

Abstract
Aims
Naturally occurring transmissible spongiform encephalopathies (TSEs) accumulate disease specific forms of prion protein on cell membranes in association with pathognomonic lesions. We wished to determine whether synthetic prion protein disorders recapitulated these and other sub-cellular TSE specific changes.
Methods
SSLOW is a TSE initiated with refolded synthetic prion protein. Five terminally sick hamsters previously intra-cerebrally inoculated with 3rd passage SSLOW were examined using light and immunogold electron microscopy.
Results
SSLOW affected hamsters showed widespread abnormal prion protein (PrPSSLOW) and amyloid plaques. PrPSSLOW accumulated on plasma-lemmas of neurites and glia without pathological changes. PrPSSLOW also co-localised with increased coated vesicles and pits, coated spiral membrane invaginations and membrane microfolding. PrPSSLOW was additionally observed in lysosomes of microglial cells but not of neurons or astrocytes.
Conclusions
PrPSSLOW is propagated by cell membrane conversion of normal PrP and lethal disease may be linked to the progressive growth of amyloid plaques. Cell membrane changes present in SSLOW are indistinguishable from those of naturally occurring TSEs. However, some lesions found in SSLOW are absent in natural animal TSEs and vice versa. SSLOW may not entirely recapitulate neuropathological features previously described for natural disease. End–stage neuropathology in SSLOW, particularly the nature and distribution of amyloid plaques may be significantly influenced by the early re-distribution of seeds within the inoculum and its recirculation following interstitial, perivascular and other drainage pathways. The way in which seeds are distributed and aggregate into plaques in SSLOW has significant overlap with murine APP overexpressing mice challenged with Aβ.
Keywords: Transmissible spongiform encephalopathy, prion disease, synthetic prion protein, membrane pathology, amyloid, cerebral amyloid angiopathy
Introduction
The transmissible spongiform encephalopathies (TSEs) or prion diseases include Creutzfeldt-Jakob disease of man, scrapie of sheep and bovine spongiform encephalopathy of cattle. The infectious agent or prion is thought to be mainly, or exclusively, composed of abnormal, infectious conformational variants of a normal host-coded cell surface sialoglycoprotein called prion protein (PrPc) [1]. Where sufficient compatibility exists between the infecting prions and the host PrPc the former may template the conversion of PrPc into new prions. Recent studies have also shown that aggregated proteins found in other chronic neurodegenerative diseases of man such as Alzheimer’s disease, Huntington’s disease and Parkinson’s disease may also induce misfolding of native proteins leading some to suggest that prion-like disease mechanisms may be much more widespread than previously thought [2].
Recent experiments in which prion disorders were created in the laboratory and in the absence of infectious TSE brain homogenates, have been influential in reinvigorating comparisons between prion diseases and other chronic neurodegenerative disease. Novel disease inducing paradigms include generation of transgenic mice expressing mutant forms of PrP corresponding to human genetic forms of prion disease [3,4] and inoculation of rodents with PrP sources created or amplified in vitro from non-infectious sources [5–8]. In the latter categories of experiments, protein misfolding cyclic amplification (PMCA) has been used to create infectious prions from normal or purified brain homogenates [5,9,10] or from synthetic mutant PrP peptides [11] while in other approaches, chemical or physical treatments of synthetic PrP have created fibrillar or aggregated forms that may also cause disease when inoculated intra-cerebrally [6–8]. Interpretation of these experiments can sometimes be problematic where insufficient clinical, physiological and pathological data on aged transgenic mouse lines are available. Mice that carry normal or mutant PrP at physiological levels remain healthy through life span [12, 13,14] but some transgenic lines of rodents that express excess wild-type [7] or mutated [11] PrP peptides, may develop spontaneous disease in old age. When brain homogenates from these latter mice are used to challenge other mice carrying lower expression levels of the same mutant PrP [14,15] an accelerated disease is reported [7,14]. Moreover, experimental prion disorders often lack typical clinical signs of scrapie and/or have a highly protracted clinical disease course with long incubation periods that do not show a typical pattern of adaptation on serial passage. In addition, atypical pathological features and/or molecular forms of PrP are described [3,5–7,16]. Thus, the interpretation is often constrained by limitations in the clinical, physiological and pathological information available on both the donors and transgenic lines used for bioassay readout. In contrast, studies that have induced scrapie-like disease phenotypes in wild type rodents challenged with re-folded synthetic PrP sources [8,17,18] eliminate some of these ambiguities.
In addition to spongiform changes of neuropil, TSE specific lesions of membranes are common to classical TSEs circulating in domestic and wild animals, man and in their laboratory rodent counterparts. However, they are absent from some rodent models of familial prion disorders and recombinant PrP sources that accumulate amyloid but do not cause clinical disease. In the present study we have sought to determine whether sub-cellular lesions found in naturally occurring TSEs are also features of fatal transmissible TSEs initiated with synthetic PrP. SSLOW [8,17], (Synthetic Strain Leading to Over-Weight) is one such laboratory generated prion disorder resulting from inoculation of a recombinant PrP source. To induce SSLOW a full length wild-type recombinant hamster PrP was converted into fibrillar aggregates using denaturing agents and cycles of heat exposure. Challenge of wild-type hamsters with this synthetic aggregated PrP did not cause clinical disease but evidence of PrP aggregation was found using both Western blotting (WB) and PMCA. When these brain homogenates were further passaged in wild-type hamsters a pathological and molecular phenotype was observed that was consistent with TSE infection [8]. Our observations suggest that SSLOW has membrane pathology that is in common with other TSEs as well as some disease features similar to mice overexpressing APP and which generate Aβ plaques following challenge with Alzheimer’s disease brain homogenates. However, there were also some differences in the pathology of SSLOW when comparing it with classical TSE sources, which may point to differences in pathogenesis.
Materials and methods
For the purposes of this paper we will use the term TSE to describe prion diseases that are found naturally, along with their experimental rodent counterparts, and the term experimental prion disease for all laboratory created prion disorders originating from bacterially derived synthetic PrP sources. We will further refer to the disease associated PrP detected in the brains of TSEs as PrPd and the abnormal PrP found in SSLOW as PrPSSLOW.
Details of the creation of SSLOW scrapie have been published previously [8]. Briefly, a recombinant full length hamster PrP was dissolved in 6M guanidine hydrochloride and fibrilised in the presence of 1M guanidine hydrochloride, 3M urea and EDTA. These recombinant PrP fibrils were annealed either in the presence of normal hamster brain homogenate or bovine serum albumin using five alternate one min. intervals of exposure to heat at 80°C followed by exposure at 37°C. Weanling Syrian hamsters were intra-cerebrally inoculated with the annealed recombinant PrP. At 661 days hamsters were sacrificed and one of six showed presence of protease resistant PrP (PrPres) by WB while four of 13 showed PrPres after one round of PMCA. When brain homogenates from WB or PMCA positive hamsters were used for second passage, clinical disease was observed after incubation periods of 481 ± 4 days and 565± 14 days, respectively.
For a third passage, groups of hamsters were challenged with a second passage SSLOW homogenate. At third passage hamsters developed clinical disease at 322 ± 8 days or 325± 8 days post inoculation for 1% and 0.1% brain homogenates respectively. The clinical course of disease was protracted and five hamsters were euthanized approximately 200 days after initial clinical signs (503 days post inoculation) and perfusion-fixed using 4% paraformaldehyde / 0.1% gluteraldehyde. Each of the five brains was placed in a brain-mould and serially sliced at 1mm. Alternate 1mm sections were processed and embedded in paraffin wax for immunohistochemistry (IHC) to detect PrPSSLOW, using primary PrP antibodies 1C5 (J-S Kim, Hallym University, Republic of Korea), which recognizes the region 119–130 corresponding to the GXXXG motif, the glycine zipper region, conserved in all mammals [19], and Saf84 (Bioquote, York, UK). Incubation with these antibodies was followed by avidin-biotin amplification and DAB visualization (Vector Laboratories Ltd, Peterborough, UK), and haematoxylin counterstaining. Additional IHC was carried out with antibodies to ubiquitin, glial fibrillary acidic protein (GFAP), (Dako, Ely, Cambridgeshire, UK) coronin (to label microglial cells) (Abcam, Cambridge, UK) and to hyperphosphorylated tau (antibody AT8, Fisher Scientific, Loughborough, UK) by standard procedures.
From the remaining alternate 1mm thick tissue slices, 1mm3 blocks were cut to provide representation of cerebral cortex, hippocampus, cerebellum, thalamus and area postrema. These blocks were post fixed in osmium tetroxide, dehydrated and embedded in araldite. Serial 1μm sections were stained by toluidine blue or were etched, pre-treated and subjected to IHC for disease specific PrP with 1C5 or 1A8 PrP antibodies [20] .
Selected blocks with appropriate immunolabelled areas were then taken for ultrastructural studies. For immunogold labelling, ultrathin 65 nm sections were etched and alternate sections pre-treated with formic acid. 1A8, 1C5 or pre-immune serum were applied routinely. Sections were incubated with a 5nm gold probe (BBI International, Cardiff UK), enhanced and counterstained with lead citrate and uranyl acetate.
Tubulovesicular bodies (TVBs) are invariably found in rodent forms of scrapie such as hamster 263K or mouse ME7 where they are often abundant [21]. These structures are less commonly encountered in some naturally occurring ruminant TSEs [22]. To determine whether TVBs were present in this study, 65nm tissue sections were taken from 6 blocks of the thalamus of the five SSLOW affected hamsters and from single thalamic blocks from two 263K hamster scrapie, two CH22 hamster scrapie and two ME7 scrapie mouse brains. These were stained using uranyl acetate and lead citrate and randomised. Coded samples were then examined and the presence of TVB or otherwise recorded, after which samples were decoded.
Hamster 263K and murine ME7 scrapie sections were also included within each IHC run as controls, while other controls used included sections from J20 mouse brains [23] for amyloid plaques formed from mutant Alzheimer’s precursor protein (APP), and normal, aged rodent brain tissue.
All animal experiments were carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocol was approved by the Institutional Animal Care and Use Committee of the University of Maryland, Baltimore (Assurance Number A32000-01; Permit Number: 0309001).
RESULTS
Light microscopy
All five SSLOW-affected hamster brains showed vacuolation (Fig 2a) and PrPSSLOW accumulations at all levels of the brain. Diffuse punctuate PrPSSLOW accumulation was present throughout the hippocampus (Fig 1a), mid-cortical layers of the cerebral cortex and thalamus. Widespread PrPSSLOW amyloid plaques were associated with the glial limitans of the pia (Fig 1f) and ventricles (Fig 1c, 1d). Less common forms of accumulation included a peri-neuronal type in the striatum and glial cells with intra-cytoplasmic peri-nuclear granular PrPSSLOW accumulations at several locations.
Fig 2. Vacuolation.
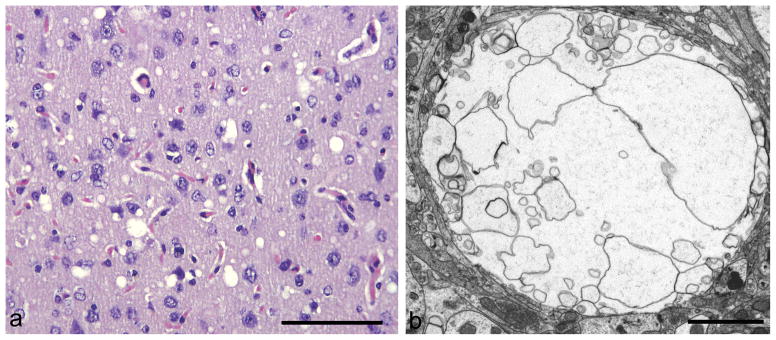
a) Ovoidal vacuoles of the neuropil in the hypothalamus.
b) Vacuole in a dendrite with intact plasma-lemma and numerous intra-vacuolar membrane bound profiles.
a: HE; b: uranyl acetate / lead citrate staining.
Mag bars: a = 100 μm; b = 2 μm.
Fig 1. Light microscopic immunohistochemistry showing different types of PrPSSLOW labelling patterns and astrocytic gliosis.

a) Diffuse PrPSSLOW accumulation in hippocampus with increased intensity of labelling in the dentate gyrus (d), stratum lacunosum-moleculare (S-L) and in the corpus callosum (CC). S-R; stratum radiatum; S-O; stratum oriens.
b) GFAP immunolabelling showing gliosis predominantly in the dentate gyrus (d) and stratum lacunosum-moleculare (S-L). The stratum radiatum (S-R) of the hippocampus, which has mild-moderate punctuate PrPSSLOW labelling (see Fig 1a), does not show conspicuous astrocyte hypertrophy or hyperplasia.
c) PrPSSLOW accumulation within the ventricular space of the aqueduct of Sylvius (Aq); in the subjacent glial limitans and coating the ventricular surface of ependymal cells (arrows).
d) Marked PrPSSLOW accumulation within the area postrema (AP) and the central canal (arrow) at the level of the obex.
e) PrPSSLOW immunolabelling within the walls and surrounding blood vessels of the corpus callosum.
f) Marked thickening of the sub-pial glial limitans (GL) associated with marked PrPSSlow accumulation.
g) Marked PrPSSLOW accumulation associated with an old infarct.
a, c, e, f, g, h: PrPSSLOW immunolabelling; b, d: GFAP immunolabelling.
Mag bars: a = 500 μm, b = 500 μm, c =100 μm, d = 100 μm, e = 50 μm, f = 50 μm, g = 100 μm.
Mild or moderate, diffuse, punctuate accumulations of PrPSSLOW such as occurred in the stratum radiatum and dentate gyrus of the hippocampus (Fig 1a), were not associated with gliosis (Fig 1b). More intense diffuse punctuate PrPSSLOW accumulations were accompanied by a florid gliosis, such as in the stratum lacunosum-moleculare of the hippocampus (Fig 1b). In addition, marked astrocytic gliosis was seen in association with amyloid accumulations of the glial limitans of the pia and ependyma and in the area postrema. The area postrema is one of the circumventricular organs (CVOs) of the brain and was consistently represented in tissue sections of the caudal medulla. Other circumventricular organs were inconsistently represented but when available they showed high levels of PrPSSLOW labelling (Fig 1e) together with gliosis, particularly in the median eminence and in the sub-fornical organ. Abundant PrPSSLOW was also found lying free within the ventricular space, particularly the 3rd ventricle and aqueduct (Fig 1c, 1d). Generally, increases in reactive astrocytes were accompanied by a proportionate, though often smaller, increase in reactive microglia. In neuropil surrounding sub-pial and periventricular amyloid deposits, astrocytosis conspicuously predominated over microgliosis (Fig S1). Numerous small tau positive puncta and fine irregular processes were also present in areas of intense PrPSSLOW labelling but were infrequent or absent around amyloid plaques and vessels with angiopathy (Fig S2).
Within the corpus callosum (Fig 1e) and the stratum lacunosum-moleculare of the hippocampus, a prominent vasculopathy was associated with PrPSSLOW accumulation. Around some of these vessels amyloid plaques were found and irregular accumulations of amyloid were also found in the sub-ventricular and sub-ependymal neuropil.
An old needle tract infarct with marked neo-vascularisation (Fig 1g) also showed prominent PrPSSLOW accumulation.
Electron microscopy
Table 1 shows a list of selected sub-cellular pathological features and their presence or absence in SSLOW compared with several previously described TSEs.
Table 1.
showing presence of selected lesions in SSLOW compared with representative natural and rodent TSEs and a transgenic mouse line.
| Lesions that do not co-localise with PrP d/SSLOW | SSLOW | 263K | Me7 | BSE | Scrapie | TG3* |
|---|---|---|---|---|---|---|
| Vacuoles | yes | yes | yes | yes | yes | yes |
| Tubulovesicular bodies | no | yes | yes | yes | yes | yes |
| Abnormal and increased multivesicular bodies | rare | yes | yes | yes | yes | yes |
| Autophagic vacuoles | rare | yes | yes | yes | yes | yes |
| Wallerian-type degeneration | yes | yes | yes | yes | yes | yes |
| Dystrophic neurites | yes | yes | yes | yes | yes | yes |
| Abnormal and enlarged mitochondria | yes | rare | no | no | no | no |
| Abnormal pial membrane laminations | yes | no | no | no | no | no |
| Autophagy | no | yes | yes | yes | yes | yes |
| Membrane lesions that do co- localise with PrPd/SSLOW | ||||||
| Neuritic plasma-lemmal PrPd /SSLOW accumulation | yes | yes | yes | yes | yes | yes |
| Increased coated vesicles and pits of neurites | yes | yes | yes | yes | yes | yes |
| Spiral dendritic and axonal membrane invaginations | yes | yes | yes | yes | yes | yes |
| Membrane microfolds | yes | yes | yes | yes | yes | yes |
| Non-membrane lesions that do co-localise with PrPd/SSLOW | ||||||
| Microglial lysosomal PrPd/SSLOW accumulation | yes | yes | yes | yes | yes | yes |
| Ependymocyte lysosomal PrPd/SSLOW accumulation | no | yes | no | no | no | no |
| Astrocytic lysosomal PrPd/SSLOW accumulation | no | yes | yes | yes | yes | yes |
| Neuronal lysosomal PrPd/SSLOW accumulation | no | yes | yes | yes | yes | no |
| Amyloid fibrils in neuropil | yes | yes | yes | rare | rare | yes |
| Amyloid plaques | yes | yes | yes | no | rare | rare |
| Cerebral amyloid angiopathy | yes | no | no | no | no | no |
Expresses hamster PrP on astrocytes on a PrP null neuronal background.
Pathology of SSLOW typical of previously described TSEs
Sub-cellular features of SSLOW that have previously been described in animal TSEs included vacuoles and membrane alterations. Neuropil vacuoles in SSLOW were typical of vacuoles found in other TSEs, being predominantly in dendrites and often containing membrane fragments that were unbounded or bounded by the plasma-membrane of neurites (Fig 2b). Dendritic membranes frequently showed invaginations (Fig 3a – d) that were sometimes spiral (Fig 3a), occasionally branched and sometimes covered by a clathrin-like coat (Fig 3b, 3c). These were often associated with a marked increase in coated pits and vesicles (Fig 3c). All were heavily labelled for PrPSSLOW (Fig 3b – d) and less abundantly with ubiquitin (not shown). Other PrPSSLOW and ubiquitin associated membrane changes (Fig 4a – c) consisted of small folds or polyp like protrusions (microfolding) that extended from larger hypertrophic processes of astrocytes recognised by prominent bundles of intermediate filaments (Fig 4a, 4b) and rarely, dendrites. Astrocyte membrane micro-folding was the most widespread and conspicuous change seen by electron microscopy, considerably more so than even the vacuolation. PrPSSLOW was also found on cell membranes of microvilli of ependymocytes (Fig 4d) a feature not encountered in naturally occurring animal TSEs but occasionally seen in 263K hamster scrapie (personal observation).
Fig 3. Plasma-lemmal membrane invaginations.

a) A dendrite (D) contains two plasma-lemmal membrane invaginations (arrows) the longer of which has a spiral arrangement. The dendrite lacks other sub-cellular organelles such as microtubules.
b) A tangentially sectioned dendrite (D) shows PrPSSLOW accumulation associated with coated tubular structures (arrowheads) and small membrane invaginations. PrPSSLOW is also present on membranes of adjacent processes.
c) A cross section of a dendrite (D) shows, at one pole, a marked increase in PrPSSLOW labelled coated pits and sub-plasma-lemmal fused tubular membrane complexes.
d) A large uncoated dendritic (D) membrane invagination (arrow) showing PrPSSLOW accumulation.
a: Uranyl acetate / lead citrate staining; b–d: immunogold labelling for PrPSSLOW
Mag bars: a = 1 μm, b = 0.5 μm, c = 1 μm, d = 0.5 μm.
Fig 4. Plasma-lemmal membrane microfolding.

a) An early focus of plasma-membrane changes of astrocyte processes (A) showing ruffling of surface contours (bounded by dotted line) and irregular polyp like outfolds (P) of the plasma-membrane. Some cross or tangentially sectioned polyp-like folds have irregular ovoidal or elliptical profiles.
b) Prominent PrPSSLOW labelling of microfolded and polyp like extensions of astrocytic plasma-membranes. Larger astrocytic processes (A) from which these membrane changes arise are filled with intermediate filaments. PrPSSLOW is predominantly associated with membranes of microfolds.
c) An area of hyperplastic glial limitans of the pia showing parallel arrays of thin unlabelled astrocytic processes (arrowheads) with small areas of astrocytic microfolding showing PrPSSLOW accumulation.
d) PrPSSLOW labelling on microvilli (arrows) of ependymocytes (E) and on amyloid fibrils (arrowheads) within the ventricular lumen.
a: Uranyl acetate / lead citrate staining; b, c, d: immunogold labelling for PrPSSLOW Mag bars: a = 0.5 μm, b = 1 μm c = 1 μm, d = 1 μm.
Abundant, closely packed but irregularly arranged amyloid fibrils were present throughout the area postrema (Fig S3), the sub-pial glial limitans and in association with the ventricles including fibrils lying free within the ventricular space. Both well defined and small poorly defined amyloid fibrils labelled heavily for PrPSSLOW (Fig 5a, 5b and Fig S3).
Fig 5. Amyloid fibrils and plaques.

a) Sparse fibril accumulation in grey matter. Short PrPSSLOW amyloid fibrils (arrowheads) are present in an expanded extracellular space surrounding largely neuritic processes.
b) Mature plaque adjacent to a blood vessel. The plaque has a dense central core from which radiate bundles of amyloid fibrils. Amyloid fibrils are labelled for PrPSSLOW.
c) An immature amyloid plaque adjacent to the ventricles, showing abnormal irregular glial processes, many lacking organelles- associated with irregular plasma-lemmas, and ill-defined, short and irregularly arranged amyloid fibrils (arrows) in close proximity to folded membranes. It is difficult to separately distinguish incipient fibril from astrocytic plasma-membrane in many parts of the figure. Only a minority of glial processes possess intermediate filaments (IF).
a, b: Immunogold labelling for PrPSSLOW c: uranyl acetate / lead citrate staining.
Mag bars: a = 0.5 μm, b = 2 μm, c = 0.5 μm.
The first evidence of aggregation of PrPSSLOW into amyloid fibrils was found in association with the convoluted and microfolded astrocytic processes described above (Fig 5c) suggesting astrocyte membranes were the major source of extra-cellular PrPSSLOW amyloid. Loosely arranged amyloid fibrils and dense amyloid plaques were abundant in association with the ventricular system (both within the ventricular lumen (Fig 4d) and beneath the ependyma) and beneath the glial limitans of the pia, the area postrema, and around areas of cerebral amyloid angiopathy.
Brains from naturally occurring TSEs and rodent adapted scrapie, consistently show PrPd accumulation in lysosomes of neurons, astrocytes and microglia. In SSLOW affected hamsters, PrPSSLOW was detected within lysosomes of a minority of microglial cells (not shown), but not within lysosomes of neurons or astrocytes. In agreement with other TSEs no PrPSSLOW co-localisation was found for vacuoles, tubulovesicular bodies, abnormal multivesicular bodies, mitochondria, auto-phagosomes or dystrophic neurites.
A number of sub-cellular lesions that lack co-localisation with PrPd in naturally occurring TSEs were also present in SSLOW (Table 1). Such PrPSSLOW negative lesions included increased numbers of markedly enlarged multivesicular bodies (late endosomes), activated microglia, apoptotic neurons, autophagic vacuoles, and dystrophic neurites. However, SSLOW tissues did not show these features with the same frequency as is found in rodent TSEs and abnormally enlarged multivesicular bodies were rare. As with the distribution of astrocytosis detected by light microscopy, areas of neuropil with mild or moderate PrPSSLOW accumulation had mostly a morphologically normal neuropil and the changes listed above were confined to neuroanatomic areas of high PrPSSLOW concentration.
Tubulovesicular bodies (TBVs) are characteristic lesions of rodent and animal TSEs. TBVs are approximately 30–35 nm diameter ovoidal or short tubular particles of unknown molecular composition that accumulate in high concentration in neurites in all animal TSEs, in CJD and some Gerstmann-Sträussler-Scheinker (GSS) brains [21]. TBVs were not recognised in any SSLOW brains either on initial inspection or in the blind coded trial, although once de-coded, all six samples from ME7, 263K or 22CH rodent scrapie were identified as having TBVs.
Sub-cellular lesions of SSLOW not encountered in animal TSEs
Lesions not previously encountered in animals included cerebral amyloid angiopathy (CAA) of the corpus callosum and stratum lacunosum and an unusual reaction of the glial limitans of the pia and ependyma.
PrPSSLOW was prominently associated with blood vessels and cerebral amyloid angiopathy of the corpus callosum and the stratum lacunosum-moleculare (Fig 6a – e). Lesions of cerebral amyloid angiopathy (Fig 6a, 6b) included hypertrophy of the perivascular glial limitans (Fig 6a, 6c), displacement or disorganisation of basement membranes (Fig 6a, 6b) and infiltration and replacement of smooth muscle cells by amyloid fibrils (Fig 6a, 6b). Where severe vascular lesions were present, PrPSSLOW labelled basement membranes of endothelial cells and smooth muscle cells (Fig 6c – e) and was also found on intra-mural and peri-vascular amyloid fibrils. Less severe vascular involvement showed diminished amyloid accumulation and some vessels showed basement membrane labelling alone.
Fig 6. Cerebral amyloid angiopathy.

a) Abnormal blood vessel of the stratum lacunosum–moleculare showing disorganised media and marked perivascular astrocytic gliosis (A).
b) Detail of blood vessel in (a) showing marked amyloid (Am) replacement of the tunica media.
c) Perivascular PrPSSLOW accumulation and early deposition of PrPSSLOW in a part of the ablumenal basement membrane (arrow). L: lumen of blood vessel.
d) Blood vessel showing localised perivascular PrPSSLOW infiltration and accumulation of PrPSSLOW in vascular basement membranes (arrow).
e) Detail of arteriolar media showing PrPSSLOW in several basement membranes (arrows). The vessel lumen is bottom left.
a, b, c: Uranyl acetate / lead citrate staining; d, e, f: immunogold labelling for PrPSSLOW
Mag bars: a = 5 μm, b = 1 μm, c = 5 μm, d = 2 μm, f = 1 μm.
The markedly thickened glial limitans of the pia-mater and ependyma seen at light microscopy corresponded to markedly hypertrophic astrocytic processes. Hypertrophic processes surrounding the ventricles were arranged as parallel stacks of processes (Fig 4c). Between these parallel stacks clear zones of astrocytic membrane microfolding as described above were present (Fig 4c). PrPSSLOW immunolabelling was not found on the laminar stacks of astrocyte processes (Fig 4c).
Unusual enlarged mitochondria often with irregular arrangements of christae were abundant in SSLOW (Fig S4). Similar mitochondria were a rare feature of hamster 263K scrapie but were absent from other murine scrapie sources or ruminant TSEs.
Discussion
It has previously been shown that intra-cerebral inoculation of a refolded synthetic PrP did not cause disease at primary passage but resulted in a lethal disease (SSLOW) on secondary passage, with pathological and biochemical properties consistent with rodent TSEs [8]. In the present study we show that sub-cellular membrane and vacuolar lesions of SSLOW at third passage are indistinguishable from those of naturally occurring TSEs and their laboratory rodent counterparts.
PrPc conversion, membrane pathology and amyloid
All naturally occurring and experimental animal TSE strains show distinctive plasma-lemmal membrane invaginations, increased coated pits and vesicles, and membrane microfolding that co-localise with PrPd and ubiquitin (reviewed in [24]). Similar lesions have been described in sporadic CJD [25]. The membrane lesions putatively arise from stable PrPd-protein complexes within membranes that resist excision and endocytotic re-cycling [24]. As closely similar membrane features were also found in SSLOW, we infer that synthetic PrP sources can also propagate and form stable membrane complexes that convert PrPc to PrPSSLOW and perturb membrane morphology after intra-cerebral injection in rodents.
The molecular phenotype and distribution of PrPSSLOW is unique compared to previously described TSEs. In some TSEs and prion diseases aggregated PrP is released from membranes to form amyloid plaques. Uni-centric stellate (kuru-type) plaques are a feature of kuru, sporadic CJD, [26], bovine amyloidotic spongiform encephalopathy [27] and some murine adapted scrapie strains; multicentric plaques are often found in GSS disease [26, 28] and micro-plaques of poorly defined filamentous structures are seen in variably protease sensitive prionopathy [29]. Amyloid plaques or loose amyloid fibril accumulations are rare or absent in most naturally occurring TSEs of wild or domestic animals [24]. In contrast, amyloid plaques are a diagnostic feature of vCJD, a fraction of sCJD patients [26] and are the sole or major pathology feature in some GSS patients [26]. Generalised cerebro-vascular amyloidosis occurs in a sub-population of familial prion diseases in which C terminal stop mutations eliminate the glyco-phospho-inositol (GPI) anchor on PrP [30] and in scrapie infected mice transgenic for a GPI anchorless form of PrP. [12, 31]. Stellate plaques form around individual dendrites [32] while multicentric plaques appear to be formed by multifocal recruitment from many different localised sources of extracellular aggregated PrP [33]. In comparison to the spectrum of established distributions of PrPd, the plaques in SSLOW are distinct both with respect to their location in the brain, which is predominantly in association with the glial limitans, and in cells where fibrils are found embedded in microfolded astrocytic processes. The neuroanatomically restricted cerebro-vascular amyloid of SSLOW has parallels with the predominant location of amyloid in some transgenic rodents expressing APP following challenge with Aβ [ 34–36 ] (see below) .
Sub-cellular features of TSEs compared with SSLOW
Table 1 lists a selection of TSE associated lesions and their subjectively assessed abundance in previously described TSEs compared to SSLOW. In addition to the type and distribution of plaques as described other differences include the absence of intra-neuronal and intra-glial lysosomal PrPSSLOW accumulations and TVBs in SSLOW. Although of unknown molecular composition, TBVs are considered by some to be a pathognomonic feature of prion disease. While some magnitude differences in lesions might be attributed to strain effects [37], qualitative differences in lesions may point to differences in disease pathogenesis. The unusual nature and wide distribution of amyloid fibrils and plaques in SSLOW might be explained by a wide distribution of seeds by the inoculum followed by multiple pathways of propagation.
Impact of high volume inoculates
Relatively large volumes are used for the intra-cranial inoculation of TSEs into rodents. This inoculate is re-distributed in varying proportions into the interstitial fluid, perivascular spaces, the cerebrospinal fluid and blood.
Inoculum reaching the interstitial fluid is removed along basement membranes of capillaries and arterioles [38 ] and via the meningeal blood vessels to cervical lymph nodes [39]. However, particulates will not pass though these spaces. When particulate tracers such as Indian ink are injected into the hippocampus and neocortex of the mouse, they are rapidly distributed along perivascular spaces to the ventral corpus callosum, the hippocampal fissure, around blood vessels of the stratum lacunosum and the ventromedial sub-surface of the hippocampus [34]. Over-expressing APP transgenic mice intra-cerebrally inoculated with Alzheimer’s disease brain show accumulations of Aβ at approximately 5 months post challenge with this same distribution pattern [34–36] as do transgenic L101L mice intracerebrally challenged with 8kDa GSS [33 ]. Thus, the distribution of CAA in SSLOW brains is consistent with a high concentration of PrPSLOW seeds being drained or trapped at these sites at or around the time of intra-cerebral challenge.
SSLOW plaques and amyloid fibrils are abundant in association with the glial limitans of the ventricles and pia. Experimental studies show that under conditions of increased pressure created by high volume inoculates, CSF may enter the brain via perivascular pathways and solutes in a molecular weight range consistent with multimeric complexes of PrP may thus concentrate in the perivascular sub-pial neuropil [40]. Under these conditions CSF and solutes may also cross tight junctions lacking periventricular white matter [40, 41]. Thus, following high volume intracerebral injection, amyloid seeds within the SSLOW brain homogenate will rapidly distribute to the sub-pia and CSF within the ventricular and sub-arachnoid spaces. From these compartments high concentrations of seeds will be present in association with the glial limitans and corresponds to sites at which amyloid plaques are observed in terminal disease affected SSLOW brains.
Excess SSLOW homogenate will ultimately drain to the vascular system [41] and recirculate to reach the CVOs. Amyloid seeds in blood can readily amplify in CVOs because of its incomplete blood brain barrier [42]. Similarly, newly-formed blood vessels arising from needle tract infarcts are also susceptible to penetration by amyloid seeds circulating in the blood [43–44]. Thus, all major sites at which amyloid plaques and fibrils were detected in SSLOW corresponds to those sites that would have received the maximum concentration and earliest exposure to seeds within the original inocula as dispersed by interstitial fluid, perivascular flow and via the blood.
In summary, the disease SSLOW provides proof of principle that a refolded synthetic PrP can initiate a fatal transmissible prion protein disorder when passaged in hamsters and, furthermore, that the resulting disease shows the pathognomonic membrane changes and amyloid fibril aggregation common to most naturally occurring animal TSEs. The distribution of amyloid fibrils in SSLOW is consistent with amyloid seeds in the inocula being disseminated by different routes and mechanisms more or less simultaneously to widely separate neuroanatomical sites at the time of inoculation. This same level of dispersal might be expected for any intracranial inoculation of rodents, raising the question of whether inoculation route and volume influence patterns of PrPd accumulation generally, and whether the patterns observed can rightfully be interpreted as strain phenotypes. The unusually protracted clinical phase of disease is consistent with slow propagation of PrPSSLOW and subsequent amyloid accumulation. The study provides some evidence to suggest that molecules or aggregates that convert PrPc on cell membranes may differ from other aggregated forms which assemble into fibrils within the interstitial spaces. The wide distribution of PrPSSLOW in interstitial fluids and perivascular spaces is similar to that seen for Aβ plaques in mice over-expressing APP that have been challenged with Alzheimer’s disease brain homogenate. Closer study of the molecular nature of aggregates with converting and fibrillising properties could be important in understanding disease mechanisms in protein misfolding generally. Although many aspects of natural TSE pathology were reproduced by SSLOW further studies are necessary to determine whether synthetic PrP sources reproduce all characteristics of naturally occurring TSEs.
Supplementary Material
Figure S1. Astrocytic and microglial activation:
a) Stratum lacunosum moleculare (SL) showing moderate astrocytosis and mild vacuolation. Bar = 200 μm. Immunohistochemistry for GFAP.
b) Stratum lacunosum moleculare showing mild microglial activation and mild vacuolation. Bar = 100 μm. Immunohistochemistry for coronin.
c) Severe astrocytosis in the superior colliculus associated with marked vacuolation. Bar = 200 μm. Immunohistochemistry for GFAP.
d) Severe microgliosis associated with marked vacuolation in the midbrain tectum. Bar = 100 μm. Immunohistochemistry for coronin.
e) Marked astrocytosis is present around several sub-ependymal plaques (arrowheads). Bar = 100 μm. Immunohistochemistry for GFAP.
f) Small numbers of activated microglia are present in the neuropil adjacent to amyloid plaques (arrowheads) in the peri-ventricular glial limitans. Bar = 100 μm. Immunohistochemistry for coronin.
Figure S2. Tau immunolabelling:
Cerebral cortex showing moderate vacuolation and punctuate tau immunolabelled structures. Bar = 50 μm. Immunohistochemistry for tau.
Figures S3. Area postrema:
a) A large areas of neuropil surrounding a blood vessel shows loss of normal architecture, increased extracellular space and a dystrophic neurite (d). Bar = 3 μm. Uranyl acetate / lead citrate.
b) Detail of an area similar to the above, showing loss of normal cellular processes and replacement by poorly defined fibrillar (arrowheads) PrPSSLOW amyloid fibrils. Bar = 0.5 μm. Immunogold for PrPSSLOW.
c) Accumulation of PrPSSLOW in association with convoluted and microfolded processes, and poorly defined fibrils (arrowheads) surrounding a large astrocytic process (A). Bar = 1 μm. Immunogold for PrPSSLOW.
Figures S4. Enlarged mitochondria:
Enlarged mitochondria with abnormal christae were mostly located in glial cell cytoplasm. A normal mitochondrion is present at M. Bar = −0.5 μm. Uranyl acetate/lead citrate.
Acknowledgments
MJ, IVB and RGR conceived and designed the study: NM and IVB annealed synthetic PrP and NM, IVB and RGR carried out animal studies: MJ, LG, GM J-SK prepared and characterised reagents and carried out pathology: MJ wrote the paper with contributions by GM, IVB and RGR. This work was supported by NIH grant NS045585 to IVB and a Veterans Affairs Merit Award to RGR.
Footnotes
The authors have no conflicts of interest with the data presented in this manuscript.
Reference List
- 1.Prusiner SB. Development of the prion concept. In: Prusiner SB, editor. Prion Biology and Diseases. Cold Spring Harbor Laboratory Press; 1999. pp. 67–112. [Google Scholar]
- 2.Brundin P, Melki R, Kopito R. Prion-like transmission of protein aggregates in neurodegenerative diseases. Nat Rev Mol Cell Biol. 2010;11:301–307. doi: 10.1038/nrm2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hsiao K, Groth D, Scott M, Yang S-L, Serban H, Rapp D, Foster D, Torchia M, Dearmond SJ, Prusiner SB. Serial transmission in rodents of neurodegeneration from transgenic mice expressing mutant prion protein. Proc Natl Acad Sci USA. 1994;91:9126–9130. doi: 10.1073/pnas.91.19.9126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Telling GC, Haga T, Torchia M, Tremblay P, DeArmond SJ, Prusiner SB. Interactions between wild-type and mutant prion proteins modulate neurodegeneration transgenic mice. Genes Dev. 1996;10:1736–1750. doi: 10.1101/gad.10.14.1736. [DOI] [PubMed] [Google Scholar]
- 5.Barria MA, Mukherjee A, Gonzalez-Romero D, Morales R, Soto C. De novo generation of infectious prions in vitro produces a new disease phenotype. PLoS Pathog. 2009;5:e1000421. doi: 10.1371/journal.ppat.1000421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Colby DW, Giles K, Legname G, Wille H, Baskakov IV, DeArmond SJ, Prusiner SB. Design and construction of diverse mammalian prion strains. Proc Natl Acad Sci U S A. 2009;106:20417–20422. doi: 10.1073/pnas.0910350106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Colby DW, Wain R, Baskakov IV, Legname G, Palmer CG, Nguyen HO Lemus A, Cohen FE, DeArmond SJ, Prusiner SBI. Protease-sensitive synthetic prions. PLoS Pathog. 2010;6:e1000736. doi: 10.1371/journal.ppat.1000736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Makarava N, Kovacs GG, Bocharova O, Savtchenko R, Alexeeva I, Budka H Rohwer RG, Baskakov IV. Recombinant prion protein induces a new transmissible prion disease in wild-type animals. Acta Neuropathol. 2010;119:177–187. doi: 10.1007/s00401-009-0633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104:9741–9746. doi: 10.1073/pnas.0702662104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deleault NR, Kascsak R, Geoghegan JC, Supattapone S. Species-dependent differences in cofactor utilization for formation of the protease-resistant prion protein in vitro. Biochemistry. 2010;49:3928–3934. doi: 10.1021/bi100370b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kaneko K, Ball HL, Wille H, Zhang H, Groth D, Torchia M, Tremblay P, Safar J, Prusiner SB, DeArmond SJ, Baldwin MA, Cohen FE. A synthetic peptide initiates Gerstmann-Straussler-Scheinker [GSS] disease in transgenic mice. J Mol Biol. 2000;295:997–1007. doi: 10.1006/jmbi.1999.3386. [DOI] [PubMed] [Google Scholar]
- 12.Chesebro B, Race B, Meade-White K, Lacasse R, Race R, Klingeborn M, Striebel J, Dorward D, McGovern G, Jeffrey M. Fatal transmissible amyloid encephalopathy: a new type of prion disease associated with lack of prion protein membrane anchoring. PloS Pathog. 2010;6:e1000800. doi: 10.1371/journal.ppat.1000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manson JC, Jamieson E, Baybutt H, Tuzi NL, Barron R, McConnell I, Somerville R, Ironside J, Will R, Sy MS, Melton DW, Hope J, Bostock C. A single amino acid alteration [101L] introduced into murine PrP dramatically alters incubation time of transmissible spongiform encephalopathy. Embo J. 1999;18:6855–6864. doi: 10.1093/emboj/18.23.6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nazor KE, Kuhn F, Seward T, Green M, Zwald D, Pürro M, Schmid J, Biffiger K, Power AM, Oesch B, Raeber AJ, Telling GC. Immunodetection of disease-associated mutant PrP, which accelerates disease in GSS transgenic mice. Embo J. 2005;24:2472–2480. doi: 10.1038/sj.emboj.7600717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tremblay P, Ball HL, Kaneko K, Groth D, Hegde RS, Cohen FE, DeArmond SJ, Prusiner SB, Safar JG. Mutant PrPSc conformers induced by a synthetic peptide and several prion strains. J Virol. 2004;78:2088–2099. doi: 10.1128/JVI.78.4.2088-2099.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jackson WS, Borkowski AW, Faas H, Steele AD, King OD, Watson N, Jasanoff A, Lindquist S. Spontaneous generation of prion infectivity in fatal familial insomnia knockin mice. Neuron. 2009;63:438–450. doi: 10.1016/j.neuron.2009.07.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Makarava N, Kovacs GG, Savtchenko R, Alexeeva I, Budka H, Rohwer RG, Baskakov IV. Genesis of mammalian prions: from non-infectious amyloid fibrils to a transmissible prion disease. PLoS Pathog. 2011;7:e1002419. doi: 10.1371/journal.ppat.1002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Makarava N, Kovacs GG, Savtchenko R, Alexeeva I, Ostapchenko VG, Budka H, Rohwer RG, Baskakov IV. A new mechanism for transmissible prion diseases. J Neurosci. 2012;32:7345–7355. doi: 10.1523/JNEUROSCI.6351-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Choi JK, Park SJ, Jun YC, Oh JM, Jeong BH, Lee HP, Park SN, Carp RI, Kim YS. Generation of monoclonal antibody recognized by the GXXXG motif [glycine zipper] of prion protein. Hybridoma [Larchmt] 2006;25:271–277. doi: 10.1089/hyb.2006.25.271. [DOI] [PubMed] [Google Scholar]
- 20.Farquhar CF, Somerville RA, Ritchie LA. Post-mortem immunodiagnosis of scrapie and bovine spongiform encephalopathy. J Virol Methods. 1989;24:215–222. doi: 10.1016/0166-0934(89)90023-2. [DOI] [PubMed] [Google Scholar]
- 21.Liberski PP, Budka H, Yanagihara R, Gibbs CJ, Gajdusek DC. Tubulovesicular structures. In: Liberski PP, editor. Light and electron microscopic neuropatholgy of slow virus disorders. Boca Raton: CRC Press; 1993. pp. 373–392. [Google Scholar]
- 22.Ersdal C, Goodsir CM, Simmons MM, McGovern G, Jeffrey M. Abnormal prion protein is associated with changes of plasma membranes and endocytosis in bovine spongiform encephalopathy [BSE]-affected cattle brains. Neuropathol Appl Neurobiol. 2009;35 :259–271. doi: 10.1111/j.1365-2990.2008.00988.x. [DOI] [PubMed] [Google Scholar]
- 23.Mucke L, Masliah E, Yu GQ, Mallory M, Rockenstein EM, Tatsuno G, Hu K, Kholodenko D, Johnson-Wood K, McConlogue L. High-level neuronal expression of Aβ 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J Neurosci. 2000;20:4050–4058. doi: 10.1523/JNEUROSCI.20-11-04050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jeffrey M, McGovern G, Siso S, Gonzalez L. Cellular and sub-cellular pathology of animal prion diseases: relationship between morphological changes, accumulation of abnormal prion protein and clinical disease. Acta Neuropathol. 2011;121:113–134. doi: 10.1007/s00401-010-0700-3. [DOI] [PubMed] [Google Scholar]
- 25.Bastian FO. Spiroplasma-like inclusions in Creutzfeldt-Jakob disease. Arch Pathol Lab Med. 1979;103:665–669. [PubMed] [Google Scholar]
- 26.Ironside JW, Ghetti B, Head MW, Piccardo P, Will RG. Prion Diseases. In: Love S, Louis DN, Ellison DW, editors. Greenfield’s Neuropathology. London: Hodder Arnold; 2009. pp. 1197–1273. [Google Scholar]
- 27.Casalone C, Zanusso G, Acutis P, Ferrari S, Capucci L, Tagliavini F, Monaco S, Caramelli M. Identification of a second bovine amyloidotic spongiform encephalopathy: Molecular similarities with sporadic Creutzfeldt-Jakob disease. Proc Nat Acad Sci USA. 2004;101:3065–3070. doi: 10.1073/pnas.0305777101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sikorska B, Liberski PP, Sobow T, Budka H, Ironside JW. Ultrastructural study of florid plaques in variant Creutzfeldt-Jakob disease: a comparison with amlyoid plaqes in kuru, Creutzfeldt-Jakob disease and Gerstman-Sträussler-Scheinker disease. Neuropathol Appl Neurobiol. 2009;35 :46–59. doi: 10.1111/j.1365-2990.2008.00959.x. [DOI] [PubMed] [Google Scholar]
- 29.Gambetti P, Dong Z, Yuan J, Xiao X, Zheng M, Alshekhlee A, Castellani R, Cohen M, Barria MA, Gonzalez-Romero D, Belay ED, Schonberger LB, Marder K, Harris C, Burke JR, Montine T, Wisniewski T, Dickson DW, Soto C, Hulette CM, Mastrianni JA, Kong Q, Zou WQ. A novel human disease with abnormal prion protein sensitive to protease. Ann Neurol. 2008;63:697–708. doi: 10.1002/ana.21420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Revesz T, Holton JL, Lashley T, Plant G, Frangione B, Rostagno A, Ghiso J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009;118:115–13. doi: 10.1007/s00401-009-0501-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Raymond GJ, Race B, Hollister JR, Offerdahl DK, Moore RA, Kodali R, Raymond LD, Hughson AG, Rosenke R, Long D, Dorward DW, Baron GS. Isolation of novel synthetic prion strain by amplification in transgenic mice co-expressing wild-type and anchorless prion proteins. J Virol. 2012;86:11763–11778. doi: 10.1128/JVI.01353-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jeffrey M, Goodsir CM, Bruce ME, McBride PA, Farquhar C. Morphogenesis of amyloid plaques in 87v murine scrapie. Neuropathol Appl Neurobiol. 1994;20:535–542. doi: 10.1111/j.1365-2990.1994.tb01007.x. [DOI] [PubMed] [Google Scholar]
- 33.Jeffrey M, McGovern G, Chambers EV, King D, González L, Manson JC, Ghetti B, Piccardo P, Barron RM. Mechanism of PrP-amyloid formation in mice without transmissible spongiform encephalopathy. Brain Pathol. 2012;22:58–66. doi: 10.1111/j.1750-3639.2011.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Walker LC, Callahan MJ, Bian F, Durham RA, Roher AE, Lipinski WJ. Exogenous induction of cerebral beta-amyloidosis in beta APP- transgenic mice. Peptides. 2002;23:1241–1247. doi: 10.1016/s0196-9781(02)00059-1. [DOI] [PubMed] [Google Scholar]
- 35.Kane MD, Lipinski WJ, Callahan MJ, Bian F, Durham RA, Schwarz RD, Roher AE, Walker LC, et al. Evidence for seeding of beta-amyloid by intracerebral infusion of Alzheimer brain extracts in beta-amyloid precursor protein-transgenic mice. J Neurosci. 2000;20:3606–3611. doi: 10.1523/JNEUROSCI.20-10-03606.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M. Soluble Aβ seeds are potent inducers of cerebral β amyloid deposition. J Neurosci. 2011;31(41):14488–14495. doi: 10.1523/JNEUROSCI.3088-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jeffrey M, McGovern G, Goodsir CM, Gonzalez L. Strain-associated variations in abnormal PrP trafficking of sheep scrapie. Brain Pathol. 2009;19:1–11. doi: 10.1111/j.1750-3639.2008.00150.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Carare RO, Bernardes-Silva M, Newman TA, Page AM, Nicoll JA, Perry VH, Weller RO. Solutes, but not cells, drain from the brain parenchyma along basement membranes of capillaries and arteries: significance for cerebral amyloid angiopathy and neuroimmunology. Neuropathol Appl Neurobiol. 2012;34:131–144. doi: 10.1111/j.1365-2990.2007.00926.x. [DOI] [PubMed] [Google Scholar]
- 39.Weller RO, Djuanda E, Yow HY, Carare RO. Lymphatic drainage of the brain and the pathophysiology of neurological disease. Acta Neuropathol. 2009;117:1–14. doi: 10.1007/s00401-008-0457-0. [DOI] [PubMed] [Google Scholar]
- 40.Iliff JJ, Wang M, Liao Y, Plogg BA, Peng W, Gundersen GA, Benveniste H, Vates GE, Deane R, Goldman SA, Nagelhus EA, Nedergaard M. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including Amyloid β. Sci Transl Med. 2012;4:147ra111. doi: 10.1126/scitranslmed.3003748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Weller RO, Subash M, Preston SD, Mazanti I, Carare RO. Perivascular drainage of amyloid-beta peptides from the brain and its failure in cerebral amyloid angiopathy and Alzheimer’s disease. Brain Pathol. 2008;18:253–266. doi: 10.1111/j.1750-3639.2008.00133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schroder R, Linke RP. Cerebrovascular involvement in systemic AA and AL amyloidosis: a clear haematogenic pattern. Virchows Arch. 1999;434:551–560. doi: 10.1007/s004280050383. [DOI] [PubMed] [Google Scholar]
- 43.Siso S, Jeffrey M, Gonzalez L. Neuroinvasion in sheep transmissible spongiform encephalopathies: the role of the haematogenous route. Neuropathol Appl Neurobiol. 2009;35:232–246. doi: 10.1111/j.1365-2990.2008.00978.x. [DOI] [PubMed] [Google Scholar]
- 44.Siso S, Jeffrey M, Martin S, Houston F, Hunter N, Gonzalez L. Pathogenetical significance of porencephalic lesions associated with intracerebral inoculation of sheep with the bovine spongiform encephalopathy [BSE] agent. Neuropathol Appl Neurobiol. 2009;35:247–258. doi: 10.1111/j.1365-2990.2009.01013.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Astrocytic and microglial activation:
a) Stratum lacunosum moleculare (SL) showing moderate astrocytosis and mild vacuolation. Bar = 200 μm. Immunohistochemistry for GFAP.
b) Stratum lacunosum moleculare showing mild microglial activation and mild vacuolation. Bar = 100 μm. Immunohistochemistry for coronin.
c) Severe astrocytosis in the superior colliculus associated with marked vacuolation. Bar = 200 μm. Immunohistochemistry for GFAP.
d) Severe microgliosis associated with marked vacuolation in the midbrain tectum. Bar = 100 μm. Immunohistochemistry for coronin.
e) Marked astrocytosis is present around several sub-ependymal plaques (arrowheads). Bar = 100 μm. Immunohistochemistry for GFAP.
f) Small numbers of activated microglia are present in the neuropil adjacent to amyloid plaques (arrowheads) in the peri-ventricular glial limitans. Bar = 100 μm. Immunohistochemistry for coronin.
Figure S2. Tau immunolabelling:
Cerebral cortex showing moderate vacuolation and punctuate tau immunolabelled structures. Bar = 50 μm. Immunohistochemistry for tau.
Figures S3. Area postrema:
a) A large areas of neuropil surrounding a blood vessel shows loss of normal architecture, increased extracellular space and a dystrophic neurite (d). Bar = 3 μm. Uranyl acetate / lead citrate.
b) Detail of an area similar to the above, showing loss of normal cellular processes and replacement by poorly defined fibrillar (arrowheads) PrPSSLOW amyloid fibrils. Bar = 0.5 μm. Immunogold for PrPSSLOW.
c) Accumulation of PrPSSLOW in association with convoluted and microfolded processes, and poorly defined fibrils (arrowheads) surrounding a large astrocytic process (A). Bar = 1 μm. Immunogold for PrPSSLOW.
Figures S4. Enlarged mitochondria:
Enlarged mitochondria with abnormal christae were mostly located in glial cell cytoplasm. A normal mitochondrion is present at M. Bar = −0.5 μm. Uranyl acetate/lead citrate.