

Abstract
Protein-protein interactions (PPIs) are important in all aspects of cellular function and there is interest in finding inhibitors of these contacts. However, PPIs with weak affinities and/or large interfaces have traditionally been more resistant to the discovery of inhibitors, partly because it is more challenging to develop high throughput screening (HTS) methods that permit direct measurements of these physical interactions. Here, we explored whether the functional consequences of a weak PPI might be used as a surrogate for binding. As a model, we used the bacterial ATPase DnaK and its partners DnaJ and GrpE. Both DnaJ and GrpE bind DnaK and catalytically accelerate its ATP cycling, so we used stimulated nucleotide turnover to indirectly report on these PPIs. In pilot screens, we identified compounds that block activation of DnaK by either DnaJ or GrpE. Interestingly, at least one of these molecules blocked binding of DnaK to DnaJ, while another compound disrupted allostery between DnaK and GrpE without altering the physical interaction. These findings suggest that the activity of a reconstituted multi-protein complex might be used in some cases to identify allosteric inhibitors of challenging PPIs.
Keywords: allostery, molecular chaperones, co-chaperone, ATPase assay, Hsp70, Hsp40
Multi-protein complexes are critical to cellular functions.1–4 These complexes are typically assembled from a combination of enzymes and non-enzymes: the enzymes, such as a demethylase, protease or ATPase, often conduct the “work” associated with the system, while the non-enzymes regulate this activity – either by dictating subcellular location, guiding the selection of binding partners, or controlling enzyme turnover rates. Thus, the protein-protein interactions (PPIs) between enzymes and non-enzymes are critical for the overall function of the complexes and inhibitors of these PPIs are important chemical probes.5, 6 More recently, there has also been renewed interest in targeting PPIs in the treatment of disease.7–10
While there has been tremendous progress in the general area of PPI inhibitors, it has become clear that some types of PPIs are more challenging to target than others.11, 12 In particular, PPIs involving weak (KD >200 nM) interactions that occur over large contact surfaces (>2,500 Å2) tend to be more difficult to inhibit.11 One challenge in finding inhibitors of weak interactions is that it is difficult to develop robust, high throughput screening (HTS) methodology to directly measure the physical interactions between transient partners. Accordingly, many groups, including our own, have been interested in exploring new HTS platforms that are specifically designed for use against these types of challenging PPIs.13, 14 These methods, such as fragment-based screens and high content screening (HCS), are promising to open the number of “druggable” PPIs to include even the challenging targets.11, 12
As an attractive model system, we have been studying the Escherichia coli chaperone complex, which is composed of an enzyme (DnaK) and multiple non-enzymes (DnaJ, GrpE, peptide substrate).15 DnaK is a member of the highly conserved heat shock protein 70 kDa (Hsp70) family of molecular chaperones, which are important in protein quality control.16, 17 Like other Hsp70s, DnaK is an ATP-driven enzyme that has a nucleotide-binding domain (NBD) and a substrate-binding domain (SBD) (Fig 1A). ATP is hydrolyzed in the NBD, while the SBD binds to hydrophobic segments of polypeptides, such as those exposed in misfolded proteins.18, 19 Allosteric communication between the two domains modulates the affinity of DnaK for peptides; DnaK binds loosely in the ATP-bound state, while it binds tightly in the ADP-bound form.20, 21 A major role of DnaK's non-enzyme partners, DnaJ and GrpE, is to regulate this ATP cycling. Specifically, DnaJ and peptides stimulate the rate of nucleotide hydrolysis in DnaK,22, 23 while GrpE accelerates release of ADP and peptide.24 Together, the components of the DnaK-DnaJ-GrpE-peptide complex work together to coordinate ATP hydrolysis and regulate dynamic binding to misfolded proteins.
Fig 1.

High throughput screens identify selective inhibitors of individual multi-protein complexes. (A) Schematic of the DnaK-DnaJ-GrpE-substrate system. Nucleotide hydrolysis by DnaK is stimulated by DnaJ and peptide substrate, while GrpE stimulates ADP and peptide substrate release. (B) Results of eight parallel, pilot HTS campaigns. The indicated non-enzyme partner was added at an amount that either saturated steady state ATP hydrolysis or at the half maximal amount (Km, app). Confirmed actives = repeated in triplicate, dose response < 75 μM. Unique actives = compounds found with a specific non-enzyme but not the others. (C) Comparison of the actives from screening 3,880 molecules against the DnaK-DnaJ and DnaK-GrpE combinations in 384-well plates. In these screens, DnaJ was used at Km, app and GrpE at saturation. The chemical structures of representative unique actives are shown.
Each of the components of the DnaK-DnaJ-GrpE-peptide complex is thought to play an important role in chaperone functions in vivo and this system is highly conserved in mammals.15 Thus, inhibitors of the individual PPIs are expected to be powerful chemical probes and these molecules may even find use in the treatment of bacterial infections, cancer and neurodegenerative diseases.25 However, DnaJ and peptides each bind DnaK with weak, micromolar affinities,26, 27 while GrpE binds DnaK over a large and topologically complex surface (~2800 Å2).24 These partners interact with DnaK transiently (e.g. fast on - fast off), acting as catalysts rather than stable binding partners. As evidence of this mechanism, substoichiometric amounts of DnaJ are sufficient to convert DnaK from its ATP to ADP-bound state under single turnover conditions.28 Further, structural studies on DnaK-DnaJ have provided insight into the possible mechanism of this transient interaction, as the protein-protein contact surface is shallow and almost entirely electrostatic,26 suggesting that the two proteins form dynamic complexes that are able to form and dissolve rapidly. In E. coli, the levels of DnaK are approximately 10-fold greater than the concentration of DnaJ or GrpE, suggesting that this weak interaction is physiologically relevant.
As discussed above, it has proven especially challenging to find inhibitors of weak, transient PPIs, such as those between DnaK-DnaJ.5 In this work, we sought to use the enzymatic activities of the reconstituted DnaK-DnaJ, DnaK-GrpE and DnaK-peptide complexes as a possible surrogate for the physical, bi-molecular interactions. We considered this approach potentially feasible because, despite their moderate to weak affinities for DnaK, each of the non-enzyme partners (DnaJ, GrpE and peptide substrates) produce dramatic effects on ATP cycling, enhancing steady state hydrolysis by approximately 10-fold, 2-fold and 3-fold, respectively.23, 29 Thus, even though they bind transiently, these non-enzyme “catalysts” produce potent effects on nucleotide turnover.
In this study we measured phosphate release from eight distinct, reconstituted E. coli DnaK complexes and screened a pilot chemical library for possible inhibitors. Strikingly, we found that both the identity of the non-enzyme (e.g. DnaJ or GrpE) and its stoichiometry relative to DnaK (e.g. maximal or half-maximal) affected the number and types of inhibitors that were identified. At least one of these molecules had the characteristics of a direct inhibitor of the DnaK interaction with DnaJ, while another molecule operated at an allosteric site in DnaK to block stimulation by GrpE. These results suggest that PPI inhibitors with interesting mechanisms-of-action can be identified via screening reconstituted multi-protein complexes in vitro. This approach might contribute to a growing arsenal of HTS methods for finding inhibitors of challenging PPIs.
RESULTS
Design of HTS campaigns to identify inhibitors of the DnaK-DnaJ-GrpE-peptide system
We reasoned that one way to screen for inhibitors of weak PPIs might be to monitor the functional consequences of the interactions (e.g. ATP hydrolysis), rather than measuring the physical binding events themselves. This approach might be particularly well suited for weak contacts, such as the one between DnaK and DnaJ, because these interactions are technically challenging to directly measure using typical, HTS-compatible formats, such as flow cytometry, FP, AlphaLisa or surface plasmon resonance (SPR).30, 31 Yet, the transient PPIs between DnaK and DnaJ provide robust and readily measured changes in enzymatic turnover.22, 23
Accordingly, a series of small-scale pilot screens was performed to better understand the potential feasibility of such an approach. We first expressed and purified E. coli DnaK, DnaJ and GrpE and synthesized a model peptide substrate with the sequence NRLLLTG.32 Using an adaptation of a malachite green assay for detecting release of inorganic phosphate,33 we confirmed that DnaJ, GrpE and the NRLLLTG peptide all stimulated the steady state ATPase activity of DnaK. During these experiments, we also determined the stoichiometry of each partner that was required to maximally and half-maximally promote hydrolysis (Suppl. Fig 1A). For example, DnaJ stimulated the ATPase activity of DnaK (0.4 μM) with a half-maximal concentration of 0.05 μM and reached full stimulation at ~6 μM, values consistent with literature precedent. We then reconstituted DnaK with each of these partners to establish a series of 8 different screening targets (screens A–H; Fig 1B). All of the screens used the same amount of DnaK (0.4 μM) and varied only in the identity and stoichiometry of the non-enzyme. In selecting this series of targets, we focused on exploring the effects of molar ratio by either saturating the levels of the partners (screens A, C and E) or using half-maximal amounts (screens B, D and F) (Fig 1B). We hypothesized that high levels of non-enzyme partner might yield better signal:noise and Z' values, while half-maximal levels might facilitate discovery of PPI inhibitors by decreasing competition between the test molecules and the partner proteins. In addition to the binary complexes, we also assembled ternary complexes of DnaK-DnaJ-peptide (screen G) and DnaK-GrpE-peptide (screen H). Screen G was included because DnaJ and peptide are known to use synergistic allosteric pathways to stimulate ATP hydrolysis,34 while screen H was included because GrpE alone has a relatively modest effect on ATPase activity and we suspected that the signal:noise in the DnaK-GrpE screens (screens C and D) may not be sufficient to achieve good screening characteristics.
Parallel chemical screens yield inhibitors of distinct DnaK complexes
The series of reconstituted targets was screened against a pilot library of ~300 molecules.13 This library was composed of commercially available compounds and was assembled at the University of Michigan's Center for Chemical Genomics. Guided by previous observations,35 we specifically selected a library that is enriched with plant natural products because these molecules are expected to yield relatively high “hit rates” (up to 3 or 4 % in some DnaK screens)35, allowing us to rapidly and cost effectively test the performance of this HTS approach on a relatively small number of compounds. On each plate, 12 wells were assigned to a positive control (e.g. lacking only the enzyme, DnaK) and 12 wells served as negative controls (1% DMSO). Active molecules were defined as those that reduced the signal by at least three standard deviations from the negative controls with intrinsic fluorescence values less than 500 AFUs. Compounds that met these criteria were then subject to dose response in triplicate and were considered “confirmed actives” if they had IC50 values less than 75 μM (Fig 1B). Of the eight screens, only the DnaK-GrpE screen with low GrpE levels (screen D) failed to give a Z' factor greater than the 0.5 cutoff due to a poor signal:noise; thus, it was removed from subsequent analyses.
From the pilot screening results, a number of interesting observations were made. First, seven compounds were identified that were inhibitors of all the reconstituted complexes, regardless of their composition. The broad activity of these molecules suggests that they may be competitive inhibitors of ATP binding in DnaK or that they interfere with the assay (e.g. strong aggregators). More interesting were the compounds that acted on only specific multi-protein complexes, but not others (“unique actives”; Fig 1B and 1C). For example, 10 compounds were identified as active in only the DnaK-DnaJ screen (screen B), but not the screens involving GrpE or peptide (screens C, E and F). Likewise, 4 compounds were inhibitors in the DnaK-GrpE screen (screen C), but not in the screens involving any of the other non-enzymes. These results suggest that the combination of components chosen for the screen may favor discovery of molecules exclusive for that pair. This is an interesting result because the same enzyme, DnaK, is used in all of the parallel pilot screens. We speculate that conformational changes, which occur as a consequence of the individual PPIs, might create new opportunities for inhibitor binding. For example, the ADP-bound form of DnaK is not heavily populated in the absence of DnaJ, because the rate limiting step, ATP hydrolysis rate is slow.36 Thus, molecules that bind the ADP-bound state of DnaK might only become potent when this state becomes significantly populated by the DnaJ-DnaK interac. Another interesting observation was that screens of ternary combinations did not reveal new compounds that were not already found in the relevant binary complexes (Fig 1C), although this result may be limited by the small size of the pilot library.
We also found that “saturating” the amount of non-enzyme (especially DnaJ or peptide) tended to suppress the identification of inhibitors, consistent with the idea that half-maximal levels are more permissive to inhibitor discovery (Fig 1B and Suppl. Fig 1B). For example, dropping the level of DnaJ to its half-maximal concentration (0.05 μM) increased the number of confirmed actives from 11 to 23 (Fig. 1B). This observation is interesting because HTS campaigns, at least in our experience, typically start with the goal of optimizing the signal:noise in order to obtain the best possible screening statistics (e.g. Z' factor, etc). Thus, while this approach is technically sound (and in some cases required), maximizing the signal in a PPI assay may, in some cases, create a disadvantage for the discovery of inhibitors. Together, these studies provided insights into the design principles and implementation strategies for screens against reconstituted multi-protein complexes.
Next, we wanted to explore this HTS concept in studies of larger and more diverse chemical collections. In these studies, we focused on the DnaK-DnaJ (screen B) and DnaK-GrpE (screen C) combinations for re-screening against an expanded collection of ~3880 known bioactive molecules, including the MS2000 and NCC libraries. These compounds were screened at ~50 μM in 384-well plate format using a quinaldine red-based modification of the malachite green assay.37 The Z' factors from these screens were between 0.6 and 0.7 and CV values were between 6 and 9% (Fig 1B). The primary actives were subject to the same triage criteria as in the pilot screens, yielding 31 confirmed hits against DnaK-DnaJ and 18 against DnaK-GrpE. Of these compounds, 10 were common to both DnaJ and GrpE, leaving 21 unique hits for DnaK-DnaJ and 8 for DnaK-GrpE (Fig 1D). The unique inhibitors of DnaK-DnaJ included myricetin (Myr) and zafirlukast (Zaf), which were previously identified as inhibitors of DnaK-DnaJ.14, 35 In addition, these screens revealed a number of additional molecules, including pancuronium bromide (PaBr) and telmisartan (Tel), which appeared as actives in the DnaK-GrpE screen but not the DnaK-DnaJ screen (Fig 1D).
Using re-purchased compounds, we confirmed that Myr and Zaf are only inhibitors of the DnaK-DnaJ combination (Fig 2A), while Tel and PaBr were only inhibitors of the DnaK-GrpE combination (Fig 2B). For example, Zaf inhibited DnaJ-stimulated ATPase activity (IC50 = 37 ± 1.1 μM) but it did not have a pronounced effect on GrpE-stimulation (IC50 > 200 μM). Because PaBr is weakly soluble and the activity of Myr has already been reported,35 we selected Zaf and Tel as test molecules for further characterization. Specifically, we measured the activity of these molecules against each of the possible binary combinations (DnaK-DnaJ, DnaK-GrpE and DnaK-peptide) and against DnaK's intrinsic ATPase activity. In these studies, we varied the levels of each non-enzyme and tested if compounds could interfere with the individual stimulatory activities. These results showed that Zaf is able to suppress the activity of DnaK-DnaJ, but that it had weak or no activity against DnaK-GrpE or DnaK alone (Fig 2C). In contrast, Tel had little activity against DnaK alone or the complexes containing DnaJ or peptide, but it significantly inhibited the DnaK-GrpE combination (Fig 2D).
Fig 2.

Active compounds identified in the binary HTS experiments are selective for either DnaJ- or GrpE-stimulated ATPase activity. (A) Zaf and Myr inhibit ATPase activity of the DnaK-DnaJ complex. Results are the representative averages of triplicate of three independent experiments. Error bars represent SEM. (B) Tel and PaBr inhibit the DnaK-GrpE complex. (C and D) The ATPase activity of either DnaK alone or DnaK stimulated by DnaJ, GrpE or peptide substrate (NRLLLTG) was measured at three concentrations of Zaf (C) or Tel (D). Zaf has activity against the DnaK-DnaJ and DnaK-substrate combinations, with weak activity against the DnaK alone or DnaK-GrpE combinations. Conversely, Tel inhibited the DnaK-GrpE pair, but had weak activity against the others. All experiments are the representative averages of triplicate of three independent experiments, and the error bars represent SEM. Curves were fit to the Michaelis-Menten equation.
It is worth noting that these HTS “hits” are unlikely to be selective for the DnaK system in cells. In fact, both Zaf and Tel are already FDA-approved drugs with previously known targets: Zaf is a leukotriene receptor antagonist used in the treatment of asthma,38 while Tel is an angiotensin II receptor antagonist and selective modulator of peroxisome proliferator-activated receptor gamma (PPAR-γ).39 Although we did not consider Zaf or Tel to be particularly strong leads for further development, we wanted to explore their mechanisms-of-action to begin defining the general ways that compounds might interfere with the functions of the DnaK multi-protein systems. Specifically, we were interested in whether these molecules might directly compete with non-enzyme partners for binding to DnaK (“orthosteric” inhibitors) or whether they might impact the communication between DnaK and the non-enzymes without disrupting the PPI itself (e.g. by binding to an important allosteric site).
Zafirlukast preferentially binds ADP-DnaK and enhances DnaK's affinity for substrate
To explore the mechanism by which Zaf inhibits the DnaK-DnaJ combination, we first tested whether it interacted with DnaK using intrinsic tryptophan fluorescence. DnaK has a single tryptophan located at the NBD-SBD interface (Fig 3A), and this residue is commonly used to probe structural changes in DnaK.40 When DnaK (5 μM) was incubated with 25 μM or 100 μM of Zaf, the fluorescence intensity at 342 nm decreased by ~25% and the peak shifted by ~ 2 nm (Suppl. Fig 2A), suggesting that Zaf binds to DnaK. Using this approach, dose-dependent changes in tryptophan fluorescence were measured and, interestingly, we found that the apparent affinity (KD) was dependent on nucleotide: Zaf bound DnaK with a KD of 52 ± 12 μM in the presence of ADP (Fig 3A) but its KD was greater than 100 μM for ATP-bound DnaK (Suppl. Fig 2B). The ADP-bound form of DnaK is known to have a better affinity for peptide substrates.19 Thus, to test whether Zaf could stabilize the “tight binding” form of DnaK, we measured the affinity of DnaK for a fluorescent 10-mer peptide derived from the MHC class I antigen HLA-B2702 (FITC-HLA). We first confirmed that FITC-HLA binds to DnaK with low micromolar affinity using a fluorescence polarization (FP) assay (Suppl. Fig 2D). This affinity is similar to what had been previously found for binding of FITC-HLA to human Hsp70.41 Addition of Zaf enhanced the apparent affinity of DnaK for FITC-HLA (Fig 3B), suggesting that it stabilizes the tight-binding form of DnaK. To better understand the relationship between Zaf and nucleotide binding, we performed additional FP studies with a fluorescent nucleotide analog (FAM-ATP). In this assay, Zaf was unable to compete with FAM-ATP for binding to DnaK (Suppl. Fig 2C), suggesting that it binds outside the ATP-binding cleft to stabilize the ADP-bound state. Finally, we tested whether Zaf might block binding of labeled DnaJ to DnaK, using a fluorescence-quenching assay.35 In this platform, Zaf slightly weakened binding of DnaJ to DnaK by ~1.8 fold (Fig 3C). Although this effect did not reach statistical significance it was nonetheless reproducible, suggesting that Zaf might partially block this PPI. Together, these studies suggest that Zaf binds the ADP-bound form of DnaK, stabilizes binding to peptides and partially inhibits physical interactions with DnaJ.
Fig 3.
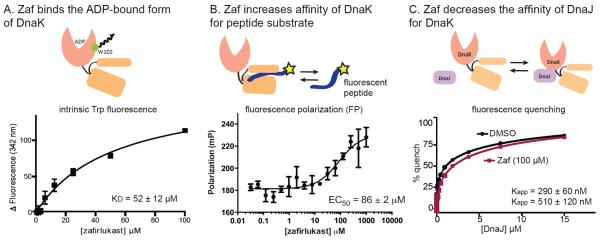
Zaf binds the ADP-bound form of DnaK and enhances the apparent affinity of DnaK for substrates. (A) Intrinsic Trp fluorescence of DnaK in the presence of ADP (1 mM) and Zaf. Zaf had no effect on Trp fluorescence in the ATP- bound states (p = 0.06) (Suppl. Fig 2). Results are the representative averages of triplicate of three independent experiments, and the error bars represent SEM. (B) Zaf enhances the apparent affinity of DnaK for a model peptide substrate (FITC-HLA), as measured by fluorescence polarization (FP). (C) Zaf partially inhibits binding of DnaJ to DnaK. DnaK and DnaJ were labeled with a fluorescence quench pair, as described in the Materials and Methods. Zaf weakened the interaction by ~2-fold (p = 0.07). Binding curves were fit to the Langmuir binding equation; dose response curves were fit to the Hill equation.
Telmisartan interacts with the IB subdomain of DnaK and allosterically inhibits nucleotide affinity
To elucidate the mechanism of Tel inhibition, we first tested whether the molecule might bind to DnaK using the tryptophan fluorescence assay described above. Unfortunately, Tel interfered with the Trp fluorescence signature, preventing interpretation of those studies (data not shown). However, a recent mutagenesis study suggested a pocket in DnaK that might be involved in Tel-mediated inhibition of GrpE function.42 Specifically, it was recently found that mutations in the IB and IIB subdomains of DnaK, including Phe67, Arg71, Phe91 and Lys263, suppresses the ability of GrpE to stimulate DnaK's ATPase activity. Because the behavior of these mutants was similar to what was seen with Tel addition, we hypothesized that the compound might also bind in this region. To test this model, we used induced fit docking to generate a model of Tel bound to the putative binding pocket in the NBD of DnaK (see the Materials and Methods). This simulation suggested that Tel might bind between the IB and IIB subdomains and, in the two best energy orientations, Tel was predicted to make hydrophobic contacts with a series of residues (Fig 4A). To test this prediction, we titrated Tel into a sample of 15N DnaKNBD (residues 1–388) and performed the TROSY-HSQC NMR experiment (Fig 4B). Analysis of the results suggested a number of strong (two standard deviations, 2σ) and intermediate (at least one standard deviation, 1σ) chemical shift perturbations. Mapping these chemical shifts onto the DnaKNBD crystal structure (1DKG) supported the idea that Tel binds to the 1B subdomain of the chaperone (Fig 4B and Suppl. Fig 3). Residues with the largest change in chemical shift were found in the site predicted by computational docking to bind Tel. Additional residues were clustered in surface-exposed regions of the NBD, which could arise from allosteric interactions. We did not observe any binding of Tel to either DnaJ or GrpE by isothermal calorimetry (ITC) (KD > 100 μM) (Suppl. Fig 4A, B). Together with the NMR data, the results confirm that Tel binds to DnaK in the NBD, but not to either of the co-chaperones.
Fig 4.

Tel binds subdomain IB in DnaK to allosterically block GrpE activity. (A) Results of docking Tel to the IB subdomain, showing the two lowest energy conformations (see the Materials and Methods for details). GrpE is removed from the structure (PDB 1DKG) for clarity. (B) Titration of Tel into the nucleotide-binding domain of DnaK (15N DnaKNBD) provided NMR chemical shifts that support the binding of Tel to DnaK. Red = > 2σ shift; Yellow > 1σ shift; Green < 1 s shift; Gray = unassigned or overlapped (see Suppl. Fig. 3). (C) Mutation of residues in the predicted docking site supports the proposed Tel-binding site. Tel inhibits GrpE stimulation of wild type and a control mutant (D233A), but mutations near the proposed binding site (R56A and M89A) were resistant. Results of the ATPase assays are the average of triplicates and error bars represent SEM. (D) Overlay of Tel-sensitive residues on the co-crystal structure of DnaK's NBD in complex with GrpE. Red = mutations that block GrpE stimulation. Blue = mutations that block Tel activity. Yellow = residues predicted to bind Tel by docking.
To further explore the binding site suggested by the docking and NMR studies, we mutated some of the nearby residues (Arg56 and Met89) in the pocket and an unrelated residue, Asp23342 and measured the ability of Tel to block GrpE-stimulation of these mutants using ATPase assays. These studies showed that both R56A and M89A were resistant to Tel, while the control mutant (D233A) was identical to wild type (~1.5-fold increase in Km) (Fig 4C). Together, these results suggest that Tel might bind in a pocket between the IB and IIB subdomains of DnaK. Interestingly, this predicted binding site does not overlap with the surface of DnaK that is normally bound to GrpE24 (Fig 4D), suggesting that Tel acts through an allosteric mechanism. In fact, Tel had no effect on the physical interaction between DnaK and GrpE, as measured by the fluorescence-quenching assay (Suppl. Fig 4C). Together, these data suggest that Tel may interrupt allosteric conformational changes that occur in DnaK upon binding of GrpE, without blocking their physical interaction.
Although both Tel and Zaf were identified as inhibitors of DnaK's ATPase activity in the primary HTS experiments, the subsequent mechanistic studies showed that they had very different mechanisms. For example, while Zaf dramatically enhanced binding of DnaK to FITC-HLA in the FP assay and had no effect on nucleotide affinity, Tel had no effect on FITC-HLA binding (Suppl. Fig 4D) and it interfered with binding to FAM-ATP (Suppl. Fig 4E). Thus, although both Tel and Zaf might be considered “inhibitors” of DnaK, they have distinct mechanisms and they target different co-chaperone activities.
DISCUSSION
Chemical screens yield molecules with distinct inhibitory mechanisms
There is growing interest in targeting PPIs and an emerging realization that not all PPIs are equally amenable to HTS-based methods. We performed pilot screens using eight different combinations of DnaK with its various non-enzyme partners to explore whether stimulated enzymatic activity might be used as a surrogate for transient or challenging PPIs. An interesting observation from the pilot screens was that changing the identity of the non-enzyme component (e.g. switching DnaJ for GrpE) allowed discovery of “unique actives” (e.g. those some compounds that inhibit one combination and not others). On first glance, this finding is counterintuitive, because the same enzyme, DnaK, was used in all of the screens. Why might changing the identity of the non-enzyme favor discovery of unique actives? It would seem unlikely that these compounds could be competitive with nucleotide, because such molecules would be expected to be inhibitors of all the combinations. Rather, our limited follow-up studies on Zaf and Tel (see Figs 3 and 4) suggest that the molecules identified using this HTS approach may be more likely to disrupt specific PPIs or PPI-induced conformational changes. Another theoretical way that unique actives might emerge from these types of screens is through the action of the compounds on the non-enzyme (e.g. DnaJ or GrpE) itself. It is important to note that we cannot fully discount the possibility that Tel or Zaf might weakly bind to GrpE or DnaJ, although we were unable to measure such an interaction. However, it seems logical that such mechanisms will be identified in screens of larger chemical collections.
Following the pilot screens, we examined ~3,800 compounds for their ability to inhibit ATPase activity of either the DnaK-DnaJ or DnaK-GrpE complexes. These studies confirmed the results of the pilot screens and led to the identification of a number of molecules that targeted one complex without influencing the other. To understand what types of mechanisms these molecules might have, we explored the activity of Zaf and Tel in a series of secondary assays. These assays were designed to reveal effects on PPIs and the biochemical activities of the DnaK systems. Interestingly, we found that Zaf only inhibited the ATPase activity of the DnaK-DnaJ combination and that it weakened the physical interaction between DnaK and DnaJ (see Fig 3). Also, this molecule bound the ADP-bound form of DnaK and stabilized substrate-DnaK complexes. Based on these findings, a likely mechanism is that DnaJ first promotes ATP hydrolysis in DnaK, followed by binding of Zaf to ADP-DnaK, which traps this nucleotide state. This “dead-end” complex appears to have a weak ability to re-bind to DnaJ, but a strong ability to remain bound to peptide substrates. It is known that DnaJ binds poorly to DnaK in the ADP-bound form.19 Thus, the effects of Zaf on the DnaK-DnaJ interaction are likely due to trapping of the “dead-end” ADP-bound complex. Interestingly, stabilization of the ADP-bound form of Hsp70s reduces accumulation of proteotoxic proteins in cellular and animal models of neurodegenerative disease,43, 44 so this step in the ATPase cycle appears to be especially important in protein quality control. Molecules with a mechanism-of-action (MoA) similar to Zaf might be useful in those settings and, more importantly, this HTS approach might be a good platform for identifying compounds with this MoA.
In contrast to Zaf, Tel was identified as an inhibitor of the DnaK-GrpE combination, with little effect on the DnaJ-DnaK or other combinations. Interestingly, Tel appeared to block GrpE activity without impacting the physical interaction between these partners. Rather, NMR, mutagenesis and modeling results suggest that Tel might bind between the IB and IIB subdomains, on the opposite face of DnaK than the one involved in GrpE binding (see Fig 4). How might binding in this region impact GrpE function without impacting its affinity for DnaK? GrpE normally rotates the IIB subdomain relative to IB and opens the nucleotide-binding cleft.45 Thus, one possibility is that Tel might interfere with the conformational transitions needed to couple GrpE binding with its effects on ADP release, perhaps by limiting mobility of the IIB subdomain. Tel also had a mild (2-fold) effect on FAM-ATP binding (see Suppl. Fig 3), but it isn't yet clear how this reduced nucleotide affinity might relate to its inhibition of GrpE stimulation.
CONCLUSIONS
A growing number of studies have reported potent inhibitors of PPIs, including both small molecules and protein mimics that either directly7 or allosterically46 inhibit the formation of protein complexes. These molecules have great promise as chemical probes for better understanding the biology and “druggability” of multi-protein complexes. Against this backdrop, the studies described here provide an HTS approach that appears to be particularly well suited for finding orthosteric and/or allosteric inhibitors of challenging PPIs, especially those in which the interaction produces a measurable change in enzyme turnover rates.
METHODS
Reagents and general methods
Myricetin (Myr) was purchased from Sigma (St. Louis, MO), zafirlukast (Zaf) from Cayman Chemical (Ann Arbor, MI), pancuronium bromide (PaBr) from Santa Cruz Biotechnology (Santa Cruz, CA), and telmisartan (Tel) from AK Scientific (Union City, CA). The identities and purities (> 90%) of all compounds were confirmed by NMR and mass spectrometry. Alexa Fluor 488 was purchased from Invitrogen, and BHQ-10 carboxylic acid was obtained from Biosearch Technologies. All other biological reagents were obtained from Sigma (St. Louis, MO) unless otherwise noted. All spectroscopic measurements were obtained using a SpectraMax M5 microplate reader (Molecular Devices).
Peptide synthesis
The peptide FITC-HLA (RENLRIARLY) was synthesized on Wang resin using microwave-assisted DIC/HOBt solid-phase peptide synthesis. It was capped with two β-alanine residues and labeled on resin via the N-terminus with fluorescein-5-isothiocyanate (Anaspec). Crude TFA-cleaved peptide (> 90% purity) was extracted with ether and stored in DMSO as a concentrated stock at −20 °C until use. The NR peptide (NRLLLTG) was synthesized on Wang resin, cleaved with TFA, precipitated with ether, and purified with reverse-phase HPLC using 0.1% TFA/CH3CN solvent system (>95% purity). The masses of the peptides were verified using electrospray ionization mass spectrometry.
Protein expression and purification
DnaK, DnaJ, and GrpE were expressed and purified as previously described,14 using a His column and subsequent cleavage of the His tag by TEV protease. DnaK was further purified using an ATP column, while both DnaJ and GrpE were subjected to final purification on a Superdex 200 gel filtration column (GE Healthcare). All proteins were concentrated and stored in 25 mM HEPES buffer containing 10 mM KCl and 5 mM MgCl2 (pH 7.5) until use. Protein purities were estimated at greater than 90% by SDS-2 PAGE. The BCA (bicinchoninic acid) assay kit (Thermo Fisher Scientific Inc) was used to measure total protein concentration and the activity of the purified proteins was verified with the described ATPase assays.
High-throughput screening
The high-throughput screening methodology was developed following previously published protocols.13, 14 The libraries used were a natural product library,35 the NCC collection of ~500 bioactive molecules and the MicroSource MS2000 library containing ~2000 bioactives. The quinaldine red (QR) reagent was prepared fresh for each experiment by mixing stock solutions of 0.05% QR, 2% polyvinyl alcohol, 6% ammonium heptamolybdate tetrahydrate in 6 M HCl, and water in a 2:1:1:2 ratio. DnaK at 0.4 μM and the indicated concentrations of co-chaperones (DnaJ, GrpE, or NRLLLTG) were diluted into assay buffer (100 mM Tris-HCl, 20 mM KCl, 6 mM MgCl2, 0.01% Triton-X, pH 7.4), and 5 μL of this solution was added to each well of a white, opaque, low-volume 384-well plate (Greiner Bio-One). To this solution, 200 nL of compound stocks (2 μM) or DMSO was added to each well for a final screening concentration of ~55 μM. ATP (1 mM) was added to begin the reaction, followed by incubation for 3 hours at 37 °C. QR reagent (15 μL) was added and the reaction was quenched with 2 μL of 32% sodium citrate after 2 minutes. Following incubation at 37 °C for 15 minutes, the fluorescence intensity (excitation 430 nm, emission 530 nm) was measured on a PHERAstar plate reader. Standard curves were obtained using stock solutions of dibasic potassium phosphate. Z' scores were calculated using no DnaK solutions as the positive control (100% “inhibited”) and DMSO-treated samples as the negative control (0% inhibited).
ATPase assays
ATPase assays were performed as described.13, 14 Stock solutions of DnaK, DnaJ, or GrpE were made in assay buffer (100 mM Tris-HCl, 20 mM KCl, 6 mM MgCl2, pH 7.4). Unless otherwise noted, the DnaK concentration was 0.6 μM, while DnaJ and GrpE concentrations are indicated. If applicable, stock solutions of compound were made in DMSO, then diluted into 15 μL of assay buffer and protein in clear, flat-bottom 96-well plates (Thermo Fisher Scientific Inc) to the final concentrations noted. Absorbance was measured at 620 nm. Data were fit to the Michaelis-Menten equation (Y = VmaxX/[Km + X]) in GraphPad PRISM.
Tryptophan fluorescence
Tryptophan fluorescence was measured as previously described.13, 14 DnaK was diluted to 5 μM in storage buffer containing 1 mM of nucleotide and Zaf at the indicated concentrations, with a total volume of 25 μL in black, flat-bottom 96-well plates (Costar). The mixture was incubated for 30 minutes at 37 °C in the dark, and either the emission spectrum between 300 and 450 nm or emission at 340 nm (excitation 290 nm) was measured. Binding data were fit to a form of the Langmuir isotherm (Y = BmaxX/[Kd + X]).
Fluorescence quenching
DnaK and DnaJ/GrpE were labeled with Alexa Fluor 488 and BHQ-10 carboxylic acid, respectively, and their binding affinity was measured by FRET as previously described.13, 14 Briefly, compound was diluted to the final indicated assay concentration from a concentrated DMSO stock into 20 μL of assay buffer (50 mM HEPES, 75 mM NaCl, pH 7.2) containing 50 nM labeled DnaK, labeled DnaJ at the noted concentrations, and 1 mM ATP. Following incubation for 1 hour at 37 °C, fluorescence at 525 nm (excitation 480 nm, cutoff 515 nm) was measured. The compounds did not affect either the fluorescence of Alexa-labeled DnaK or the absorbance spectra of BHQ-10 labeled partner. Binding data were fit to the Langmuir isotherm, as described above. Statistical significance was determined using an unpaired t-test.
Fluorescence polarization
Binding of fluorescent peptide (FITC-HLA) to DnaK was carried out using the method of Ricci and Williams,41 with minor modifications. In a black, round-bottom, low-volume 384-well plate (Corning), 5 μM DnaK and 1 mM ATP in 10 μL of assay buffer (25 mM HEPES, 150 mM KCl, pH 7.2) were incubated with the indicated compound concentrations or a solvent control for 30 minutes at room temperature. A stock of FITC-HLA was diluted to 25 nM into each well, for a total volume of 20 μL. The plate was incubated in the dark for 10 minutes at room temperature before reading fluorescence polarization (excitation 494 nm, emission 519 nm). The dose response data was fit to the Hill equation (Y = Emax/[1 + (EC50/X)nH]), providing EC50 values.
We also measured binding of a fluorescent ATP analog, N6-(6-Amino)hexyl-ATP-5-FAM (FAM-ATP) (Axxora LLC), to DnaK using a fluorescence polarization binding assay. An aqueous stock of FAM-ATP was diluted to 20 nM in assay buffer (100 mM Tris-HCl, 20 mM KCl, 6 mM MgCl2, pH 7.4) and titrated with DnaK in a black, round-bottom, low-volume, 384-well plate (Corning) in a total volume of 20 μL. The plate was incubated in the dark for 10 minutes at room temperature before reading fluorescence polarization (excitation 485 nm, emission 535 nm). Binding data were fit to the Langmuir equation as described above. Competition data were fit to the Hill equation.
Docking
We used AutoDock 4 to simulate binding of Tel to DnaKNBD. First, GrpE was removed from the crystal structure (PDB: 1DKG). For the computations, we used published parameters.47 The grid box was located between the IB and IIB subdomains, near the top of the nucleotide-binding cleft, with 0.2Å resolution. Docked conformations were evaluated using PyMOL. The calculations were performed on an Apple MacBook5.1 running Mac OS X 10.6.8.
Isothermal titration calorimetry
Isothermal calorimetric titrations were performed on a VP-ITC MicroCalorimeter (MicroCal, Inc.) at 25 °C. DnaJ or GrpE were diluted into buffer containing 25 mM HEPES (pH 7.5), 5 mM MgCl2, 10 mM KCl, 5 mM TCEP, and 0.5% DMSO to a final concentration of 10 μM. Protein samples were extensively dialyzed and added into the calorimetric cell (cell volume = 1.43 mL). DnaJ and GrpE were individually titrated with 100 μM Tel in 30 × 10 μL increments. Injections were performed at 2 μL/s. Data were analyzed using Microcal Origin (v2.9).
NMR
Binding of Tel to DnaKNBD was measured by 2D HSQC-TROSY NMR on a Varian/Agilent 800 MHz NMR system, using methods that were previously described.47 Briefly, small aliquots of compound solution (100 mM in DMSO) were added to 15N-labeled DnaKNBD(1-388) (100 μM) in NMR buffer (25 mM Tris, 10 mM MgCl2, 5 mM KCl, 10% 2H2O, 0.01% sodium azide, pH 7.1, 5 mM ADP, 10 mM potassium phosphate). Identical aliquots of DMSO without compound were added to the protein sample in NMR control experiments. Residues were selected as significantly affected if the compound-induced chemical shift, correcting for the shift with DMSO alone, were above one standard deviation (1σ; see Suppl. Fig. 3).
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants from the NIH (NS059690) and NSF (MCB-0844512) and a gift from the Michigan Alzheimer's Disease Center (MADC). The authors acknowledge the expert assistance of L. Chang, S. Vander Roest and T. McQuade.
Footnotes
Supporting Information. Additional supplemental figures are available is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interests.
REFERENCES
- (1).Alber F, Dokudovskaya S, Veenhoff LM, Zhang W, Kipper J, Devos D, Suprapto A, Karni-Schmidt O, Williams R, Chait BT, Rout MP, Sali A. Determining the architectures of macromolecular assemblies. Nature. 2007;450:683–694. doi: 10.1038/nature06404. [DOI] [PubMed] [Google Scholar]
- (2).Chari A, Fischer U. Cellular strategies for the assembly of molecular machines. Trends in biochemical sciences. 2010;35:676–683. doi: 10.1016/j.tibs.2010.07.006. [DOI] [PubMed] [Google Scholar]
- (3).Peterson-Kaufman KJ, Carlson CD, Rodriguez-Martinez JA, Ansari AZ. Nucleating the assembly of macromolecular complexes. Chembiochem. 2010;11:1955–1962. doi: 10.1002/cbic.201000255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Good MC, Zalatan JG, Lim WA. Scaffold proteins: Hubs for controlling the flow of cellular information. Science. 2011;332:680–686. doi: 10.1126/science.1198701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Thompson AD, Dugan A, Gestwicki JE, Mapp AK. Fine-tuning multiprotein complexes using small molecules. ACS Chem Biol. 2012;7:1311–1320. doi: 10.1021/cb300255p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Gordo S, Giralt E. Knitting and untying the protein network: Modulation of protein ensembles as a therapeutic strategy. Protein Sci. 2009;18:481–493. doi: 10.1002/pro.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wells JA, McClendon CL. Reaching for high-hanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- (8).Jubb H, Higueruelo AP, Winter A, Blundell TL. Structural biology and drug discovery for protein-protein interactions. Trends Pharm Sci. 2012;33:241–248. doi: 10.1016/j.tips.2012.03.006. [DOI] [PubMed] [Google Scholar]
- (9).Meireles LM, Mustata G. Discovery of modulators of protein-protein interactions: Current approaches and limitations. Curr Top Med Chem. 2010 doi: 10.2174/156802611794072632. [DOI] [PubMed] [Google Scholar]
- (10).Arkin MR, Whitty A. The road less traveled: Modulating signal transduction enzymes by inhibiting their protein-protein interactions. Curr Opin Chem Biol. 2009;13:284–290. doi: 10.1016/j.cbpa.2009.05.125. [DOI] [PubMed] [Google Scholar]
- (11).Smith MC, Gestwicki JE. Features of protein-protein interactions that translate into potent inhibitors: Topology, surface area and affinity. Expert Rev Molec Med. 2012;14:e16. doi: 10.1017/erm.2012.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Jochim AL, Arora PS. Assessment of helical interfaces in protein-protein interactions. Molec BioSystems. 2009;5:924–926. doi: 10.1039/b903202a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Chang L, Bertelsen EB, Wisén S, Larsen EM, Zuiderweg ER, Gestwicki JE. High-throughput screen for small molecules that modulate the atpase activity of the molecular chaperone dnak. Anal Biochem. 2008;372:167–176. doi: 10.1016/j.ab.2007.08.020. [DOI] [PubMed] [Google Scholar]
- (14).Miyata Y, Chang L, Bainor A, McQuade TJ, Walczak CP, Zhang Y, Larsen MJ, Kirchhoff P, Gestwicki JE. High-throughput screen for escherichia coli heat shock protein 70 (hsp70/dnak): ATPase assay in low volume by exploiting energy transfer. J Biomol Screen. 2010;15:1211–1219. doi: 10.1177/1087057110380571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lindquist S, Craig EA. The heat-shock proteins. Annu Rev Genet. 1988;22:631–677. doi: 10.1146/annurev.ge.22.120188.003215. [DOI] [PubMed] [Google Scholar]
- (16).Daugaard M, Rohde M, Jaattela M. The heat shock protein 70 family: Highly homologous proteins with overlapping and distinct functions. FEBS Lett. 2007;581:3702–3710. doi: 10.1016/j.febslet.2007.05.039. [DOI] [PubMed] [Google Scholar]
- (17).Mayer MP, Bukau B. Hsp70 chaperones: Cellular functions and molecular mechanism. Cell Mol Life Sci. 2005;62:670–684. doi: 10.1007/s00018-004-4464-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Rudiger S, Buchberger A, Bukau B. Interaction of hsp70 chaperones with substrates. Nature Struct Biol. 1997;4:342–349. doi: 10.1038/nsb0597-342. [DOI] [PubMed] [Google Scholar]
- (19).Mayer MP, Schroder H, Rudiger S, Paal K, Laufen T, Bukau B. Multistep mechanism of substrate binding determines chaperone activity of hsp70. Nat Struct Biol. 2000;7:586–593. doi: 10.1038/76819. [DOI] [PubMed] [Google Scholar]
- (20).Zuiderweg ER, Bertelsen EB, Rousaki A, Mayer MP, Gestwicki JE, Ahmad A. Allostery in the hsp70 chaperone proteins. Topics Curr Chem. 2012 doi: 10.1007/128_2012_323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Swain JF, Dinler G, Sivendran R, Montgomery DL, Stotz M, Gierasch LM. Hsp70 chaperone ligands control domain association via an allosteric mechanism mediated by the interdomain linker. Mol Cell. 2007;26:27–39. doi: 10.1016/j.molcel.2007.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Gassler CS, Buchberger A, Laufen T, Mayer MP, Schroder H, Valencia A, Bukau B. Mutations in the dnak chaperone affecting interaction with the dnaj cochaperone. Proc Natl Acad Sci U S A. 1998;95:15229–15234. doi: 10.1073/pnas.95.26.15229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Wittung-Stafshede P, Guidry J, Horne BE, Landry SJ. The j-domain of hsp40 couples atp hydrolysis to substrate capture in hsp70. Biochemistry. 2003;42:4937–4944. doi: 10.1021/bi027333o. [DOI] [PubMed] [Google Scholar]
- (24).Harrison CJ, Hayer-Hartl M, Di Liberto M, Hartl F, Kuriyan J. Crystal structure of the nucleotide exchange factor grpe bound to the atpase domain of the molecular chaperone dnak. Science. 1997;276:431–435. doi: 10.1126/science.276.5311.431. [DOI] [PubMed] [Google Scholar]
- (25).Patury S, Miyata Y, Gestwicki JE. Pharmacological targeting of the hsp70 chaperone. Curr Top Med Chem. 2009;9:1337–1351. doi: 10.2174/156802609789895674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ahmad A, Bhattacharya A, McDonald RA, Cordes M, Ellington B, Bertelsen EB, Zuiderweg ER. Heat shock protein 70 kda chaperone/dnaj cochaperone complex employs an unusual dynamic interface. Proc Natl Acad Sci USA. 2011;108:18966–18971. doi: 10.1073/pnas.1111220108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Slepenkov SV, Witt SN. Kinetic analysis of interdomain coupling in a lidless variant of the molecular chaperone dnak: Dnak's lid inhibits transition to the low affinity state. Biochemistry. 2002;41:12224–12235. doi: 10.1021/bi0263208. [DOI] [PubMed] [Google Scholar]
- (28).Pierpaoli EV, Sandmeier E, Schonfeld HJ, Christen P. Control of the dnak chaperone cycle by substoichiometric concentrations of the co-chaperones dnaj and grpe. J Biol Chem. 1998;273:6643–6649. doi: 10.1074/jbc.273.12.6643. [DOI] [PubMed] [Google Scholar]
- (29).Packschies L, Theyssen H, Buchberger A, Bukau B, Goody RS, Reinstein J. Grpe accelerates nucleotide exchange of the molecular chaperone dnak with an associative displacement mechanism. Biochemistry. 1997;36:3417–3422. doi: 10.1021/bi962835l. [DOI] [PubMed] [Google Scholar]
- (30).Makley LN, Gestwicki JE. Expanding the number of 'druggable' targets: Non-enzymes and protein-protein interactions. Chem Biol & Drug Des. 2013;81:22–32. doi: 10.1111/cbdd.12066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Berggard T, Linse S, James P. Methods for the detection and analysis of protein-protein interactions. Proteomics. 2007;7:2833–2842. doi: 10.1002/pmic.200700131. [DOI] [PubMed] [Google Scholar]
- (32).Pellecchia M, Montgomery DL, Stevens SY, Vander Kooi CW, Feng HP, Gierasch LM, Zuiderweg ER. Structural insights into substrate binding by the molecular chaperone dnak. Nat Struct Biol. 2000;7:298–303. doi: 10.1038/74062. [DOI] [PubMed] [Google Scholar]
- (33).Geladopoulos TP, Sotiroudis TG, Evangelopoulos AE. A malachite green colorimetric assay for protein phosphatase activity. Anal Biochem. 1991;192:112–116. doi: 10.1016/0003-2697(91)90194-x. [DOI] [PubMed] [Google Scholar]
- (34).Laufen T, Mayer MP, Beisel C, Klostermeier D, Mogk A, Reinstein J, Bukau B. Mechanism of regulation of hsp70 chaperones by dnaj cochaperones. Proc Natl Acad Sci U S A. 1999;96:5452–5457. doi: 10.1073/pnas.96.10.5452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Chang L, Miyata Y, Ung PM, Bertelsen EB, McQuade TJ, Carlson HA, Zuiderweg ER, Gestwicki JE. Chemical screens against a reconstituted multiprotein complex: Myricetin blocks dnaj regulation of dnak through an allosteric mechanism. Chem Biol. 2011;18:210–221. doi: 10.1016/j.chembiol.2010.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).McCarty JS, Buchberger A, Reinstein J, Bukau B. The role of atp in the functional cycle of the dnak chaperone system. J Mol Biol. 1995;249:126–137. doi: 10.1006/jmbi.1995.0284. [DOI] [PubMed] [Google Scholar]
- (37).Zuck P, O'Donnell GT, Cassaday J, Chase P, Hodder P, Strulovici B, Ferrer M. Miniaturization of absorbance assays using the fluorescent properties of white microplates. Anal Biochem. 2005;342:254–259. doi: 10.1016/j.ab.2005.04.029. [DOI] [PubMed] [Google Scholar]
- (38).Scow DT, Luttermoser GK, Dickerson KS. Leukotriene inhibitors in the treatment of allergy and asthma. Amer Family Phys. 2007;75:65–70. [PubMed] [Google Scholar]
- (39).Benson SC, Pershadsingh HA, Ho CI, Chittiboyina A, Desai P, Pravenec M, Qi N, Wang J, Avery MA, Kurtz TW. Identification of telmisartan as a unique angiotensin ii receptor antagonist with selective ppargamma-modulating activity. Hypertension. 2004;43:993–1002. doi: 10.1161/01.HYP.0000123072.34629.57. [DOI] [PubMed] [Google Scholar]
- (40).Buchberger A, Theyssen H, Schroder H, McCarty JS, Virgallita G, Milkereit P, Reinstein J, Bukau B. Nucleotide-induced conformational changes in the atpase and substrate binding domains of the dnak chaperone provide evidence for interdomain communication. J Biol Chem. 1995;270:16903–16910. doi: 10.1074/jbc.270.28.16903. [DOI] [PubMed] [Google Scholar]
- (41).Ricci L, Williams KP. Development of fluorescence polarization assays for the molecular chaperone hsp70 family members: Hsp72 and dnak. Current chemical genomics. 2008;2:90–95. doi: 10.2174/1875397300802010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Chang L, Thompson AD, Ung P, Carlson HA, Gestwicki JE. Mutagenesis reveals the complex relationships between atpase rate and the chaperone activities of escherichia coli heat shock protein 70 (hsp70/dnak) J Biol Chem. 2010;285:21282–21291. doi: 10.1074/jbc.M110.124149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Wang AM, Miyata Y, Klinedinst S, Peng HM, Chua JP, Komiyama T, Li X, Morishima Y, Merry DE, Pratt WB, Osawa Y, Collins CA, Gestwicki JE, Lieberman AP. Activation of hsp70 reduces neurotoxicity by promoting polyglutamine protein degradation. Nature Chem Biol. 2013;9:112–118. doi: 10.1038/nchembio.1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Abisambra J, Jinwal UK, Miyata Y, Rogers J, Blair L, Li X, Seguin SP, Wang L, Jin Y, Bacon J, Brady S, Cockman M, Guidi C, Zhang J, Koren J, Young ZT, Atkins CA, Zhang B, Lawson LY, Weeber EJ, Brodsky JL, Gestwicki JE, Dickey CA. Allosteric heat shock protein 70 inhibitors rapidly rescue synaptic plasticity deficits by reducing aberrant tau. Biol Psych. 2013 doi: 10.1016/j.biopsych.2013.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Zhuravleva A, Gierasch LM. Allosteric signal transmission in the nucleotide-binding domain of 70-kda heat shock protein (hsp70) molecular chaperones. Proc Natl Acad Sci USA. 2011;108:6987–6992. doi: 10.1073/pnas.1014448108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Berg T. Allosteric switches: Remote controls for proteins. Angew Chem Int Ed Engl. 2009;48:3218–3220. doi: 10.1002/anie.200806169. [DOI] [PubMed] [Google Scholar]
- (47).Rousaki A, Miyata Y, Jinwal UK, Dickey CA, Gestwicki JE, Zuiderweg ER. Allosteric drugs: The interaction of antitumor compound mkt-077 with human hsp70 chaperones. J Mol Biol. 2011;411:614–632. doi: 10.1016/j.jmb.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.