

Abstract
Hypoxia-inducible factor (HIF)-1 is a transcription factor known to play an important role in regulating the innate immune response to infection. Under baseline conditions, cellular HIF-1 levels in leukocytes are scarce, but levels rise rapidly in response to hypoxia or molecular signals of infection or inflammation such as microbial surface molecules and host-derived cytokines. Innate immune cells such as macrophages, neutrophils and mast cells exhibit increased microbicidal activity when HIF-1 levels are increased, and mice lacking HIF-1 are more susceptible to invasive bacterial infection. In this study, we used genetic and pharmacologic means to determine whether HIF-1 also plays an important role in the adaptive immune response to infection. HIF-1α/Tie-2 Cre+ mice harboring a >90% knockdown of HIF-1 in myeloid cells were studied. We found antigen-presenting cells from these mice expressed lower levels of MHC-II and the costimulatory molecules CD80 and CD86, and were less able to induce T cell proliferation. These differences were present at baseline and persisted after activation. Increasing HIF-1 levels in WT cells by using the prolyl hydroxylase inhibitor drug AKB-4924 had the opposite effect, increasing MHC and co-stimulatory molecule expression and T cell proliferation. In experimental vaccination, HIF-1α/Tie-2 Cre+ mice exhibited a weaker T cell response and lower antibody levels in response to vaccination than WT mice, while WT mice treated with a drug to elevate HIF-1 responded more strongly to vaccination. Thus HIF-1 participates in bridging the innate and adaptive immune responses, and may merit further exploration as an adjuvant target.
INTRODUCTION
Antibiotics and childhood vaccinations drastically lowered the burden of infectious diseases in the 20th century, but in recent decades progress in combating infectious diseases has slowed. A better understanding of the factors which control the immune response to infection would help us design treatments or vaccine adjuvants that would improve our ability to control infectious diseases. Hypoxia-inducible factor (HIF) has been called a “master regulator” of innate immune function [1] because it plays a crucial role in enhancing the bactericidal activity of myeloid cells such as macrophages and neutrophils [2]. Its role in the adaptive immune response to infection has to date received less attention.
HIF is a basic helix-loop-helix transcription factor [3] first identified because of its role in erythropoietin regulation [4]. The transcriptional factor is a heterodimer composed of HIF-α and HIF-β subunits [3]. HIF-α is actually a family of three genes: HIF-1α, HIF-2α, and HIF-3α, of which HIF-1α is the best understood. HIF-3α is distantly related to HIF-1α and HIF-2α [5] and little is known about its function, although it may inhibit the activity of HIF-1α and HIF-2α [6]. The HIF-1α and HIF-2α subunits are closely related7, although they differ in tissue distribution: HIF-1α is ubiquitously expressed, whereas HIF-2α is most abundantly expressed in vascular endothelial cells during embryonic development [7].
Both HIF-α and HIF-β are constitutively produced, but in the presence of oxygen, HIF-α is rapidly degraded by oxygen-dependent mechanisms [8]. In the absence of oxygen, HIF-α is not degraded and instead translocates into the nucleus where it dimerizes with HIF-β and binds to hypoxia-response elements (HRE) in the promoter regions of more than 70 genes9. HIF has been shown to regulate genes involved in glycolysis, angiogenesis, cell differentiation, apoptosis, and others [8, 9].
Sites of infection and inflammation are hypoxic microenvironments, with oxygen tensions reported under 1% [10]. The low oxygen tension is due to the combination of reduced perfusion due to physical damage and a high density of activated inflammatory cells that place a high metabolic load on the area. Consequently, innate immune cells must execute their key antimicrobial functions under hypoxic conditions. HIF1-deficient myeloid cells exhibit reduced levels of cellular ATP (15–40% of normal), emphasizing the essential role of the transcriptional regulator for energy production through glycolysis in these immune cells [2]. Through HIF-1 induction under conditions of hypoxia, myeloid cells release more nitric oxide (NO), granule proteases and antimicrobial peptides, survive longer because of reduced apoptosis, and kill Gram-positive and -negative bacterial pathogens more efficiently than at normoxia [11, 12]. HIF activation is apparent during the differentiation of circulating monocytes into tissue macrophages [13], and may promote phagocytic uptake of bacteria under hypoxia [14].
Myeloid cells such as macrophages and dendritic cells (DC) play a crucial role in triggering the adaptive response to infection through their function as antigen-presenting cells (APCs), but our understanding of the role of HIF-1 in this critical aspect of host defense is much less developed. Research to examine potential roles of HIF in APCs has yielded contradictory results (reviewed by [15]), and much of the previous work has focused more closely upon the effects of hypoxia per se, rather than HIF transcriptional regulation, on DC activities. While some investigators have produced data that would indicate that hypoxia inhibits DC differentiation, maturation markers and antigen capture [16–18], others have come to exactly the opposite conclusion, namely that hypoxia promoted DC maturation both alone [19] and in combination with lipopolysaccharide (LPS) stimulus [17].
Hypoxia and HIF induction cannot be approached as functional equivalents in studies of the immune responses, because HIF is also induced at the transcriptional level by a variety of signals other than hypoxia, including markers of infection and inflammation such as cytokines [20, 21] and viral proteins [22, 23]. Furthermore, when HIF is activated by hypoxia, its contribution to transcriptional regulation comprises a different set of target genes than when it is activated by a TLR ligand such as LPS [24].
Here we contribute a new approach to our understanding of HIF in DC biology, by employing the genetic system of myeloid HIF-1α-null deficient mice. Coupled with pharmacological studies with a potent HIF-1 specific pharmacological agonist, we examine antigen presentation function in vitro and in vivo, lending support to a significant role of HIF regulation in these phenotypes.
Methods
Cell Culture
Bone marrow (BM) was obtained from the hind legs of mice age 8–16 weeks and grown in RPMI with 10% endotoxin-free FBS, 50 U/ml penicillin, 50 μg/ml streptomycin, 50 μg/ml gentamicin, 50 μM β2-mercaptoethanol, 10% conditioned media from GM-CSF-expressing B16 cells, and 20 ng/ml IL-4 (Peprotech). Media was changed on day 3 and 5 and the nonadherent cells were used between days 8–11. RAW264.7 macrophages were grown in RPMI with 10% FBS, 50 U/ml penicillin, and 50 μg/ml streptomycin.
Surface Marker Analysis
BM-derived DC from HIF-1α/Tie-2 Cre+ or Cre− mice differentiated for 8–11 d were plated in triplicate at 106 cells/well in 12-well plates and treated with LPS (10 or 100 ng/ml, as indicated) TNF (500 U/ml), or media alone, overnight. RAW264.7 macrophages were also plated in triplicate at 106 cells/well in 12-well plates and treated with AKB-4924 (10 or 100 μM, as indicated), the vehicle for AKB-4924 (cyclodextrin, at equivalent concentration), both LPS (100 ng/ml) and 4924, or vehicle alone overnight. The following day, the nonadherent DC or RAW macrophages were harvested, washed, and stained with the fluorescent anti-mouse antibodies CD11c-FITC, MHC-II-PE, CD86-PECy7, and CD80-APC. Fluorescence was measured by flow cytometry (FacsCalibur, BD Biosciences) and analyzed using FlowJo (TreeStar). Mean fluorescence was calculated on cells gated on CD11c+ cells.
Cytokine Protein Measurement
BM-derived DC from HIF-1α/Tie-2 Cre+ or Cre− mice differentiated for 8–11 d were plated in triplicate at 106 cells/well in 12-well plates and treated with LPS (10 or 100 ng/ml, as indicated) or media alone, overnight. For some experiments, BM-derived DC from C57BL/6 mice (Charles River) between days 8–11 of differentiation were treated with AKB-4924 (10 or 100 μM, as indicated), the vehicle for AKB-4924 (cyclodextrin, at equivalent concentration), both LPS (10 ng/ml) and AKB-4924, or vehicle alone overnight. The following day, the supernatant was harvested and protein levels measured by ELISA (IL-6, BD Biosciences #555240; IL-10, BD Biosciences #555252; IL-12 p70, BD Biosciences #555256; TNFα R&D Biosciences #DY410).
Cytokine RT-PCR
BM-derived DC from HIF-1α/Tie-2 Cre+ or Cre− mice that had been differentiated for 8 to11 days were plated in triplicate at 106 cells/well in 12-well plates and treated with LPS (10 or 100 ng/ml, as indicated) or media alone for 4 h. For some experiments, bone-marrow-derived dendritic cells from C57BL/6 mice (Charles River) between day 8 and day 11 of differentiation were treated with AKB-4924 (10 or 100 μM, as indicated), the vehicle for AKB-4924 (cyclodextrin, at equivalent concentration), both LPS (10 ng/ml) and 4924, or vehicle alone for four hours. RNA was obtained using the RNEasy Mini kit (Qiagen, Cat #74104), reverse transcribed to DNA using the iScript cDNA synthesis kit (BioRad, Cat#170-8890), and RT-PCR run using iQ SYBR Green Supermix (BioRad, Cat#170-8882). RT-PCR reaction conditions: 50°C for 10 min, 95°C for 5 min, 40 cycles of 95°C for 10 sec followed by 56°C for 30 sec, 95°C for 10 sec, and a melt curve from 55°C to 90°C in increments of 0.5°C for 5 sec each. Data was normalized using β-actin. Primer sequences and annealing temperatures used for RT-PCR analysis of β-actin (housekeeping control), IL-6, IL-8, IL-10, IL-12 and TNF-α are available on request.
T Cell Proliferation Assay
BM-derived DC from HIF-1α/Tie-2 Cre+ or Cre− mice differentiated for 8–11 days were harvested, washed, plated at 2 × 104 cells/well in a 96-well plate, and incubated overnight with the OT-I peptide SIINFEKL at 20 μg/ml or 0 μg/ml, or with the OT-II peptide OVA323–337 at 20 μg/ml or 0 μg/ml. The next day, CD8 T cells were obtained from the spleen and lymph nodes of naïve OT-I mice, and CD4+ T cells obtained from the spleen and lymph nodes of naïve OT-II mice using MACS purification kits (Miltenyi Biotec, CD4+ Cat #130-095-248; CD8α+ Cat#130-095-236). The T cells were added at 2 × 105 cells/well (a 1:10 DC:T cell ratio) in RPMI with 10% FBS, 2 mM L-glutamine, 50 U/ml penicillin, 50 μg/ml streptomycin, 50 μM β-mercaptoethanol, 10 mM HEPES and 100 μM MEM-NEAA. After 3 d incubation, 0.25 μCi/well of 3H-thymidine was added and the plate was incubated for an additional 8–16 h, at which point the cells were lysed by freezing at −80°C and read on an automated harvester (Becton Dickinson).
Vaccination Experiments
HIF-1α/Tie-2 Cre+ mice and age- and sex-matched Cre− controls were immunized subcutaneously (s.c.) on the back with 50 μg antigen (SIINFEKL or OVA) in 50 μl PBS emulsified with 50 μl IFA, for a total volume of 100 μl. In other experiments, wild-type C57BL/6 mice were treated with 5 mg/kg AKB-4924 or vehicle control by intraperitoneal (i.p.) injection 24 h before, 1 h before, and 16 h after immunization as above. The SIINFEKL-immunized mice were sacrificed on day 8, spleens were harvested, plated at 2 × 106 cells/well in a 96-well ELISPOT plate pre-coated with anti-IFNγ antibody, then re-stimulated with SIINFEKL at 2.5 μg/ml or with irrelevant OT-II peptide as a negative control. The number of IFNγ-producing cells in the whole splenocyte population was measured by ELISPOT (Mabtech). The OVA-immunized mice were bled on day 14 after immunization. ELISA plates were coated with 2 μg/ml OVA in PBS, and the OVA-specific IgG serum antibody titer was measured using anti-mouse IgG-HRP linked antibodies.
Statistical Analysis
Statistical significance was calculated by t test or two-way ANOVA, as appropriate for the data, using GraphPad Prism.
RESULTS
DC provide three signals to T cells: the antigen, presented in the context of MHC-I or MHC-II; costimulatory signals, achieved through ligation of surface molecules; and lastly, the release of cytokines and other soluble mediators. The combination of signals alerts the T cells to the foreign antigen, activates them, and modulates the strength and polarization of the adaptive immune response. To find out what role HIF plays in the ability of antigen-presenting cells (APCs) to stimulate the adaptive immune response, we used HIF-1α/Tie-2 Cre mice. These mice harbor a targeted deletion of HIF-1α in endothelial cells and hematopoietic precursors; HIF-1α/Tie2-Cre line exhibits 98% deletion efficiency in bone-marrow cells [25].
We first looked at the effect of HIF-1α ablation on the DC lineage itself. BM-derived DC from HIF-1α KO mice and Cre- controls showed decreased expression of key surface molecules. Immature DC lacking HIF expressed less MHC-II and the costimulatory molecules CD80 and CD86 at baseline, and when they were activated by overnight exposure to LPS the deficit persisted (Fig. 1A). Likewise, DC that had been matured by exposure to TNFα also expressed lower levels of MHC-II, CD80, and CD86 (Fig. 1B). These findings suggest that HIF-1α is important for the ability of activated DC to deliver the first and second activation signals to T cells.
Figure 1. Dendritic cells (DC) that lack HIF-1α express lower levels of MHC-II and costimulatory molecules.
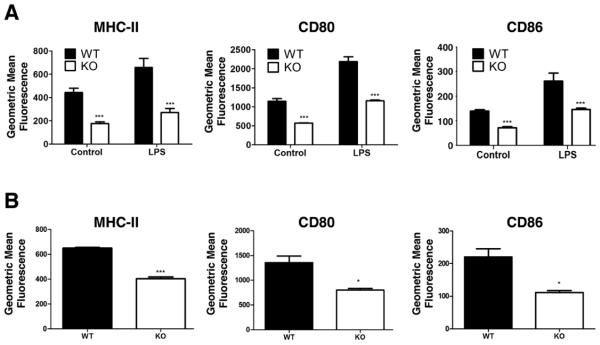
Immature BM-derived DC stimulated with 100 ng/ml LPS (A) or matured with 500 U/ml TNF-α (B) overnight and were evaluated for MHC-II, CD80 and CD86 expression using specific antibodies and FACS analysis. Statistical significance was calculated using (A) two-way ANOVA and (B) unpaired, two-tailed t-test; *P < .05, ***P < .001.
In contrast, we did not identify a requirement for HIF-1α in the LPS-stimulated release of cytokines by DC. Key cytokines known to play a role in the adaptive immune response were examined. IL-6 is a B cell differentiation factor, and it also may play a role in polarizing the T helper cell response, while IL-12 is a proinflammatory cytokine that promotes Th1 polarization and IL-10 is an anti-inflammatory cytokine that promotes Th2 polarization [26, 27]. TNFα enhances the proliferation of T cells. No differences were found for any of these cytokines at the level of protein (Fig. 2A) or mRNA transcript (Fig. 2B). This result distinguishes DC from other myeloid cells such as macrophages and neutrophils, in which we reported that deletion of HIF-1α modulated the release of certain pro-inflammatory cytokines [11].
Figure 2. HIF-1 is not essential for release of key cytokines from DC.
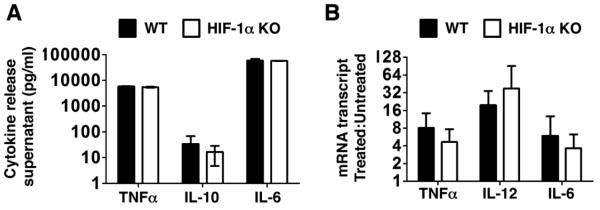
(A) BM-derived dendritic cells (DC) from WT and HIF-1α myeloid null mice were stimulated with LPS (10 ng/ml) overnight; cytokine release was quantified by ELISA. Data is representative experiment with triplicate samples, representative of 5 experiments with similar results. (B) DC were stimulated with LPS (10 ng/ml) for 4 h and cytokine transcripts monitored by real-time RT-PCR. Data shown is the pooled results of 5 experiments. Statistical significance was calculated using two-tailed t tests.
As a primary function of DC is to activate T cells, we next asked whether DC lacking HIF-1α were able to stimulate T cell proliferation. BM-derived DC were coated with the MHC-I restricted peptide SIINFEKL or the MHC-II restricted peptide OVA323–337, and then incubated with CD8+ T cells or CD4+ T cells from OT-I or OT-II mice, respectively. Both CD4+ and CD8+ T cells incubated with KO DC exhibited significantly less proliferation than those incubated with Cre− control DC (Fig. 3A, B), indicating that HIF-1α plays an important role in the ability of DC to trigger T cell proliferation.
Figure 3. HIF-1α is important for DC-triggered T cell proliferation and HIF-1α myeloid-KO mice have weakened responses to vaccination.

BM-derived DC were coated with the MHC II-restricted OVA subpeptide OVA323–329 and incubated with CD4+ T cells from OT-II mice (A) or coated with the MHC-I restricted OVA subpeptide SIINFEKL and incubated with CD8+ T cells from OT-I mice (B) for 3 days. T cell proliferation was measured by the incorporation of 3H thymidine. Data represent the normalized results of 3 (A) or 4 (B) experiments. (C) Six myeloid HIF-1α Tie-2 Cre+ and 6 Cre− littermate controls were vaccinated with 50 μg/mouse SIINFEKL peptide. Eight days after vaccination, mice were sacrificed and specific T cells in their spleens measured by IFNγ ELISPOT. (D) Myeloid HIF-1α Tie-2 Cre+ and Cre− mice were vaccinated with 50 μg/mouse OVA protein. Blood was collected 14 days after vaccination and OVA titer was measured using a specific ELISA. Data shown is the combined results of two separate experiments, for a total of 11 WT and 9 KO mice. Statistical significance was calculated by two-tailed t test. *P < .05, **P < .01, ***P < .001.
Since the loss of HIF-1α impairs the ability of APCs to present antigen, provide costimulatory signals, and stimulate T cell proliferation in vitro, we hypothesized that it should also impair the in vivo response to vaccination. Indeed, when HIF-1α KO mice were vaccinated with the ovalbumin subpeptide SIINFEKL, they produced fewer antigen-specific T cells than Cre− control mice (Fig. 3C). However, when serum antibody titers were examined, the effect was weaker. Although a trend of diminished response could be discerned, the effect of HIF-1α ablation on serum IgG titer in response to vaccination with OVA did not reach statistical significance (Fig. 3D).
Our evidence in the genetic model systems to this point suggested that loss of HIF-1α impairs the function of DC in promoting adaptive immune response. For the purpose of preventing or treating infectious disease, one could contemplate whether pharmacological agents that increase cellular HIF-1α levels could improve DC function in the immune response. Proof-of-principle studies were performed with RAW264.7 macrophages, showing that when HIF levels are increased with HIF-1α boosting drug AKB-4924 [28], the macrophages express higher levels of MHC-II, CD80, and CD86 (Fig. 4A), a result in agreement with the genetic studies in Figure 1. AKB-4924 inhibits the prolyl hydroxylases [28] that negatively regulate HIF-1α [29], and preferentially elevates HIF-1α over HIF-1β[28]. Also in agreement with our data from the knockout mice, we found that AKB-4924 had no effect on cytokine release from WT BM-derived DC (Fig. 4B), but did strengthen both the cell-mediated (Fig. 4C) and humoral (Fig. 4D) response to OVA immunization.
Figure 4. Pharmacological augmentation of HIF-1α increases DC activation and response to vaccination.

(A) RAW murine macrophages were treated with 100 μM AKB-4924 or vehicle (cyclodextrin) control overnight and surface marker expression measured by flow cytometry. (B) WT BM-derived DC were treated with 100 μM AKB-4924, 10 ng/ml LPS, both AKB-4924 and LPS, or vehicle control, and cytokine release measured by ELISA. (C and D) WT C57BL/6 mice (5 per group) were pretreated with AKB-4924 (5 mg/kg intraperitoneal) or vehicle control before immunization with 50 μg of (C) SIINFEKL peptide or (D) OVA protein. (C) Splenic T cell response was measured by IFNγ ELISPOT 8 days after immunization; (D) Anti-OVA serum IgG was measured by ELISA 14 days after immunization. Statistical significance was determined by two-tailed t-test; *P <.05, ***P < .001.
DISCUSSION
The data presented above indicates that HIF-1α plays a modest but discernable role in the ability of DC to trigger the adaptive immune response to infection. Loss of HIF-1α reduces the surface expression of major histocompatibility and costimulatory molecules on DC, leading to an impaired ability to stimulate both CD4+ and CD8+ T cell proliferation, without significant effects upon the release of cytokines. These changes correlate to a weaker cell-mediated and humoral response to vaccination. Elevating HIF with the drug AKB-4924 produces the opposite effects, enhancing DC antigen presentation and T cell stimulatory functions.
Our results provide new information relevant to a somewhat confounding literature regarding the influence of reduced oxygen tension on DC activity. Using murine [19] or human [30] DC, two groups separately found that while hypoxia alone did not affect antigen presentation or T cell activation, it did synergize with LPS to create a stronger effect than LPS alone. Along those lines, it was reported that hypoxic macrophages were better at activating T cells while releasing more of the Th1-polarizing cytokine IL-12 [31]; however, other groups arrived at opposite conclusions, suggesting hypoxic DC had impaired antigen presentation and T cell activation [32] and promoted a Th2 phenotype [33]. Still others have reported a mixed phenotype [16]. Differences experimental approaches used with regard to source and purity of DC precursors, differentiation/maturation protocols, degree and duration of the hypoxic stimulus, have been suggested as potential explanations for these discrepancies [15]. Our research was the first to couple genetically modified mice and pharmacological tools to distinguish the effect of the transcription factor HIF-1α specifically from more global, hypoxia-dependent effects.
The compound AKB-4924 has been found to stabilize HIF-1α activity in macrophages and neutrophils and to promote their direct bactericidal activity against drug-resistant pathogens [28]. This project was motivated in part by an interest in determining whether a similar approach to pharmacologically augment HIF-1α could be contemplated as an adjuvant strategy for vaccines. Drugs related to AKB-4924 that modulate prolyl hydroxylases have advanced in clinical trials of extended therapy to boost erythropoietin levels in anemia, showing a favorable safety profile to date [34]; thus their short term use in vaccines would likewise be feasible. Our preliminary results show that pharmacologically increasing HIF-1 levels has a modest effect to improve the response to experimental vaccination. Whether this effect could be harnessed and optimized to prove useful in adjuvant formulations merits further exploration.
ACKNOWLEDGEMENTS
We would like to thank Stephen Hedrick for providing us with the OT-I and OT-II mice, Robert Shalwitz and Anna Kotsakis of Aerpio Therapeutics for providing AKB-4924, and Davorka Messmer and Rebecca Saenz for their invaluable advice on experimental design. Research was provided by NIH/NIAID grants 093451 and 090863 to VN. TB was supported through the UCSD NIH/NIGMS Training Program in Molecular and Cellular Pharmacology (T32 007752) and the UCSD NIH/NIAMS Dermatology Investigator Training Program (T32 062496).
Footnotes
DISCLOSURE Randall Johnson, who provided the KO mice, is on SAB of Aerpio Therapeutics, who provided AKB-4924 HIF agonist. Victor Nizet and Aerpio Therapeutics have collaborated on NIH and Department of Defense Grants. Aerpio Therapeutics had no interest in design or reporting of the present study.
REFERENCES
- 1.Zarember KA, Malech HL. HIF-1α: a master regulator of innate host defenses? J Clin Invest. 2005;115:1702–1704. doi: 10.1172/JCI25740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cramer T, Yamanishi Y, Clausen BE, Forster I, Pawlinski R, Mackman N, Haase VH, Jaenisch R, Corr M, Nizet V, et al. HIF-1α is essential for myeloid cell-mediated inflammation. Cell. 2003;112:645–657. doi: 10.1016/s0092-8674(03)00154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci U S A. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Semenza GL, Wang GL. A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol. 1992;12:5447–5454. doi: 10.1128/mcb.12.12.5447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gu YZ, Moran SM, Hogenesch JB, Wartman L, Bradfield CA. Molecular characterization and chromosomal localization of a third alpha-class hypoxia inducible factor subunit, HIF3α. Gene Expr. 1998;7:205–213. [PMC free article] [PubMed] [Google Scholar]
- 6.Hara S, Hamada J, Kobayashi C, Kondo Y, Imura N. Expression and characterization of hypoxia-inducible factor (HIF)-3alpha in human kidney: suppression of HIF-mediated gene expression by HIF-3alpha. Biochem Biophys Res Commun. 2001;287:808–813. doi: 10.1006/bbrc.2001.5659. [DOI] [PubMed] [Google Scholar]
- 7.Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A. 1997;94:4273–4278. doi: 10.1073/pnas.94.9.4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weidemann A, Johnson RS. Biology of HIF-1α. Cell Death Differ. 2008;15:621–627. doi: 10.1038/cdd.2008.12. [DOI] [PubMed] [Google Scholar]
- 9.Wenger RH, Stiehl DP, Camenisch G. Integration of oxygen signaling at the consensus HRE. Sci STKE. 2005;2005:re12. doi: 10.1126/stke.3062005re12. [DOI] [PubMed] [Google Scholar]
- 10.Silver IA. Tissue PO2 changes in acute inflammation. Adv Exp Med Biol. 1977;94:769–774. doi: 10.1007/978-1-4684-8890-6_106. [DOI] [PubMed] [Google Scholar]
- 11.Peyssonnaux C, Datta V, Cramer T, Doedens A, Theodorakis EA, Gallo RL, Hurtado-Ziola N, Nizet V, Johnson RS. HIF-1α expression regulates the bactericidal capacity of phagocytes. J Clin Invest. 2005;115:1806–1815. doi: 10.1172/JCI23865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mecklenburgh KI, Walmsley SR, Cowburn AS, Wiesener M, Reed BJ, Upton PD, Deighton J, Greening AP, Chilvers ER. Involvement of a ferroprotein sensor in hypoxia-mediated inhibition of neutrophil apoptosis. Blood. 2002;100:3008–3016. doi: 10.1182/blood-2002-02-0454. [DOI] [PubMed] [Google Scholar]
- 13.Oda T, Hirota K, Nishi K, Takabuchi S, Oda S, Yamada H, Arai T, Fukuda K, Kita T, Adachi T, et al. Activation of hypoxia-inducible factor 1 during macrophage differentiation. Am J Physiol Cell Physiol. 2006;291:C104–113. doi: 10.1152/ajpcell.00614.2005. [DOI] [PubMed] [Google Scholar]
- 14.Anand RJ, Gribar SC, Li J, Kohler JW, Branca MF, Dubowski T, Sodhi CP, Hackam DJ. Hypoxia causes an increase in phagocytosis by macrophages in a HIF-1α-dependent manner. J Leukoc Biol. 2007;82:1257–1265. doi: 10.1189/jlb.0307195. [DOI] [PubMed] [Google Scholar]
- 15.Sica A, Melillo G, Varesio L. Hypoxia: a double-edged sword of immunity. J Mol Med (Berl) 2011;89:657–665. doi: 10.1007/s00109-011-0724-8. [DOI] [PubMed] [Google Scholar]
- 16.Mancino A, Schioppa T, Larghi P, Pasqualini F, Nebuloni M, Chen IH, Sozzani S, Austyn JM, Mantovani A, Sica A. Divergent effects of hypoxia on dendritic cell functions. Blood. 2008;112:3723–3734. doi: 10.1182/blood-2008-02-142091. [DOI] [PubMed] [Google Scholar]
- 17.Elia AR, Cappello P, Puppo M, Fraone T, Vanni C, Eva A, Musso T, Novelli F, Varesio L, Giovarelli M. Human dendritic cells differentiated in hypoxia down-modulate antigen uptake and change their chemokine expression profile. J Leukoc Biol. 2008;84:1472–1482. doi: 10.1189/jlb.0208082. [DOI] [PubMed] [Google Scholar]
- 18.Ricciardi A, Elia AR, Cappello P, Puppo M, Vanni C, Fardin P, Eva A, Munroe D, Wu X, Giovarelli M, Varesio L. Transcriptome of hypoxic immature dendritic cells: modulation of chemokine/receptor expression. Mol Cancer Res. 2008;6:175–185. doi: 10.1158/1541-7786.MCR-07-0391. [DOI] [PubMed] [Google Scholar]
- 19.Jantsch J, Chakravortty D, Turza N, Prechtel AT, Buchholz B, Gerlach RG, Volke M, Glasner J, Warnecke C, Wiesener MS, et al. Hypoxia and hypoxia-inducible factor-1α modulate lipopolysaccharide-induced dendritic cell activation and function. J Immunol. 2008;180:4697–4705. doi: 10.4049/jimmunol.180.7.4697. [DOI] [PubMed] [Google Scholar]
- 20.Hellwig-Burgel T, Rutkowski K, Metzen E, Fandrey J, Jelkmann W. Interleukin-1β and tumor necrosis factor-alpha stimulate DNA binding of hypoxia-inducible factor-1. Blood. 1999;94:1561–1567. [PubMed] [Google Scholar]
- 21.Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L. IL-1beta-mediated up-regulation of HIF-1α via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J. 2003;17:2115–2117. doi: 10.1096/fj.03-0329fje. [DOI] [PubMed] [Google Scholar]
- 22.Wakisaka N, Kondo S, Yoshizaki T, Murono S, Furukawa M, Pagano JS. Epstein-Barr virus latent membrane protein 1 induces synthesis of hypoxia-inducible factor 1α. Mol Cell Biol. 2004;24:5223–5234. doi: 10.1128/MCB.24.12.5223-5234.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sodhi A, Montaner S, Patel V, Zohar M, Bais C, Mesri EA, Gutkind JS. The Kaposi's sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1α. Cancer Res. 2000;60:4873–4880. [PubMed] [Google Scholar]
- 24.Jantsch J, Wiese M, Schodel J, Castiglione K, Glasner J, Kolbe S, Mole D, Schleicher U, Eckardt KU, Hensel M, et al. Toll-like receptor activation and hypoxia use distinct signaling pathways to stabilize hypoxia-inducible factor 1 α (HIF1α) and result in differential HIF1α-dependent gene expression. J Leukoc Biol. 2011;90:551–562. doi: 10.1189/jlb.1210683. [DOI] [PubMed] [Google Scholar]
- 25.Tang N, Wang L, Esko J, Giordano FJ, Huang Y, Gerber HP, Ferrara N, Johnson RS. Loss of HIF-1α in endothelial cells disrupts a hypoxia-driven VEGF autocrine loop necessary for tumorigenesis. Cancer Cell. 2004;6:485–495. doi: 10.1016/j.ccr.2004.09.026. [DOI] [PubMed] [Google Scholar]
- 26.Blanco P, Palucka AK, Pascual V, Banchereau J. Dendritic cells and cytokines in human inflammatory and autoimmune diseases. Cytokine Growth Factor Rev. 2008;19:41–52. doi: 10.1016/j.cytogfr.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Moore KW, de Waal Malefyt R, Coffman RL, O'Garra A. Interleukin-10 and the interleukin-10 receptor. Annu Rev Immunol. 2001;19:683–765. doi: 10.1146/annurev.immunol.19.1.683. [DOI] [PubMed] [Google Scholar]
- 28.Okumura CY, Hollands A, Tran DN, Olson J, Dahesh S, von Kockritz-Blickwede M, Thienphrapa W, Corle C, Jeung SN, Kotsakis A, et al. A new pharmacological agent (AKB-4924) stabilizes hypoxia inducible factor-1 (HIF-1) and increases skin innate defenses against bacterial infection. J Mol Med (Berl) 2012;90:1079–1089. doi: 10.1007/s00109-012-0882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG., Jr. HIFα targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science. 2001;292:464–468. doi: 10.1126/science.1059817. [DOI] [PubMed] [Google Scholar]
- 30.Spirig R, Djafarzadeh S, Regueira T, Shaw SG, von Garnier C, Takala J, Jakob SM, Rieben R, Lepper PM. Effects of TLR agonists on the hypoxia-regulated transcription factor HIF-1α and dendritic cell maturation under normoxic conditions. PLoS One. 2010;5:e0010983. doi: 10.1371/journal.pone.0010983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Acosta-Iborra B, Elorza A, Olazabal IM, Martin-Cofreces NB, Martin-Puig S, Miro M, Calzada MJ, Aragones J, Sanchez-Madrid F, Landazuri MO. Macrophage oxygen sensing modulates antigen presentation and phagocytic functions involving IFN-gamma production through the HIF-1α transcription factor. J Immunol. 2009;182:3155–3164. doi: 10.4049/jimmunol.0801710. [DOI] [PubMed] [Google Scholar]
- 32.Wang Q, Liu C, Zhu F, Liu F, Zhang P, Guo C, Wang X, Li H, Ma C, Sun W, Zhang Y, Chen W, Zhang L. Reoxygenation of hypoxia-differentiated dentritic cells induces Th1 and Th17 cell differentiation. Mol Immunol. 2010;47:922–931. doi: 10.1016/j.molimm.2009.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang M, Ma C, Liu S, Sun J, Shao Q, Gao W, Zhang Y, Li Z, Xie Q, Dong Z, Qu X. Hypoxia skews dendritic cells to a T helper type 2-stimulating phenotype and promotes tumour cell migration by dendritic cell-derived osteopontin. Immunology. 2009;128:e237–249. doi: 10.1111/j.1365-2567.2008.02954.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bernhardt WM, Wiesener MS, Scigalla P, Chou J, Schmieder RE, Gunzler V, Eckardt KU. Inhibition of prolyl hydroxylases increases erythropoietin production in ESRD. J Am Soc Nephrol. 2010;21:2151–2156. doi: 10.1681/ASN.2010010116. [DOI] [PMC free article] [PubMed] [Google Scholar]