H, Jiang C, Kang KS, Lee YS, Kim SH, Lu J. Involvement of c-Jun N-terminal kinase in G2/M arrest and caspase-mediated apoptosis induced by sulforaphane in DU145 prostate cancer cells. Nutr Cancer. 2005;52:213–24. doi: 10.1207/s15327914nc5202_11. [DOI] [PubMed] [Google Scholar]
- 175.Jackson SJ, Singletary KW. Sulforaphane inhibits human MCF-7 mammary cancer cell mitotic progression and tubulin polymerization. J Nutr. 2004;134:2229–36. doi: 10.1093/jn/134.9.2229. [DOI] [PubMed] [Google Scholar]
- 176.Kuroiwa Y, Nishikawa A, Kitamura Y, Kanki K, Ishii Y, Umemura T, Hirose M. Protective effects of benzyl isothiocyanate and sulforaphane but not resveratrol against initiation of pancreatic carcinogenesis in hamsters. Cancer Lett. 2006;241:275–80. doi: 10.1016/j.canlet.2005.10.028. [DOI] [PubMed] [Google Scholar]
- 177.Shen G, Xu C, Chen C, Hebbar V, Kong AN. p53-independent G1 cell cycle arrest of human colon carcinoma cells HT-29 by sulforaphane is associated with induction of p21CIP1 and inhibition of expression of cyclin D1. Cancer Chemother Pharmacol. 2006;57:317–27. doi: 10.1007/s00280-005-0050-3. [DOI] [PubMed] [Google Scholar]
- 178.Singh AV, Xiao D, Lew KL, Dhir R, Singh SV. Sulforaphane induces caspase-mediated apoptosis in cultured PC-3 human prostate cancer cells and retards growth of PC-3 xenografts in vivo. Carcinogenesis. 2004;25:83–90. doi: 10.1093/carcin/bgg178. [DOI] [PubMed] [Google Scholar]
- 179.Zanichelli F, Capasso S, Cipollaro M, Pagnotta E, Carteni M, Casale F, Iori R, Galderisi U. Dose-dependent effects of R-sulforaphane isothiocyanate on the biology of human mesenchymal stem cells, at dietary amounts, it promotes cell proliferation and reduces senescence and apoptosis, while at anti-cancer drug doses, it has a cytotoxic effect. Age (Dordr) 2012;34:281–93. doi: 10.1007/s11357-011-9231-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Srivastava RK, Tang SN, Zhu W, Meeker D, Shankar S. Sulforaphane synergizes with quercetin to inhibit self-renewal capacity of pancreatic cancer stem cells. Front Biosci (Elite Ed) 2011;3:515–28. doi: 10.2741/e266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Zhou W, Kallifatidis G, Baumann B, Rausch V, Mattern J, Gladkich J, Giese N, Moldenhauer G, Wirth T, Buchler MW, Salnikov AV, Herr I. Dietary polyphenol quercetin targets pancreatic cancer stem cells. Int J Oncol. 2010;37:551–61. doi: 10.3892/ijo_00000704. [DOI] [PubMed] [Google Scholar]
- 182.Kallifatidis G, Labsch S, Rausch V, Mattern J, Gladkich J, Moldenhauer G, Buchler MW, Salnikov AV, Herr I. Sulforaphane increases drug-mediated cytotoxicity toward cancer stem-like cells of pancreas and prostate. Mol Ther. 2011;19:188–95. doi: 10.1038/mt.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Rausch V, Liu L, Kallifatidis G, Baumann B, Mattern J, Gladkich J, Wirth T, Schemmer P, Buchler MW, Zoller M, Salnikov AV, Herr I. Synergistic activity of sorafenib and sulforaphane abolishes pancreatic cancer stem cell characteristics. Cancer Res. 2010;70:5004–13. doi: 10.1158/0008-5472.CAN-10-0066. [DOI] [PubMed] [Google Scholar]
- 184.Li Y, Zhang T, Korkaya H, Liu S, Lee HF, Newman B, Yu Y, Clouthier SG, Schwartz SJ, Wicha MS, Sun D. Sulforaphane, a dietary component of broccoli/broccoli sprouts, inhibits breast cancer stem cells. Clin Cancer Res. 2010;16:2580–90. doi: 10.1158/1078-0432.CCR-09-2937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Baur JA, Sinclair DA. Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov. 2006;5:493–506. doi: 10.1038/nrd2060. [DOI] [PubMed] [Google Scholar]
- 186.Ahmad A, Farhan AS, Singh S, Hadi SM. DNA breakage by resveratrol and Cu(II): reaction mechanism and bacteriophage inactivation. Cancer Lett. 2000;154:29–37. doi: 10.1016/s0304-3835(00)00351-7. [DOI] [PubMed] [Google Scholar]
- 187.Shankar S, Nall D, Tang SN, Meeker D, Passarini J, Sharma J, Srivastava RK. Resveratrol inhibits pancreatic cancer stem cell characteristics in human and KrasG12D transgenic mice by inhibiting pluripotency maintaining factors and epithelial–mesenchymal transition. PLoS One. 2011;6:e16530. doi: 10.1371/journal.pone.0016530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Pandey PR, Okuda H, Watabe M, Pai SK, Liu W, Kobayashi A, Xing F, Fukuda K, Hirota S, Sugai T, Wakabayashi G, Koeda K, Kashiwaba M, Suzuki K, Chiba T, Endo M, Fujioka T, Tanji S, Mo YY, Cao D, Wilber AC, Watabe K. Resveratrol suppresses growth of cancer stem-like cells by inhibiting fatty acid synthase. Breast Cancer Res Treat. 2011;130:387–98. doi: 10.1007/s10549-010-1300-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Zhang W, Sviripa V, Kril LM, Chen X, Yu T, Shi J, Rychahou P, Evers BM, Watt DS, Liu C. Fluorinated N, N-dialkylaminostilbenes for Wnt pathway inhibition and colon cancer repression. J Med Chem. 2011;54:1288–97. doi: 10.1021/jm101248v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Lampe JW, Nishino Y, Ray RM, Wu C, Li W, Lin MG, Gao DL, Hu Y, Shannon J, Stalsberg H, Porter PL, Frankenfeld CL, Wahala K, Thomas DB. Plasma isoflavones and fibrocystic breast conditions and breast cancer among women in Shanghai, China. Cancer Epidemiol Biomarkers Prev. 2007;16:2579–86. doi: 10.1158/1055-9965.EPI-07-0368. [DOI] [PubMed] [Google Scholar]
- 191.Adlercreutz H, Markkanen H, Watanabe S. Plasma concentrations of phytooestrogens in Japanese men. Lancet. 1993;342:1209–10. doi: 10.1016/0140-6736(93)92188-y. [DOI] [PubMed] [Google Scholar]
- 192.Mills PK, Beeson WL, Phillips RL, Fraser GE. Cohort study of diet, lifestyle, and prostate cancer in Adventist men. Cancer. 1989;64:598–604. doi: 10.1002/1097-0142(19890801)64:3<598::aid-cncr2820640306>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- 193.Jacobsen BK, Knutsen SF, Fraser GE. Does high soy milk intake reduce prostate cancer incidence? The Adventist Health Study (United States) Cancer Causes Control. 1998;9:553–7. doi: 10.1023/a:1008819500080. [DOI] [PubMed] [Google Scholar]
- 194.Banerjee S, Li Y, Wang Z, Sarkar FH. Multi-targeted therapy of cancer by genistein. Cancer Lett. 2008;269:226–42. doi: 10.1016/j.canlet.2008.03.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Chen Y, Zaman MS, Deng G, Majid S, Saini S, Liu J, Tanaka Y, Dahiya R. MicroRNAs 221/222 and genistein-mediated regulation of ARHI tumor suppressor gene in prostate cancer. Cancer Prev Res (Phila) 2011;4:76–86. doi: 10.1158/1940-6207.CAPR-10-0167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Khan N, Mukhtar H. Multitargeted therapy of cancer by green tea polyphenols. Cancer Lett. 2008;269:269–80. doi: 10.1016/j.canlet.2008.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Liu L, Lai CQ, Nie L, Ordovas J, Band M, Moser L, Meydani M. The modulation of endothelial cell gene expression by green tea polyphenol-EGCG. Mol Nutr Food Res. 2008;52:1182–92. doi: 10.1002/mnfr.200700499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Kim J, Zhang X, Rieger-Christ KM, Summerhayes IC, Wazer DE, Paulson KE, Yee AS. Suppression of Wnt signaling by the green tea compound (−)-epigallocatechin 3-gallate (EGCG) in invasive breast cancer cells. Requirement of the transcriptional repressor HBP1. J Biol Chem. 2006;281:10865–75. doi: 10.1074/jbc.M513378200. [DOI] [PubMed] [Google Scholar]
- 199.Van Aller GS, Carson JD, Tang W, Peng H, Zhao L, Copeland RA, Tummino PJ, Luo L. Epigallocatechin gallate (EGCG), a major component of green tea, is a dual phosphoinositide-3-kinase/mTOR inhibitor. Biochem Biophys Res Commun. 2011;406:194–9. doi: 10.1016/j.bbrc.2011.02.010. [DOI] [PubMed] [Google Scholar]
- 200.Fix LN, Shah M, Efferth T, Farwell MA, Zhang B. MicroRNA expression profile of MCF-7 human breast cancer cells and the effect of green tea polyphenon-60. Cancer Genomics Proteomics. 2010;7:261–77. [PubMed] [Google Scholar]
- 201.Siddiqui IA, Asim M, Hafeez BB, Adhami VM, Tarapore RS, Mukhtar H. Green tea polyphenol EGCG blunts androgen receptor function in prostate cancer. FASEB J. 2011;25:1198–207. doi: 10.1096/fj.10-167924. [DOI] [PMC free article] [PubMed] [Google Scholar]