

Abstract
Smad4 loss occurs frequently in human skin squamous cell carcinoma (SCC), but it is unknown if this loss increases ultraviolet-induced (UV) carcinogenesis, a major etiological factor in skin cancer. In the present study, mice with keratinocyte-specific Smad4 deletion (K14.Smad4−/−) and wildtype (WT) littermates were chronically UV-irradiated. Compared to WT, K14.Smad4−/− mice exhibited increased DNA damage and increased susceptibility to UV-induced skin cancer. Among genes involved in repairing UV-induced DNA damage, Excision repair cross-complementation group1 (Ercc1) mRNA was significantly reduced in UV treated K14.Smad4−/− skin compared to WT skin. Further analysis revealed that Smad4 loss confers reduced Snail binding to the Ercc1 regulatory elements, resulting in reduced Ercc1 transcription. Consistently, transient transfection of Snai1 into Smad4−/− keratinocytes led to increased repair of UV-induced DNA lesions. Transfection of Ercc1 into Smad4−/− keratinocytes restored repair of UV-induced DNA damage. Further, immunostaining revealed that the presence of Smad4 protein is associated with the presence of Snail and Ercc1 proteins in human skin SCC and precancerous actinic keratoses (AK). Collectively, Smad4 loss associated Snail reduction compromises Ercc1-mediated DNA repair, contributing to increased UV-induced skin carcinogenesis. Thus we identified a role for Snail in UV-induced DNA repair.
Introduction
Solar ultraviolet (UV) radiation is the primary cause of skin cancer. Non-melanoma skin cancer, including basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), is the most common form of skin cancer (Foster et al., 2008). Solar UV radiation is comprised of UVA (315–400nm), UVB (280–315nm) and UVC (100–280nm), wherein UVB is a strong carcinogen (de Gruijl and Van der Leun, 1994; Narayanan et al., 2010).
The transcription factor Smad4 is a signaling mediator for transforming growth factor β (TGFβ), activin, and bone morphogenetic protein (BMP) (Moustakas et al., 2001). Smad4 down regulation, at the gene level through loss of heterozygosity (LOH) or at the protein level, is frequent in human skin SCCs (Hoot et al., 2008). Mice with keratinocyte-specific Smad4 deletion develop spontaneous SCCs (Qiao et al., 2006; Yang et al., 2005). We have found that Smad4 loss in the oral mucosa causes functional defects in the Fanconi Anemia (FA) and BRCA1 DNA repair pathways, and spontaneous head and neck SCC (Bornstein et al., 2009). It remains to be determined if Smad4 loss affects repair of other types of DNA damage. Because UV-induced carcinogenesis is primarily initiated by DNA damage, and not all DNA repair genes are in the FA pathway, we sought to determine if Smad4 plays a role in the repair of UV-induced DNA damage and thus Smad4 loss increases susceptibility to UV-induced skin carcinogenesis.
UV radiation causes DNA damage primarily due to generation of cyclobutane pyrimidine dimers (CPD) and pyrimidine pyrimidone (6-4PP) photoproducts. CPD and 6-4PP are repaired primarily by the nucleotide excision repair (NER) pathway (Leibeling et al., 2006). Xeroderma pigmentosum (XP) patients with defective NER exhibit 1,000 fold increased susceptibility to skin cancer (Kraemer et al., 1987). There are seven XP genes involved in this pathway, XPA through XPG (Leibeling et al., 2006). Ercc1 is an endonuclease that, along with XPF, excises a short oligonucleotide encompassing a UV-induced lesion, which is followed by replicative DNA polymerases filling in the gap, using the complementary strand as the template (Nouspikel, 2009). Keratinocyte specific deletion of Ercc1 in mice causes hypersensitivity to UV-induced skin cancer, demonstrating the critical role of Ercc1 in repairing UV-induced DNA damage (Doig et al., 2006). Genetic mutation or loss of Ercc1 is rare in humans. In this report, we examined the effect of keratinocyte specific Smad4 deletion on UV-induced skin carcinogenesis and focused our molecular analysis on repair of UV-induced DNA damage. We provide evidence that Smad4 regulates Ercc1 expression through Snail-mediated transcription, thus Smad4 deletion contributed to increased susceptibility to UV carcinogenesis.
Results
Epidermal Smad4 deletion causes increased susceptibility to UV-induced SCC
We generated mice with epidermal specific deletion of Smad4 by mating K14.CrePR1 mice with Smad4 floxed mice (Smad4f/f) (Berton et al., 2000). Mice with Cre-mediated Smad4 deletion in keratinocytes were defined as K14.Smad4−/− mice (n=29). Monogenic K14CrePR1 and Smad4f/f mice (n=25) with normal Smad4 expression level (Figure 1a), were used as controls, (Figure 1b). All mice are in the C57BL/6 background. Both groups of mice were shaved and subjected to UVA and UVB irradiation three times a week for 40 weeks. We started with a dose of 1 J/cm2 of UVA and 0.12 J/cm2 of UVB, which did not induce erythema in either group. Therefore, we increased UV irradiation to 10 J/cm2 of UVA and 1.2 J/cm2 of UVB to induce erythema and gradually reduced UV doses (detailed in Figure S1) to enable long-term UV irradiation without morbidity due to severe UV-induced damage, ending with 4.3 J/cm2 of UVA and 1.74 J/cm2 of UVB. Without UV exposure, K14.Smad4−/− mice did not develop spontaneous skin tumors, unlike previously reports for MMTV.Smad4−/− or K5.Smad4−/− mice (Qiao et al., 2006; Yang et al., 2005) presumably due to patchy and weak Cre activity in K14.CrePR1 mice (Vassar et al., 1989; Wagner et al., 2001; Wang et al., 1997; Zhou et al., 2002). Interestingly, within 10 weeks of UV irradiation, K14.Smad4−/− mice started to develop skin tumors, and by 40 weeks, 24 out of 29 mice developed skin tumors (Figure 1b). In contrast, none of the control mice developed tumors during the study period (Figure1b), consistent with previous reports that C57BL/6 mice are extremely resistant to UV carcinogenesis (Strickland and Swartz, 1987). Histological examination revealed that UV-induced K14.Smad4−/− tumors were SCCs (Figure 1c).
Figure 1. Smad4−/− mice developed UV-induced skin tumors.
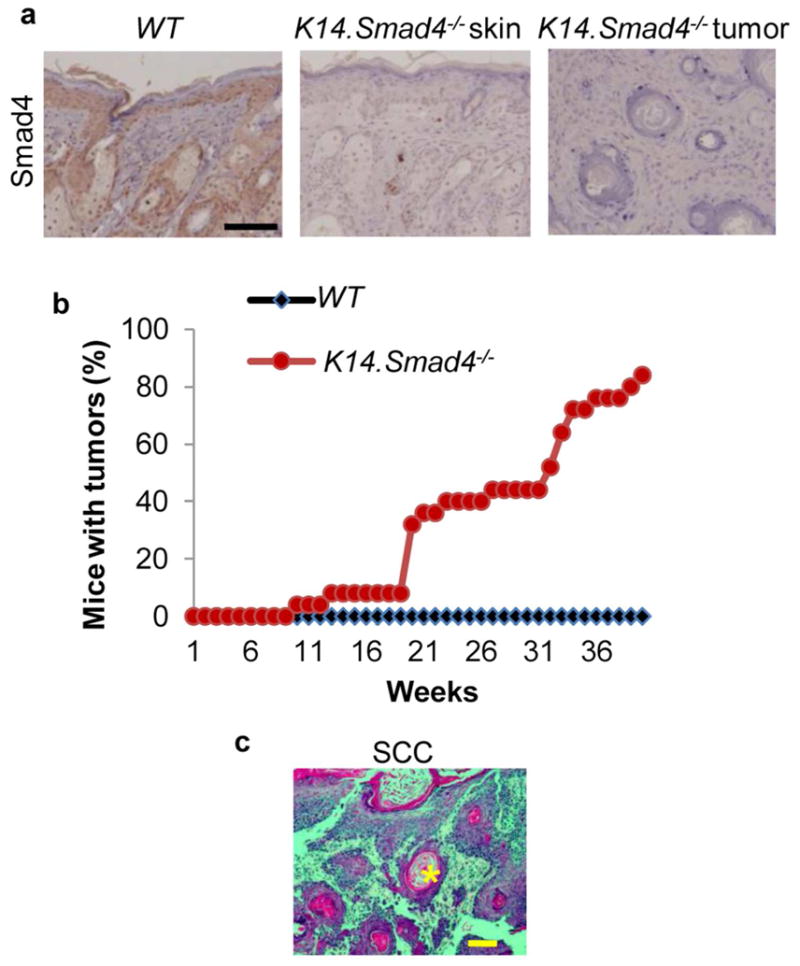
(a) IHC staining of Smad4 in skin samples from UV-irradiated WT and Smad4−/− mice. (b) Kinetics of tumor formation: data points represent mice that developed tumors, expressed as a percentage of mice with tumors among the total number of mice in that genotype group. (c) H&E staining showing a typical UV-induced K14.Smad4−/− SCC morphology. Keratin pearl is highlighted with an asterisk. Scale bars: 100μm.
Smad4−/− keratinocytes exhibit defects in repairing UV-induced DNA damage
We analyzed expression of phosphorylated H2AX (p-H2AX), a marker of DNA damage (Hanasoge and Ljungman, 2007; Marti et al., 2006), in UV-irradiated WT skin and K14.Smad4−/− skin and tumors. K14.Smad4−/− skin and tumors showed a 68% and a 192% increase, respectively, in the number of p-H2AX positive cells compared to WT skin (Figure 2a, 2b). This data suggested that DNA repair of UV-induced lesions may be reduced in the skin of K14.Smad4−/− mice. To determine if increased DNA damage in UV-irradiated K14.Smad4−/− skin reflects inherent defects in DNA repair after Smad4 loss, we assessed levels of spontaneous DNA damage in primary keratinocytes from neonatal K14.Smad4−/− and WT skin by alkaline comet assay (Figure 2c). Smad4−/− keratinocytes exhibit a 16.5% increase in comet tail DNA compared to WT keratinocytes, representing higher numbers of double and single strand DNA breaks (DSB and SSB) (Figure 2d).
Figure 2. Increased DNA damage in UV-irradiated K14.Smad4−/− skin, and reduced repair of CPD in Smad4−/− keratinocytes.

(a) Immunofluorescence staining of p-H2AX in UV-irradiated mouse skin and tumors. Keratin 14 was used as counterstain. Scale bar is 100μm. (b) Quantification of p-H2AX staining, n=6, 8 and 4 for WT skin, Smad4−/− skin, Smad4−/− tumors, respectively. (c) Alkaline comet assay using WT and Smad4−/− keratinocytes. (d) Quantification of % DNA in comet tail. n=44. (e) CPD levels in UVB-irradiated WT and Smad4−/− keratinocytes was measured by ELISA. CPD level in Smad4−/− keratinocytes at 10 minutes was set at 100%. P ≥ 0.1 at 5, 10 and 30 minutes, respectively. *denotes p ≤ 0.0002, and ** denotes p ≤ 0.003, n=3. Error bars are S.E.M. (all panels).
To focus on UV-induced DNA damage, we measured CPD levels in WT and Smad4−/− keratinocytes irradiated with 60 mJ/cm2 of UVB (Figure 2e). Consistent with a previous report (Nguyen et al., 2010), CPD levels within the first 30 minutes reflected the UV dose and there was no statistically significant difference between the two cell types. Afterwards, CPD levels began to decline in WT cells presumably due to DNA repair. In contrast, CPD levels remain unchanged in Smad4−/− keratinocytes. These results suggest that accumulated DNA damage is a direct effect of Smad4 loss in keratinocytes.
Reduced expression of Ercc1 mRNA and protein in UV-irradiated K14.Smad4−/− skin
To dissect the mechanism underlying reduced DNA repair in Smad4−/− keratinocytes, we compared gene expression in WT and Smad4−/− keratinocytes using a mouse DNA damage signaling pathway RT-PCR array and found reduced expression of Ercc1, Checkpoint homolog (Hus1), Checkpoint kinase 1 (Chk1), Exonuclease 1 (Exo1), Chromatin assembly factor 1a (Chaf1a) and Meiotic recombination 11 homolog A (Mre11) genes in Smad4−/− cells (Figure 3a). Among DNA repair genes down regulated in Smad4−/− keratinocytes, none are documented direct Smad4 targets (Koinuma et al., 2009). Hence, the effect of Smad4 on these genes requires additional mediators, which could vary among these targets. We began with examination of Ercc1, which has an indispensable role in excisional repair of UV-induced damage (Chipchase and Melton, 2002) and was the most down regulated gene in Smad4−/− keratinocytes (Figure 3a). We measured Ercc1 mRNA levels in UV-irradiated WT and K14.Smad4−/− skin and observed an 81% reduction of Ercc1 mRNA level in Smad4−/− skin compared to WT skin (Figure 3b). Ercc1 immunostaining of UV-irradiated WT skin was localized to the nucleus, it was dramatically reduced in K14.Smad4−/− epidermis and in K14.Smad4−/− SCCs (Figure 3c).
Figure 3. Reduced expression of DNA repair genes in Smad4−/− keratinocytes, reduced Ercc1 in K14.Smad4−/− skin and tumors.

(a) qRT-PCR measured mRNA levels of indicated genes in WT and Smad4−/− keratinocytes. The mRNA level of each gene in WT keratinocytes was set at 1. * p≤ 0.002. Error bars are S.E.M., n=3. (b) mRNA levels of Smad4 and Ercc1 in UV-irradiated WT and K14.Smad4−/− skin. * p≤ 0.02. Error bars are S.E.M., n=3 for WT skin and n=7 for Smad4−/− skin. (c) Immunohistochemical staining of Ercc1 was performed on UV-irradiated WT and K14.Smad4−/− skin and tumors.
Reduced expression of Ercc1 mRNA and protein in K14.Smad4−/− skin is a result of reduced Snail expression
To further study mechanisms of Ercc1 reduction we searched for putative transcription factor binding sites in the 2kb promoter region of the mouse Ercc1 gene using P-Match software. We did not find putative Smad4 binding sites, but instead found several putative binding sites (CANNTG) for the transcription factor Snail in the promoter and near the transcription start site (TSS) of Ercc1 (Figure S4a). Snail, a protein encoded by the SNAI1 gene, is a direct Smad4 target (Hoot et al., 2008; Peinado et al., 2003) so we investigated the possibility of Smad4 regulation of Ercc1 through Snail. Next, we examined Snai1 expression level by qRT-PCR in UV-irradiated WT and Smad4−/− keratinocytes (Figure 4a). Snai1 mRNA was induced by UV irradiation in both keratinocytes, although the level in Smad4−/− keratinocytes continued to be lower than WT at all time points (Figure 4a). Similarly, we found an 86% reduction in Snai1 level in UV-irradiated K14.Smad4−/− skin compared to UV-irradiated WT skin (Figure 4b). We also examined Ercc1 mRNA levels in these samples and, consistent with a previous report (Murakami et al., 2001), it was induced by UV irradiation in WT and Smad4−/− keratinocytes, but the level was 2-fold lower in Smad4−/− keratinocytes than in WT keratinocytes with or without UV irradiation (Figure S2).
Figure 4. Snail regulates Ercc1 expression, Snail and Ercc1 mediate CPD repair in keratinocytes.

(a) Snai1 mRNA level in UV-irradiated keratinocytes, the level in untreated WT keratinocytes was set at 1. (b) Snai1 mRNA level in UV-irradiated mice skin, the level in UV-irradiated WT skin was set at 1, n=4 for WT, n=7 for Smad4−/−. * denotes p= 0.027 (c) Ercc1 mRNA level in human Snai1-transfected keratinocytes, the level in empty vector transfected WT keratinocytes was set at 1. ** denotes p≤ 0.002. (d) Snail Chromatin Immunoprecipitation from WT and Smad4−/− keratinocyte extracts with/without UV irradiation. * p<0.03, ** p< 0.005. (e) CPD level in UVB-irradiated Snai1 transfected keratinocytes *p = 5.09E−5, ** p=0.005, *** p=0.01. (f) CPD level in UVB-irradiated Ercc1 transfected keratinocytes, *=0.026, **=0.046, *** p=0.10, n=3. Error bars: S.E.M.
To further test if reduced Snail causes reduced Ercc1 expression in Smad4−/− keratinocytes, we overexpressed human Snail in WT and Smad4−/− keratinocytes and observed that Ercc1 transcripts were up regulated in Snai1 transfected WT and Smad4−/− keratinocytes (Figure 4c). Expression of human Snai1 mRNA was equivalent in the two cell lines upon transfection (Figure S3). Because Snail has been shown to induce Ercc1 expression in head and neck cancer (Hsu et al., 2010), we examined if Smad4 loss causes reduced Snail binding to Ercc1 regulatory elements. Using chromatin immunoprecipitation assays, we detected direct Snail binding to the Ercc1 regulatory region encompassing the putative Snail binding sites at 124–129 bp (Site1, Sup. Figure 4a), 276–281 bp, and 300–305 bp (Site 2, Figure S4a) from the TSS of the mouse Ercc1 gene. The locations of the Snail binding sites in the mouse Ercc1 gene are similar to those in the human Ercc1 gene, where they are important for Snail-mediated transcription (Hsu et al., 2010). Snail binding to the Ercc1 regulatory region was decreased to 35% at site 1 and 32% at site 2 in Smad4−/− keratinocytes prior to UV irradiation (Figure 4d, Figure S4b). In UV-irradiated WT keratinocytes, similar to Snail mRNA levels (Figure 4a), Snail binding to both sites was significantly increased at 12 and 24 hours, compared to non-irradiated keratinocytes (Figure 4d). Although Snail binding at both sites was also increased in UV-irradiated Smad4−/− keratinocytes compared to non-irradiated Smad4−/− keratinocytes, its binding levels were significantly lower than in WT keratinocytes at both 12 and 24 hours (Figure 4d). Collectively, these results suggest that reduced Snail levels in K14.Smad4−/− skin lead to reduced Ercc1 transcription.
Restoration of Snail or Ercc1 expression in Smad4−/− keratinocytes rescues DNA repair defect
We assessed if Snail restoration in Smad4−/− keratinocytes could attenuate defects in repair of UV-induced DNA damage. WT and Smad4−/− keratinocytes were transfected with a human Snail expression plasmid or empty vector, irradiated with UVB, and CPD level was measured by ELISA at different time points during recovery of these cells. Remarkably, we observed a dramatic decrease in CPD adducts upon Snai1 transfection in Smad4−/− cells at 1 and 3 hour time points following UV irradiation (Figure 4e).
Subsequently, we examined the contribution of reduced Ercc1 expression to the DNA repair defect in Smad4−/− cells. Transfection of human Ercc1 expression vector into Smad4−/− keratinocytes led to a dramatic reduction of CPD level at 1 and 3 hours following UV irradiation, compared to empty vector transfected keratinocytes (Figure 4f), suggesting that reduced Ercc1 is a major contributor to the DNA repair defect observed in these cells (Figure 4f).
Expression of Smad4 is associated with expression of Snail and Ercc1 proteins in human skin SCC and actinic keratoses (AK)
Data from our mouse model suggest that Smad4 mediated Snail expression leads to Ercc1 expression. If this finding applies to human SCCs, we would expect the presence of Smad4 protein to be associated with the presence of Snail and Ercc1 proteins, and conversely the absence of Smad4 to be associated with the absence of Snail and Ercc1. Immunostaining in a tissue array with 76 skin SCC samples showed Smad4 expression was lost in 47/76 (62%) cases, consistent with our previous report (Hoot et al., 2008), and 43/76 (57%) cases expressed Ercc1 (Figure 5a, b, Table S1). Consistent with our previous report (Hoot et al., 2008), a majority of cases (57/76, 75%) were Snail positive. We observed a statistically significant association between expression of Smad4 and Snail proteins in SCC. Further, most cases positive for Snail (67%) were also positive for Ercc1 and conversely, most cases negative for Snail (74%) were also negative for Ercc1. Finally, we found a statistically significant association between expression of Smad4 and Ercc1 proteins. Most Smad4 positive cases (86%) were also positive for Ercc1 and conversely, the majority of Smad4 negative cases (60%) were Ercc1 negative.
Figure 5. Expression of Smad4 is associated with expression of Snail and Ercc1 proteins in human skin SCC and AK.

Smad4, Snail and Ercc1 were stained in a human skin SCC tissue array (a, b) and human actinic keratosis (c, d) Representative images of a Smad4, Snail and Ercc1 triple negative case (a, c) and triple positive case (b, d). Scale bar: 100μm for all panels.
To further determine if the above associations occur in the early stages of skin carcinogenesis thereby suggesting a causal role of Smad4 loss-mediated DNA damage in skin carcinogenesis, we performed the above immunostaining in 28 human AK samples (Figure 5c, d, Table S1). We found loss of Smad4 protein in 16/28 (57%) AK cases, suggesting Smad4 loss is an early event during UV-induced skin carcinogenesis, similar to our findings in head and neck cancer (Bornstein et al., 2009). Analogous to our above observations in SCC, we observed statistically significant associations between expression of i) Smad4 and Snail ii) Snail and Ercc1 and iii) Smad4 and Ercc1 proteins.
Discussion
In the present study we examined the contribution of Ercc1 to UV-induced skin cancer. Among molecules related to UV-induced DNA repair, Ercc1 was the most down regulated in Smad4−/− keratinocytes, and transfection of an Ercc1 plasmid into these cells rescued their defective repair of UV-induced CPDs, suggesting that Ercc1 is an important contributor to the UV-susceptibility of K14.Smad4−/− mice. Several other DNA repair genes that are down regulated in Smad4−/− keratinocytes have also been reported to regulate DNA damage signaling, cell cycle checkpoints, chromatin assembly after DNA damage, and DSB repair (Chen and Sanchez, 2004; Martini et al., 1998; Niida and Nakanishi, 2006; Olson et al., 2007; Sertic et al., 2011). It remains to be determined whether reduced levels of these genes contribute to the increased susceptibility to UV-induced skin cancer in K14.Smad4−/− mice.
Our data suggest that Smad4-dependent Snai1 expression is one of the major mechanisms inducing Ercc1 expression during UV carcinogenesis. First, loss of Smad4 led to reduced Snai1 expression and reduced Snail binding to the Ercc1 regulatory binding sites; second, Snail activated Ercc1 expression in keratinocytes. Third, we observed co-expression of Smad4, Snail and Ercc1 proteins in human skin SCCs and precancerous AKs. Similar expression patterns of Snail and Ercc1 have also been observed in head and neck cancer, and bladder cancer (Hsu et al., 2010; Kawashima et al., 2012). Thus, Snail regulated Ercc1 expression may occur broadly in human cancers.
Snail is an important mediator of epithelial to mesenchymal transition (EMT) and thereby plays key roles in normal morphogenetic movements in the embryo and cancer cell migration promoting metastasis in a variety of tumor types, including skin cancer (Batlle et al., 2000; Cano et al., 2000). Additionally, Snail is shown to promote skin cancer through inducing inflammation (Du et al., 2010). Thus, Snail is considered an oncogenic molecule. Our finding that Snai1 expression and Snail binding to the Ercc1 promoter and induction of Ercc1 expression are stimulated by UV-irradiation suggests Snail plays a dual role in UV-induced skin carcinogenesis. On one hand, it promotes Ercc1 mediated DNA repair, thereby inhibiting mutagenesis caused by UV radiation. Consistent with our observation, embryonic fibroblasts, from a Snail transgenic mouse expressing Snail at a level 20% higher than the endogenous level, are more resistant to DNA damaging γ radiation (Perez-Mancera et al., 2005). On the other hand, in later stages of cancer, when cancer cells have accumulated multiple mutations, Snail-induced DNA repair may trigger resistance to DNA damaging therapeutic agents. Supporting this notion, co-expression of Ercc1 and Snail correlates with resistance to the chemotherapeutic agent cisplatin, and poor prognosis in head and neck cancer (Hsu et al., 2010). Similarly, overexpression of Snail in breast cancer cells confers protection against the DNA damaging effect of topoisomerase inhibitor (Kajita et al., 2004).
In summary, we identified a role of Smad4 in regulation of Ercc1 expression through Snail-mediated transcriptional regulation of Ercc1. We also ascertained a role of Snail in promoting repair of UV-induced DNA damage via regulation of Ercc1 expression. Loss of Smad4 expression in the epidermis leads to a reduced level of Ercc1, resulting in defective DNA repair in the epidermis after UV irradiation. Abrogation of DNA repair is likely to cause accumulation of UV-induced mutations in K14.Smad4−/− skin and ultimately lead to carcinogenesis. Our work necessitates further investigation of the stage-specific effects of Smad4, Snail and Ercc1-associated DNA repair in cancer initiation and progression, and its influence on the response of cancer cells to therapies using DNA damaging agents.
Materials and Methods
Mouse model
The generation of K14.CrePR1 and Smad4fl/fl mice has been described previously (Berton et al., 2000; Yang et al., 2002). Bigenic mice K14.CrePR1/Smad4fl/fl were generated by mating K14.CrePR1 mice with Smad4fl/fl mice. Genotype was determined by PCR as described previously (Berton et al., 2000; Yang et al., 2002). Keratinocyte specific Smad4 deletion was achieved in K14.Cre/Smad4fl/fl mice by topical application of RU486 for 5 days, when mice were 8 weeks old (Owens et al., 2008). Animal experiments and care were approved by the Institutional Animal Care and Use Committee at the University of Colorado Denver, Anschutz Medical Campus.
UV irradiation of mice
Mice were exposed to UV radiation beginning 3 months of age in a cabinet (Daavlin, Bryan) for 40 weeks and the dosage was increased gradually as depicted in Supplementary Figure 1.
Tumor histology
Skin and tumor samples were fixed in neutral buffered formalin overnight, embedded, sectioned and stained with Hematoxylin and Eosin (H&E) as previously described (Lu et al., 2006). Tumor types were determined by H&E analysis using criteria defined previously (Aldaz et al., 1987).
Immunohistochemistry (IHC) and Immunofluorescence
Immunostaining was performed as previously described (Han et al., 2005). Sections were incubated with an antibody overnight at 4°C as follows: anti-Phospho-Histone2AX (1:100, Cell Signaling, Danvers), anti-Ercc1 (1:100, Santa Cruz Biotechnology, Santa Cruz), anti-Smad4 (1:20, Santa Cruz Biotechnology, Santa Cruz) for mouse sections, anti-Smad4 (1:50, Epitomics, Burlingame) for human tissue sections, anti-Snail (1:50, Abcam, Cambridge). The human skin SCC tumor array SK802 (U.S. Biomax, Rockville) was used. The Department of Dermatopathology, Shanghai Skin Diseases Hospital provided human AK samples as de-identified archived paraffin sections. For IHC, sections were incubated with biotinylated secondary antibodies (1:400) and avidin-peroxidase (Vector Laboratories, Burlingame). Immunostaining was visualized using diaminobenzidine (DAKO, Denmark). For immunofluorescence, Alexa Fluor 488 and 594 labeled secondary antibodies (Life Technologies, Grand Island) were used.
Keratinocyte culture
Primary WT and Smad4−/− keratinocytes were generated from the above mice as described previously (Han et al., 2006). Keratinocytes in culture were irradiated with 60 mJ/cm2 of UVB, culture medium (Lonza, Switzerland) was replaced with fresh medium and cells were incubated in a 5% CO2 incubator for the indicated time period.
Comet assay
Oxiselect Comet assay kit (Cell Biolabs, San Diego) was used according to the manufacturer’s instructions. DNA was stained with Vista Green, and quantification of the comet tail moment was done using CASP software.
RNA Extraction and Quantitative PCR
Total RNA was extracted from mouse skin and tumors in Trizol (Life Technologies, Grand Island) as described previously (Hoot et al., 2008). RNA extraction from cultured keratinocytes was performed using the RNAeasy kit (Qiagen, Valencia). Total RNA was reverse transcribed using Superscript III First Strand Synthesis System (Life Technologies, Grand Island) followed by quantitative PCR using SYBR Green master mix (Life Technologies, Grand Island) using primers listed in Supplementary table 2. Alternatively, RT-qPCR was done using primers listed in Supplementary table 3 and Brilliant qRTPCR mix (Agilent technologies, Santa Clara). A mouse DNA damage signaling pathway (SA Biosciences, Valencia) PCR array was used. For each genotype, three to seven samples were analyzed and gene expression was normalized to GAPDH or 18S rRNA.
Chromatin immunoprecipitation (ChIP)
ChIP assays were performed as previously described (Hoot et al., 2010). Briefly, cultured WT and Smad4−/− keratinocytes were crosslinked with 1% formaldehyde, genomic DNA was sheared in cell lysate by sonication, immunoprecipitation was performed at 4°C overnight with 4Pg of antibody to Snail (Abcam, Cambridge) or normal rabbit IgG (Santa Cruz Biotechnology, Santa Cruz). This was followed by crosslink reversal, DNA purification and quantitative PCR using primers listed in Supplementary Table 4. Enrichment of Snail at the Ercc1 regulatory region was obtained by normalizing the relative level of the Ercc1 regulatory region in the output (immunoprecipitated DNA) to the relative level of an intergenic region (lacking Snail binding sites) in the output. The relative level of the Ercc1 regulatory region in the output was determined by normalizing the level of the Ercc1 regulatory region in the output to that in the input.
Transfection of keratinocytes
WT and Smad4−/− keratinocytes were transfected with 5μg of pEGFPC2-SNAI1 (Addgene, 16225) or pEGFP (Lonza, Switzerland) or pCMVFlagErcc1 using the Epithelial Cell Nucleofection kit (Lonza) following the Amaxa nucleofector program M-005 (Lonza, Switzerland). Cells were harvested 72 hours post transfection, RNA was extracted and quantitative RTPCR was performed as above.
CPD ELISA
WT and Smad4−/− keratinocytes were exposed to UVB radiation at 60mJ/cm2, and samples were harvested at different time points after treatment. Genomic DNA was extracted using the DNeasy kit (Qiagen, Valencia) and ELISA was performed using the Oxiselect UV-induced DNA damage ELISA kit for CPD quantitation (Cell Biolabs, San Diego).
Statistics
The student’s t test was used to analyze data from qRT-PCR, comet assay and CPD ELISA to determine statistical significance of results. Fisher’s exact test was used to analyze data from IHC staining of human skin SCC and AK samples.
Supplementary Material
Acknowledgments
This work was supported by NIH grants CA87849 and DE15953 to X.-J. Wang. L.B. was a visiting scholar supported by a grant (No. 81060189) from The National Natural Science Foundation of China (NSFC). N.S. was supported by Shanghai Natural Science Foundation (11ZR1432900). F.L. was a visiting scholar supported by The National Natural Science Foundation of China (NSFC,81102596). We are grateful to Drs. Lei Li and Mien Chie Hung (MD Anderson Cancer Center) for the Ercc1 and Snail expression plasmids, respectively, Dr. Chuxia Deng (NIH) for providing Smad4fl/fl mice, and Pamela Garl for editing the manuscript.
Abbreviations used
- WT
wild type
- AK
actinic Keratosis
- TSS
transcription start site
Footnotes
The authors declare no conflict of interest.
Note: Supplementary data for this article are available at Journal of Investigative Dermatology Online.
References
- Aldaz CM, Conti CJ, Klein-Szanto AJ, Slaga TJ. Progressive dysplasia and aneuploidy are hallmarks of mouse skin papillomas: relevance to malignancy. Proceedings of the National Academy of Sciences of the United States of America. 1987;84:2029–32. doi: 10.1073/pnas.84.7.2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batlle E, Sancho E, Franci C, Dominguez D, Monfar M, Baulida J, et al. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nature cell biology. 2000;2:84–9. doi: 10.1038/35000034. [DOI] [PubMed] [Google Scholar]
- Berton TR, Wang XJ, Zhou Z, Kellendonk C, Schutz G, Tsai S, et al. Characterization of an inducible, epidermal-specific knockout system: differential expression of lacZ in different Cre reporter mouse strains. Genesis. 2000;26:160–1. [PubMed] [Google Scholar]
- Bornstein S, White R, Malkoski S, Oka M, Han G, Cleaver T, et al. Smad4 loss in mice causes spontaneous head and neck cancer with increased genomic instability and inflammation. The Journal of clinical investigation. 2009;119:3408–19. doi: 10.1172/JCI38854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano A, Perez-Moreno MA, Rodrigo I, Locascio A, Blanco MJ, del Barrio MG, et al. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nature cell biology. 2000;2:76–83. doi: 10.1038/35000025. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sanchez Y. Chk1 in the DNA damage response: conserved roles from yeasts to mammals. DNA repair. 2004;3:1025–32. doi: 10.1016/j.dnarep.2004.03.003. [DOI] [PubMed] [Google Scholar]
- Chipchase MD, Melton DW. The formation of UV-induced chromosome aberrations involves ERCC1 and XPF but not other nucleotide excision repair genes. DNA repair. 2002;1:335–40. doi: 10.1016/s1568-7864(02)00010-1. [DOI] [PubMed] [Google Scholar]
- de Gruijl FR, Van der Leun JC. Estimate of the wavelength dependency of ultraviolet carcinogenesis in humans and its relevance to the risk assessment of a stratospheric ozone depletion. Health physics. 1994;67:319–25. doi: 10.1097/00004032-199410000-00001. [DOI] [PubMed] [Google Scholar]
- Doig J, Anderson C, Lawrence NJ, Selfridge J, Brownstein DG, Melton DW. Mice with skin-specific DNA repair gene (Ercc1) inactivation are hypersensitive to ultraviolet irradiation-induced skin cancer and show more rapid actinic progression. Oncogene. 2006;25:6229–38. doi: 10.1038/sj.onc.1209642. [DOI] [PubMed] [Google Scholar]
- Du F, Nakamura Y, Tan TL, Lee P, Lee R, Yu B, et al. Expression of snail in epidermal keratinocytes promotes cutaneous inflammation and hyperplasia conducive to tumor formation. Cancer research. 2010;70:10080–9. doi: 10.1158/0008-5472.CAN-10-0324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster PJ, Dunn EA, Karl KE, Snir JA, Nycz CM, Harvey AJ, et al. Cellular magnetic resonance imaging: in vivo imaging of melanoma cells in lymph nodes of mice. Neoplasia. 2008;10:207–16. doi: 10.1593/neo.07937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han G, Li AG, Liang YY, Owens P, He W, Lu S, et al. Smad7-induced beta-catenin degradation alters epidermal appendage development. Developmental cell. 2006;11:301–12. doi: 10.1016/j.devcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- Han G, Lu SL, Li AG, He W, Corless CL, Kulesz-Martin M, et al. Distinct mechanisms of TGF-beta1-mediated epithelial-to-mesenchymal transition and metastasis during skin carcinogenesis. The Journal of clinical investigation. 2005;115:1714–23. doi: 10.1172/JCI24399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanasoge S, Ljungman M. H2AX phosphorylation after UV irradiation is triggered by DNA repair intermediates and is mediated by the ATR kinase. Carcinogenesis. 2007;28:2298–304. doi: 10.1093/carcin/bgm157. [DOI] [PubMed] [Google Scholar]
- Hoot KE, Lighthall J, Han G, Lu SL, Li A, Ju W, et al. Keratinocyte-specific Smad2 ablation results in increased epithelial-mesenchymal transition during skin cancer formation and progression. The Journal of clinical investigation. 2008;118:2722–32. doi: 10.1172/JCI33713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoot KE, Oka M, Han G, Bottinger E, Zhang Q, Wang XJ. HGF upregulation contributes to angiogenesis in mice with keratinocyte-specific Smad2 deletion. The Journal of clinical investigation. 2010;120:3606–16. doi: 10.1172/JCI43304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu DS, Lan HY, Huang CH, Tai SK, Chang SY, Tsai TL, et al. Regulation of excision repair cross-complementation group 1 by Snail contributes to cisplatin resistance in head and neck cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:4561–71. doi: 10.1158/1078-0432.CCR-10-0593. [DOI] [PubMed] [Google Scholar]
- Kajita M, McClinic KN, Wade PA. Aberrant expression of the transcription factors snail and slug alters the response to genotoxic stress. Molecular and cellular biology. 2004;24:7559–66. doi: 10.1128/MCB.24.17.7559-7566.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawashima A, Takayama H, Kawamura N, Doi N, Sato M, Hatano K, et al. Co-expression of ERCC1 and Snail is a prognostic but not predictive factor of cisplatin-based neoadjuvant chemotherapy for bladder cancer. Oncology letters. 2012;4:15–21. doi: 10.3892/ol.2012.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koinuma D, Tsutsumi S, Kamimura N, Imamura T, Aburatani H, Miyazono K. Promoter-wide analysis of Smad4 binding sites in human epithelial cells. Cancer science. 2009;100:2133–42. doi: 10.1111/j.1349-7006.2009.01299.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraemer KH, Lee MM, Scotto J. Xeroderma pigmentosum. Cutaneous, ocular, and neurologic abnormalities in 830 published cases. Archives of dermatology. 1987;123:241–50. doi: 10.1001/archderm.123.2.241. [DOI] [PubMed] [Google Scholar]
- Leibeling D, Laspe P, Emmert S. Nucleotide excision repair and cancer. Journal of molecular histology. 2006;37:225–38. doi: 10.1007/s10735-006-9041-x. [DOI] [PubMed] [Google Scholar]
- Lu SL, Herrington H, Reh D, Weber S, Bornstein S, Wang D, et al. Loss of transforming growth factor-beta type II receptor promotes metastatic head-and-neck squamous cell carcinoma. Genes & development. 2006;20:1331–42. doi: 10.1101/gad.1413306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marti TM, Hefner E, Feeney L, Natale V, Cleaver JE. H2AX phosphorylation within the G1 phase after UV irradiation depends on nucleotide excision repair and not DNA double-strand breaks. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:9891–6. doi: 10.1073/pnas.0603779103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martini E, Roche DM, Marheineke K, Verreault A, Almouzni G. Recruitment of phosphorylated chromatin assembly factor 1 to chromatin after UV irradiation of human cells. The Journal of cell biology. 1998;143:563–75. doi: 10.1083/jcb.143.3.563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustakas A, Souchelnytskyi S, Heldin CH. Smad regulation in TGF-beta signal transduction. Journal of cell science. 2001;114:4359–69. doi: 10.1242/jcs.114.24.4359. [DOI] [PubMed] [Google Scholar]
- Murakami T, Fujimoto M, Ohtsuki M, Nakagawa H. Expression profiling of cancer-related genes in human keratinocytes following non-lethal ultraviolet B irradiation. Journal of dermatological science. 2001;27:121–9. doi: 10.1016/s0923-1811(01)00124-4. [DOI] [PubMed] [Google Scholar]
- Narayanan DL, Saladi RN, Fox JL. Ultraviolet radiation and skin cancer. International journal of dermatology. 2010;49:978–86. doi: 10.1111/j.1365-4632.2010.04474.x. [DOI] [PubMed] [Google Scholar]
- Nguyen TA, Slattery SD, Moon SH, Darlington YF, Lu X, Donehower LA. The oncogenic phosphatase WIP1 negatively regulates nucleotide excision repair. DNA repair. 2010;9:813–23. doi: 10.1016/j.dnarep.2010.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niida H, Nakanishi M. DNA damage checkpoints in mammals. Mutagenesis. 2006;21:3–9. doi: 10.1093/mutage/gei063. [DOI] [PubMed] [Google Scholar]
- Nouspikel T. DNA repair in mammalian cells: So DNA repair really is that important? Cellular and molecular life sciences: CMLS. 2009;66:965–7. doi: 10.1007/s00018-009-8734-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olson E, Nievera CJ, Lee AY, Chen L, Wu X. The Mre11-Rad50-Nbs1 complex acts both upstream and downstream of ataxia telangiectasia mutated and Rad3-related protein (ATR) to regulate the S-phase checkpoint following UV treatment. The Journal of biological chemistry. 2007;282:22939–52. doi: 10.1074/jbc.M702162200. [DOI] [PubMed] [Google Scholar]
- Owens P, Bazzi H, Engelking E, Han G, Christiano AM, Wang XJ. Smad4-dependent desmoglein-4 expression contributes to hair follicle integrity. Developmental biology. 2008;322:156–66. doi: 10.1016/j.ydbio.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peinado H, Quintanilla M, Cano A. Transforming growth factor beta-1 induces snail transcription factor in epithelial cell lines: mechanisms for epithelial mesenchymal transitions. The Journal of biological chemistry. 2003;278:21113–23. doi: 10.1074/jbc.M211304200. [DOI] [PubMed] [Google Scholar]
- Perez-Mancera PA, Perez-Caro M, Gonzalez-Herrero I, Flores T, Orfao A, de Herreros AG, et al. Cancer development induced by graded expression of Snail in mice. Human molecular genetics. 2005;14:3449–61. doi: 10.1093/hmg/ddi373. [DOI] [PubMed] [Google Scholar]
- Qiao W, Li AG, Owens P, Xu X, Wang XJ, Deng CX. Hair follicle defects and squamous cell carcinoma formation in Smad4 conditional knockout mouse skin. Oncogene. 2006;25:207–17. doi: 10.1038/sj.onc.1209029. [DOI] [PubMed] [Google Scholar]
- Sertic S, Pizzi S, Cloney R, Lehmann AR, Marini F, Plevani P, et al. Human exonuclease 1 connects nucleotide excision repair (NER) processing with checkpoint activation in response to UV irradiation. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:13647–52. doi: 10.1073/pnas.1108547108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strickland PT, Swartz RP. Inheritance of susceptibility to phototumorigenesis and persistent hyperplasia in F1 hybrids between SENCAR mice and BALB/c or C57BL/6 mice. Cancer research. 1987;47:6294–6. [PubMed] [Google Scholar]
- Vassar R, Rosenberg M, Ross S, Tyner A, Fuchs E. Tissue-specific and differentiation-specific expression of a human K14 keratin gene in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 1989;86:1563–7. doi: 10.1073/pnas.86.5.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner KU, McAllister K, Ward T, Davis B, Wiseman R, Hennighausen L. Spatial and temporal expression of the Cre gene under the control of the MMTV-LTR in different lines of transgenic mice. Transgenic research. 2001;10:545–53. doi: 10.1023/a:1013063514007. [DOI] [PubMed] [Google Scholar]
- Wang X, Zinkel S, Polonsky K, Fuchs E. Transgenic studies with a keratin promoter-driven growth hormone transgene: prospects for gene therapy. Proceedings of the National Academy of Sciences of the United States of America. 1997;94:219–26. doi: 10.1073/pnas.94.1.219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Mao C, Teng Y, Li W, Zhang J, Cheng X, et al. Targeted disruption of Smad4 in mouse epidermis results in failure of hair follicle cycling and formation of skin tumors. Cancer research. 2005;65:8671–8. doi: 10.1158/0008-5472.CAN-05-0800. [DOI] [PubMed] [Google Scholar]
- Yang X, Li C, Herrera PL, Deng CX. Generation of Smad4/Dpc4 conditional knockout mice. Genesis. 2002;32:80–1. doi: 10.1002/gene.10029. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Wang D, Wang XJ, Roop DR. In utero activation of K5.CrePR1 induces gene deletion. Genesis. 2002;32:191–2. doi: 10.1002/gene.10064. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.