

Abstract
Myocardial inflammation and damage can lead to lethal acute or chronic cardiac failure. A variety of regulatory mechanisms limit the magnitude and duration of T cell responses in the heart. Insights into these regulatory mechanisms have come from studies of specific deficiencies in central or peripheral T cell tolerance which cause or enhance the severity of myocarditis. Under non-inflammatory conditions, constitutive DC presentation of cardiac peptides to naïve T cells in cardiac draining lymph nodes tolerizes recirculating naïve T cells specific for these antigens. Cardiac antigen-specific naïve T cells, especially those specific of α-myosin heavy chain peptides, become activated and differentiate into expanded clones of effector T cells under various conditions, such as cardiac infection and/or genetic variations in peripheral tolerance. The pathology that these effector cells cause in the myocardium is limited by PD-L1 expressed on myocardial cells in response to inflammatory cytokines, and by CTLA-4 dependent mechanisms. The PD-1:PD-L1 pathway works together with other pathways to keep the heart safe, a combined genetic deficiencies of PD-1 and other regulatory genes synergize to cause myocarditis. T cell derived IFNγ contributes to the inflammatory damage to the heart in autoimmune myocarditis, but it also engages regulatory mechanisms that limit disease, including upregulation of PD-L1, and differentiation of TNF and iNOS expressing DCs from monocytes. iNOS derived from these DCs and other IFNγ stimulated cells inhibits expansion of T cells that cause myocarditis. Regulatory T cells also appear to be critical for suppression of effector T cells specific for myocardial antigens.
Keywords: Autoimmunity, Heart, Myocarditis, Myosin, T cells, Tolerance
Introduction
Adaptive immune responses directed to antigens in the myocardium occur in the setting of viral or parasitic infection of heart cells, when self-tolerance to cardiac antigens is broken leading to autoimmune myocarditis, and in cardiac allograft rejection. Immune responses initially specific for microbes that infect the heart may lead to autoimmune myocarditis because of antigenic mimicry. T cell and antibody responses to antigens within the myocardium, whether they are microbial, self or alloantigens, result in acute and chronic inflammatory processes that impair cardiac function, cause permanent damage to the tissue, and often lead to dilated cardiomyopathy with congestive heart failure. In all these cases, there is strong evidence that T lymphocytes play a central role in the pathogenesis of the cardiac disease, either by direct cytotoxicity, by enhancing the inflammatory functions of other cells, and by helping B cells produce pathogenic antibodies. On the flip side, healthy myocardium is remarkably devoid of inflammatory cells, including T lymphocytes, even in aging but cardiovascular disease-free individuals who usually have chronic inflammatory infiltrates in many other tissues. Because inflammation, even without irreversible cell injury can fatally disrupt cardiac function, it is likely that the bar for inciting adaptive immune responses and inflammation in the heart has been set higher by evolution than in many other tissues. This review will discuss different mechanisms that regulate T cell responses in the heart. These regulatory pathways become evident from the finding of T cell-mediated myocarditis when they are impaired. The discussion will focus on autoimmune noninfectious myocarditis, as well as examples of T cell regulation in infectious myocarditis that appear to have an autoimmune component, including Coxsackie 3B virus (CV3B) and Trypanosoma cruzi (T cruzi) infection.
The role of α-myosin specific T cells and the failure of central tolerance in autoimmune myocarditis
Patients and rodents with myocarditis or dilated cardiomyopathy are more likely to have circulating autoantibodies specific for certain cardiac antigens including β-adrenergic receptor and alpha myosin, and these antibodies include T-dependent IgGs, reviewed in [1]. Rodent models have been key to defining the role and specificity of T cells in autoimmune myocarditis, and have led to an understating of the T cell specifies in human disease as well. CVB3 infection of certain strains of mice, including A/J and BALB/c, develop a chronic myocarditis and dilated cardiomyopathy, recapitulating human cardiac disease associated with enterovirus-infection, and increased titers of autoantibodies specific for cardiac myosin are detected in the mice. Huber and colleague showed that T cells taken from CVB3 infected mice that are susceptible to myocarditis can lyse cardiomyocytes in vitro, and can induce myocarditis after transfer to uninfected mice [2, 3]. These data supported the hypothesis that chronic cardiac disease after acute viral myocarditis may be mediated by autoimmune T cells specific for cardiac antigens. Following up on these observations, Rose and colleagues showed that immunization of some strains of mice with cardiac myosin in adjuvant cause an autoimmune myocarditis [4]. This model disease is called experimental autoimmune myocarditis (EAM), and the strains that are susceptible to EAM are the same as those that develop chronic myocarditis and dilated cardiomyopathy after CVB3 infection [5]. EAM was demonstrated to be T cell-mediated [6], and the myocarditic peptide epitopes that drive the autoimmune T cell response were identified [7, 8]. The peptides are derived from heart-specific α-myosin heavy chain (α-MHC) but not from β-myosin found in both skeletal and heart muscle. Going back to viral myocarditis, the specificity of myocarditic CD4+ T cells isolated from CVB3 infected mice was shown to be for a cardiac myosin peptide (αMHC334–352) that causes EAM upon injection in susceptible mice [9].
Recently, the work of Lipes and colleagues has elucidated the basis for mouse and human susceptibility to autoimmune myocarditis mediated by α-MHC specific T cells. They showed that transgenic NOD mice expressing the human type 1 diabetes class II MHC risk allele HLA-DQ8 in place of I-Ag7 develop myocarditis mediated by α-MHC-specific CD4+ T cells, without any immunization [10]. Central T cell tolerance to many peripheral tissue restricted protein antigens requires the expression of these self-proteins by thymic medullary epithelial cells under the control of a specialized transcription factors such as AIRE [11]. The Lipes group found that α-MHC is not expressed in the thymus of mice or humans and that α-MHC reactive CD4+ T cells are found in the blood of healthy subjects. Notably, significantly more off these self-reactive T cells are found in patients with myocarditis [10]. The same group also reported that myocardial infarction in NOD mice and Type 1 diabetic patients precipitates autoimmune responses to α-MHC and myocarditis, while myocardial infarction in wild-type mice or Type 2 diabetics does not [12]. Overall this body of work indicates that there is a lack of central T cell tolerance to α-MHC, which in combination with a genetic predisposition to autoimmunity and additional inflammatory insults to the heart, conspires to cause autoimmune myocarditis (Figure 1).
Figure 1. The roles of central and peripheral tolerance in the development and protection against T cell mediated autoimmune myocarditis.
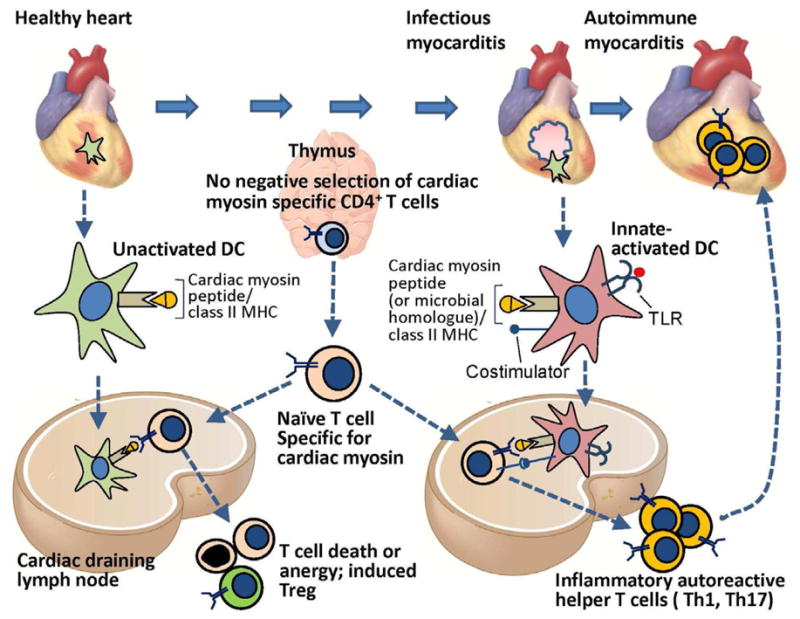
Central T cell tolerance to many peripheral tissue-specific protein antigens occurs by AIRE-dependent expression of these proteins in thymic epithelial cells (TMEC) and presentation of peptides derived from the proteins to developing antigen-specific T cells, leading to deletion of the T cells. Cardiac myosin (α-myosin heavy chain) is not expressed by TMEC, and therefore α-myosin heavy chain-specific T cells emerge from the thymus. These myocardial-antigen specific naïve T cells and are found in the blood of mice and humans, and during their recirculation, migrate through the cardiac draining lymph node (CDLN). Dendritic cells that display α myosin peptides in complex with class II MHC are present in the healthy heart, and these resting tolerogenic DCs also traffic to the CDLN, where they present the α myosin peptides to the naïve α-myosin heavy chain-specific CD4+ T cells, leading to deletion, anergy or Treg induction. If the heart is infected (e.g. by coxsackievieus B virus, T Cruzi) or is damaged by ischemic infarction, myocardial DCs are activated by TLR ligands, become capable of productively activating cardiac myosin-specific naïve T cells in the CDLN, leading to differentiation of inflammatory effector CD4+ T cells that migrate back to the heart and cause autoimmune myocarditis. It is possible that T cells specific for microbial peptides that are homologous to myocardial antigens are also involved in autoimmune myocarditis. The susceptibility to developing T cell mediated myocarditis depends on inheritance of certain alleles of MHC genes as well as other autoimmune susceptibility genes.
There is also evidence that the pathogenesis chronic cardiac disease caused by Trypanosoma cruzi (T Cruzi) infection in Chagas heart disease involves T cell responses against cardiac myosin, in addition to responses to antigens expressed by persistent microbes, reviewed in [13]. T cell rich infiltrates, including CD8+ CTL and IFNγ producing CD4+ T cells are present in the affected human hearts [14, 15, 16]. CD4+ T cells from hearts of Chagas disease patients respond to both T cruzi B13 protein and cardiac myosin [17, 18]. Studies of experimental Chagas disease in mice have reinforced the concept that T cell-driven autoimmunity exists due to antigen mimicry between T cruzi and cardiac antigens. For example, CD4+ T cells derived from T cruzi–infected mice can induce myocarditis when injected into syngeneic but uninfected hosts [19]. These observations indicate that mechanisms of regulation of T cell responses in the heart may be important in maintaining he balance between control of parasitic infection vs. harmful T cell responses induced by the parasites that cause myocyte damage.
Myocardial antigen presenting cells and T cell activation in the cardiac draining lymph node
Class II major histocompatibility complex molecule (MHC) expressing bone marrow derived antigen presenting cells are abundant in normal hearts, and in mice many of these cells display α myosin heavy chain peptide 614–629/class II MHC complexes [20, 21]. We described a paratracheal cardiac draining lymph node (CDLN) in mice which becomes enlarged in the setting of myocarditis [22, 23]. When effector T cells are transferred into cMy-mOva mice, they accumulate and proliferate in this lymph node prior to any evidence of myocarditis in the heart [22]. When CFSE-labeled naïve OT-1 T cells are transferred into cMy-mOva mice, they proliferate in the CDLN and undergo apoptosis, but they do not proliferate in other lymph nodes (unpublished data). Subsequently, this lymph node has been shown to be the site of expansion of α-myosin heavy chain specific CD4+ T cells in spontaneous and post-infarction models of myocarditis in NOD mice [10, 12, 24]. These findings are consistent with the hypothesis that cardiac antigens are constitutively processed and derivative peptides are displayed by class II MHC on cardiac DCs, and these DCs are transported to draining lymph nodes in healthy individuals. Mechanisms of peripheral tolerance may determine the fate of heart antigen specific T cells in the heart draining node. With the right combination of genetic predisposition to autoimmunity (e.g. NOD background in mice, diabetes susceptibility genes in humans), infection (cardiotropic viral infection) or injury (e.g. myocardial infarction), together with a failure of central tolerance (e.g. failure of T cell tolerance for cardiac myosin), the result will be activation and effector differentiation of myocardial antigen specific T cells. Once these cells enter and cause inflammatory damage to the myocardium, a continuing cycle of injury, cardiac antigen presentation by activated DCs and T cell activation may ensue [25].
Regulation of T cell-mediated cardiac inflammation by B7-CD28 family inhibitory pathways
The B7-CD28 family of costimulatory molecules and receptors includes the B7 CD80 (B7-1) and CD86 (B7-2) and their activating receptor CD28 on T cells, and ICOS-ligand (CD275) which binds to ICOS (CD278) on T cells. In addition, these proteins families include regulatory molecules that counteract activation of T cells by antigen recognition and costimulation [26, 27]. The CD28-family regulatory molecules are CTLA-4 (CD152) and PD-1 (CD279), and their expression is upregulated upon T cell activation. CTLA-4 binds CD80 and CD86, with higher affinity than does CD28, while PD-1 binds the B7 family proteins PD-L1 (CD274) and PD-L2 (CD273). Data from various studies discussed next indicate that PD-L1:PD-1 and B7:CTLA-4 interactions appear to be important in suppressing T cell responses in the heart.
High cardiac expression of PD-L1 mRNA relative to most other tissues in the mouse was described in one of the first papers characterizing the protein [28]. We subsequently showed that PD-L1 could be significantly upregulated on cardiac microvascular endothelial cells in vitro by IFN-γ, and this induced expression suppressed CD8+ T cell activation by and killing of the endothelial cells [29]. We explored the role of the PD-1: PD-L1/PD-L2 pathway in protecting myocardium from T cell injury in a transgenic model of CD8+ T cell myocarditis. In this model, developed in our laboratory, ovalbumin peptide specific CD8+ T cells from OT-1 TCR transgenic mice are adoptively transferred into C57BL/6 mice expressing a transgene encoding membrane bound ovalbumin under the control of the α-myosin heavy chain promoter (cMy-mOva mice) [22]. When effector OT-1 CTL are transferred into cMy-mOva mice, myocarditis develops within 4–6 days, of varying severity depending on the dose of T cells transferred. PD-L1 serves a significant protective role in this model. A given dose of OT-1 effectors that causes self-limiting nonlethal disease in cMy-mOva mice causes severe and lethal disease in PD-L1/L2 deficient cMy-mOva mice [30]. In cMy-mOva with self-limiting myocarditis, PD-L1 but not PD-L2 gene expression is upregulated in the heart, and PD-L1 protein is expressed widely on myocardial microvascular endothelial cells. Furthermore the severity of myocarditis in chimeric PD-L1 deficient cMy-mOva mice with wild type bone marrow is equivalent to globally PD-L1 deficient mice [30]. Thus PD-L1 on non-hematopoietic cells, most likely endothelium, is providing the protection against CTL in the heart. We have also shown that transfer of PD-1 deficient OT-1 cells results in increased severity of myocarditis in cMy-mOva mice compared PD-1 expressing OT-1 cells [31]. This later finding confirms the importance of PD-1 as a relevant receptor of PD-L1 in cardiac protection, although the possibility that B7-PD-L1 interactions, which have been shown to inhibit T cell activation [32] are not ruled out. The data from these studies of cMy-mOva mice clearly demonstrate that PD-L1 expression induced by T cell IFNγ protects against CD8+ T cell mediated injury and inflammation in the heart. This may be relevant in the setting of infections, autoimmunity, allograft rejection, but the cMy-mOva model does not address the possibility of a constitutive role of the PD-1:PD-L1 pathway in peripheral self-tolerance to cardiac antigens.
An important role for the PD-1:PD-L1 pathway in maintaining peripheral tolerance to cardiac antigens becomes evident from work with the autoimmune-prone MRL mouse strain, which is best known as a model of SLE when it carries the lpr FAS mutation. PD-L1 deficiency on the MRL background (with or without the FAS mutation) results in a severe, lethal lymphocytic myocarditis with abundant CD8+ T cells and less but significant numbers of CD4+ T cells [33]. These mice also develop a lymphocytic pneumonitis. Heart-specific autoantibodies are also found in the serum of PD-L1 deficient MRLlpr/lpr mice. Chimeric MRL mice with PD-L1 deficiency restricted only to bone marrow derived cells still develop severe myocarditis, indicating that hematopoietic cell expression of PD-L1 is major checkpoint in maintaining self tolerance to cardiac antigens. This finding does not rule out a possible regulatory role for endothelial PD-L1 as well in the MRL mice. A similar severe lethal myocarditis is reported to occur in PD-1 deficient MRL mice[34], and is characterized by marked CD4+ and CD8+ T cell infiltrates as well as anti-myosin autoantibodies. The specificities of the cardiac infiltrating T cells in either the PD-L1 or PD-1 deficient MRL mice were not determined.
Spontaneous lethal T cell mediated myocarditis also occurs in BALB/c mice deficient in bot LAG-3 and PD-1[35]. LAG-3 a membrane protein homologous to CD4 that is expressed on T cells, and has incompletely characterized inhibitory functions, including some related to Treg function [36]. Nonetheless, a Treg defect was not observed in the LAG-3/PD-1 deficient myocarditic mice. The fact that PD-1 deficiency in combination with either of two unrelated immunoregulatory defects (MRL background or LAG-3 deficiency) result in severe spontaneous myocarditis indicates the central importance of PD-1 in maintaining peripheral T cell tolerance to myocardial antigens. Further exploration of the synergies between PD-1 pathway deficiency and other conditions that may put the heart at risk are warranted.
EAM induced by immunization of BALB/c mice with α–MHC peptide in complete Freund’s adjuvant (CFA) is usually a self-limiting CD4+ helper T cell mediated disease [1], involving both Th1 and Th17 cells. We have shown that PD-1 deficiency increases severity of EAM [31]. PD-1 deficiency on the BALB/c background was reported to result in a dilated cardiomyopathy mediated by anti-troponin IgG autoantibodies [37, 38]. Although such an autoantibody response suggests that autoreactive helper T cells are involved, no evidence for such responses was documented in these reports, nor was there evidence of active myocarditis preceding the dilated cardiomyopathy.
The PD-1 pathway also protects the myocardium form T cell-mediated damage in the setting of infectious myocarditis. Blocking anti-PD-1 or anti-PD-L1 antibodies enhanced cardiac inflammation in C3H/He mice infected with Coxsackievirus B3 [39]. In that study, the authors demonstrated that PD-L1 expression was upregulated on cardiac myocytes in the infected mice. T cell mediated cardiac damage in the setting of T cruzi infection of C57Bl/6 mice is also controlled by the PD-1 pathway [40]. PD-1 was significantly upregulated on CD4+ and CD8+ T cells infiltrating the myocardium of infected mice, which is typical of exhausted T cells in the setting of chronic viral infection [41]. Antibody blockade of PD-1 or PD-L1, or genetic deficiency PD-1, resulted in enhanced myocardial inflammation, with increased inflammatory cells and expression of IFNγ and iNOS. This enhanced T cell response was associated with marked increase in mortality, even though the impairments of the T cell inhibitory pathway resulted in decreased blood and tissue parasite content. In other words, an intact PD-1:PD-PL-1 pathway was permissive for chronic parasitic infection, but also critical to prevent lethal T cell-mediated myocarditis related o to the infection.
CTLA-4 clearly has a critical role in self tolerance and homeostasis, most dramatically shown by the phenotype of CTLA-4 deficient mice, which develop a diffuse lethal multi-organ lymphoproliferative disease within the first few weeks of life [42, 43]. A striking T cell myocarditis is present in these mice, which likely contributes to the lethality of the phenotype. The specificities of these T cells are not known, however myocarditis as well as other organ involvement is reduced in TCR transgenic/RAG deficient background [26], suggesting the disease is driven by antigens. The way CTLA-4 regulates T cell responses is not fully understood, but likely involves both effector T cell intrinsic inhibitory signaling, competition with CD28 for B7-1/2 binding, and regulatory T cell function. Blocking anti-CTLA-4 antibody is reported to enhance severity of EAM [44]. We explored the impact of CTLA-4 deficiency in CD8+ T cells on myocarditis, with a focus on T cell intrinsic mechanism of CTLA-4 function. For this purpose, we bred the CTLA-4 null allele into OT-1 Rag1−/− mice, and transferred CTLA-4 null OT-1 effectors into the cMy-mOva mice. Remarkably, mice receiving the CTLA-4 deficient CTL uniformly died by day 10 after transfer of fulminant myocarditis, while transfer of the same dose of CTLA-4 expressing OT-1 caused only 20% mortality through 30 days [45]. The enhanced pathogenicity of the CTLA-4 null cells reflects enhanced proliferation of these cells, rather than enhanced cytotoxicity of each cell. Transfer of naïve OT-1 cells did not result in spontaneous myocarditis, whether or not the T cells were CTLA-4 null or wild type. The contribution of CTLA-4 in regulating T cells in the context of infectious myocarditis is not well documented. IFNγ producing T cells from blood and heart of chronically infected patients with Chagas heart disease express high levels of CTLA-4 and the leukocyte immunoglobulin like receptor 1 (LIR-1), suggesting an exhausted phenotype of these cells [46].
The paradoxical regulatory role of interferon-γ in regulating T cell mediated pathology in the heart
Interferon γ (IFNγ), which is produced by CD4+ TH1 cells, CD8+ CTL, NK cells, and NKT cells, has several well defined inflammatory effects on various cell types, including endothelium and macrophages. Many of the pathological effects of TH1 cells in autoimmune disease are attributable to IFNγ. In the case of T cell mediated inflammation in the heart, however, the influence of IFNγ is a complex combination of pro-inflammatory effects and anti-inflammatory effects, including a feedback inhibition of the effector T cells that produce the cytokine (Figure 2). TH1 cells are clearly involved in the pathogenesis of EAM [1]. α-MHC reactive T cells in NOD DQ8+ mice that spontaneously develop myocarditis and in the blood of myocarditic patents are also IFNγ producers {Lv, 2011 #269}. Furthermore, IFNγ transgenic mice on a C57Bl/6 background develop a chronic active myocarditis with no other inflammatory organ pathology [47]. Nonetheless, there is robust evidence that IFNγ also serves a protective role against myocarditis. For example, BALB/c mice with IFNγ or IFNγ receptor deficiencies develop markedly more severe and lethal EAM than wild type controls [48–50]. T-bet deficient mice develop more severe EAM compared to controls, because of lack of IFNγ production by heart-infiltrating CD8+ T cells, leading to an exaggerated Th17 response [51]. Likewise, IFNγ-induced chemokine expression is part of the inflammatory pathology in the CD8+ T cell-mediated myocarditis in the cMy-mOva model [23], yet myocarditis is more severe in cMy-mOva mice when IFNγ-deficient OT-1 cells are transferred or the cMy-Ova mice lack IFNγ receptor [30].
Figure 2. Interferon γ-dependent regulation of pro-inflammatory T cell responses in the heart.
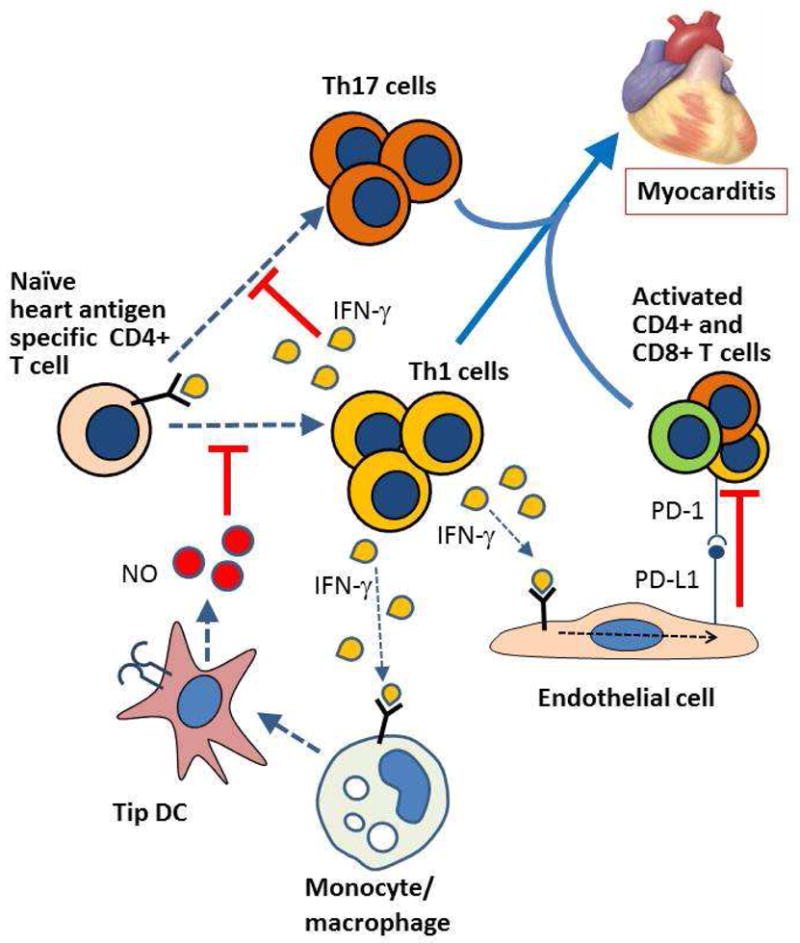
Although TH1 cells, as well as TH17 cells, contribute to acute and chronic inflammatory damage to the myocardium in infectious and autoimmune myocarditis, IFNγ production by TH1 cells exerts several feedback regulatory effects that dampen T cell activation and effector functions. IFNγ induces PD-L1 on myocardial endothelial cells (and possibly myocytes) which inhibits effector T cell activation by binding to PD-1 on the T cells. IFNγ together with TLR ligands promote the differentiation of TNF and iNOS producing DCs (Tip DCs) which, through the action of NO, inhibit effector T cell proliferation. IFNγ also blocks the differentiation of TH17 cells. The absence of IFNγ or its receptor exacerbates disease in experimental infectious and autoimmune myocarditis.
The finding that myocarditis is enhanced when IFNγ signaling is genetically absent implies that IFNγ can inhibit the T cells that cause the myocarditis. Several different IFNγ dependent mechanisms of T cell inhibition appear to be involved (Figure 2). In EAM, IFNγ produced by TH1 cells, in combination with TLR2 ligands induces the differentiation of monocytes into TNF and iNOS producing DCs (Tip DCs). The iNOS released by the Tip DCs inhibits expansion of α–MHC specific T cells and therefore limits the progression of myocarditis [48, 52]. Furthermore, IFNγ inhibits EAM by other iNOS-independent effects on T cell apoptosis and activation [53]. In the cMy-mOva model of CD8+ T cell mediated myocarditis, we have shown that OT-1 derived IFNγ and cMy-mOva IFNγR are essential for induction of PD-L1 in the heart, which is required to prevent lethal disease [30]. It is likely that these various T cell inhibitory mechanisms are at work in both EAM and the cMy-mOva models. Therefore, T cells that respond to myocardial antigens and initiate myocarditis produce IFNγ which contributes to the inflammatory pathology, but is also required to limit further T cell activation and T cell mediated inflammatory damage to the heart. It is perhaps surprising that in the studies cited above severe myocarditis could develop in the absence of IFNγ, the major inflammatory cytokine produced by TH1 cells and CTLs. There are however likely explanations. Both TH17 and TH1 cells are involved in EAM [1, 54], and in the absence of IFNγ producing, enhanced Th7 responses may develop [51]. Th17 cells produce IL-17 and TNF, cytokines that are potent inducers of neutrophilic inflammation, which is typically seen in EAM. In the case of the CTL-mediated myocarditis in cMy-mOva mice, the INFγ deficient OT-1 cells will retain their cytotoxic function, and CTL killing of myocytes can outpace local efferocytosis and lead to inflammatory necrosis of myocytes. The CTLs also secrete chemokines and TNF, which likely contribute to the inflammatory response.
IFNγ-producing cells are abundant in failing hearts of Chagas disease patients [55], and these cells appear to be important for limiting disease severity [56]. In mice, IFNγ expressing CD8+ T cells have a protective effect against T. Cruzi associated cardiac injury [57].
Regulatory T cells and peripheral tolerance induction to treat or prevent myocarditis
The fundamental importance of regulatory T cells in preventing T cell-mediated damage to tissues is widely known, and if the heart is relatively more resistant to T cell-mediated responses than other tissues, it is likely that Treg play a role. Nonetheless, there remains a scarcity of robust data about Treg mediated protection against effector T cells in the healthy or myocarditic heart. Correlative data indicate the predictable finding that milder T cell-mediated inflammatory disease is associated with more Treg numbers or more suppressive function. For example, the suppressive activity of peripheral blood Treg (identified as CD4+CD25+ cells) from Chagas disease was significantly higher if the patients had mild or no cardiomyopathy compared to patients with moderate and severe cardiomyopathy [58]. Comparisons of two MHC identical strains of mice which differ in their susceptibility to EAM have revealed a lower proportion of Treg and higher proportion of TH17 cells among CD4+ T cells in the susceptible strains compared to the resistant strain [59]. Genetic deficiencies or treatments that result in enhanced myocardial inflammation in the setting of CVB3 infection are also associated with reduced Treg. For example, lack of either thrombospondin-2 or β2-integrins in BALB/c mice results in enhanced CVB3-induced myocarditis [60, 61]. In addition, antibody blockade of the immunoregulatory molecule Tim-3 during acute CVB3 infection of BALB/c mice enhanced severity of myocarditis, which may be a result of reduced expression of CTLA-4 dependent-regulatory T cell responses [62].
The concept of treating autoimmune disease by interventions that enhance Treg suppression of autoreactive T cells, which has been widely adopted in translational immunology research, is a developing area of research in myocarditis. Several approaches can be taken to enhance Treg responses, including pharmacological agents that promote Treg differentiation, delivery of autoantigens in a tolerogenic form, and adoptive therapy with tolerogenic DCs. An example of a pharmacologic approach is Galectin-9, a ligand for the immunoinhibitory receptor TIM-3. Galectin-9 treatment is reported to reduce severity of CVB3-induced myocarditis, through Treg activation, as well as selective TH2 and not TH1 activation [63]. Delivery of myocardial antigens in a tolerogenic has been described. Treatment of rats with a myosin peptide in incomplete Freund’s adjuvant (IFA) protected against EAM by inducing IL-10 producing T cells, and similar protection was achieved by transfer of DCs from the tolerized rats into naïve rats [64]. Nasal administration of cardiac myosin peptides to BALB/c mice to induce mucosal tolerance was shown to be effective in reducing severity of the autoimmune phase of CVB3 myocarditis, though enhancement of Treg and bystander cell IL-10 production [65]. IL-10 producing T cells were also implicated in reducing severity of EAM in BALB/c mice treated with DCs infected with herpes virus entry mediator (HVEM) expressing adenovirus [66].
The identification of well defined auto antigens in experimental and human autoimmune myocarditis together with the emerging understanding of mechanism of peripheral tolerance required to suppress T cell specific for these antigens should facilitate the development of highly specific tolerance based therapeutic approaches to myocarditis.
Final comments
This manuscript is part of a dedicated issue devoted to Professor Abul Abbas and his many contributions in teaching and research. It is part of the Journal of Autoimmunity’s yearly themes devoted to critical issues and contributions by outstanding immunologists; in the past these have included Drs. Ian Mackay, Noel Rose, Chella Davis, Harry Moutsopoulos and Pierre Youinou [67–70]. I count myself extremely lucky to have had so many opportunities to work with Abul. He has been my mentor and advisor, colleague and coauthor, and a reliable and trusted career long friend. Abul has become iconic ambassador for immunology research and education worldwide. He has had a large impact on peers and trainees because of his own research on immune regulation, his journal editing (a founding editor of Immunity), his textbook authoring (major titles in both immunology and pathology), his teaching (at Harvard and UCSF, and in postgraduate courses all over the world) and his leadership in translational immunology (President of FOCiS). His intellect, enthusiasm, and generosity with time and ideas have been inspirational and sustaining for a great many people in at all stages of their careers.
Research Highlights.
Myocardial infections with viruses or parasites may lead to a failure of peripheral tolerance of cardiac antigen-specific T cells, leading to T cell mediated autoimmune myocarditis.
Interferon γ produced by pro-inflammatory T cells induces regulatory mechanisms, including iNOS and PD-L1 expression, that limit T cell mediated cardiac damage.
Induction of specific tolerance to cardiac antigens such as α-myosin heavy chain is a therapeutic goal for the treatment and prevention of autoimmune myocarditis.
Acknowledgments
A.H.L. is supported by National Institutes of Health grant R01HL087282 and P50HL56985
Footnotes
Disclosure statement
The author has declared no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cihakova D, Rose NR. Pathogenesis of myocarditis and dilated cardiomyopathy. Adv Immunol. 2008;99:95–114. doi: 10.1016/S0065-2776(08)00604-4. [DOI] [PubMed] [Google Scholar]
- 2.Lodge PA, Herzum M, Olszewski J, Huber SA. Coxsackievirus B-3 myocarditis. Acute and chronic forms of the disease caused by different immunopathogenic mechanisms. Am J Path. 1987;128:455–63. [PMC free article] [PubMed] [Google Scholar]
- 3.Estrin M, Huber SA. Coxsackievirus B3-induced myocarditis. Autoimmunity is L3T4+ T helper cell and IL-2 independent in BALB/c mice. Am J Path. 1987;127:335–41. [PMC free article] [PubMed] [Google Scholar]
- 4.Neu N, Rose NR, Beisel KW, Herskowitz A, Gurri-Glass G, Craig SW. Cardiac myosin induces myocarditis in genetically predisposed mice. J Immunol. 1987;139:3630–6. [PubMed] [Google Scholar]
- 5.Li HS, Ligons DL, Rose NR. Genetic complexity of autoimmune myocarditis. Autoimmunity reviews. 2008;7:168–73. doi: 10.1016/j.autrev.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Smith SC, Allen PM. Myosin-induced acute myocarditis is a T cell-mediated disease. J Immunol. 1991;147:2141–7. [PubMed] [Google Scholar]
- 7.Donermeyer DL, Beisel KW, Allen PM, Smith SC. Myocarditis-inducing epitope of myosin binds constitutively and stably to I-Ak on antigen-presenting cells in the heart. J Exp Med. 1995;182:1291–300. doi: 10.1084/jem.182.5.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pummerer CL, Luze K, Grassl G, Bachmaier K, Offner F, Burrell SK, et al. Identification of cardiac myosin peptides capable of inducing autoimmune myocarditis in BALB/c mice. J Clin Invest. 1996;97:2057–62. doi: 10.1172/JCI118642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gangaplara A, Massilamany C, Brown DM, Delhon G, Pattnaik AK, Chapman N, et al. Coxsackievirus B3 infection leads to the generation of cardiac myosin heavy chain-alpha-reactive CD4 T cells in A/J mice. Clin Immunol. 2012;144:237–49. doi: 10.1016/j.clim.2012.07.003. [DOI] [PubMed] [Google Scholar]
- 10.Lv H, Havari E, Pinto S, Gottumukkala RV, Cornivelli L, Raddassi K, et al. Impaired thymic tolerance to alpha-myosin directs autoimmunity to the heart in mice and humans. J Clin Invest. 2011;121:1561–73. doi: 10.1172/JCI44583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Anderson MS, Su MA. Aire and T cell development. Curr Opin Immunol. 2011;23:198–206. doi: 10.1016/j.coi.2010.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gottumukkala RVSRKLvH, Cornivelli L, Wagers AJ, Kwong RY, Bronson R, et al. Myocardial Infarction Triggers Chronic Cardiac Autoimmunity in Type 1 Diabetes. Science Trans Med. 2012;4:138ra80. doi: 10.1126/scitranslmed.3003551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marin-Neto JA, Cunha-Neto E, Maciel BC, Simões MV. Pathogenesis of Chronic Chagas Heart Disease. Circulation. 2007;115:1109–23. doi: 10.1161/CIRCULATIONAHA.106.624296. [DOI] [PubMed] [Google Scholar]
- 14.Gomes JA, Bahia-Oliveira LM, Rocha MO, Martins-Filho OA, Gazzinelli G, Correa-Oliveira R. Evidence that development of severe cardiomyopathy in human Chagas’ disease is due to a Th1-specific immune response. Infect Immun. 2003;71:1185–93. doi: 10.1128/IAI.71.3.1185-1193.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fonseca SG, Moins-Teisserenc H, Clave E, Ianni B, Nunes VL, Mady C, et al. Identification of multiple HLA-A*0201-restricted cruzipain and FL-160 CD8+ epitopes recognized by T cells from chronically Trypanosoma cruzi-infected patients. Microbes Infect. 2005;7:688–97. doi: 10.1016/j.micinf.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 16.Cunha-Neto E, Bilate AM, Hyland KV, Fonseca SG, Kalil J, Engman DM. Induction of cardiac autoimmunity in Chagas heart disease: a case for molecular mimicry. Autoimmun. 2006;39:41–54. doi: 10.1080/08916930500485002. [DOI] [PubMed] [Google Scholar]
- 17.Cunha-Neto E, Coelho V, Guilherme L, Fiorelli A, Stolf N, Kalil J. Autoimmunity in Chagas’ disease. Identification of cardiac myosin-B13 Trypanosoma cruzi protein crossreactive T cell clones in heart lesions of a chronic Chagas’ cardiomyopathy patient. J Clin Invest. 1996;98:1709–12. doi: 10.1172/JCI118969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Iwai LK, Juliano MA, Juliano L, Kalil J, Cunha-Neto E. T-cell molecular mimicry in Chagas disease: identification and partial structural analysis of multiple cross-reactive epitopes between Trypanosoma cruzi B13 and cardiac myosin heavy chain. J Autoimmun. 2005;24:111–7. doi: 10.1016/j.jaut.2005.01.006. [DOI] [PubMed] [Google Scholar]
- 19.Ribeiro-Dos-Santos R, Mengel JO, Postol E, Soares RA, Ferreira-Fernandez E, Soares MB, et al. A heart-specific CD4+ T-cell line obtained from a chronic chagasic mouse induces carditis in heart-immunized mice and rejection of normal heart transplants in the absence of Trypanosoma cruzi. Parasite Immunol. 2001;23:93–101. doi: 10.1046/j.1365-3024.2001.00368.x. [DOI] [PubMed] [Google Scholar]
- 20.Spencer SC, Fabre JW. Characterization of the tissue macrophage and the interstitial dendritic cell as distinct leukocytes normally resident in the connective tissue of rat heart. J Exp Med. 1990;171:1841–51. doi: 10.1084/jem.171.6.1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith SC, Allen PM. Expression of myosin-class II major histocompatibility complexes in the normal myocardium occurs before induction of autoimmune myocarditis. Proc Natl Acad Sci U S A. 1992;89:9131–5. doi: 10.1073/pnas.89.19.9131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Grabie N, Delfs MW, Westrich JR, Love VA, Stavrakis G, Ahmad F, et al. IL-12 is required for differentiation of pathogenic CD8+ T cell effectors that cause myocarditis. J Clin Invest. 2003;111:671–80. doi: 10.1172/JCI16867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Taqueti VR, Grabie N, Colvin R, Pang H, Jarolim P, Luster AD, et al. T-bet controls pathogenicity of CTLs in the heart by separable effects on migration and effector activity. J Immunol. 2006;177:5890–901. doi: 10.4049/jimmunol.177.9.5890. [DOI] [PubMed] [Google Scholar]
- 24.Nindl V, Maier R, Ratering D, De Giuli R, Zust R, Thiel V, et al. Cooperation of Th1 and Th17 cells determines transition from autoimmune myocarditis to dilated cardiomyopathy. Eur J Immunol. 2012;42:2311–21. doi: 10.1002/eji.201142209. [DOI] [PubMed] [Google Scholar]
- 25.Lv H, Lipes MA. Role of impaired central tolerance to alpha-myosin in inflammatory heart disease. Trends Cardiovasc Med. 2012;22:113–7. doi: 10.1016/j.tcm.2012.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greenwald RJ, Freeman GJ, Sharpe AH. The B7 family revisited. Annu Rev Immunol. 2005;23:515–48. doi: 10.1146/annurev.immunol.23.021704.115611. [DOI] [PubMed] [Google Scholar]
- 27.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and Its Ligands in Tolerance and Immunity. Annu Rev Immunol. 2008 doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freeman GJ, Long AJ, Iwai Y, Bourque K, Chernova T, Nishimura H, et al. Engagement of the PD-1 immunoinhibitory receptor by a novel B7 family member leads to negative regulation of lymphocyte activation. J Exp Med. 2000;192:1027–34. doi: 10.1084/jem.192.7.1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rodig N, Ryan T, Allen JA, Pang H, Grabie N, Chernova T, et al. Endothelial expression of PD-L1 and PD-L2 down-regulates CD8+ T cell activation and cytolysis. Eur J Immunol. 2003;33:3117–26. doi: 10.1002/eji.200324270. [DOI] [PubMed] [Google Scholar]
- 30.Grabie N, Gotsman I, DaCosta R, Pang H, Stavrakis G, Butte MJ, et al. Endothelial programmed death-1 ligand 1 (PD-L1) regulates CD8+ T-cell mediated injury in the heart. Circulation. 2007;116:2062–71. doi: 10.1161/CIRCULATIONAHA.107.709360. [DOI] [PubMed] [Google Scholar]
- 31.Tarrio ML, Grabie N, Bu DX, Sharpe AH, Lichtman AH. PD-1 protects against inflammation and myocyte damage in T cell-mediated myocarditis. J Immunol. 2012;188:4876–84. doi: 10.4049/jimmunol.1200389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–22. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lucas JA, Menke J, Rabacal WA, Schoen FJ, Sharpe AH, Kelley VR. Programmed death ligand 1 regulates a critical checkpoint for autoimmune myocarditis and pneumonitis in MRL mice. J Immunol. 2008;181:2513–21. doi: 10.4049/jimmunol.181.4.2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang J, Okazaki IM, Yoshida T, Chikuma S, Kato Y, Nakaki F, et al. PD-1 deficiency results in the development of fatal myocarditis in MRL mice. Int Immunol. 2010;22:443–52. doi: 10.1093/intimm/dxq026. [DOI] [PubMed] [Google Scholar]
- 35.Okazaki T, Okazaki IM, Wang J, Sugiura D, Nakaki F, Yoshida T, et al. PD-1 and LAG-3 inhibitory co-receptors act synergistically to prevent autoimmunity in mice. J Exp Med. 2011;208:395–407. doi: 10.1084/jem.20100466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Okamura T, Fujio K, Sumitomo S, Yamamoto K. Roles of LAG3 and EGR2 in regulatory T cells. Ann Rheum Dis. 2012;71 (Suppl 2):i96–100. doi: 10.1136/annrheumdis-2011-200588. [DOI] [PubMed] [Google Scholar]
- 37.Okazaki T, Tanaka Y, Nishio R, Mitsuiye T, Mizoguchi A, Wang J, et al. Autoantibodies against cardiac troponin I are responsible for dilated cardiomyopathy in PD-1-deficient mice. Nature Med. 2003;9:1477–83. doi: 10.1038/nm955. [DOI] [PubMed] [Google Scholar]
- 38.Nishimura H, Okazaki T, Tanaka Y, Nakatani K, Hara M, Matsumori A, et al. Autoimmune dilated cardiomyopathy in PD-1 receptor-deficient mice. Science. 2001;291:319–22. doi: 10.1126/science.291.5502.319. [DOI] [PubMed] [Google Scholar]
- 39.Seko Y, Yagita H, Okumura K, Azuma M, Nagai R. Roles of programmed death-1 (PD-1)/PD-1 ligands pathway in the development of murine acute myocarditis caused by coxsackievirus B3. Cardiovasc Res. 2007;75:158–67. doi: 10.1016/j.cardiores.2007.03.012. [DOI] [PubMed] [Google Scholar]
- 40.Gutierrez FR, Mariano FS, Oliveira CJ, Pavanelli WR, Guedes PM, Silva GK, et al. Regulation of Trypanosoma cruzi-induced myocarditis by programmed death cell receptor 1. Infect Immun. 2011;79:1873–81. doi: 10.1128/IAI.01047-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sharpe AH, Wherry EJ, Ahmed R, Freeman GJ. The function of programmed cell death 1 and its ligands in regulating autoimmunity and infection. Nat Immunol. 2007;8:239–45. doi: 10.1038/ni1443. [DOI] [PubMed] [Google Scholar]
- 42.Waterhouse P, Penninger JM, Timms E, Wakeham A, Shahinian A, Lee KP, et al. Lymphoproliferative disorders with early lethality in mice deficient in Ctla-4. Science. 1995;270:985–8. doi: 10.1126/science.270.5238.985. [DOI] [PubMed] [Google Scholar]
- 43.Tivol EA, Borriello F, Schweitzer AN, Lynch WP, Bluestone JA, Sharpe AH. Loss of CTLA-4 leads to massive lymphoproliferation and fatal multiorgan tissue destruction, revealing a critical negative regulatory role of CTLA-4. Immunity. 1995;3:541–7. doi: 10.1016/1074-7613(95)90125-6. [DOI] [PubMed] [Google Scholar]
- 44.Ying H, Yang L, Qiao G, Li Z, Zhang L, Yin F, et al. Cutting edge: CTLA-4--B7 interaction suppresses Th17 cell differentiation. J Immunol. 2010;185:1375–8. doi: 10.4049/jimmunol.0903369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Love VA, Grabie N, Duramad P, Stavrakis G, Sharpe A, Lichtman A. CTLA-4 ablation and interleukin-12 driven differentiation synergistically augment cardiac pathogenicity of cytotoxic T lymphocytes. Circ Res. 2007;101:248–57. doi: 10.1161/CIRCRESAHA.106.147124. [DOI] [PubMed] [Google Scholar]
- 46.Argüello RJ, Albareda MC, Alvarez MG, Bertocchi G, Armenti AH, Vigliano C, et al. Inhibitory Receptors Are Expressed by Trypanosoma cruzi-Specific Effector T Cells and in Hearts of Subjects with Chronic Chagas Disease. PLoS ONE. 2012;7:e35966. doi: 10.1371/journal.pone.0035966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Reifenberg K, Lehr HA, Torzewski M, Steige G, Wiese E, Kupper I, et al. Interferon-gamma induces chronic active myocarditis and cardiomyopathy in transgenic mice. Am J Path. 2007;171:463–72. doi: 10.2353/ajpath.2007.060906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eriksson U, Kurrer MO, Bingisser R, Eugster HP, Saremaslani P, Follath F, et al. Lethal autoimmune myocarditis in interferon-gamma receptor-deficient mice: enhanced disease severity by impaired inducible nitric oxide synthase induction. Circulation. 2001;103:18–21. doi: 10.1161/01.cir.103.1.18. [DOI] [PubMed] [Google Scholar]
- 49.Eriksson U, Kurrer MO, Sebald W, Brombacher F, Kopf M. Dual role of the IL-12/IFN-gamma axis in the development of autoimmune myocarditis: induction by IL-12 and protection by IFN-gamma. J Immunol. 2001;167:5464–9. doi: 10.4049/jimmunol.167.9.5464. [DOI] [PubMed] [Google Scholar]
- 50.Afanasyeva M, Wang Y, Kaya Z, Stafford EA, Dohmen KM, Sadighi Akha AA, et al. Interleukin-12 receptor/STAT4 signaling is required for the development of autoimmune myocarditis in mice by an interferon-gamma-independent pathway. Circulation. 2001;104:3145–51. doi: 10.1161/hc5001.100629. [DOI] [PubMed] [Google Scholar]
- 51.Rangachari M, Mauermann N, Marty RR, Dirnhofer S, Kurrer MO, Komnenovic V, et al. T-bet negatively regulates autoimmune myocarditis by suppressing local production of interleukin 17. J Exp Med. 2006;203:2009–19. doi: 10.1084/jem.20052222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kania G, Siegert S, Behnke S, Prados-Rosales R, Casadevall A, Luscher TF, et al. Innate Signalling Promotes Formation of Regulatory Nitric Oxide-Producing Dendritic Cells Limiting T Cell Expansion in Experimental Autoimmune Myocarditis. Circulation. 2013 doi: 10.1161/CIRCULATIONAHA.112.000434. [DOI] [PubMed] [Google Scholar]
- 53.Barin JG, Talor MV, Baldeviano GC, Kimura M, Rose NR, Cihakova D. Mechanisms of IFNgamma regulation of autoimmune myocarditis. Exp Mol Pathol. 2010;89:83–91. doi: 10.1016/j.yexmp.2010.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Baldeviano GC, Barin JG, Talor MV, Srinivasan S, Bedja D, Zheng D, et al. Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ Res. 2010;106:1646–55. doi: 10.1161/CIRCRESAHA.109.213157. [DOI] [PubMed] [Google Scholar]
- 55.Rocha Rodrigues DB, dos Reis MA, Romano A, Pereira SA, de Teixeira VP, Tostes S, Jr, et al. In situ expression of regulatory cytokines by heart inflammatory cells in Chagas’ disease patients with heart failure. Clin Dev Immunol. 2012;2012:361730. doi: 10.1155/2012/361730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Laucella SA, Postan M, Martin D, Hubby Fralish B, Albareda MC, Alvarez MG, et al. Frequency of interferon- gamma -producing T cells specific for Trypanosoma cruzi inversely correlates with disease severity in chronic human Chagas disease. J Infect Dis. 2004;189:909–18. doi: 10.1086/381682. [DOI] [PubMed] [Google Scholar]
- 57.Silverio JC, Pereira IR, da Cipitelli MC, Vinagre NF, Rodrigues MM, Gazzinelli RT, et al. CD8+ T-cells expressing interferon gamma or perforin play antagonistic roles in heart injury in experimental Trypanosoma cruzi-elicited cardiomyopathy. PLoS Pathog. 2012;8:e1002645. doi: 10.1371/journal.ppat.1002645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guedes PM, Gutierrez FR, Silva GK, Dellalibera-Joviliano R, Rodrigues GJ, Bendhack LM, et al. Deficient regulatory T cell activity and low frequency of IL-17-producing T cells correlate with the extent of cardiomyopathy in human Chagas’ disease. PLoS Neg Trop Dis. 2012;6:e1630. doi: 10.1371/journal.pntd.0001630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chen P, Baldeviano GC, Ligons DL, Talor MV, Barin JG, Rose NR, et al. Susceptibility to autoimmune myocarditis is associated with intrinsic differences in CD4(+) T cells. Clin Exp Immunol. 2012;169:79–88. doi: 10.1111/j.1365-2249.2012.04598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tipping PG, Cornthwaite LJ, Holdsworth SR. Beta 2 integrin independent neutrophil recruitment and injury in anti-GBM glomerulonephritis in rabbits. Immunol Cell Biol. 1994;72:471–9. doi: 10.1038/icb.1994.71. [DOI] [PubMed] [Google Scholar]
- 61.Papageorgiou AP, Swinnen M, Vanhoutte D, VandenDriessche T, Chuah M, Lindner D, et al. Thrombospondin-2 prevents cardiac injury and dysfunction in viral myocarditis through the activation of regulatory T-cells. Cardiovasc Res. 2012;94:115–24. doi: 10.1093/cvr/cvs077. [DOI] [PubMed] [Google Scholar]
- 62.Frisancho-Kiss S, Nyland JF, Davis SE, Barrett MA, Gatewood SJ, Njoku DB, et al. Cutting edge: T cell Ig mucin-3 reduces inflammatory heart disease by increasing CTLA-4 during innate immunity. J Immunol. 2006;176:6411–5. doi: 10.4049/jimmunol.176.11.6411. [DOI] [PubMed] [Google Scholar]
- 63.Lv K, Xu W, Wang C, Niki T, Hirashima M, Xiong S. Galectin-9 administration ameliorates CVB3 induced myocarditis by promoting the proliferation of regulatory T cells and alternatively activated Th2 cells. Clin Immunol. 2011;140:92–101. doi: 10.1016/j.clim.2011.03.017. [DOI] [PubMed] [Google Scholar]
- 64.Li Y, Heuser JS, Kosanke SD, Hemric M, Cunningham MW. Protection against experimental autoimmune myocarditis is mediated by interleukin-10-producing T cells that are controlled by dendritic cells. Am J Path. 2005;167:5–15. doi: 10.1016/S0002-9440(10)62948-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fousteri G, Dave A, Morin B, Omid S, Croft M, von Herrath MG. Nasal cardiac myosin peptide treatment and OX40 blockade protect mice from acute and chronic virally-induced myocarditis. J Autoimmun. 2011;36:210–20. doi: 10.1016/j.jaut.2011.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cai G, Wang H, Qin Q, Zhang J, Zhu Z, Liu M, et al. Amelioration of myocarditis by HVEM-overexpressing dendritic cells through induction of IL-10-producing cells. Cardiovasc Res. 2009;84:425–33. doi: 10.1093/cvr/cvp219. [DOI] [PubMed] [Google Scholar]
- 67.Tzioufas AG, Kapsogeorgou EK, Moutsopoulos HM. Pathogenesis of Sjogren’s syndrome: what we know and what we should learn. J Autoimmun. 2012;39:4–8. doi: 10.1016/j.jaut.2012.01.002. [DOI] [PubMed] [Google Scholar]
- 68.Gershwin ME, Shoenfeld Y. Chella David: a lifetime contribution in translational immunology. J Autoimmun. 2011;37:59–62. doi: 10.1016/j.jaut.2011.05.011. [DOI] [PubMed] [Google Scholar]
- 69.Selmi C, Leung PS, Sherr DH, Diaz M, Nyland JF, Monestier M, Rose NR, Gershwin ME. Mechanisms of environmental influence on human autoimmunity: a National Institute of Environmental Health Sciences expert panel workshop. J Autoimmun. 2012;39:272–84. doi: 10.1016/j.jaut.2012.05.007. [DOI] [PubMed] [Google Scholar]
- 70.Jamin C, Renaudineau Y, Pers JO. Pierre Youinou: when intuition and determination meet autoimmunity. J Autoimmun. 2012;39:117–20. doi: 10.1016/j.jaut.2012.05.004. [DOI] [PubMed] [Google Scholar]