

Abstract
Parathyroid hormone (PTH) signaling via PTH 1 receptor (PTH1R) involves mitogen-activated protein kinase (MAPK) pathways. MAPK phosphatase 1 (MKP1) dephosphorylates and inactivates MAPKs in osteoblasts, the bone-forming cells. We previously showed that PTH1R activation in differentiated osteoblasts upregulates MKP1 and downregulates pERK1/2–MAPK and cyclin D1. In this study, we evaluated the skeletal phenotype of Mkp1 knockout (KO) mice and the effects of PTH in vivo and in vitro. Microcomputed tomography analysis of proximal tibiae and distal femora from 12-week-old Mkp1 KO female mice revealed osteopenic phenotype with significant reduction (8–46%) in bone parameters compared with wild-type (WT) controls. Histomorphometric analysis showed decreased trabecular bone area in KO females. Levels of serum osteocalcin (OCN) were lower and serum tartrate-resistant acid phosphatase 5b (TRAP5b) was higher in KO animals. Treatment of neonatal mice with hPTH (1–34) for 3 weeks showed attenuated anabolic responses in the distal femora of KO mice compared with WT mice. Primary osteoblasts derived from KO mice displayed delayed differentiation determined by alkaline phosphatase activity, and reduced expressions of Ocn and Runx2 genes associated with osteoblast maturation and function. Cells from KO females exhibited attenuated PTH response in mineralized nodule formation in vitro. Remarkably, this observation was correlated with decreased PTH response of matrix Gla protein expression. Expressions of pERK1/2 and cyclin D1 were inhibited dramatically by PTH in differentiated osteoblasts from WT mice but much less in osteoblasts from Mkp1 KO mice. In conclusion, MKP1 is important for bone homeostasis, osteoblast differentiation and skeletal responsiveness to PTH.
Introduction
Osteoporosis, which is characterized by reduced bone mass, is a major health concern around the world. Parathyroid hormone (PTH), secreted from the parathyroid glands, is involved in calcium homeostasis and is a critical mediator of skeletal development and remodeling (Hock et al. 2002). Daily injections of PTH increase bone mass and reduce fracture incidence in osteoporotic patients (Neer et al. 2001, Pietrogrande 2010) and accelerate fracture healing in animal models (Jahng & Kim 2000, Andreassen et al. 2001). However, prolonged use of PTH can cause hypercalciuria and hypercalcemia in man (Pietrogrande 2010), and osteosarcoma in rodents (Alves de Oliveira et al. 2010); these side effects limit its safe dose and duration of therapy. Dissecting the molecular mechanisms of PTH actions may uncover novel therapeutic targets for the prevention and reversal of osteoporosis and bone-related diseases and minimizing adverse effects of PTH.
PTH and PTH-related peptide (PTHrP) bind to the PTH 1 receptor (PTH1R) and stimulate multiple signaling cascades including adenylate cyclase/protein kinase A, phospholipase C/protein kinase C, and mitogen-activated protein kinases (MAPKs); leading to pleiotropic anabolic and catabolic actions in bone (for review see Datta & Abou-Samra (2009)). The mechanism of PTH action is complex and the pathways involved in the anabolic actions of PTH are yet to be fully delineated (Jilka 2007, Datta & Abou-Samra 2009). Depending on the duration and frequency of exposure the net effect of PTH can be either anabolic with increased bone mass, improved skeletal structure, biomechanical strength, and reduced fracture risk (Dempster et al. 2001, Zhou et al. 2001, Yang et al. 2007) or catabolic showing decreased bone mass and hypercalcemia (Dobnig & Turner 1997, Schiller et al. 1999, Marx 2000). The control of bone mass involves osteoblasts, the bone-forming cells, osteoclasts, the bone-resorbing cells, and osteocytes. Overexpression and knockout (KO) studies suggest that PTH, PTHrP, and PTH1R are important physiologically for the maturation and differentiation of osteoblasts (Karaplis et al. 1994, Weir et al. 1996, Lanske et al. 1999, Calvi et al. 2001, Miao et al. 2002). Increased proliferation and differentiation of bone-forming osteoblastic cells in vitro and in vivo, decreased osteoblast apoptosis, and activation of bone lining cells, are among the several mechanisms that have been proposed for PTH anabolic effects (for review see Datta & Abou-Samra (2009)). MAPKs have been shown to regulate multiple cellular activities in osteoblasts (Schindeler & Little 2006). PTH shows both up-and downregulations of MAPK during osteoblast proliferation and differentiation (Swarthout et al. 2001, Higuchi et al. 2002, Franceschi & Xiao 2003, Nakayama et al. 2003). The mammalian MAPKs include extracellular signal-regulated kinases (ERK1/2), p38-MAPKs, c-Jun N-terminal kinases, and ERK5 (Pearson et al. 2001). Using MC3T3-E1 osteoblastic cell line and primary osteoblasts, we have demonstrated that PTHrP increases pERK1/2–MAPK and cyclin D1 in undifferentiated cells but decreases pERK1/2– MAPK and cyclin D1 in differentiated osteoblasts, suggesting that such regulation by PTH1R could be an important determinant of the life span and the bone-forming activity of osteoblasts (Chen et al. 2004, Datta et al. 2005, 2007b). Recent work has also established an important role for the MAPK pathway in RUNX2 phosphorylation and transcriptional activity in skeletal development (Ge et al. 2007).
MAPKs are activated by a cascade of phosphorylation, and, inactivated by dephosphorylation. Several protein phosphatases can dephosphorylate MAPKs including tyrosine, serine/threonine, and dual-specificity phosphatases (Cobb 1999, English et al. 1999, Keyse 2000). The dual-specificity MAPK phosphatases (MKPs) are known to be involved in cell survival, proliferation, differentiation, and apoptosis (Keyse 2000, Theodosiou & Ashworth 2002, Wu & Bennett 2005). ERK–MAPK is primarily dephosphorylated/deactivated by MKP1 (Keyse 2000). Among the 11 members of MKP family MKP1 is activated by PTH or PTHrP in pancreatic β-cells (Zhang et al. 2003), murine osteoblastic cells (Aghaloo et al. 2006), and UMR cells (Homme et al. 2004, Qin et al. 2005). MKP1 may play an important role in immune responsiveness (Chi et al. 2006, Salojin et al. 2006), in osteoclasts, and anti-inflammatory bone loss (Carlson et al. 2009, Sartori et al. 2009). Whether MKP1 plays a role in osteoblasts and PTH1R anabolic action in bone is yet to be established. Using an engineered model of bone tissue growth we showed that the anabolic effects of PTH involve upregulation of MKP1, inactivation of pERK1/2, downregulation of cyclin D1 and induction of osteoblast cell growth arrest (Datta et al. 2010a). These data suggested that MKP1 is a possible mediator of the anabolic actions of PTH1R in osteoblasts. Consistent with our observation, PTH was reported to downregulate pERK1/2 in UMR 106–01 osteoblastic cells via upregulation of MKP1 and inhibition of c-Raf (Lai & Mitchell 2009). In this study, we demonstrate that female mice deficient in MKP1 have a decreased bone mass. Furthermore, we show that deletion of Mkp1 attenuates PTH anabolic action in bone in vivo. Primary calvarial osteoblasts derived from female Mkp1 KO mice display decreased and delayed differentiation and disparate PTH response compared with wild-type (WT) controls. Therefore, Mkp1 is an important gene that physiologically regulates bone mass and may become a therapeutic target for the treatment of bone diseases associated with low bone mass.
Materials and Methods
Animals
Cryopreserved embryos of Mkp1 KO mice were provided by Bristol–Myers Squibb Pharmaceutical Research Institute and regenerated into mice on C57Bl6/129 mixed background at the Jackson Laboratory (Bar Harbor, ME, USA; Dorfman et al. 1996, Zhao et al. 2005). We obtained these mice through Material Transfer Agreement from Bristol–Myers Squibb. These mice, containing a disruption within exon 2 of Mkp1, were bred in-house either inter-crossing heterozygous or homozygous KO breeders to yield both WT and KO mice. In this study, the experiments were performed with neonatal, 3–5 week- or 11–16-week-old WT and KO female mice and were fed with rodent chow (Lab Diet, Bentwood, MD, USA). For genotyping, real-time PCR analysis was performed by Transnetyx (Cordova, TN, USA). All animals were maintained in facilities operated by Wayne State University, and all animal experimental procedures were approved by the Institutional Animal Care and Use Committee for the Use and Care of Animals (IACUC).
PTH treatment in vivo
Three to four days old Mkp1 KO and WT controls were administered once daily injection s.c. of hPTH (1–34) (Bachem, Torrance, CA, USA; 50 μg/kg), or vehicle (0 9% sodium chloride), 5–6 days/week for 21 days. Mice were killed 24 h after the last injection, and bone tissues were collected.
Skeletal phenotyping, and microcomputed tomography analysis
Femora and tibiae were dissected free of soft tissue, fixed in 10% neutral buffered formalin for 48 h, and analyzed by microcomputed tomography (microCT or mCT, eXplore Locus SP, GE Healthcare Pre-Clinical Imaging, London, ON, Canada) at the Orthopedic Research Laboratory of University of Michigan. Specimens were immersed in water and scanned four at a time by the Parker method (180° + 20° fan angle) of rotation at 80 kVp and 80 μA and added filtration in the form of both an acrylic beam flattener and a 0 02 in. aluminum filter. Images were reconstructed at an isotropic voxel size of 18 μm and calibrated for densitometry. A metaphyseal region of interest was isolated from the distal femora and proximal tibiae starting at the interface between the growth plate and metaphyseal trabecular bone and extending a standardized percentage of the bone length into the metaphysis using a spinning algorithm in the transverse plane (MicroView version 2.2, GE Healthcare Pre-Clinical Imaging). Trabecular bone volume/total volume (BV/TV), bone surface/bone volume (BS/BV), trabecular thickness (Tb.Th), trabecular number (Tb.N), and trabecular spacing (Tb.Sp) along with bone mineral content (BMC) and tissue mineral density (TMD) were calculated after applying a uniform threshold of 1200 Hounsfield Units (HU). For cortical analysis, a diaphyseal region of interest was isolated using a standardized percentage of the bone length located in the center of the diaphysis of the femur and immediately proximal to the tibia–fibular junction of the tibia. Cortical thickness, bending moment of inertia, inner (endosteal) and outer (periosteal) perimeters, cross-sectional area, BMC, and TMD were calculated after applying a uniform threshold of 2000 HU.
Staining and bone histomorphometry
Femora were fixed with 4% paraformaldehyde for 24 h, decalcified with 10% EDTA (pH 6 5) in PBS, dehydrated, infiltrated, and paraffin-embedded. Five micrometer thick longitudinal sections were stained with hematoxylin and eosin, and digital photomicrographs of the distal femurs were captured under 5 × magnification using a Zeiss Axioplan 2 microscope (Zeiss, Gottingen, Germany) equipped with a software controlled (Axiovision version 4.8.2; Zeiss) digital camera. The percentage of the area occupied by trabecular bone in the region proximal to the epiphyseal growth plate was calculated based on the measurement in square micrometer (μm2) obtained with the software. Cuboidal osteoblasts lining trabecular bone surface were counted in the same area. To quantitate osteoclasts, sections immediately adjacent to those described above were stained for tartrate-resistant acid phosphatase (TRAP) using an acid phosphatase leukocyte kit from Sigma–Aldrich, as instructed by the manufacturer. TRAP-positive multinucleated cells, evidenced by a dark purple staining in the cytoplasm, were counted as number of osteoclasts per unit length of bone trabeculae in the same areas analyzed above. All histomorphometrical analyses were performed by investigators blinded to the genotype of the mice.
Biochemical markers of bone turnover
Serum levels of osteocalcin (OCN) and TRAP5b were measured using mouse sandwich ELISA kits from Biomedical Technologies (Stoughton, MA, USA) and Immuno-diagnostic System (Fountain Hill, AZ, USA) respectively. The manufacturer’s protocols were followed, and all samples were assayed in duplicate. A standard curve was generated from each kit, and absolute concentrations were extrapolated from the standard curve. The coefficients of variations for inter- and intra-assay measurements were 10% for all assays.
Primary osteoblast culture
Primary osteoblasts were isolated from calvaria by serial digestion (Datta et al. 2005). Briefly, calvaria were dissected, isolated and subjected to sequential digestions in collagenase A (2 mg/ml) and trypsin (0 25%) for 20, 40, and 90 min. Cells from the third digest were rinsed, counted, and plated in αMEM containing 10% FBS, 100 U/ml penicillin, and 1 μg/ml streptomycin. Primary cultures were used without passage.
Alkaline phosphatase and von Kossa staining
Primary cells were plated in six-well plates with a density of 2×105 cells/well. The medium was changed to osteogenic medium with the addition of ascorbic acid (50 μg/ml) and β-glycerophosphate (10 mM). The cells were cultured for 3–21 days, and the medium was replaced every 2–3 days with or without 100 nM PTH. Histochemical staining and alkaline phosphatase (ALKP) activity was determined at different days. One set of cells was fixed and stained for ALKP by incubating the cells with p-nitrophenyl phosphate substrate 5 using Sigma Fast BCIP/NBT tablets (B5655) (Sigma–Aldrich) at room temperature for 30 min. Another set of cells was lysed with buffer containing 150 mM Tris–HCl pH 9, 0 1 mM MgCl2, and 1% Triton X-100. The lysates were incubated with Sigma Fast tablets at 37°8C and the optical absorbance was read at 405 nm. After the absorbance was taken, protein concentrations from these wells were measured with the Bio-Rad DC protein assay kit (Bio-Rad) using BSA as standard. The ALKP activities were normalized to protein content and plotted. The mineralization assays were performed by the von Kossa method. At the end of 21 days culture period, the cells were fixed with 95% EtOH and stained with AgNO3 to detect phosphate deposits in bone nodules as described (Marsh et al. 1995). The deposits of calcium were shown by the formation of opaque mineralized nodules. The number of nodules were counted and plotted as percent relative expression compared with untreated WT cells. Undifferentiated proliferating osteoblasts were used as a negative control.
RNA preparation and real-time PCR
Total RNA was extracted from primary osteoblasts using TRIZOL reagent (Invitrogen), and cDNAs were prepared using the TaqMan RT assay system (Applied Biosystems, Foster City, CA, USA). The yield and purity of RNA was estimated spectrophotometrically using A260/A280 ratio. Real-time PCR was performed by StepOne Plus system (Applied Biosystems) using FAM-labeled primers (cyclin D1 #Mm00432359; Gapdh #Mm99999915; Alkp #Mm01187117; Ocn #AIAAA61; On #Mm0470030; Runx2 #Mm0501584; matrix Gla protein (Mgp) #Mm00485009; Pth1R, Mm00441046; Applied Biosystems). Gapdh was used as an internal control. After calculating cycle threshold (Ct) and ∆Ct values for each sample, the relative differences from the control sample were calculated.
SDS–PAGE and western analysis
SDS–PAGE and western blot analysis were performed as described previously (Datta et al. 2005). Calvarial osteoblasts were differentiated for 7 days with ascorbic acid (50 μg/ml) and β-glycerophosphate (10 mM) as above and induced with 100 nM PTH or PTHrP for 10 min to 5 h for subsequent experiments. Following induction cells were washed twice with cold PBS, scraped and lysed for 30 min at 4°C following sonication with RIPA buffer containing protease inhibitors (Sigma). Cell lysates were cleared by centrifugation at 14 000 g for 45 min. An aliquot of each lysate was removed for protein concentration determination. SDS–PAGE was performed in 10–12% polyacrylamide. Each lane was loaded with 40–80 μg protein of cell lysates. For a given western blot analysis all lanes received equal protein loads. Prestained molecular weight standards were run in parallel lanes. After electrophoresis, the proteins were transferred to a polyvinylidene difluoride membrane (Bio-Rad Laboratories, Inc.) in 25 mM Tris–HCl, 192 mM glycine, 20% v/v methanol, 0 01% SDS (pH 8 5) using a semi-dry transfer system (Hoeffer Inc, Holliston, MA, USA). Residual protein binding sites on the membrane were blocked by TBST (20 mM Tris–HCl, pH7 6, 137 mM NaCl, 0 5% Tween-20) containing 5% nonfat dry milk for 3–12 h. The membranes were then incubated with the primary antiserum at room temperature. After washing with TBST, a HRP-conjugated secondary antibody was added for 20–60 min. The protein bands were visualized by autoradiography using an enhanced chemiluminescence detection system (Pierce, Rockford, IL, USA). The protein band intensities on the autoradiograms (all with exposures within the linear range of the film) were quantified using Scion Software (Frederick, MD, USA).
Statistical analysis
All values are expressed as mean±S.E.M. or S.D. Comparisons between the groups were made by either unpaired two-tailed Student’s t-test or ANOVA with Instat Biostatistics Program (GraphPad Software, San Diego, CA, USA) to determine significance between the groups. P values <0 05 were considered statistically significant.
Results
Bone phenotype in Mkp1 KO female mice
To determine the role of MKP1 in bone mass, tibiae and femora of 12-week-old littermate or age-matched WT and Mkp1 KO mice were examined by mCT (Fig. 1A; Tables 1 and 2). Compared with their respective values in WT bones, the femoral trabecular bone mass (BV/TV) was 40% lower in KO female mice. These changes were associated with 28% increase in BS/BV, and 23% decrease in Tb.Th, 24% decrease in Tb.N, 8% decrease in mean thickness, and 14% decrease in moment of inertia (Iyy). In addition, the cortical inner perimeter, the outer perimeter, and the cortical area were decreased by 4, 4 and 9%, respectively, in the KO mice compared with WT controls. The trabecular BMC, trabecular TMD, and cortical BMC were decreased by 30, 4 and 10%, respectively; and an increase in BS/BV (30%) and Tb.Sp (64%) were also noted in the female KO mice (Table 1).
Figure 1.
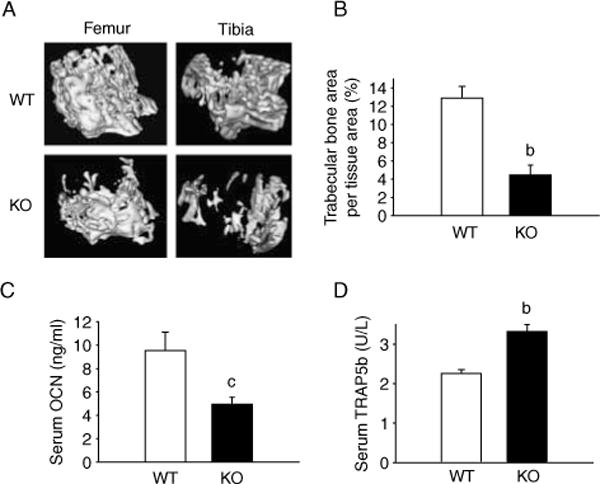
Mkp1 deletion results in decreased bone phenotype of female mice. (A) Representative 3D microCT images from 12-week-old wild-type (WT) and Mkp1 knockout (KO) mice tibiae and femora are shown. Mkp1 KO females show osteopenia compared with WT controls. (B) Histomorphometric analysis, (C) serum osteocalcin (OCN), and (D) serum TRAP5b assays were performed and plotted as mean±S.E.M. (n = 4–5). b, P<0.01 and c, P<0.05 versus respective WT control.
Table 1.
Trabecular and cortical microcomputed tomography (mCT) analyses of femora from 12-week-old wild-type (WT) and mitogen-activated protein kinase phosphatase 1 (Mkp1) knockout (KO) female mice. Data are presented as group mean±S.E.M.
| WT | KO | |
|---|---|---|
|
|
|
|
| Measurements | ||
| Trabecular BV/TV (%) | 0.366±0.017 | 0.217±0.011a |
| Trabecular BS/BV (mm/mm2) | 37.33±1.56 | 47.87±0.95a |
| Tb.Th (μm) | 0.054±0.002 | 0.042±0.001a |
| Tb.N (1/mm) | 6.78±0.06 | 5.174±0.19a |
| Tb.Sp (mm) | 0.093±0.03 | 0.1527±0.008a |
| Cortical mean thickness (mm) | 0.18±0.004 | 0.166±0.004c |
| Cortical Iyy (mm4) | 0.14±0.004 | 0.12±0.007c |
| Cortical inner perimeter (mm) | 3.88±0.03 | 3.74±0.05c |
| Cortical outer perimeter (mm) | 4.99±0.03 | 4.78±0.07c |
| Cortical area (mm2) | 0.75±0.015 | 0.68±0.02c |
| Trabecular BMC (mg) | 0.857±0.02 | 0.595±0.03a |
| Trabecular TMD (mg/cc) | 505.46±4.34 | 485.62±5.54c |
| Cortical BMC (mg) | 2.89±0.06 | 2.59±0.10c |
| Cortical TMD (mg/cc) | 1009.1±6.98 | 1011.3±6.72 |
|
|
||
BV, bone volume; BS, bone surface; TV, total volume; Tb.N, trabecular number; Tb.Th, trabecular thickness; Tb.Sp, trabecular spacing; Iyy, bending moment of inertia for the medial–lateral axis; BMC, bone mineral content; TMD, tissue mineral density.
P<0 001 and
P<0 05 versus WT(n=4–6).
Table 2.
Trabecular and cortical microcomputed tomography (mCT) analyses of tibiae from 12-week-old wild-type (WT) and mitogen-activated protein kinase phosphatase 1 (Mkp1) knockout (KO) female mice. Data are presented as group mean±S.E.M.
| WT | KO | |
|---|---|---|
|
|
|
|
| Measurements | ||
| Trabecular BV/TV (%) | 0.344±0.017 | 0.217±0.008a |
| Trabecular BS/BV (mm/mm2) | 38.64±1.64 | 47.11±1.37b |
| Tb.Th (μm) | 0.052±0.002 | 0.043±0.001b |
| Tb.N (1/mm) | 6.59±0.07 | 5.1±0.17a |
| Tb.Sp (mm) | 0.099±0.003 | 0.155±0.006a |
| Cortical mean thickness (mm) | 0.231±0.003 | 0.22±0.003c |
| Cortical Iyy (mm4) | 0.056±0.002 | 0.048±0.002b |
| Cortical inner perimeter (mm) | 1.98±0.03 | 1.98±0.03 |
| Cortical outer perimeter (mm) | 3.42±0.024 | 3.35±0.03 |
| Cortical area (mm2) | 0.60±0.007 | 0.57±0.01b |
| Trabecular BMC (mg) | 0.46±0.02 | 0.30±0.01a |
| Trabecular TMD (mg/cc) | 540.35±7.52 | 514.23±10.91 |
| Cortical BMC (mg) | 1.3±0.02 | 1.18±0.03b |
| Cortical TMD (mg/cc) | 1130.6±1.95 | 1097±10.05b |
| Cortical BVF | 0.309±0.006 | 0.301±0.008 |
|
|
||
BV, bone volume; TV, total volume; BS, bone surface; Tb.N, trabecular number; Tb.Th, trabecular thickness; Tb.Sp, trabecular spacing; Iyy, bending moment of inertia for the medial–lateral axis; BMC, bone mineral content; TMD, tissue mineral density; BVF, bone volume fraction.
P<0 001,
P<0 01, and
P<0 05 versus WT (n=4–6).
mCT analyses of tibiae from 12-week-old WT and Mkp1 KO female mice (Table 2) showed changes that were similar to those observed in the femora from KO mice displaying decrease in trabecular BV/TV (36%), Tb.Th (18%), Tb.N (23%), mean thickness (4%), Iyy (14%), cortical area (5%), trabecular BMC (35%); and increase in BS/BV (22%) and Tb.Sp (56%). Tibial cortical parameters were also significantly affected in the KO mice showing decrease in cortical BMC (10%), and TMD (3%).
Bone histomorphometry revealed a reduced trabecular bone formation in Mkp1 KO compared with WT female mice (KO 4 5±1 1 versus WT 12 9±1 3%, mean trabecular bone area per tissue area, Fig. 1B). This 65% decrease in trabecular bone found in Mkp1 KO female mice was statistically significant (P<0 01). The number of osteoclasts and osteoblasts remained unchanged in Mkp1 KO mice (data not shown), however, serum OCN, a bone formation parameter, was reduced by 48% (KO 4 95±0 062 versus WT 9 53±1 587 ng/ml; P<0 05, Fig. 1C) and serum TRAP5b, a bone resorption parameter, was increased by 47% (KO 3 319±0 176 versus WT 2 255±0 10 U/l; P<0 01, Fig. 1D) compared with WT animals. These data suggest that Mkp1 KO female mice develop osteopenia with reduced osteoblast and increased osteoclast activities.
Mkp1 deletion attenuates the increase in bone mass following PTH treatment that is observed in WT controls
To examine the effects of PTH in the Mkp1 KO mice in vivo, we selected growing mice in which PTH administration causes dramatic bone anabolic effects in vivo (Rihani-Bisharat et al. 1998, Demiralp et al. 2002). As expected, in neonatal female WT mice treated with PTH for 21 days, bone mass was significantly increased with higher femoral BV/TV (87%), Tb.Th (33%), Tb.N (27%), and lower Tb.Sp (46%) than in the vehicle-treated WT group (Fig. 2). In contrast, PTH treated Mkp1 KO female had significantly less pronounced effects with only 51% increase in BV/TV, 24% increase in Tb.Th, and 22% decrease in Tb.Sp compared with vehicle-treated KO group (Fig. 2). The difference in Tb.N between vehicle and PTH-treated KO animals did not reach statistical significance.
Figure 2.
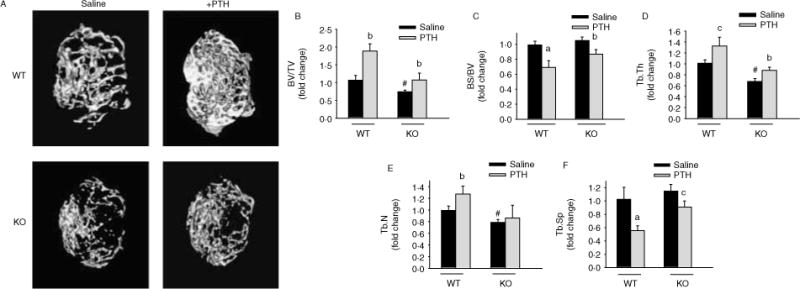
Mkp1 deletion attenuates PTH anabolic response in growing female mice. (A) Representative microCT images of distal femoral trabecular bone from 3-week-old wild-type (WT) and Mkp1 knockout (KO) female mice with vehicle (0 9% saline) and PTH treatment. (B) Bone volume/total volume (BV/TV), (C) bone surface/bone volume (BS/BV), (D) trabecular thickness (Tb.Th), (E) trabecular number (Tb.N), and (F) trabecular spacing (Tb.Sp) were analyzed and plotted as fold changes with respect to saline-treated WT control group. The values for saline-treated WT control are BV/TV (%), 0 235±0 031; BS/BV (mm/mm2), 64 75±3 40; Tb.Th (μm), 0 033±0 005; Tb.N (1/mm), 7 45±0.61; Tb.Sp (mm), 0.118±0.017. All data are expressed as mean±S.D. (n=4–5). a, P<0.001; b, P<0.01; and c, P<0.05 versus respective saline control; # P<0 01 versus WT saline.
Deletion of Mkp1 alters osteoblast differentiation and PTH response in primary osteoblast cultures
To evaluate the effects of systemic Mkp1 deletion on osteoblastogenesis in vitro, primary calvarial osteoblasts from 4- to 5-week-old WT and KO mice were cultured in osteogenic media for 2 weeks. We also assessed PTH1R expression in Mkp1 null osteoblasts by real-time PCR analysis and compared it to that in WT cells. Our results demonstrate that both WTand Mkp1 null osteoblasts express similar levels of Pth1R mRNA (Fig. 3A). The primary cultures from WT mice showed increased ALKP staining and activity from day 3 to 7 compared with undifferentiated controls; which then decline by more than 50% thereafter. In cultures from KO mice the ALKP staining and activity were observed at day 5 and did not decline until later time points; this suggests delayed osteoblast differentiation (Fig. 3B and C). PTH treatment significantly increased ALKP activity at days 3–5 in osteoblastic cells derived from WT mice. In contrast, PTH did not influence ALKP activity in osteoblasts derived from the KO group (Fig. 3C). Alkp mRNA expression was increased by four- to eight-fold in differentiated osteoblasts from WT mice at days 5–7; then declined, almost to undetectable levels between days 15 and 20. On the other hand, the expression of Alkp in differentiated osteoblasts from KO mice began to rise significantly around day 15 and remained increased. The stimulatory or inhibitory effects of PTH on Alkp mRNA expression in osteoblasts derived from WT mice were not seen in cells from KO mice (Fig. 3D). Similar results were noted in osteoblasts derived from younger (3-week-old) or older (11–16-week-old) mice (data not shown). These data suggest that MKP1 is required for osteoblast differentiation and regulation by PTH.
Figure 3.
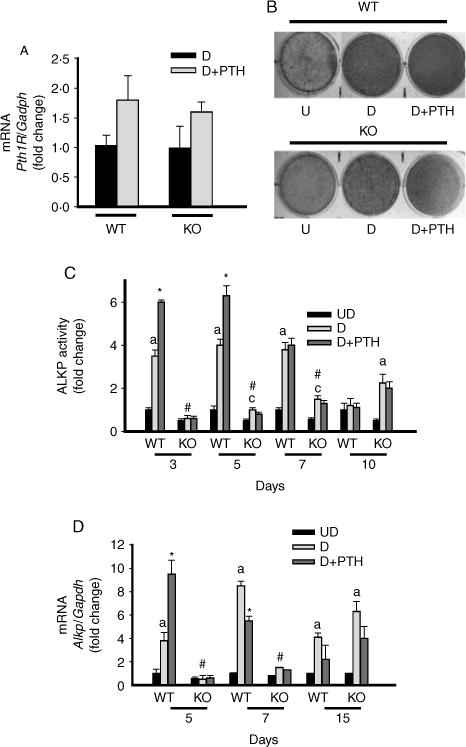
Altered osteoblast differentiation in calvarial cells from Mkp1 knockout (KO) female mice with and without PTH treatment. Cells were isolated from calvaria of 4–5-week-old WT and Mkp1 KO mice. The cells were differentiated with ascorbic acid and β-glycerophosphate. (A) Pth1R mRNA expression in differentiated primary calvarial osteoblasts derived from WT and Mkp1 KO mice at day 7 with and without 100 nM PTH treatment. Total RNA was isolated from triplicate independent cultures. Results are graphically represented after normalization with Gapdh as fold change (mean±S.D.) with respect to PTH untreated differentiated cells from WT cultures. (B) Histochemical staining of ALKP. Representative wells of 4–6 individual experiments with similar results for ALKP staining at day 5, with or without 100 nM PTH, are shown. (C) Time course of ALK activity in primary calvarial osteoblasts with or without PTH treatment. Results are expressed as fold change with respect to undifferentiated wild-type (WT) control cells at day 3. The basal ALKP activity for undifferentiated WT control cells at day 3 is 1 15±0 18 units/mg protein per min. (D) Time course of Alkp mRNA expression in primary calvarial osteoblasts with or without PTH treatment. Total RNA was isolated from triplicate independent cultures from day 5 to day 15. Results are graphically represented after normalization with Gapdh as fold change with respect to undifferentiated wild-type (WT) control cells at day 5. UD, undifferentiated cells; D, differentiated cells. Values are expressed as mean±S.D. from 4 to 6 individual experiments. a, P<0 001 versus UD; c, P<0 05 versus UD; *P<0 01 versus D without PTH, # P<0 001 versus WT-D cells at same time point.
The effects of continuous PTH administration on in vitro bone matrix mineralization are attenuated in osteoblasts derived from female Mkp1 KO mice
To determine the effects of Mkp1 deletion on osteoblastogenesis and cell maturation, calvarial osteoblasts from 11-week-old female WT and KO mice were differentiated in the presence or absence of PTH and mineralized nodule formation was assessed by von Kossa staining. PTH inhibited mineralization in osteoblast cultures from WT mice to background levels (Fig. 4A). In contrast, PTH showed only 15–20% inhibition of mineralization in osteoblasts derived from KO mice. Similar results were obtained in osteoblasts derived from younger (3–5-week-old) or older (16-week-old) KO animals (data not shown). Involvement of MGP in PTH-mediated inhibition of mineralization in osteoblastic cells has been previously described (Gopalakrishnan et al. 2001). Therefore, we measured the effect of PTH on the steady-state level of the Mgp transcript. As previously reported (Gopalakrishnan et al. 2001), PTH dramatically increased the expression of Mgp mRNA in osteoblasts from WT controls. In contrast, PTH had no effect on Mgp mRNA expression in osteoblasts derived from KO mice (Fig. 4B).
Figure 4.
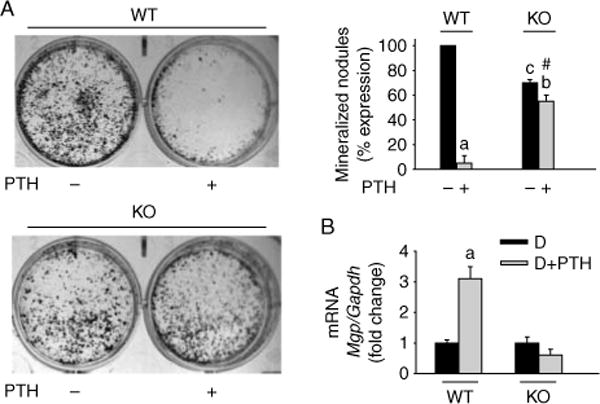
Effects of Mkp1 deletion on osteoblast mineralization and MGP mRNA expression with or without PTH treatment. Cells were isolated from calvaria of 11-week-old wild-type (WT) and Mkp1 knockout (KO) mice. The cells were differentiated with ascorbic acid and β-glycerophosphate for 21 days with or without 100 nM PTH. Mineralized nodule formation was examined by von Kossa staining as described in the Materials and Methods section. (A) Representative wells of 5–7 individual experiments with similar results for nodule formation are shown. The number of nodules was counted and plotted as percentage (%) expression of nodules with respect to PTH untreated WT cells from 5 to 6 individual experiments. Values are expressed as mean±S.D. a, P<0 001 versus untreated cells; b, P<0 01 versus untreated cells; c, P<0 01 versus untreated WT control cells; # P<0 001 versus PTH-treated WT control cells. The number of mineralized nodules in osteoblasts derived from KO animals without PTH treatment was 30% lower compared with PTH untreated WT cultures. (B) Mgp expression in primary calvarial osteoblasts derived from WT and Mkp1 KO mice at day 15 with and without PTH treatment. Total RNA was isolated from triplicate independent cultures. Results are graphically represented after normalization with Gapdh as fold change (mean±S.D.) with respect to PTH untreated differentiated cells from WT cultures. D, differentiated cells; a, P<0 001 versus D.
Effects of Mkp1 deletion on osteoblast gene expression and PTH action
The role of MKP1 in osteoblast differentiation/maturation and PTH action was further evaluated by expressions of selected osteogenic marker genes (Ocn, On, and Runx2) at the basal state and after continuous PTH treatment. In calvarial osteoblasts isolated from KO mice, the expressions of Ocn and Runx2 were significantly lower in earlier time points compared with osteoblasts derived from WT mice and increased gradually from day 7 to 15, again suggesting decreased and delayed differentiation of Mkp1 null osteoblasts (Fig. 5A–C). PTH downregulation of Ocn (Fig. 5A) and Runx2 (Fig. 5C) gene expression observed in WTosteoblasts was not evident in osteoblasts derived from Mkp1 KO animals. The level of On mRNA during osteoblast differentiation was not significantly different between WT and KO mice. In contrast to WT osteoblasts, a twofold increase in On expression was noted in cells from KO animals following PTH treatment (Fig. 5B). These data further support an important role for MKP1 on osteoblast differentiation and PTH molecular action.
Figure 5.
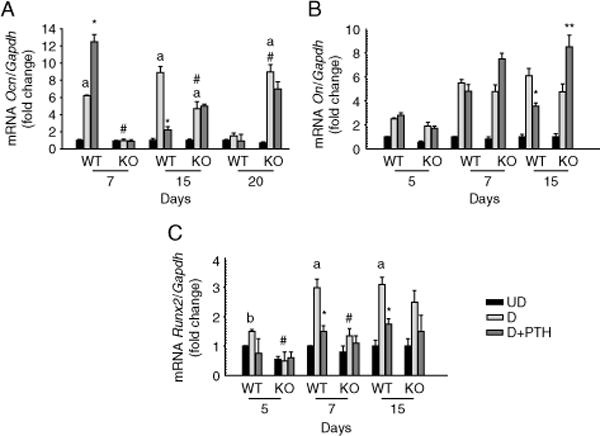
Temporal expressions of osteoblast differentiation marker genes from 11-week-old female wild-type (WT) and Mkp1 knockout (KO) mice. Time course of (A) osteocalcin (Ocn), (B) osteonectin (On), and (C) Runx2 mRNA expressions in primary calvarial osteoblasts, with or without PTH treatment. Total RNA was isolated from triplicate independent cultures from day 5 to day 15. Results are graphically represented after normalization with Gapdh as fold change (mean±S.D.) with respect to undifferentiated cells at day 7 (A) or day 5 (B and C) from WT control. UD, undifferentiated cells; D, differentiated cells; a, P<0 001 versus UD; b, P<0 01 versus UD; # P<0 05 versus WT-D cells at the same time point; *P<0 05 versus WT-D without PTH; **P<0 02 versus KO-D without PTH.
MKP1 deletion reduces the effect of PTH on ERK1/2 phosphorylation and cyclin D1 expression in primary osteoblast cultures
We have previously shown that overexpression of MKP1 induces osteoblastic cell growth arrest (Datta et al. 2010a). Using Mkp1 null calvarial osteoblasts, we observe approximately twofold increase in osteoblast number during the first week of culture (data not shown). This is consistent with the previous report by Ma et al. (2009) using bone marrow stromal cells from Mkp1 KO animals. Our previous study reported that PTH1R activation downregulates pERK1/2 and cyclin D1 in differentiated osteoblastic cells (Chen et al. 2004, Datta et al. 2005, 2007b, 2010c). To determine the role of MKP1 in PTH effects on pERK1/2 and cyclin D1 expressions we evaluated these responses in calvarial osteoblasts from female WT and Mkp1 KO mice. Cyclin D1 mRNA was significantly increased more than twofold in Mkp1 null osteoblasts compared with osteoblasts from WT mice (Fig. 6C). The basal levels of pERK1/2 and cyclin D1 protein showed an increased tendency in osteoblasts derived from Mkp1 null animals but did not reach statistical significance (Fig. 6). PTH significantly decreased pERK1/2 (Fig. 6A), cyclin D1 (Fig. 6B) protein levels, and cyclin D1 mRNA (Fig. 6C) in osteoblasts from WT mice. These effects were attenuated in cells derived from KO animals indicating that MKP1 is involved in PTH-regulated ERK–MAPK and cyclin D1 inactivation and osteoblast proliferation/differentiation.
Figure 6.
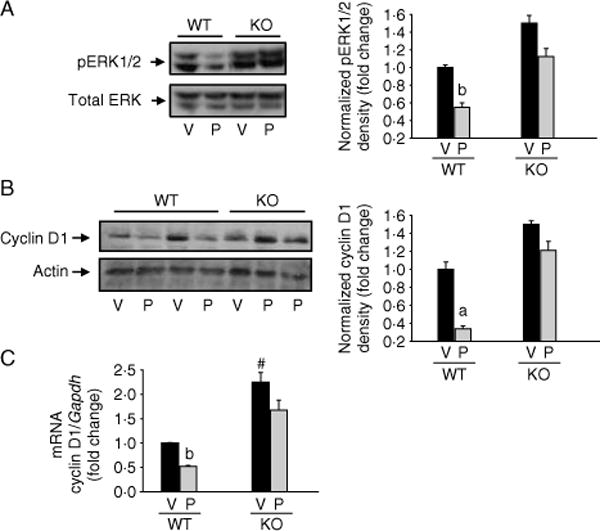
Effect of Mkp1 deletion on PTH-mediated pERK1/2 and cyclin D1 expression in osteoblasts derived from wild-type (WT) and Mkp1 knockout (KO) female mice. Seven days differentiated calvarial osteoblasts from either WT or KO female mice were treated with 100 nM PTH (P) or vehicle (V) for (A) 10 min, (B) 5 h, or (C) 1 h. Total cellular protein or RNA was harvested. Western blot analyses were performed for (A) pERK1/2 and total ERK and (B) cyclin D1 and actin. Total ERK and actin were used as loading controls. Densitometric values were normalized and plotted as fold change with respect to WT-V treated cells. (C) Real-time PCR analysis was performed for cyclin D1 mRNA, normalized with Gapdh as described in the Materials and Methods section and plotted as fold changes with respect to WT-V treated cells. Representative data from at least 3 to 4 independent experiments are shown. Data are expressed as mean±S.D. a, P<0 001 versus V; b, P<0 05 versus V; # P<0 05 versus WT-V.
Discussion
Despite intensive investigation, the physiological role of MAPK pathway in bone formation and PTH action remains incompletely understood, especially regarding the regulation in osteoblasts. PTH is a known bone anabolic agent because it increases osteoblast maturation and activity (Hock et al. 2002). An important role for ERK–MAPK pathway in osteoblast differentiation and skeletal development involving stimulation of RUNX2 phosphorylation and transcriptional activity (Ge et al. 2007), and a role for MKP1, a primary phosphatase for regulating MAPK signaling, in osteoclasts and bone homeostasis has been recently reported (Carlson et al. 2009). The data reported here demonstrate that deletion of Mkp1 in female mice results in reduced bone parameters, and reduced bone acquisition associated with altered osteoblast molecular action and decreased osteoblast activity. When Mkp1 KO mice are treated with PTH, we found that PTH had less anabolic effects on bone. These results suggest that MKP1 is required for the anabolic actions of PTH in bone. Our observation that Mkp1 KO females have osteopenic phenotype in the basal state is consistent with those of Carlson et al. (2009). A preliminary report by Ma et al. (2009) suggested a reduced bone phenotype in Mkp1 KO males with no difference in bone phenotype in KO females. This controversy may be attributed to differences in genetic background (Beamer et al. 2002). Several inbred strains of mice show skeletal differences, such as crossing C57Bl/6 with C3H/HeJ or 129S1/SvlmJ, as well as mice with spontaneous mutations resulting in skeletal abnormalities. These inbred strains of mice with allelic differences revealed sex-specific genetic regulation for genotype–phenotype correlations (Turner et al. 2003, Delahunty et al. 2006, Beamer et al. 2007, DeMambro et al. 2010). Current evidence also describes that skeletal sexual dimorphism is not just the end result of differences in sex steroid secretion between the sexes, but depends on gender differences in GH–insulin-like growth factor 1 (IGF1) and mechanical sensitivity to loading as well (Callewaert et al. 2010). Association of aging, calorie restriction and MAPK activity has been recently suggested (Chung et al. 2009). Therefore, genetic background, age, cage condition, breeding program, and experimental conditions and settings may play a role in these observations. Despite this inconsistency all these findings suggest an important role for MKP1 in bone mass homeostasis. Our initial studies also revealed sexual dimorphism in the bone phenotype of the Mkp1 KO animals showing osteopetrosis in KO males (Datta et al. 2010b), implying involvement of MKP1 in regulating bone mass in males and females. It is to be investigated if the sexually dimorphic bone phenotype is secondary to sex hormones or to genetic differences between males and female Mkp1 KO mice.
Bone formation involves the recruitment of osteoprogenitors that mature into bone-forming osteoblasts that actively synthesize and mineralize the bone matrix. In this study, we report that MKP1 plays a role in the control of bone mass through osteoblast differentiation and function. Osteoblast differentiation can be monitored by examining the time course of expression of Alkp (an early osteoblast marker) and Ocn (a late osteoblast marker). Osteoblast differentiation in primary cultures proceeds in two stage developmental processes. During the ‘initiation phase’, 1–2 weeks in culture, the cells slowly proliferate, express Alkp and other bone-specific genes, and produce and assemble extracellular matrix. During the ‘maturation phase’, occurring after 2 or 3 weeks of culture, matrix mineralization is observed (Cheng et al. 1994, Marsh et al. 1995, Wang et al. 1999). Thus, mineralization is considered a functional end point reflecting advanced cell differentiation in osteogenic cultures in vitro. In this study, we demonstrated that Mkp1 deletion delays osteoblast differentiation in terms of ALKP activity and gene expression. ALKP activity and expression was sustained for a longer time in osteoblasts derived from KO mice. These data suggest an increased proliferative effect during postconfluence allowing a periodic rise in the pool of osteoprogenitors and a decrease in bone-forming mature osteoblasts in KO animals. This was further supported by decreased Ocn and Runx2 mRNA expression; and less mineralized nodules in osteoblasts from KO females compared with WT controls. PTH inhibition of Alkp, Ocn, and Runx2 mRNA expression observed in WT osteoblasts was not evident in cells derived from KO animals. This observation may raise an important question, that is, if PTH1R are expressed in Mkp1 null osteoblasts and whether they are functional. In this study, the expression of Pth1R mRNA was found to be similar in cells derived from WTand KO animals. Furthermore, in contrast to WTosteoblasts, On expression was increased in Mkp1 null osteoblasts following PTH treatment indicating that PTH or PTH1R was functional and biologically active, but in an opposite manner than in WT cells. The significance of PTH induced On upregulation is not clear. There are a limited number of studies describing the role of ON in bone remodeling with variable findings. On-null animals display enhanced bone formation and decreased osteoclast formation with PTH treatment (Kitahara et al. 2003) and ovariectomy of On-deficient mice show decreased bone resorption (Yoshitake et al. 1999) compared with WT. On the other hand, increased osteoclast response to PTH in On-null mice has been reported (Machado do Reis et al. 2008). At the present time, our results imply that ON levels may play a role in modulating the balance between bone formation and resorption in response to PTH in Mkp1 null mice.
Using different osteogenic cells it has been shown that ALKP activity is not always proportional to in vitro mineralization (Hoemann et al. 2009). Therefore, additional factors, other than proliferation and differentiation, may play a role in this process. Inhibition of osteoblast-mediated mineralization is one of the major catabolic effects of PTH on bone. Our data that PTH increased Ocn and Alkp expression in pre-confluent, less mature cultures are consistent with previous observations using cultured cell line or primary bone marrow stromal cells (Nishida et al. 1994, Isogai et al. 1996, Jiang et al. 2004). In contrast, PTH had no effects on Alkp or Ocn expression in cultures from female Mkp1 KO mice. This suggests that the effect of PTH on osteoblast maturation is MKP1 dependent. Our finding that PTH treatment decreased mineralized nodule formation in osteoblasts from WT female mice is consistent with a previously published report (Koh et al. 1999). To characterize the mechanism of altered PTH response of nodule formation in cultures from KO females, we measured Mgp mRNA expression. MGP functions as an inhibitor of mineralization both in vitro and in vivo (Luo et al. 1995, 1997). It is a vitamin K-dependent protein found in hypertrophic chondrocytes, cementum, vascular smooth muscle cells, and bone (Price 1989, Sato et al. 1998). Mgp-deficient mice display abnormal calcification of cartilage and extensive mineralization of arteries (Luo et al. 1997). Previously, it was shown that PTH induces Mgp expression and established that this induction is critical for PTH-mediated inhibition of osteoblast mineralization in MC3T3-E1 osteoblastic cells (Gopalakrishnan et al. 2001). In this study, we demonstrate that in contrast to WT animals, osteoblasts from KO females show significantly reduced Mgp mRNA expression following PTH treatment. A recent study showed that PTH regulates Mgp expression via both PKA and ERK–MAPK pathways in osteoblastic cells (Suttamanatwong et al. 2007). Taking into account our previous observations and those of others that MKP1 dephosphorylates ERK1/2 in osteoblasts (Datta et al. 2010a) it is not unreasonable to propose that the lack of PTH effect on in vitro osteoblast mineralization is at least partially dependent on MKP1 via Mgp.
Our previous study showed that PTH1R activation by PTH and PTHrP inhibits pERK1/2 and cyclin D1 expression and induces cell growth arrest in differentiated mature osteoblasts (Datta et al. 2005, 2010c). Interestingly, PTH and PTHrP (data not shown) failed to downregulate pERK1/2 and cyclin D1 in primary osteoblastic cells derived from female Mkp1 KO mice. Furthermore, PTH decreased expression of cyclin D1 mRNA in osteoblasts from WT female whereas this effect was attenuated in cells from Mkp1 KO females. Our data indicate that MKP1 signaling is important for the inhibitory effects of PTH1R on cyclin D1 in differentiated cells, osteoblastic growth arrest, and bone formation. It remains to be determined if endogenous Mkp1 deletion influences PTH regulation of other PTH1R target genes involved in its differential effects on osteoblast bone formation. Nevertheless, both altered osteoblastic (this study) and osteoclastic activities (this study, Carlson et al. (2009)) are likely to be responsible for the osteopenic bone phenotype of Mkp1 KO females. Our data do not preclude a role for other downstream targets of MKP1, such as stress responsive p38-MAPK and c-Jun N-terminal kinases (JNK) (Wu & Bennett 2005). Involvement of p38-MAPK associated with increased MKP1 in osteoblasts has been previously suggested by us (Datta et al. 2007a) and a role of both p38- and JNK– MAPK has been described in bone development (Greenblatt et al. 2010, Zhong et al. 2011).
In summary, our results provide evidence for the roles of MKP1 in osteoblast differentiation, pERK1/2 and cell cycle regulatory protein cyclin D1 expression, and PTH response to bone formation. To our knowledge, this study is the first to show an important role for MKP1 in mineralization involving Mgp. On the basis of this study and other findings, we propose that MKP1 plays a critical role in bone homeostasis, osteoblast differentiation, and in mediating bone anabolic effects of PTH.
Acknowledgments
We thank Sonali Sharma and Allen D Saliganan for technical assistance. MKP1 KO mice were obtained through Material Transfer Agreement between Bristol–Myers Squibb and N S D (Wayne State University). Acknowledgment is also due to animal care facilities, Division of Laboratory and Animal Research, WSU, for taking care of our mice.
Funding
This work was supported by Institutional funding and NIH DK087848 to N S D. K L K was supported by R01DE018290.
Footnotes
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
References
- Aghaloo TL, Pirih FQ, Shi A, Bezouglaia O, Tetradis S. Parathyroid hormone induces mitogen-activated kinase phosphatase 1 in murine osteoblasts primarily through cAMP-protein kinase A signaling. Journal of Periodontology. 2006;77:21–30. doi: 10.1902/jop.2006.77.1.21. [DOI] [PubMed] [Google Scholar]
- Alves de Oliveira EC, Szejnfeld VL, Pereira da Silva N, Coelho Andrade LE, Heldan de Moura Castro C. Intermittent PTH1–34 causes DNA and chromosome breaks in osteoblastic and nonosteoblastic cells. Calcified Tissue International. 2010;87:424–436. doi: 10.1007/s00223-010-9396-6. [DOI] [PubMed] [Google Scholar]
- Andreassen TT, Fledelius C, Ejersted C, Oxlund H. Increases in callus formation and mechanical strength of healing fractures in old rats treated with parathyroid hormone. Acta Orthopaedica Scandinavica. 2001;72:304–307. doi: 10.1080/00016470152846673. [DOI] [PubMed] [Google Scholar]
- Beamer WG, Donahue LR, Rosen CJ. Genetics and bone. Using the mouse to understand man. Journal of Musculoskeletal & Neuronal Interactions. 2002;2:225–231. [PubMed] [Google Scholar]
- Beamer WG, Shultz KL, Ackert-Bicknell CL, Horton LG, Delahunty KM, Coombs HF, III, Donahue LR, Canalis E, Rosen CJ. Genetic dissection of mouse distal chromosome 1 reveals three linked BMD QTLs with sex-dependent regulation of bone phenotypes. Journal of Bone and Mineral Research. 2007;22:1187–1196. doi: 10.1359/jbmr.070419. [DOI] [PubMed] [Google Scholar]
- Callewaert F, Sinnesael M, Gielen E, Boonen S, Vanderschueren D. Skeletal sexual dimorphism: relative contribution of sex steroids, GH–IGF1, and mechanical loading. Journal of Endocrinology. 2010;207:127–134. doi: 10.1677/JOE-10-0209. [DOI] [PubMed] [Google Scholar]
- Calvi LM, Sims NA, Hunzelman JL, Knight MC, Giovannetti A, Saxton JM, Kronenberg HM, Baron R, Schipani E. Activated parathyroid hormone/parathyroid hormone-related protein receptor in osteoblastic cells differentially affects cortical and trabecular bone. Journal of Clinical Investigation. 2001;107:277–286. doi: 10.1172/JCI11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson J, Cui W, Zhang Q, Xu X, Mercan F, Bennett AM, Vignery A. Role of MKP-1 in osteoclasts and bone homeostasis. American Journal of Pathology. 2009;175:1564–1573. doi: 10.2353/ajpath.2009.090035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Koh AJ, Datta NS, Zhang J, Keller ET, Xiao G, Franceschi RT, D’Silva NJ, McCauley LK. Impact of the mitogen-activated protein kinase pathway on parathyroid hormone-related protein actions in osteoblasts. Journal of Biological Chemistry. 2004;279:29121–29129. doi: 10.1074/jbc.M313000200. [DOI] [PubMed] [Google Scholar]
- Cheng SL, Yang JW, Rifas L, Zhang SF, Avioli LV. Differentiation of human bone marrow osteogenic stromal cells in vitro: induction of the osteoblast phenotype by dexamethasone. Endocrinology. 1994;134:277–286. doi: 10.1210/en.134.1.277. [DOI] [PubMed] [Google Scholar]
- Chi H, Barry SP, Roth RJ, Wu JJ, Jones EA, Bennett AM, Flavell RA. Dynamic regulation of pro- and anti-inflammatory cytokines by MAPK phosphatase 1 (MKP-1) in innate immune responses. PNAS. 2006;103:2274–2279. doi: 10.1073/pnas.0510965103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung HY, Cesari M, Anton S, Marzetti E, Giovannini S, Seo AY, Carter C, Yu BP, Leeuwenburgh C. Molecular inflammation: underpinnings of aging and age-related diseases. Ageing Research Reviews. 2009;8:18–30. doi: 10.1016/j.arr.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobb MH. MAP kinase pathways. Progress in Biophysics and Molecular Biology. 1999;71:479–500. doi: 10.1016/S0079-6107(98)00056-X. [DOI] [PubMed] [Google Scholar]
- Datta NS, Abou-Samra AB. PTH and PTHrP signaling in osteoblasts. Cellular Signalling. 2009;21:1245–1254. doi: 10.1016/j.cellsig.2009.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta NS, Chen C, Berry JE, McCauley LK. PTHrP signaling targets cyclin D1 and induces osteoblastic cell growth arrest. Journal of Bone and Mineral Research. 2005;20:1051–1064. doi: 10.1359/JBMR.050106. [DOI] [PubMed] [Google Scholar]
- Datta NS, Kolailat R, Pettway GJ, Berry JE, McCauley LK. Induction of mitogen-activated protein kinase phosphatase-1 and down-regulation of p38 and ERK1/2 phosphorylation in PTHrP stimulated differentiated osteoblasts. The FASEB Journal. 2007a;21(768):4. [Google Scholar]
- Datta NS, Pettway GJ, Chen C, Koh AJ, McCauley LK. Cyclin D1 as a target for the proliferative effects of PTH and PTHrP in early osteoblastic cells. Journal of Bone and Mineral Research. 2007b;22:951–964. doi: 10.1359/jbmr.070328. [DOI] [PubMed] [Google Scholar]
- Datta NS, Kolailat R, Fite A, Pettway G, Abou-Samra AB. a Distinct roles for mitogen-activated protein kinase phosphatase-1 (MKP-1) and ERK–MAPK in PTH1R signaling during osteoblast proliferation and differentiation. Cellular Signalling. 2010;22:457–466. doi: 10.1016/j.cellsig.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta NS, Mahalingam CD, Kreider J, Kirkwood K, Goldstein SA, Abou-Samra AB. MKP-1 knockout mice reveal sexual dimorphism in bone mass and disperate PTHrP responsiveness of primary calvarial osteoblasts; American Society of Bone and Mineral Research Annual Conference; SU0116, Toronto, Canada. 2010b. [Google Scholar]
- Datta NS, Samra TA, Mahalingam CD, Datta T, Abou-Samra AB. Role of PTH1R internalization in osteoblasts and bone mass using a phosphorylation-deficient knock-in mouse model. Journal of Endocrinology. 2010c;207:355–365. doi: 10.1677/JOE-10-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delahunty KM, Shultz KL, Gronowicz GA, Koczon-Jaremko B, Adamo ML, Horton LG, Lorenzo J, Donahue LR, Ackert-Bicknell C, Kream BE, et al. Congenic mice provide in vivo evidence for a genetic locus that modulates serum insulin-like growth factor-I and bone acquisition. Endocrinology. 2006;147:3915–3923. doi: 10.1210/en.2006-0277. [DOI] [PubMed] [Google Scholar]
- DeMambro VE, Kawai M, Clemens TL, Fulzele K, Maynard JA, Marin de Evsikova C, Johnson KR, Canalis E, Beamer WG, Rosen CJ, et al. A novel spontaneous mutation of Irs1 in mice results in hyperinsulinemia, reduced growth, low bone mass and impaired adipogenesis. Journal of Endocrinology. 2010;204:241–253. doi: 10.1677/JOE-09-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demiralp B, Chen HL, Koh AJ, Keller ET, McCauley LK. Anabolic actions of parathyroid hormone during bone growth are dependent on c-fos. Endocrinology. 2002;143:4038–4047. doi: 10.1210/en.2002-220221. [DOI] [PubMed] [Google Scholar]
- Dempster DW, Cosman F, Kurland ES, Zhou H, Nieves J, Woelfert L, Shane E, Plavetic K, Muller R, Bilezikian J, et al. Effects of daily treatment with parathyroid hormone on bone microarchitecture and turnover in patients with osteoporosis: a paired biopsy study. Journal of Bone and Mineral Research. 2001;16:1846–1853. doi: 10.1359/jbmr.2001.16.10.1846. [DOI] [PubMed] [Google Scholar]
- Dobnig H, Turner RT. The effects of programmed administration of human parathyroid hormone fragment (1–34) on bone histomorphometry and serum chemistry in rats. Endocrinology. 1997;138:4607–4612. doi: 10.1210/en.138.11.4607. [DOI] [PubMed] [Google Scholar]
- Dorfman K, Carrasco D, Gruda M, Ryan C, Lira SA, Bravo R. Disruption of the erp/mkp-1 gene does not affect mouse development: normal MAP kinase activity in ERP/MKP-1-deficient fibroblasts. Oncogene. 1996;13:925–931. [PubMed] [Google Scholar]
- English J, Pearson G, Wilsbacher J, Swantek J, Karandikar M, Xu S, Cobb MH. New insights into the control of MAP kinase pathways. Experimental Cell Research. 1999;253:255–270. doi: 10.1006/excr.1999.4687. [DOI] [PubMed] [Google Scholar]
- Franceschi RT, Xiao G. Regulation of the osteoblast-specific transcription factor, Runx2: responsiveness to multiple signal transduction pathways. Journal of Cellular Biochemistry. 2003;88:446–454. doi: 10.1002/jcb.10369. [DOI] [PubMed] [Google Scholar]
- Ge C, Xiao G, Jiang D, Franceschi RT. Critical role of the extracellular signal-regulated kinase–MAPK pathway in osteoblast differentiation and skeletal development. Journal of Cell Biology. 2007;176:709–718. doi: 10.1083/jcb.200610046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopalakrishnan R, Ouyang H, Somerman MJ, McCauley LK, Franceschi RT. Matrix gamma-carboxyglutamic acid protein is a key regulator of PTH-mediated inhibition of mineralization in MC3T3-E1 osteoblast-like cells. Endocrinology. 2001;142:4379–4388. doi: 10.1210/en.142.10.4379. [DOI] [PubMed] [Google Scholar]
- Greenblatt MB, Shim JH, Zou W, Sitara D, Schweitzer M, Hu D, Lotinun S, Sano Y, Baron R, Park JM, et al. The p38 MAPK pathway is essential for skeletogenesis and bone homeostasis in mice. Journal of Clinical Investigation. 2010;120:2457–2473. doi: 10.1172/JCI42285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi C, Myoui A, Hashimoto N, Kuriyama K, Yoshioka K, Yoshikawa H, Itoh K. Continuous inhibition of MAPK signaling promotes the early osteoblastic differentiation and mineralization of the extracellular matrix. Journal of Bone and Mineral Research. 2002;17:1785–1794. doi: 10.1359/jbmr.2002.17.10.1785. [DOI] [PubMed] [Google Scholar]
- Hock JM, Fitzpatrick LA, Bilezikian JP. Actions of parathyroid hormone. In: Bilezikian JP, Raisz LG, Rodan GA, editors. Principles of Bone Biology. 2. San Diego: Academic Press; 2002. pp. 463–482. [Google Scholar]
- Hoemann CD, El-Gabalawy H, McKee MD. In vitro osteogenesis assays: influence of the primary cell source on alkaline phosphatase activity and mineralization. Pathologie-Biologie. 2009;57:318–323. doi: 10.1016/j.patbio.2008.06.004. [DOI] [PubMed] [Google Scholar]
- Homme M, Schmitt CP, Mehls O, Schaefer F. Mechanisms of mitogen-activated protein kinase inhibition by parathyroid hormone in osteoblast-like cells. Journal of the American Society of Nephrology. 2004;15:2844–2850. doi: 10.1097/01.ASN.0000143472.13214.2C. [DOI] [PubMed] [Google Scholar]
- Isogai Y, Akatsu T, Ishizuya T, Yamaguchi A, Hori M, Takahashi N, Suda T. Parathyroid hormone regulates osteoblast differentiation positively or negatively depending on the differentiation stages. Journal of Bone and Mineral Research. 1996;11:1384–1393. doi: 10.1002/jbmr.5650111003. [DOI] [PubMed] [Google Scholar]
- Jahng JS, Kim HW. Effect of intermittent administration of parathyroid hormone on fracture healing in ovariectomized rats. Orthopedics. 2000;23:1089–1094. doi: 10.3928/0147-7447-20001001-22. [DOI] [PubMed] [Google Scholar]
- Jiang D, Franceschi RT, Boules H, Xiao G. Parathyroid hormone induction of the osteocalcin gene. Requirement for an osteoblast-specific element 1 sequence in the promoter and involvement of multiple-signaling pathways. Journal of Biological Chemistry. 2004;279:5329–5337. doi: 10.1074/jbc.M311547200. [DOI] [PubMed] [Google Scholar]
- Jilka RL. Molecular and cellular mechanisms of the anabolic effect of intermittent PTH. Bone. 2007;40:1434–1446. doi: 10.1016/j.bone.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karaplis AC, Luz A, Glowacki J, Bronson RT, Tybulewicz VL, Kronenberg HM, Mulligan RC. Lethal skeletal dysplasia from targeted disruption of the parathyroid hormone-related peptide gene. Genes and Development. 1994;8:277–289. doi: 10.1101/gad.8.3.277. [DOI] [PubMed] [Google Scholar]
- Keyse SM. Protein phosphatases and the regulation of mitogen-activated protein kinase signalling. Current Opinion in Cell Biology. 2000;12:186–192. doi: 10.1016/S0955-0674(99)00075-7. [DOI] [PubMed] [Google Scholar]
- Kitahara K, Ishijima M, Rittling SR, Tsuji K, Kurosawa H, Nifuji A, Denhardt DT, Noda M. Osteopontin deficiency induces parathyroid hormone enhancement of cortical bone formation. Endocrinology. 2003;144:2132–2140. doi: 10.1210/en.2002-220996. [DOI] [PubMed] [Google Scholar]
- Koh AJ, Beecher CA, Rosol TJ, McCauley LK. 3′,5′-Cyclic adenosine monophosphate activation in osteoblastic cells: effects on parathyroid hormone-1 receptors and osteoblastic differentiation in vitro. Endocrinology. 1999:140, 3154–3162. doi: 10.1210/en.140.7.3154. [DOI] [PubMed] [Google Scholar]
- Lai LP, Mitchell J. Parathyroid hormone inhibits phosphorylation of mitogen-activated protein kinase (MAPK) ERK1/2 through inhibition of c-Raf and activation of MKP-1 in osteoblastic cells. Cell Biochemistry and Function. 2009;27:269–275. doi: 10.1002/cbf.1568. [DOI] [PubMed] [Google Scholar]
- Lanske B, Amling M, Neff L, Guiducci J, Baron R, Kronenberg HM. Ablation of the PTHrP gene or the PTH/PTHrP receptor gene leads to distinct abnormalities in bone development. Journal of Clinical Investigation. 1999;104:399–407. doi: 10.1172/JCI6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo G, D’Souza R, Hogue D, Karsenty G. The matrix Gla protein gene is a marker of the chondrogenesis cell lineage during mouse development. Journal of Bone and Mineral Research. 1995;10:325–334. doi: 10.1002/jbmr.5650100221. [DOI] [PubMed] [Google Scholar]
- Luo G, Ducy P, McKee MD, Pinero GJ, Loyer E, Behringer RR, Karsenty G. Spontaneous calcification of arteries and cartilage in mice lacking matrix GLA protein. Nature. 1997;386:78–81. doi: 10.1038/386078a0. [DOI] [PubMed] [Google Scholar]
- Ma L, Choudhary S, Voznesensky O, Raisz L, Pilbeam C. Effects of MKP-1 deletion on osteoblast growth and differentiation; American Society of Bone and Mineral Research(ASBMR) Annual Conference; MO 0186, Denver, CO, USA. 2009. [Google Scholar]
- Machado do Reis L, Kessler CB, Adams DJ, Lorenzo J, Jorgetti V, Delany AM. Accentuated osteoclastic response to parathyroid hormone undermines bone mass acquisition in osteonectin-null mice. Bone. 2008;43:264–273. doi: 10.1016/j.bone.2008.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh ME, Munne AM, Vogel JJ, Cui Y, Franceschi RT. Mineralization of bone-like extracellular matrix in the absence of functional osteoblasts. Journal of Bone and Mineral Research. 1995;10:1635–1643. doi: 10.1002/jbmr.5650101105. [DOI] [PubMed] [Google Scholar]
- Marx SJ. Hyperparathyroid and hypoparathyroid disorders. New England Journal of Medicine. 2000;343:1863–1875. doi: 10.1056/NEJM200012213432508. [DOI] [PubMed] [Google Scholar]
- Miao D, He B, Karaplis AC, Goltzman D. Parathyroid hormone is essential for normal fetal bone formation. Journal of Clinical Investigation. 2002;109:1173–1182. doi: 10.1172/JCI14817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Tamura Y, Suzawa M, Harada S, Fukumoto S, Kato M, Miyazono K, Rodan GA, Takeuchi Y, Fujita T. Receptor tyrosine kinases inhibit bone morphogenetic protein-Smad responsive promoter activity and differentiation of murine MC3T3-E1 osteoblast-like cells. Journal of Bone and Mineral Research. 2003;18:827–835. doi: 10.1359/jbmr.2003.18.5.827. [DOI] [PubMed] [Google Scholar]
- Neer RM, Arnaud CD, Zanchetta JR, Prince R, Gaich GA, Reginster JY, Hodsman AB, Eriksen EF, Ish-Shalom S, Genant HK, et al. Effect of parathyroid hormone (1–34) on fractures and bone mineral density in postmenopausal women with osteoporosis. New England Journal of Medicine. 2001;344:1434–1441. doi: 10.1056/NEJM200105103441904. [DOI] [PubMed] [Google Scholar]
- Nishida S, Yamaguchi A, Tanizawa T, Endo N, Mashiba T, Uchiyama Y, Suda T, Yoshiki S, Takahashi HE. Increased bone formation by intermittent parathyroid hormone administration is due to the stimulation of proliferation and differentiation of osteoprogenitor cells in bone marrow. Bone. 1994;15:717–723. doi: 10.1016/8756-3282(94)90322-0. [DOI] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocrine Reviews. 2001;22:153–183. doi: 10.1210/er.22.2.153. [DOI] [PubMed] [Google Scholar]
- Pietrogrande L. Update on the efficacy, safety, and adherence to treatment of full length parathyroid hormone, PTH (1–84) in the treatment of postmenopausal osteoporosis. International Journal of Women’s Health. 2010;1:193–203. doi: 10.2147/ijwh.s4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price PA. Gla-containing proteins of bone. Connective Tissue Research. 1989;21:51–57. doi: 10.3109/03008208909049995. discussion 57–60. [DOI] [PubMed] [Google Scholar]
- Qin L, Li X, Ko JK, Partridge NC. Parathyroid hormone uses multiple mechanisms to arrest the cell cycle progression of osteoblastic cells from G1 to S phase. Journal of Biological Chemistry. 2005;280:3104–3111. doi: 10.1074/jbc.M409846200. [DOI] [PubMed] [Google Scholar]
- Rihani-Bisharat S, Maor G, Lewinson D. In vivo anabolic effects of parathyroid hormone (PTH) (28–48) and N-terminal fragments of PTH and PTH-related protein on neonatal mouse bones. Endocrinology. 1998;139:974–981. doi: 10.1210/en.139.3.974. [DOI] [PubMed] [Google Scholar]
- Salojin KV, Owusu IB, Millerchip KA, Potter M, Platt KA, Oravecz T. Essential role of MAPK phosphatase-1 in the negative control of innate immune responses. Journal of Immunology. 2006;176:1899–1907. doi: 10.4049/jimmunol.176.3.1899. [DOI] [PubMed] [Google Scholar]
- Sartori R, Li F, Kirkwood KL. MAP kinase phosphatase-1 protects against inflammatory bone loss. Journal of Dental Research. 2009;88:1125–1130. doi: 10.1177/0022034509349306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Yasui N, Nakase T, Kawahata H, Sugimoto M, Hirota S, Kitamura Y, Nomura S, Ochi T. Expression of bone matrix proteins mRNA during distraction osteogenesis. Journal of Bone and Mineral Research. 1998;13:1221–1231. doi: 10.1359/jbmr.1998.13.8.1221. [DOI] [PubMed] [Google Scholar]
- Schiller PC, D’Ippolito G, Roos BA, Howard GA. Anabolic or catabolic responses of MC3T3-E1 osteoblastic cells to parathyroid hormone depend on time and duration of treatment. Journal of Bone and Mineral Research. 1999;14:1504–1512. doi: 10.1359/jbmr.1999.14.9.1504. [DOI] [PubMed] [Google Scholar]
- Schindeler A, Little DG. Ras–MAPK signaling in osteogenic differentiation: friend or foe? Journal of Bone and Mineral Research. 2006;21:1331–1338. doi: 10.1359/jbmr.060603. [DOI] [PubMed] [Google Scholar]
- Suttamanatwong S, Franceschi RT, Carlson AE, Gopalakrishnan R. Regulation of matrix Gla protein by parathyroid hormone in MC3T3-E1 osteoblast-like cells involves protein kinase A and extracellular signal-regulated kinase pathways. Journal of Cellular Biochemistry. 2007;102:496–505. doi: 10.1002/jcb.21314. [DOI] [PubMed] [Google Scholar]
- Swarthout JT, Doggett TA, Lemker JL, Partridge NC. Stimulation of extracellular signal-regulated kinases and proliferation in rat osteoblastic cells by parathyroid hormone is protein kinase C-dependent. Journal of Biological Chemistry. 2001;276:7586–7592. doi: 10.1074/jbc.M007400200. [DOI] [PubMed] [Google Scholar]
- Theodosiou A, Ashworth A. MAP kinase phosphatases. Genome Biology. 2002;3:1–10. doi: 10.1186/gb-2002-3-7-reviews3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner CH, Sun Q, Schriefer J, Pitner N, Price R, Bouxsein ML, Rosen CJ, Donahue LR, Shultz KL, Beamer WG. Congenic mice reveal sex-specific genetic regulation of femoral structure and strength. Calcified Tissue International. 2003;73:297–303. doi: 10.1007/s00223-002-1062-1. [DOI] [PubMed] [Google Scholar]
- Wang D, Christensen K, Chawla K, Xiao G, Krebsbach PH, Franceschi RT. Isolation and characterization of MC3T3-E1 preosteoblast subclones with distinct in vitro and in vivo differentiation/mineralization potential. Journal of Bone and Mineral Research. 1999;14:893–903. doi: 10.1359/jbmr.1999.14.6.893. [DOI] [PubMed] [Google Scholar]
- Weir EC, Philbrick WM, Amling M, Neff LA, Baron R, Broadus AE. Targeted overexpression of parathyroid hormone-related peptide in chondrocytes causes chondrodysplasia and delayed endochondral bone formation. PNAS. 1996;93:10240–10245. doi: 10.1073/pnas.93.19.10240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JJ, Bennett AM. Essential role for mitogen-activated protein (MAP) kinase phosphatase-1 in stress-responsive MAP kinase and cell survival signaling. Journal of Biological Chemistry. 2005;280:16461–16466. doi: 10.1074/jbc.M501762200. [DOI] [PubMed] [Google Scholar]
- Yang D, Singh R, Divieti P, Guo J, Bouxsein ML, Bringhurst FR. Contributions of parathyroid hormone (PTH)/PTH-related peptide receptor signaling pathways to the anabolic effect of PTH on bone. Bone. 2007;40:1453–1461. doi: 10.1016/j.bone.2007.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshitake H, Rittling SR, Denhardt DT, Noda M. Osteopontin-deficient mice are resistant to ovariectomy-induced bone resorption. PNAS. 1999;96:8156–8160. doi: 10.1073/pnas.96.14.8156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B, Hosaka M, Sawada Y, Torii S, Mizutani S, Ogata M, Izumi T, Takeuchi T. Parathyroid hormone-related protein induces insulin expression through activation of MAP kinase-specific phosphatase-1 that dephosphorylates c-Jun NH2-terminal kinase in pancreatic beta-cells. Diabetes. 2003;52:2720–2730. doi: 10.2337/diabetes.52.11.2720. [DOI] [PubMed] [Google Scholar]
- Zhao Q, Shepherd EG, Manson ME, Nelin LD, Sorokin A, Liu Y. The role of mitogen-activated protein kinase phosphatase-1 in the response of alveolar macrophages to lipopolysaccharide: attenuation of proinflammatory cytokine biosynthesis via feedback control of p38. Journal of Biological Chemistry. 2005;280:8101–8108. doi: 10.1074/jbc.M411760200. [DOI] [PubMed] [Google Scholar]
- Zhong M, Carney DH, Jo H, Boyan BD, Schwartz Z. Inorganic phosphate induces mammalian growth plate chondrocyte apoptosis in a mitochondrial pathway involving nitric oxide and JNK MAP kinase. Calcified Tissue International. 2011;88:96–108. doi: 10.1007/s00223-010-9433-5. [DOI] [PubMed] [Google Scholar]
- Zhou H, Shen V, Dempster DW, Lindsay R. Continuous parathyroid hormone and estrogen administration increases vertebral cancellous bone volume and cortical width in the estrogen-deficient rat. Journal of Bone and Mineral Research. 2001;16:1300–1307. doi: 10.1359/jbmr.2001.16.7.1300. [DOI] [PubMed] [Google Scholar]