

Abstract
Background
Glarea lozoyensis is a filamentous fungus used for the industrial production of non-ribosomal peptide pneumocandin B0. In the scope of a whole genome sequencing the complete mitochondrial genome of the fungus has been assembled and annotated. It is the first one of the large polyphyletic Helotiaceae family. A phylogenetic analysis was performed based on conserved proteins of the oxidative phosphorylation system in mitochondrial genomes.
Results
The total size of the mitochondrial genome is 45,038 bp. It contains the expected 14 genes coding for proteins related to oxidative phosphorylation,two rRNA genes, six hypothetical proteins, three intronic genes of which two are homing endonucleases and a ribosomal protein rps3. Additionally there is a set of 33 tRNA genes. All genes are located on the same strand. Phylogenetic analyses based on concatenated mitochondrial protein sequences confirmed that G. lozoyensis belongs to the order of Helotiales and that it is most closely related to Phialocephala subalpina. However, a comparison with the three other mitochondrial genomes known from Helotialean species revealed remarkable differences in size, gene content and sequence. Moreover, it was found that the gene order found in P. subalpina and Sclerotinia sclerotiorum is not conserved in G. lozoyensis.
Conclusion
The arrangement of genes and other differences found between the mitochondrial genome of G. lozoyensis and those of other Helotiales indicates a broad genetic diversity within this large order. Further mitochondrial genomes are required in order to determine whether there is a continuous transition between the different forms of mitochondrial genomes or G. lozoyensis belongs to a distinct subgroup within Helotiales.
Introduction
The filamentous fungus Glarea lozoyensis (ATCC 20868) was originally isolated by plating filtrates of pond water near Madrid, Spain [1]. It came to be known as a producer of pneumocandins, non-ribosomal peptides with strong inhibitory effect on fungal glucan biosynthesis. G. lozoyensis ATCC 74030 is a mutant strain of the wild type used for the production of the antimycotic drug Caspofungin from Pneumocandin B0 [2], [3]. The taxonomy of G. lozoyensis has been revised several times [4]. It was designated Zalerion arboricola first [1], however, after thorough analysis of morphological and molecular data, it was classified as a new anamorphic genus in the order Helotiales [4]. Through sequence analysis of the ITS region of a broad array of species in Helotiales the closest relatives of G. lozoyensis were narrowed to some Cyathicula De Not species (Helotiaceae) [5].
Since mitochondrial (mt) genomes often evolve faster than nuclear genomes [6], [7], [8], they have been successfully applied as markers in evolutionary biology [9], [10]. With the emergence of next-generation sequencing in the last years, the access to whole genomes has become easy and affordable. The number of completely sequenced mt genomes of filamentous fungi has increased dramatically [11], [12], so that it is possible to use them for phylogenetic studies [13], [14], [15], [16], [17], [18]. The Fungal Mitochondrial Genome Project was launched over a decade ago [19]. Now, more than 80 fungal mitogenomes are available. Nevertheless, only four of the Helotiales order, Phialocephala subalpina, Sclerotinia sclerotiorum, Botrytis cinera and Marssonina brunnea (incomplete annotation) have been sequenced so far (Table 1).
Table 1. Selected fungus species with published mt genomes.
| Species | Class | Order | Length (bp) | Accession Nr |
| Glarea lozoyensis | Leotiomycetes | Helotiales | 45,038 | GenBank: KF169905 |
| Phialocephala subalpina | Leotiomycetes | Helotiales | 43,742 | GenBank: NC_015789 |
| Botrytis cinerea | Leotiomycetes | Helotiales | 80,799 | Broad Institute1 |
| Sclerotinia sclerotiorum | Leotiomycetes | Helotiales | 128,852 | Broad Institute2 |
| Marssonina brunnea | Leotiomycetes | Helotiales | 70,379 | Genbank: JN204424 |
| Gibberella zeae | Sordariomycetes | Hypocreales | 95,676 | GenBank: NC_009493 |
| Gibberella miniliformis | Sordariomycetes | Hypocreales | 53,753 | GenBank: NC_016687 |
| Hypocrea jecorina | Sordariomycetes | Hypocreales | 42,13 | GenBank: NC_003388 |
| Beauveria bassiana | Sordariomycetes | Hypocreales | 29,961 | GenBank: NC_010652 |
| Metarhizium anisopliae | Sordariomycetes | Hypocreales | 24,673 | GenBank: NC_008068 |
| Aspergillus niger | Eurotiomycetes | Eurotiales | 31,103 | GenBank: NC_007445 |
| Penicillium marneffei | Eurotiomycetes | Eurotiales | 35,438 | GenBank: NC_005256 |
| Emericella nidulans | Eurotiomycetes | Eurotiales | 33,227 | GenBank: NC_017896 |
| Aspergillus kawachi | Eurotiomycetes | Eurotiales | 31,222 | GenBank: AP012272 |
| Arthroderma obtusum | Eurotiomycetes | Onygenales | 24,101 | GenBank: NC_012830 |
| Trichophyton mentagrophytes | Eurotiomycetes | Onygenales | 24,297 | GenBank: NC_012826 |
| Neurospora crassa | Sordariomycetes | Sordariales | 6,484 | Broad Institute3 |
| Podospora anserina | Sordariomycetes | Sordariales | 94,192 | GenBank: NC_001329 |
| Glomerella graminicola | Sordariomycetes | Glomerellales | 39,649 | GenBank: CM001021 |
| Verticillium dahliae | Sordariomycetes | Glomerellales | 27,184 | GenBank: NC_008248 |
| Phaeosphaeria nodorum | Dothideomycetes | Pleosporales | 49,761 | GenBank: NC_009746 |
| Mycosphaerella graminicola | Dothideomycetes | Capnodiales | 43,964 | GenBank: NC_010222 |
| Candida albicans | Saccharomycetes | Saccharomycetales | 4,042 | GenBank: NC_002653 |
| Ogataea angusta | Saccharomycetes | Saccharomycetales | 41,719 | GenBank: NC_014805 |
| Pichia pastoris | Saccharomycetes | Saccharomycetales | 35,683 | GenBank: NC_015384 |
Botrytis cinerea Sequencing Project.
Sclerotinia sclerotiorum Sequencing Project.
Neurospora crassa Sequencing Project, (http://www.broadinstitute.org/).
A fungal mt genome typically contains 14 conserved protein-coding genes, 22–26 tRNA genes, and 2 rRNA genes arranged very likely in circular form [20], [21], [22], [23]. The mtDNA divergence between different fungal species is characterized by variations in intergenic regions, intronic sequences, and in the order of genes [24], [25].
We have identified the mitochondrial genome of G. lozoyensis ATCC 74030 within the whole genome of the fungus, which was assembled by combination of data from Illumina MP and PE sequencing [26]. As there are only very few mt genomes known from species of the large Helotiales order, we decided to annotate this genome in detail and to investigate its relation to the known mt genomes of Helotiales. These were also used as main reference for a thorough manual revision of the automatic annotation obtained from diverse bioinformatic tools.
Materials and Methods
Cultivation and DNA Preparation
G. lozoyensis ATCC 74030 was grown for 20 days at 25°C on a YM agar plates (yeast extract 0.1%, malt extract 1.0%, agar 1.5% in H2O). The mycelium was scraped off the plate, freeze-dried and ground to a fine powder. Total DNA was isolated with a DNeasy Plant Mini Kit (Qiagen) according to the manufacturer’s instructions.
Sequencing and Assembly
Whole genome shotgun sequencing of G. lozoyensis ATCC 74030 was performed by sequencing a paired-end library and an additional mate-pair library with an Illumina HiSeq 2000 sequencer. About 38 Gbp of reads with an average length of 75 bp were assembled to contigs using the CLC Genomics Workbench (CLCbio). Scaffold N50 is estimated to 870,933. A single 45,038 bp scaffold representing the complete mtDNA was identified by sequence similarity search to known fungal mt genomes.
Mitochondrial Genome Annotation
Potential open reading frames (ORFs) in the mtDNA sequence of G. lozoyensis were identified using Prodigal [27] and ORF Finder based on genetic code 4 (www.ncbi.nlm.nih.gov/gorf/gorf.html) performed with a Galaxy public server as a platform [28], [29]. Functional annotation was performed with BLASTp [30], BLASTx [30], Pfam [31] and MFannot (http://megasun.bch.umontreal.ca/cgi-bin/mfannot/mfannotInterface.pl). The mitochondrial genomes of P. subalpina [GenBank: JN031566] and S. sclerotiorum (http://www.broadinstitute.org) were used as references for manual annotation. tRNA genes were predicted by tRNAscan-SE [32], ARAGORN [33], RNAWEASEL [34], [35] and ARWIN [36] using the default settings for mt genomes. A potential tRNA was considered as proven when it was found by at least two of these tools. Ribosomal RNA genes were identified by sequence similarity with the corresponding genes of P. subalpina and S. sclerotiorum. Introns were predicted manually using BLASTx [30].
Phylogenetic Analysis
To determine the evolutionary background of G. lozoyensis, a concatenation of 12 OXPHOS genes (atp6, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5 and nad6) was compared with analogous sets of genes from 24 mt genomes published in GenBank. Protein sequence alignment was carried out for each protein using ClustalW with default options through a Galaxy server [37], [28], [29]. The aligned protein sequences were used to construct a maximum likelihood tree with PhyML 3.0 using LG as evolutionary model [38]. The reliability for internal branch was assessed using the aLRT test (SH-Like) as recommended in PhyML. The graphic representation was performed with Treeview (http://taxonomy.zoology.gla.ac.uk/rod/rod.html) and manual editing.
Genbank Accession Number
The G. lozoyensis ATCC 74030 mt genome sequence is deposited in GenBank under accession number KF169905.
Results and Discussion
General Features
The mitochondrial genome of G. lozoyensis ATCC 74030 comprises 45,038 bp, which is in the same range as several other mt genomes in Pezizomycotina (Table 1). 31.8% is covered with intergenic spacers of 3–1131 bp length and 15.04% with seven introns (Table 2). Among the five mt genomes reported, three are relatively large (Table 1) and only that of P. subalpina (43.7 kbp) is similar to that of G. lozoyensis (Table 1). The differences in genome size are marked by a multitude of introns and endonucleases in B. cinerea and S. sclerotiorum and a large intergenic region in M. brunnea. With 58.1% of the genome encoding structural genes, the mt genome of G. lozoyensis is rather compact (Table 2). It encodes the large and the small ribosomal RNA subunit (rnl and rns), 33 tRNAs, 14 putative proteins of the oxidative phosphorylation system (OXPHOS), 6 hypothetical proteins and 3 intronic proteins, of which one is ribosomal protein RPS3 and two are homing endonucleases (HE) (Figure 1, Table 2). As in mitochondria of most other ascomycetes, all genes and tRNAs are found on the plus strand [13], [14], [15], [16], [17], [18]. The overall G+C content of the mt genome is 29.8% (Table 2), consistent with the characteristic AT-rich nature of fungal mt genomes. The G+C content of regions encoding RNA genes is usually higher than the genome wide average. In G. lozoyensis it is 31% (Table 2), which is similar to other fungal mitochondria [39].
Table 2. General features of the mt genome of G. lozoyensis.
| Genomes features | Value |
| Genome size (bp) | 45,038 |
| G+C content (%) | 29.8 |
| No. of protein-coding genes | 20 |
| G+C content of protein-coding genes (%) | 31.0 |
| Structural proteins coding exons (%) | 38.7 |
| No. of rRNAs/tRNAs | 2/33 |
| G+C content of RNA genes (%) | 35.9 |
| rRNAs+tRNAs (%) | 14.4 |
| Coding regions (%) | 58.1 |
| Intergenic regions (%) | 31.8 |
| No. of introns | 7 |
| No. of intronic ORFs | 3 + (1)* |
| Introns (%) | 15.0 |
Eroded protein.
Figure 1. Circular mapping of the complete mt genome of G. lozoyensis.

All genes are located on the plus-strand. Protein coding genes (red; atp: ATPase synthase subunits, cob: cytochrome b, cox: cytochrome oxidase subunit, nad: NADH dehydrogenase subunit, orf: hypothetical proteins); ribosomal subunits (green; rns: small rRNA, rnl: large rRNA); tRNA genes (dark green); introns (black) and intronic proteins (purple, if protein coding). The precise positions of genes and introns are listed in table S1.
The mt genome of G. lozoyensis was compared with the fully annotated mt genomes of the Helotiales order, i.e., those of P. subalpina and S. sclerotiorum. BLASTn alignment [40] gave a sequence identity of 27% with the mt genome of P. subalpina and 19% with that of S. sclerotiorum. Moreover, DNA dot plot analysis showed that the mt genome of G. lozoyensis is colinear with that of P. subalpina (Figure S1) [40]. Considering also the similar size, the mt genome of P. subalpina was chosen as the main reference for the manual annotation of the mt genome of G. lozoyensis.
Protein Coding Genes
The following genes encoding proteins involved in respiratory chain complexes (OXPHOS) were found in the G. lozoyensis mt genome: atp6, atp8, atp9, cox1-3, cob, nad1-6 and nad4L (Figure 1, Table S1). Moreover, there are three intronic proteins, of which one is eroded (see section introns), and six hypothetical proteins (ORF1-6). All 14 OXPHOS proteins are highly conserved in P. subalpina (Table S1). Differences in sequence mainly refer to intronic proteins, which were not found in P. subalpina [13]. For instance, the large ribosomal subunits (rnl) in G. lozoyensis shares only a sequence identity of 45% with the corresponding feature in P. subalpina. This is mainly due to the presence of intron IE (2250 bp) in G. lozoyensis rnl, which also includes the gene for ribosomal protein S3 (rps3). In P. subalpina IE does not exist and rps3 forms a separate ORF [13]. ORF included in introns are presumably an excellent criterion for inferring phylogenetic relationships of fungi. In Pezizomycotina, the intronic rps-like protein may even play a role in maintaining the integrity of the mt genome [41].
The length of the cox1 gene varies widely within the investigated species. In G. lozoyensis it includes 5,412 bp, in S. sclerotiorum 12,458 bp and in P. subalpina only 1,720 bp. In contrast to the latter, S. sclerotiorum and G. lozoyensis cox1 contain several intronic proteins such as GIY-YIG and LAGLIDADG. For that reason, S. sclerotiorum cox1 was chosen as additional reference for intronic gene annotation in G. lozoyensis cox1. The G. lozoyensis GIY-YIG (842 bp) shares 87% sequence identity with a GIY-YIG of S. sclerotiorum (SS1G_20030.1; 816 bp). Since it is an eroded ORF, the DNA sequence was used for comparison instead of the protein sequence. The LAGLIDADG gene of G. lozoyensis shows only 17% identity to a LAGLIDADG from S. sclerotiorum (SS1G_20022; 323 aa, Broad Institute). However, a BLASTp search on NCBI database nr resulted a maximum identity of 76% with a LAGLIDADG of the Pezizomycotum Ajellomyces dermatitidis SLH14081. Of the six hypothetical proteins (ORF1-6) only ORF2 and ORF6 share similarities with known sequences. A part of ORF2 (132 aa) is similar to cytochrome c oxidase subunit I (cox1) of G. lozoyensis, even though only 39% of ORF2 is covered (e-value 3e−15). The sequence identity between both is 78% at the amino acid level. The occurrence of this fragment may be explained by partial gene duplication due to the presence of a transposon. In general, the evolution of gene orders in Pezizomycotina is mainly characterized by transpositions [13]. For example, in eight mt genomes of Phialocephala species, a duplication of the region around atp9 was found [13]. However, to confirm the transposon hypothesis in G. lozoyensis, more mt genomes of the same genus are required. ORF6 was found to be similar with a putative protein (ORF2) in the mt genome of P. subalpina (e-value: 7e−39, 46% identity and 90% coverage). No significant sequence similarities (BLASTp) or conserved domains (InterProScan [42]) were found for the other putative proteins.
Introns
A total of eight introns were identified in the coding genes of the G. lozoyensis mt genome by BLASTx search and sequence alignment (nr-database) [30]. Four of them are located in cox1 (coxIA, coxIB, coxIC and coxID). The others were found in the large ribosomal subunit rnl (intron IE), cox3, nad2, and nad5 (Figure 1, Table S1). coxIA and IE belong to introns group I, which is dominating in fungal mt genes, while in plant mt genes group II introns are found more frequently [43]. It is characteristic for group I introns is that all upstream exons end with a “T” and all introns end with a “G”. The conserved stems [44] were found in both genes (data not shown). Group I introns are considered to be mobile genetic elements interrupting protein-coding and structural RNA genes [45]. Most of them carry a “homing endonuclease gene” (heg) encoding a DNA endonuclease (HE), which catalyzes in the transfer and site-specific integration (“homing”) of the intron [46], [47], [48]. There are four families of DNA endonucleases (HEs) [49] denoted by the presence of conserved amino acid sequence motifs: GIY-YIG, HC-box, HNH and LAGLIDADG [50], [51]. Among these the LAGLIDADG endonucleases form the largest group. They are encountered in some bacteria and bacteriophages as well as in organelle genomes of protozoans, fungi, plants, and sometimes in early branching Metazoans [52]. Two forms can be distinguished: Proteins with a single LAGLIDADG motif which dimerize and double-motif forms derived form a gene fusion of two monomeric forms [53]. In the G. lozoyensis mt genome an intact ORF encoding a putatively functional HE referred to LAGLIDADG (342aa) was found in cox1-intron coxIA. A Pfam analysis resulted only one LAGLIDADG motive (Table S1), so that it can be considered as the single-motif form of LAGLIDADG. Moreover, in coxIB a frameshifted and inactive ( = “eroded”) heg known as GIY-YIG (835 bp) with several stop codons in the sequence was found. Despite being inactive, the conserved domain of GIY-YIG was still identified by Pfam (see section Protein coding genes and Table S1). Eroded hegs are characterized by several point and length mutations resulting in frameshifts and stop codons interrupting the ORF [54]. In species from the basal fungal lineages many introns in the long cox1-gene carry eroded hegs [55]. The erosion is generally regarded as a preliminary step before complete elimination of the intron [49]. The presence of intact and eroded hegs strengthens the hypothesis that numerous events of loss and gain have occurred during evolution. Besides the heg, coxID contains also a DUF3839 (PF12943) conserved domain, whose function is unknown. Furthermore, in the intron of the large ribosomal subunit (IE) we found a protein encoding for ribosomal protein S3 known as rps3 (519aa) (Table S1).
Genetic Code and Codon Usage
The codon usage of the G. lozoyensis mitochondrial ORFs was analyzed using genetic code four, which is common for Pezizomycotina mtDNA [56]. Most protein-coding ORFs start with the orthodox translation initiation codon ATG. Exceptions are: nad2 and cox3 with GTG, cox2 with TTA and cox1 with TTG. Six genes end with the stop codon TAG: cob, orf3, orf2, nad3, atp6, and nad5; all others with TAA, which is the preferred termination codon for fungal mt genes [22], [57]. The intronic proteins, ribosomal protein S3 (rps3) and the putative LAGLIDADG endonuclease, start with ATG but the stop codon is TAA for rps3 and TAG for LAGLIDADG. The most commonly used amino acid in the 22 protein genes is leucine followed by isoleucine (Table 3). Similar results are reported for other fungi [14], [18]. As expected from the high AT content (79%), the most frequently used codons are composed exclusively of “U” and “A”, e.g. UUA (2.78%), AUA (1.88%), AAU (1.38%), UUU (1.82%), AAA (1,27%), UAU (1.32%) and AUU (1.20%) (Table 3). The codons UGC, CGC, CUC, CGA, CGG, UCG are underrepresented, being used one to twenty times less than the GC -rich codons (Table 3).
Table 3. Codon usage of protein-coding genes in G. lozoyensis mt genome.
| Codon | AA | % | Codon | AA | % |
| GCA | A | 0.52 | CCA | P | 0.28 |
| GCC | A | 0.28 | CCC | P | 0.18 |
| GCG | A | 0.12 | CCG | P | 0.10 |
| GCU | A | 0.83 | CCU | P | 0.73 |
| UGC | C | 0.03 | CAA | Q | 0.55 |
| UGU | C | 0.29 | CAG | Q | 0.20 |
| GAC | D | 0.21 | AGA | R | 0.65 |
| GAU | D | 0.86 | AGG | R | 0.15 |
| GAA | E | 0.78 | CGA | R | 0.08 |
| GAG | E | 0.30 | CGC | R | 0.03 |
| UUC | F | 0.56 | CGG | R | 0.06 |
| UUU | F | 1.82 | CGU | R | 0.14 |
| GGA | G | 0.37 | AGC | S | 0.22 |
| GGC | G | 0.11 | AGU | S | 0.95 |
| GGG | G | 0.28 | UCA | S | 0.47 |
| GGU | G | 1.06 | UCC | S | 0.18 |
| CAC | H | 0.23 | UCG | S | 0.07 |
| CAU | H | 0.40 | UCU | S | 0.83 |
| AUA | I | 1.88 | ACA | T | 0.63 |
| AUC | I | 0.23 | ACC | T | 0.22 |
| AUU | I | 1.20 | ACG | T | 0.11 |
| AAA | K | 1.27 | ACU | T | 0.73 |
| AAG | K | 0.39 | GUA | V | 0.77 |
| CUA | L | 0.49 | GUC | V | 0.12 |
| CUC | L | 0.07 | GUG | V | 0.31 |
| CUG | L | 0.14 | GUU | V | 0.85 |
| CUU | L | 0.59 | UGG | W | 0.12 |
| UUA | L | 2.78 | UGA | W | 0.34 |
| UUG | L | 0.44 | UAC | Y | 0.43 |
| AUG | M | 0.79 | UAU | Y | 1.32 |
| AAC | N | 0.51 | UAA | Stop | 0.18 |
| AAU | N | 1.38 | UAG | Stop | 0.16 |
The overall percentage of codon usage in the protein coding genes atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6, orf 1, orf 2, orf 3, orf 4, orf 5, orf 6, rps3, intron protein (IA), intron protein (IB) and intron protein (IE) is depicted.
tRNA Genes
33 tRNA genes were identified in the G. lozoyensis mt genome, which is more than the 22–26 tRNA genes typically found in fungal mt genomes (Table 4, Figure 1) [20], [21], [22], [23], [32], [33], [36]. However, in Helotiales the numbers of tRNA genes appears to be generally higher, e.g. in S. sclerotiorum there are 33 tRNAs, in B. cinera and M. brunnea 31 tRNAs (predicted by TRNAscan tool [32]). In contrast, only 27 tRNAs were found in P. subalpina. Further mt genome sequences are required to confirm this tendency. The majority of G. lozoyensis mitochondrial tRNA genes are organized into two dense clusters. The set of 33 tRNA genes is sufficient to decode all codons in the predicted ORFs, lessening the need for tRNA import from the cytoplasm into the mitochondrium [58]. For lysine, asparagine and glycine two tRNA genes were found with the same anticodon. Furthermore, two tRNAs with different anticodons were identified for arginine, serine, phenylalanine and leucine. For methionine and isoleucine there are three tRNAs genes with identical anticodons.
Table 4. tRNAs in the mt genome of G. lozoyensis.
| AA | Anticodon | AA | Anticodon |
| Asn | GUU* | Thr | UGU |
| Arg | UCG* | Glu | UUC |
| Arg | UCU | Leu | UAG |
| Val | UAC | Leu | UAA |
| Lys | UUU* | Ala | UGC |
| Gly | UCC* | Phe | AAA* |
| Asp | GUC | Phe | GAA |
| Ser | UGA | Gln | UUG |
| Ser | GCU | His | GUG |
| Trp | UCA | Met | CAU** |
| Ile | GAU** | Cys | GCA |
| Pro | UGG | Tyr | GUA |
| Ter | UUA* |
Two tRNAs with the same anticodon.
Three tRNAs with the same anticodon.
Phylogeny and Comparative Genomics
Since mt genomes often evolve faster than nuclear genomes, especially in intergenic regions [59], [60], mitochondrial markers were successfully applied in evolutionary biology [61], [62], [63]. In Pezizomycotina, completely sequenced and annotated mt genomes are available for members of Eurotiomycetes and Sordariomycetes [11], [12], [64]. But only three genomes with complete draft annotations are available for Helotialean species (Table 1). The Helotiales is one of the most diverse fungal order with more than 350 genera and over 2,000 species including many important plant pathogens [65]. In order to gain additional evidence for the classification of G. lozoyensis, we compared the amino acid sequences of 12 OXPHOS proteins (atp6, cox1-3, cob, nad1-6, nad4L) with those from 24 other fungi to build a phylogenetic tree (Figure 2). Most nodes in this tree have high bootstrap values, which indicate the robustness of the computed tree. Five classes of filamentous ascomycetes are clearly distinguished: Dothideomycetes; Leotiomycetes, Sordariomycetes, Eurotiomycetes and Saccharomycetes (Figure 2). As found already in other studies [44], the mt genomes of yeast species cluster apart from those of filamentous fungi. G. lozoyensis is found amongst other species of the Helotiales order with high bootstrap support. This placement is in line with previous observations based on nuclear ribosomal internal transcribed spacers (ITS) [8]. The closest relative of G. lozoyensis was found to be P. subalpina. This is consistent with the high similarities found already in BLASTp analyses (see above and Table S1). A close relation between protein sequence similarity and a uniform organization of mt genomes has been found for the orders Onygenales [39] and Sordariales [18]. The arrangement of mt genes might even be used as a reference to derive a common evolutionary route in fungi. We compared the mt gene order in G. lozoyensis with that of the Helotialean species with sequenced and annotated mt genome, i.e. P. subalpina and S. sclerotiorum (Figure 3). It was found that the gene organization in G. lozoyensis deviates significantly from both fungi (Figure 3). Already the mt genomes of P. subalpina and S. sclerotiorum differ considerably [15]. Anchored genome alignments for the two adjacent species G. lozoyensis and P. subalpina show no close genome rearrangements (Figure S2A). The similar result was obtained when S. sclerotiorum was included in the analysis (Figure S2B). In contrast, there is a complete synteny in gene order between S. sclerotiorum and B. cinerea [66]. Additional mt genomes of Helotiales species are required to allow further conclusions about the considerably diverse mt gene arrangements in this order.
Figure 2. Phylogenetic analysis of G. lozoyensis based on mitochondrial protein sequences.

The evolutionary history was inferred by using the Maximum Likelihood method in PhyML 3.0 [38]. The tree was based on 12 OXPHOS proteins ATP6, COB, COX1, COX2, COX3, NAD1, NAD2, NAD3, NAD4, NAD4L, NAD5 and NAD6 from 25 fungi: G. zeae (NC_009493), G. moniliformis (NC_016687), F. oxysporum (NC_017930), H. jecorina (NC_NC003388), M. anisopliae (NC_008068), B. bassiana (NC_010652), G. graminicola (CM001021), V. dahliae (NC_008248), N. crassa (NC_001570), P. anserina (NC_001329), P. nodorum (NC_009746), M. graminicola (NC_010222), P. subalpina (NC_015789), T. rubrum (NC_012824), A. obtusum (NC_012830), P. marneffei (NC_005256), E. nidulans (NC_017896), A. niger (NC_007445), A. kawachi (AP012272), C. albicans (NC_002653), O. angusta (NC_014805), P. pastoris (NC_015384), S. sclerotiorum and B. cinera (http://www.broadinstitute.org/).
Figure 3. Mitochondrial gene order of 12 fungi species from 6 orders.
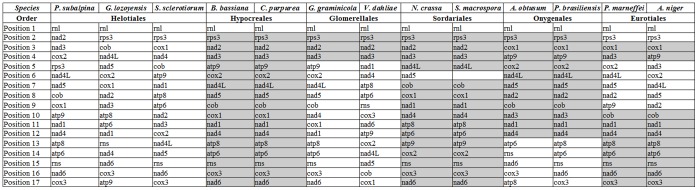
The orders are: Helotiales, Hypocreales, Glomerellales, Sordariales, Onygenales and Eurotiales. The following genes were considered for gene order comparison: atp6, atp8, atp9, cob, cox1, cox2, cox3, nad1, nad2, nad3, nad4, nad4L, nad5, nad6 and rps3. Grey columns indicate coordination in genes arrangement for each order. rnl was defined arbitrarily as the first gene in each set. (P. subalpina: Phialocephala subalpina, G. lozoyensis: Glarea lozoyensis, S. sclerotiorum: Sclerotinia sclerotiorum, B. bassiana: Beauveria bassiana, C. purpurea: Claviceps purpurea, G. graminicola: Glomerella graminicola, V. dahliae: Verticillium dahliae, N. crassa: Neurospora crassa, S. macrospora: Sordaria macrospora, A. obtusum: Arthroderma obtusum, P. brasiliensis: Paracoccidioides brasiliensis, P. marneffei: Penicillium marneffei, A. niger: Aspergillus niger.
Conclusion
We have identified the complete mt genome of G. lozoyensis on a scaffold obtained by whole-genome sequencing. Previous studies based on RAPD, microsatellite-primed PCR and nuclear ribosomal internal transcribed spacers (ITS), suggest that G. lozoyensis belongs to the order Helotiales [5]. Our mt genome analysis clearly confirms that, BLASTp analysis of G. lozoyensis mt protein sequences yielded the corresponding genes from P. subalpina (Helotiales) as closest homologs (identities 70–100%); even though, not to all genes a homolog was found in P. subalpina. In a phylogenetic analysis based on the mt proteins of 24 fungi, G. lozoyensis was clearly classified as Helotiales with P. subalpina as closest relative. However, an anchored genome alignment of the Helotialean species G. lozoyensis, P. subalpina and S. sclerotiorum revealed that there is no synteny between these three apparently closely related species. This clearly demonstrates that species within the large Helotiales order species can be highly diverse. More mt genomes of the order Helotiales are required to find out whether the gaps between the differently ordered mt genomes can be fully closed or gene arrangements are in overall very diverse within this order.
Supporting Information
G. lozoyensis mt genome is colinear with that of P. subalpina . Dotplot of mt genomes based on BLASTn analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi) with an e-value cutoff of 10−10. Sequence lengths are given along the axes in kbp. The shaded cells in the matrix indicate identical residues. a) G. lozoyensis and P. subalpina. b) G. lozoyensis and S. sclerotiorum.
(PDF)
Mauve genome comparison. Multiple alignments of four helotialean species, G. lozoyensis, P. subalpina, S. sclerotiorum and B. cinerea [67] were performed with the Mauve software package [67]. Locally collinear blocks (LCB) of the genome sequences are shown in identical colors and are connected with lines. For the genome of G. lozoyensis the annotation is displayed to allow the assignment of genes to LCBs. a) Alignment of G. lozoyensis and P. subalpina. b) Alignment of G. lozoyensis, P. subalpina and S. sclerotiorum.
(PDF)
Mitochondrial genes annotation in Glarea lozoyensis and comparison to corresponding genes/proteins in Phialocephala subalpine.
(XLS)
Acknowledgments
We sincerely thank BaseClear (Leiden, Netherlands) for sequencing the G. lozoyensis genome, Prof. Dr. Luis Corrochano and Dr. Gerald F. Bills for their generous support.
Funding Statement
L.Y. was supported by an Alexander von Humboldt Research Fellowship. The project was supported partially by the German Research Foundation (DFG). The article processing charge was funded by the open access publication fund of the Albert Ludwigs University Freiburg. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Schwartz RE, Sesin DF, Joshua H, Wilson KE, Kempf AJ, et al. (1992) Pneumocandins from Zalerion arboricola. I. Discovery and isolation. J Antibiot 45: 1853–1866. [DOI] [PubMed] [Google Scholar]
- 2. Masurekar P, Fountoulakis J, Hallada T, Sosa M, Kaplan L (1992) Pneumocandins from Zalerion arboricola. II. Modification of product spectrum by mutation and medium manipulation. The Journal of antibiotics 45: 1867–1874. [DOI] [PubMed] [Google Scholar]
- 3. Keating G, Figgitt D (2003) Caspofungin: a review of its use in oesophageal candidiasis, invasive candidiasis and invasive aspergillosis. Drugs 63: 2235–2263. [DOI] [PubMed] [Google Scholar]
- 4. Bills GF, Platas G, Fernando P, Masurekar P (1999) Reclassification of a pneumocandin-producing anamorph, Glarea lozoyensis gen. et sp. nov., previously identified as Zalerion arboricola. Mycol Res 103: 179–192. [Google Scholar]
- 5. Peláez F, Collado J, Platas G, Overy DP, Martín J, et al. (2011) Phylogeny and intercontinental distribution of the pneumocandin-producing anamorphic fungus Glarea lozoyensis. Mycology 2: 1. [Google Scholar]
- 6. Ballard JW, Whitlock MC (2004) The incomplete natural history of mitochondria. Mol Ecol 13: 729–744. [DOI] [PubMed] [Google Scholar]
- 7. Basse CW (2010) Mitochondrial inheritance in fungi. Curr Opin Microbiol 13: 712–719. [DOI] [PubMed] [Google Scholar]
- 8. Van Diepeningen AD, Goedbloed DJ, Slakhorst SM, Koopmanschap AB, Maas MF, et al. (2010) Mitochondrial recombination increases with age in Podospora anserina. Mech Ageing Dev 131: 315–322. [DOI] [PubMed] [Google Scholar]
- 9. Pantou MP, Kouvelis VN, Typas MA (2006) The complete mitochondrial genome of the vascular wilt fungus Verticillium dahliae: a novel gene order for Verticillium and a diagnostic tool for species identification. Curr Genet 50: 125–136. [DOI] [PubMed] [Google Scholar]
- 10. Kouvelis VN, Sialakouma A, Typas MA (2008) Mitochondrial gene sequences alone or combined with ITS region sequences provide firm molecular criteria for the classification of Lecanicillium species. Mycol Res 112: 829–844. [DOI] [PubMed] [Google Scholar]
- 11. O’Brien EA, Zhang Y, Wang E, Marie V, Badejoko W, et al. (2009) GOBASE: An organelle genome database. Nucleic Acids Res 37: 946–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Korab-Laskowska M, Rioux P, Brossard N, Littlejohn TG, Gray MW, et al. (1998) The Organelle Genome Database Project (GOBASE). Nucleic Acids Res 26: 138–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Duò A, Bruggmann R, Zoller S, Bernt M, Grunig CR (2012) Mitochondrial genome evolution in species belonging to the Phialocephala fortinii s.l. - Acephala applanata species complex. BMC Genomics 13: 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kouvelis VN, Ghikas DV, Typas MA (2004) The analysis of the complete mitochondrial genome of Lecanicillium muscarium (synonym Verticillium lecanii) suggests a minimum common gene organization in mtDNAs of Sordariomycetes: phylogenetic implications. Fungal Genet Biol 41: 930–940. [DOI] [PubMed] [Google Scholar]
- 15. Cardoso MA, Tambor JH, Nobrega FG (2007) The mitochondrial genome from the thermal dimorphic fungus Paracoccidioides brasiliensis. Yeast 24: 607–616. [DOI] [PubMed] [Google Scholar]
- 16. Cummings DJ, McNally KL, Domenico JM, Matsuura ET (1990) The complete DNA sequence of the mitochondrial genome of Podospora anserina. Curr Genet 17: 375–402. [DOI] [PubMed] [Google Scholar]
- 17. Woo PC, Zhen H, Cai JJ, Yu J, Lau SK, et al. (2003) The mitochondrial genome of the thermal dimorphic fungus Penicillium marneffei is more closely related to those of molds than yeasts. FEBS Lett 555: 469–477. [DOI] [PubMed] [Google Scholar]
- 18. van de Sande WWJ (2012) Phylogenetic analysis of the complete mitochondrial genome of Madurella mycetomatis confirms its taxonomic position within the order Sordariales. PLoS ONE 7: 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Paquin B, Laforest MJ, Forget L, Roewer I, Wang Z, et al. (1997) The fungal mitochondrial genome project: evolution of fungal mitochondrial genomes and their gene expression. Curr Genet 31: 380–395. [DOI] [PubMed] [Google Scholar]
- 20. Gray MW (1999) Evolution of organellar genomes. Curr Opin Genet Dev 9: 678–687. [DOI] [PubMed] [Google Scholar]
- 21. Sideris EG, Charalambous SC, Tsolomyty A, Katsaros N (1988) Mutagenesis carcinogenesis and the metal elements-DNA interaction. Prog Clin Biol Res 259: 13–25. [PubMed] [Google Scholar]
- 22. Clark-Walker GD (1992) Evolution of mitochondrial genomes in fungi. Int Rev Cytol 141: 89–127. [DOI] [PubMed] [Google Scholar]
- 23. Bertrand H (2000) Role of mitochondrial DNA in the senescence and hypovirulence of fungi and potential for plant disease control. Annu Rev Phytopathol 38: 397–422. [DOI] [PubMed] [Google Scholar]
- 24. Bullerwell CE, Gray MW (2004) Evolution of the mitochondrial genome: protist connections to animals, fungi and plants. Curr Opin Microbiol 7: 528–534. [DOI] [PubMed] [Google Scholar]
- 25. Bullerwell CE, Forget L, Lang BF (2003) Evolution of monoblepharidalean fungi based on complete mitochondrial genome sequences. Nucleic Acids Res 31: 1614–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Youssar L, Grüning BA, Erxleben A, Günther S, Hüttel W (2012) Genome sequence of the fungus Glarea lozoyensis: the first genome sequence of a species from the Helotiaceae family. Eukaryot Cell 11: 250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Giardine B, Riemer C, Hardison RC, Burhans R, Elnitski L, et al. (2005) “Galaxy: a platform for interactive large-scale genome analysis.”. Genome Research 15: 1451–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Goecks J, Nekrutenko A, Taylor J (2010) Galaxy Team (2010) Galaxy: a comprehensive approach for supporting accessible, reproducible, and transparent computational research in the life sciences. Genome Biol 11: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, et al. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, et al. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Finn RD, Mistry J, Tate J, Coggill P, Heger A, et al. (2010) The Pfam protein families database. Nucleic Acids Research Database 38: 211–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lowe TM, Eddy SR (1997) tRNAscan-SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res 25: 955–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Laslett D, Canback B (2004) ARAGORN, a program to detect tRNA genes and tmRNA genes in nucleotide sequences. Nucleic Acids Res 32: 11–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gautheret D, Lambert A (2001) Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J Mol Biol 313: 1003–1011. [DOI] [PubMed] [Google Scholar]
- 35. Eddy SR, Durbin R (1994) RNA sequence analysis using covariance models. Nucleic Acids Res 22: 2079–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arwen: Laslett D, Canbäck B (2008) ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 24: 172–175. [DOI] [PubMed] [Google Scholar]
- 37. Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, et al. (2007) ClustalW and ClustalX version 2. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- 38. Guindon S, Dufayard JF, Lefort V, Anisimova M, Hordijk W, et al. (2010) New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Systematic Biology 59: 307–21. [DOI] [PubMed] [Google Scholar]
- 39. Wu Y, Yang J, Yang F, Liu T, Leng W (2009) Recent dermatophyte divergence revealed by comparative and phylogenetic analysis of mitochondrial genomes. BMC Genomics 10: 238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang Z, Schwartz S, Wagner L, Miller W (2000) BLASTN (dotplot): “A greedy algorithm for aligning DNA sequences”. J Comput Biol 7: 203–14. [DOI] [PubMed] [Google Scholar]
- 41. Schäfer B (2003) Genetic conservation versus variability in mitochondria: the architecture of the mitochondrial genome in the petite-negative yeast Schizosaccharomyces pombe. Curr Genet 43: 311–326. [DOI] [PubMed] [Google Scholar]
- 42. Quevillon E, Silventoinen V, Pillai S, Harte N, Mulder N, et al. (2005) InterProScan: protein domains identifier. Nucleic Acids Res 33: 116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Lang BF, Laforest MJ, Burger G (2007) Mitochondrial introns: a critical view. Trends Genet 23: 119–125. [DOI] [PubMed] [Google Scholar]
- 44. Burke JM, Belfort M, Cech TR, Davies RW, Schweyen RJ, et al. (1987) Structural conventions for group I introns. Nucleic Acids Res 15: 7217–7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Edgell DR, Chalamcharla VR, Belfort M (2011) Learning to live together: mutualism between self-splicing introns and their hosts. BMC Biol 9: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lambowitz AM, Perlman PS (1990) Involvement of aminoacyl-tRNA synthetases and other proteins in group I and group II intron splicing. Trends Biochem Sci 15: 440–444. [DOI] [PubMed] [Google Scholar]
- 47. Lazowska J, Jacq C, Slonimski PP (1980) Sequence of introns and flanking exons in wild-type and box3 mutants of cytochrome b reveals an interlaced splicing protein coded by an intron. Cell 22: 333–348. [DOI] [PubMed] [Google Scholar]
- 48. Pellenz S, Harington A, Dujon B, Wolf K, Schafer B (2002) Characterization of the I-Spom I endonuclease from fission yeast: insights into the evolution of a group I intron-encoded homing endonuclease. J Mol Evol 55: 302–313. [DOI] [PubMed] [Google Scholar]
- 49. Chevalier BS, Stoddard BL (2001) Homing endonucleases: structural and functional insight into the catalysts of intron/intein mobility. Nucleic Acids Res 29: 3757–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jurica MS, Stoddard BL (1999) Homing endonucleases: structure, function and evolution. Cell Mol Life Sci 55: 1304–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Guhan N, Muniyappa K (2003) Structural and functional characteristics of homing endonucleases. Crit Rev Biochem Mol Biol 38: 199–248. [DOI] [PubMed] [Google Scholar]
- 52. Stoddard BL (2005) Homing endonuclease structure and function. Q Rev Biophys 38: 49–95. [DOI] [PubMed] [Google Scholar]
- 53. Sethuraman J, Majer A, Friedrich NC, Edgell DR, Hausner G (2009) Genes within genes: multiple LAGLIDADG homing endonucleases target the ribosomal protein S3 gene encoded within an rnl group I intron of Ophiostoma and related taxa. Mol Biol Evol 26: 2299–2315. [DOI] [PubMed] [Google Scholar]
- 54. Férandon C, Moukha S, Callac P, Benedetto JP, Castroviejo M, et al. (2010) The Agaricus bisporus cox1 gene: the longest mitochondrial gene and the largest reservoir of mitochondrial group I introns. PLoS One 18: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Goddard MR, Burt A (1999) Recurrent invasion and extinction of a selfish gene. PNAS 96: 13880–13885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fox TD (1987) Natural variation in the genetic code. Ann Rev Genet 21: 67–91. [DOI] [PubMed] [Google Scholar]
- 57. Paquin B, Lang BF (1996) The mitochondrial DNA of Allomyces macrogynus: the complete genomic sequence from an ancestral fungus. J Mol Biol 255: 688–701. [DOI] [PubMed] [Google Scholar]
- 58. Kolesnikova OA, Entelis NS, Mireau H, Fox TD, Martin RP, et al. (2000) Suppression of mutations in mitochondrial DNA by tRNAs imported from the cytoplasm. Science 289: 1931–1933. [DOI] [PubMed] [Google Scholar]
- 59. Burger G, Gray M, Lang B (2003) Mitochondrial genomes: anything goes. Trends Genet 19: 709–716. [DOI] [PubMed] [Google Scholar]
- 60. Ghikas DV, Kouvelis VN, Typas MA (2010) Phylogenetic and biogeographic implications inferred by mitochondrial intergenic region analyses and ITS1–5.8S-ITS2 of the entomopathogenic fungi Beauveria bassiana and B. brongniartii. BMC Microbiol 10: 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pantou MP, Kouvelis VN, Typas MA (2006) The complete mitochondrial genome of the vascular wilt fungus Verticillium dahliae: a novel gene order for Verticillium and a diagnostic tool for species identification. Curr Genet 50: 125–136. [DOI] [PubMed] [Google Scholar]
- 62. Seifert KA, Samson RA, Dewaard JR, Houbraken J, Lévesque CA, et al. (2007) Prospects for fungus identification using CO1 DNA barcodes, with Penicillium as a test case. Proc Natl Acad Sci 104: 3901–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Kouvelis VN, Sialakouma A, Typas MA (2008) Mitochondrial gene sequences alone or combined with ITS region sequences provide firm molecular criteria for the classification of Lecanicillium species. Mycol Res 112: 829–844. [DOI] [PubMed] [Google Scholar]
- 64. Benson DA, Karsch-Mizrachi I, Lipman DJ, Ostell J, Sayers EW (2011) Genbank. Nucleic Acids Res 39: 32–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kirk PM, Cannon PF, David JC, Stalpers JA (2001) Eds: Dictionary of the fungi. 9th edition. Oxon, UK: CAB International. 655p.
- 66. Amselem J, Cuomo CA, van Kan JAL, Viaud M, Benito EP, et al. (2011) Genomic Analysis of the Necrotrophic Fungal Pathogens Sclerotinia sclerotiorum and Botrytis cinerea. PLoS Genet 7: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Darling AE, Mau B, Nicole Perna NT (2010) progressiveMauve: Multiple Genome Alignment with Gene Gain, Loss, and Rearrangement. PLoS One 5: 11147. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
G. lozoyensis mt genome is colinear with that of P. subalpina . Dotplot of mt genomes based on BLASTn analysis (http://blast.ncbi.nlm.nih.gov/Blast.cgi) with an e-value cutoff of 10−10. Sequence lengths are given along the axes in kbp. The shaded cells in the matrix indicate identical residues. a) G. lozoyensis and P. subalpina. b) G. lozoyensis and S. sclerotiorum.
(PDF)
Mauve genome comparison. Multiple alignments of four helotialean species, G. lozoyensis, P. subalpina, S. sclerotiorum and B. cinerea [67] were performed with the Mauve software package [67]. Locally collinear blocks (LCB) of the genome sequences are shown in identical colors and are connected with lines. For the genome of G. lozoyensis the annotation is displayed to allow the assignment of genes to LCBs. a) Alignment of G. lozoyensis and P. subalpina. b) Alignment of G. lozoyensis, P. subalpina and S. sclerotiorum.
(PDF)
Mitochondrial genes annotation in Glarea lozoyensis and comparison to corresponding genes/proteins in Phialocephala subalpine.
(XLS)