

Abstract
Nonsteroidal antiinflammatory drugs have been widely used for the management of inflammation, pain and nociception. Gastric intolerance caused by most of the nonsteroidal antiinflammatory drugs used today restricts their use. Several approaches have been proposed to modify the parent nonsteroidal antiinflammatory drugs molecule in order to reduce their gastric toxicity. Oral prodrug approach is one of such approaches. In the present work three nonsteroidal antiinflammatory drugs viz. ibuprofen, diclofenac, and flurbiprofen were conjugated with sulfonamides like sulphamethoxazole and sulphanilamide via amide bond using dicyclohexylcarbodiimide coupling reaction. The synthesized prodrugs were screened for their analgesic and antiinflammatory activity using Eddy's hot plate, acetic acid-induced writhing and carrageenan-induced rat paw edema method, respectively. These prodrugs were also evaluated for their ulcerogenic potential. All synthesized prodrugs were found to be less ulcerogenic than their parent nonsteroidal antiinflammatory drugs and showed better activity profile in terms of analgesic and antiinflammatory activity as compared to their respective parent drugs.
Keywords: NSAIDs, ibuprofen, diclofenac, flurbiprofen, ulcerogenicity
Nonsteroidal antiinflammatory drugs (NSAIDs) are most widely prescribed drugs for the treatment of various inflammatory disorders including rheumatoid arthritis. However, gastrointestinal, renal and cardiovascular toxicity associated with common NSAIDs limits their usefulness[1,2,3]. All NSAIDs are believed to inhibit the biosynthesis of prostaglandins by inhibiting the group of enzymes called cyclooxygenases (COX)[3]. Gastric mucosal injury produced by NSAIDs is generally aggravated by the local irritation caused by acidic group of NSAIDs[4]. Thus temporary masking of this group gives some relief to the patient from GI irritation; hence prodrug approach is the most suitable technique for this purpose[5]. Further, many inflammatory diseases occur due to microbial infection. Sulfonamides are the candidates, which can be coupled with the free carboxylic group of NSAIDs. Because sulfonamides are proven antimicrobials and it has also been established that sulfonamides are also antiulcer, which will give relief in case of gastric ulceration induced by NSAIDs[6]. Besides, these sulfonamides increase COX-2 selectivity, because of the presence of side pocket in the structure of COX-2 enzymes where sulfonamide group can easily fit[7]. This increase in COX-2 selectivity will not only enhance the antiinflammatory activity but also decrease the inhibition of gastroprotective COX-1 enzyme.
By keeping all these aims in mind, we herein, report the synthesis of amide prodrugs of ibuprofen, diclofenac, flurbiprofen with sulfonamides (sulphamethoxazole and sulfanilamide) and their analgesic, antiinflammatory, and ulcerogenic potential.
Flurbiprofen was obtained as gift sample from Sun Pharma Pvt. Ltd., Mumbai. Sulfanilamide and sulfamethoxazole were procured from Loba Chemie, Mumbai. All other reagents and solvents used were of AR grade purchased from S. D. Fine-Chem Ltd., Mumbai, India. The Infra-red (IR) spectra were recorded in the 4000-400 cm−1ranges using KBr discs on an IR 840 spectrometer, Shimadzu, Japan. Proton nuclear magnetic (1H NMR) spectra were recorded on Varian Mercury (300 MHz) spectrometer in CDCl3 as solvent using trimethylsilane (TMS) as an internal reference standard and values are expressed in δ ppm. Mass spectrum (MS) was recorded on Hewlett Packard MS 5989 B mass spectrometer at 70 eV ionizing beam using direct insertion probe.
All the experimental procedures and protocols used in this study were reviewed and approved by the institutional animal ethical committee (IAEC), constituted as per the requirement of CPCSEA Swiss albino mice (22-25 g) and male Wistar rats (160-180 g) were used for the analgesic and antiinflammatory studies, respectively. A 12:12 light-dark cycle was followed during the experiments. Antiinflammatory activity was performed using rat paw edema method using IITC 520 Water plethysmometer. Central analgesic activity was carried out using hot plate method on Dolphin, KI 9514 Hot Plate instrument.
The amide prodrugs of ibuprofen, diclofenac and flurbiprofen with sulphamethoxazole and sulfanilamide were synthesized by coupling reactions using dicyclohexylcarbodimide (DCC)[8,9]. The weighed quantity of NSAID (0.01M) was dissolved in 40 ml of DCM and to this solution DCC (0.01M) was added. It was then stirred for half an hour at room temperature. To this solution corresponding sulfonamide (0.01M) in 20 ml DCM was added dropwise. This reaction mixture was stirred for 2 h at 0° and left overnight as such at room temperature. The status of reaction was monitored by thin layer chromatography using ethyl acetate-hexane as mobile phase. Next day after completion of reaction, reaction mixture was filtered off to remove precipitated dicyclohexyl urea. Solvent was removed and 10 ml ethyl acetate was added to the residual mass. Then it was washed with 10 ml of 10% NaHCO3, 10 ml distilled water to remove unreacted starting materials. Organic layer was dried over anhydrous magnesium sulfate and degassed to remove crude product. The crude product was further purified by column chromatography using silica gel as stationary phase and mixture of ethyl acetate and hexane as eluent. The structures of all the compounds were confirmed using spectral studies. The general scheme for synthesis is given in Scheme 1 and the characterization data for 3a-f is as follows;
Scheme 1.
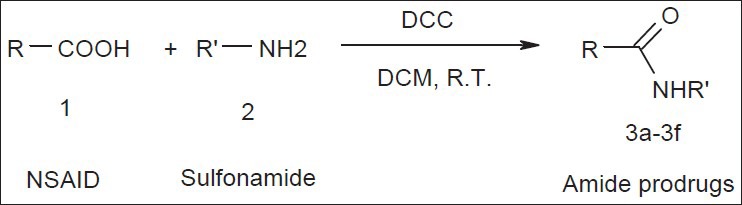
Scheme of synthesis
DCM is dichloromethane, DCC is N,N-dicyclohexylcarbodiimide. Where, R-COOH is NSAID and R’-H2 is sulfonamide. 3a, R=ibuprofen and R’=suphamethoxazole; 3b, R=ibuprofen and R’=suphanilamide; 3c, R=diclofenac and R’=suphamethoxazole; 3d, R=diclofenac and R’=suphanilamide; 3e, R=flurbiprofen and R’=suphamethoxazole; 3f, R=flurbiprofen and R’=suphanilamide;
2-(4-(2-methylpropyl) phenyl)-N-((5-methyl-1,2-oxazol 3yl) sulfamoylphenyl) propanamide (3a): Yield 76%. Mol. Wt. 441.58.1H NMR (CDCl3): δ 8.6 (s, 1H CONH); 7.70 (d, 2H, Ar (dd’); 7.50 (d, 2H, Ar (cc’); 7.26 (d, 2H, Ar (bb’); 7.14 (d, 2H, Ar (aa’); 6.2 (s, 1H, SO2NH); 3.75 (q, 1H, CH); 2.30 (s, 3H, CH3-oxazolyl); 2.50 (d, 2H, CH2); 2.50 (d, 3H, CH3); 1.90 (m, 1H, CH); 1.60 (d, 6H, CH3); Rf 0.76, ethyl acetate:hexane (1:3); IR (KBr) cm−1: 3319.26 (NH, str.), 2925.81 (Ar-CH, str.), 1679 (C=O, amide, str.), 1521 (NH, bend), 1321 (SO2NH, str.), 1163 (S=O, str.).
2-(4-(2-methylpropyl) phenyl)-N-((4-sulfamoylphenyl) propanamide (3b): Yield 70% Mol. Wt. 360.51.1H NMR (CDCl3): δ 8.12 (s, 1H CONH); 7.80 (d, 2H, Ar (dd’); 7.60 (d, 2H, Ar (cc’); 7.20 (d, 2H, Ar (bb’); 7.0 (d, 2H, Ar (aa’); 3.70 (q, 1H, CH); 2.50 (d, 2H, CH2); 2.50 (d, 3H, CH3); 1.7-1.8 (m, 1H, CH); 1.4 (d, 6H, CH3); Rf 0.79, ethyl acetate:hexane (1:3); IR (KBr) cm−1: 3325 (NH, str.), 2931.(Ar-CH, str.), 1679.88 (C=O, amide, str.), 1527.52 (NH, bend), 1305 (SO2NH, str.), 1149 (S=O, str.), 837, 590 (Ar-CH, bend).
2-{2-((2,6-dichlorophenyl) amino] phenyl}-N-((5- methyl-1,2-oxazol-3yl) 4 sulfamoylphenyl) acetamide (3c): Yield 65% Mol. Wt. 531.4 Calc. C 54.19, H 3.76, N 10.53; Found C 54.2, H 3.79, N 10.4.; Rf 0.68, ethyl acetate:hexane (1:3); IR (KBr) cm−1: 2921.96 (Ar-CH, str.), 1610.45 (C=O, amide, str.), 1564.16 (NH, bend), 1357.79 (SO2NH, str.), 1166.85 (S=O, str.), 783,669 (C-Cl, str.)
2-{2-[(2,6-dichlorophenyl) amino] phenyl}-N-(4- sulfamoylphenyl) acetamide (3d): Yield 69% Mol. Wt. 450.338. Mass m/e: 451 (M + 1, 100%); Rf 0.59, ethyl acetate:hexane (1:3); IR (KBr) cm−1: 3261.40 (NH, str.), 2933.53 (Ar-CH, str.), 1676 (C=O, amide, str.), 1523.66 (NH, bend), 1382.87,1317.29 (SO2NH, str.), 1161.07 (S=O, str.), 771, 675 (C-Cl, str.).
2-(2-fluorobiphenyl-4-yl)-N-((5-methyl-1,2-oxazol-3yl) sulfamoylphenyl] propanamide (3e): Yield 78% Mol. Wt. 479.55. Mass m/e: 479.5 (M+, 80%); Rf 0.76, ethyl acetate:hexane (1:2); IR (KBr) cm−1: 3469.70, 3282.62 (NH, str.), 2929.67, 2854.46 (Ar-CH, str.), 1699.17, 1664.45 (C=O, amide, str.), 1587.31, 1537.16 (NH, bend), 1380.94 (SO2NH, str.), 1163 (S=O, str.), 1085 (C-F, str.).
2-(2-fluorobiphenyl-4-yl)-N-((4-sulfamoylphenyl) propanamide (3f): Yield 55% Mol. Wt. 398.46, Mass m/e: 399.8 (M+1, 75%), 227.0 (M-171.48, 55%); Rf 0.87, ethyl acetate:hexane (1:3); IR (KBr) cm−1: 3282.62 (NH, str.), 2929.67, 2854.45 (Ar-CH, str.), 1662.52 (C=O, amide, str.), 1537.16 (NH, bend), 1384.79 (SO2NH, str.), 1164.92 (S=O, str.), 1072.35 (C-F, str.).
All synthesized prodrugs were evaluated for analgesic activity (central and peripheral), antiinflammatory activities and for their ulcerogenic potential. All the test compounds were relatively insoluble in water. The doses were prepared by suspending the test compounds in 0.5% aqueous sodium carboxymethyl cellulose (CMC) using a mortar and pestle. All drugs were given in a volume of 1 ml/100 g body weight of animal. Control animals received equal volume injections of 0.5% sodium CMC. Molecular equivalent doses to the doses of parent drug were calculated, In order to compare the potency of the prodrugs with that of parent drug at the same dose level. The activities of prodrugs were compared with their corresponding NSAID drug.
Peripheral analgesic activity was determined by using acetic acid-induced writhing method. Mice were divided into groups containing six animals each group. Writhing was induced by intraperitoneal (i.p.) injection of acetic acid solution (1%, 10 ml/kg, i.p.). The mice were placed individually into glass beakers and 5 min were allowed to elapse. The mice were then observed for a period of 10 min and the number of writhes was recorded for each animal. Abdominal writhing was considered as nociceptive behavior, and it was defined as an exaggerated extension of the abdomen combined with the outstretching of the hind limbs.
Central analgesic activity was performed using Eddy's hot plate method. The test compounds and standard drugs viz. ibuprofen or diclofenac or flurbiprofen and vehicle were given orally to the groups of Swiss albino mice. Sixty min after oral administration, mice were placed individually on the hot plate maintained at 55±1°. The latency of nociception response such as licking, flicking of a hind limb or jumping was noted. The experiment was terminated 20 s after their placement on the hot plate to avoid damage to the paws.
Antiinflammatory activity of the synthesized prodrugs was carried out by carrageenan-induced paw edema method using carrageenan (1%, 0.1 ml) as phlogistic agent. Wistar rats used for the study were divided into the three groups including test, standard and control each containing 6 animals per group. The initial volume of right hind paw of albino rats was measured by plethysmometer, without administration of the drug/prodrug. The drug/prodrugs were administered orally in 0.5% suspension of sodium carboxymethyl cellulose. After 1 h of administration of drug/prodrugs, carrageenan (1%, 0.1 ml solution in saline) was injected into the plantar side of right hind paw of each animal. The volume of right hind paw of Wistar rats was measured by plethysomometer after 0, 3 and 24 h. The mean difference in the volume of right hind paw of rats was compared with standard and control. Percent antiinflammatory activity was calculated using the following formula: antiinflammatory activity %=[1-(Vt-Vc)]×100, where Vc is mean change in paw volume in control group and Vt is mean change in paw volume in test group. The test drugs were considered more active if they show more percentage inhibition as compared to the standard drug.
Male Wistar rats were used for this study. The test compounds, standard drugs, and vehicle were given orally to the respective groups daily for 5 days. The rats were fasted overnight before the last dose. On the last day, the animals were sacrificed 60 min after oral administration of drugs. The stomach was removed, gently washed with normal saline and fixed in 10% formalin. The gastric mucosa was examined for lesions. The lesions were counted by visual examination using a 2×2 binocular magnifier. The ulcerative index was calculated for each animal.
For each stomach the mucosal damage was assessed according to the following scoring system: 0.5: redness, 1.0: spot ulcers, 1.5: hemorrhagic streaks, 2.0: ulcers<3, ulcers>5. The mean score of each treated minus the mean score of control group is recorded as severity index of gastric mucosal damage.
In the present study amide prodrugs of NSAIDs like ibuprofen, flurbiprofen and diclofenac were synthesized by coupling them with sulfonamides like sulphamethoxazole, sulfanilamide, using N, N-dicyclohexylcarbodiimide (DCC). DCC method of synthesis was followed because this method has an advantage of carrying reactions at room temperature and DCC itself is nonirritating and nonfuming reagent. The other method, which involves the use of thionyl chloride, was not followed as it requires high temperature and thionyl chloride is lachrymatory and irritating. NSAIDs were reacted with DCC in order to activate the carboxylic acid group. This leads to the formation of intermediate called as O-acylisourea, which on subsequent treatment with sulfonamide forms amide prodrugs. In total, six prodrugs were synthesized. The reaction yielded all the prodrugs in moderate to good quantity (55-78%).
The compounds were purified by column chromatography using suitable solvent system. All the compounds were characterized by spectroscopic techniques like IR/NMR/MASS. The characterization data supported the structure of the compounds proposed.
In order to evaluate the activity of NSAIDs after coupling them with sulfonamides; these compounds were screened for their analgesic and anti-inflammatory activity. The data for analgesic and anti-inflammatory activity is shown in Table 1.
TABLE 1.
ANALGESIC, ANTIINFLAMMATORY AND ULCEROGENIC ACTIVITY OF COMPOUNDS
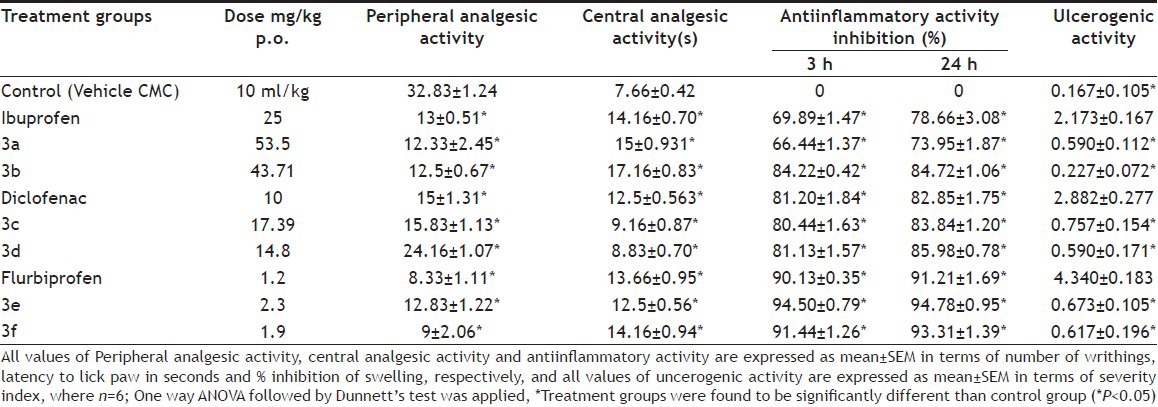
Almost all the compounds exhibited analgesic and antiinflammatory activity comparable with the parent NSAIDs. In case of peripheral analgesic activity which was performed using acetic acid-induced writhing method in mice, prodrugs of ibuprofen (3a and 3b) were found more active than the ibuprofen itself. Central analgesic activity was carried out using hot plate method by giving pain stimulus in the form of heat. Compounds 3a, 3b and 3f were found to be more active than corresponding parent NSAIDs. The antiinflammatory activity was carried out using carrageenan-induced rat paw edema method. It was evaluated by determining percentage inhibition at three and twenty four hours after drug administration. Compounds 3a, 3b and 3f exhibited more percentage inhibition than their corresponding parent drugs. This increase in the activity suggested that sulfonamides were acting synergistically with NSAIDs.
In order to determine the reduction in the ulcer formation induced by selected NSAIDs, these prodrugs were also evaluated for their ulcerogenic potential. Ulecrogenic activity is shown in Table 1 and represented as mean±SEM (n=6) of severity index. One-way ANOVA followed by Dunnett's test was applied. Treatment groups were found to be significantly (P<0.05) different than their respective parent drug groups. All prodrugs showed the drastic reduction in the ulcer formation of all the NSAIDs under study.
Possible route of hydrolysis of the synthesized prodrugs is probably by cleavage of amide bond between antiinflammatory and sulphonamide molecule by peptidases and various other amidases present in intestine, but not in stomach, where it is hypothesized to remain as intact molecule. Thus, preventing gastric side-effects produced by NSAID's.
In general, it was observed that amide prodrugs of NSAIDs with sulfanilamide showed more analgesic, antiinflammatory activity and less ulcerogenic potential than their corresponding parent NSAIDs. This may be because sulfanilamide is a prototype of sulfonamides and has sulfonamide group which increases COX-2 selectivity and thus acting synergistically. Moreover it also possesses carbonic anhydrase inhibitory activity, which may have shown inhibitory effect on gastric carbonic anhydrase enzyme thus, limiting the acid secretion. Hence, as proposed the prodrugs with greater potency and reduced gastric toxicity than the corresponding parent NSAIDs were synthesized.
However, the complete elimination of ulcer formation cannot be avoided, since the prodrug conversion avoids the gastric irritation due to direct contact of acidic group with the gastric mucosa, while the ulcer formation due to inhibition of cytoprotective prostaglandin synthesis is inevitable.
ACKNOWLEDGMENTS
Authors thank to the BCUD, Mumbai University for providing financial support.
Footnotes
Makhija, et al.: Antiinflammatory Mutual Amide Prodrugs
REFERENCES
- 1.Vane JR. Inhibition of prostaglandin synthesis as a mechanism of action for aspirin like drugs. Nat New Biol. 1971;231:232–5. doi: 10.1038/newbio231232a0. [DOI] [PubMed] [Google Scholar]
- 2.Smith JB, Willis AL. Aspirin selectively inhibits prostaglandin production in human platelets. Nat New Biol. 1971;231:235–7. doi: 10.1038/newbio231235a0. [DOI] [PubMed] [Google Scholar]
- 3.Allison MC, Howatson AG, Torrance CJ, Lee FD, Russell RI. Gastrointestinal damage associated with Nonsteroidal antiinflammmatory drugs. N Engl J Med. 1992;327:749–54. doi: 10.1056/NEJM199209103271101. [DOI] [PubMed] [Google Scholar]
- 4.Hla T, Neilson K. Human cyclooxygenase-2 c-DNA. Proc Natl Acad Sci USA. 1992;89:7384–8. doi: 10.1073/pnas.89.16.7384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Makhija DT, Somani RS. Improvement of GI tolerance of NSAID's using oral prodrug approach. Der Pharmacia Lett. 2010;2:300–9. [Google Scholar]
- 6.Subudhi BB, Panda PK, Bhatta D. Synthesis anti-ulcer activity study of 1,4-dihydropyridines and their mannich bases with sulfanilamides. Indian J Chem. 2009;48B:725–8. [Google Scholar]
- 7.Flower RJ. The development of COX-2 inhibitors. Nat Rev Drug Discov. 2003;2:179–91. doi: 10.1038/nrd1034. [DOI] [PubMed] [Google Scholar]
- 8.Doleschall G, Lempert K. On the mechanism of carboxyl condensations by carbodiimidess. Tetrahedron Lett. 1963;18:1195–9. [Google Scholar]
- 9.Smith M, Moffat JG, Khorana HG. Carbodiimdes VIII. Observations on the reactions of carbodiimides with acids and some new applications in the synthesis of phosphoric acid esters. J Am Chem Soc. 1958;80:6204–12. [Google Scholar]