

Abstract
Background:
Pre-eclampsia (PE) complicates approximately 5-7% of all pregnancies. This study investigates the effects of S-nitroso-N-acetylpenicillamine (SNAP) and S-nitrosoglutathione (GSNO) on the classical features of PE.
Materials and Methods:
On day 14 of gestation, female Sprague-Dawley rats were separated into five groups and treated intravenously for 7 days as follows: (i) 0.3 mL 0.9% saline (control, n = 11); (ii) 50 mg/kg Body Weight (BW) N-nitro-L-arginine methyl ester (L-NAME) in 0.3 mL saline (n = 10); (iii) 50 mg/kg BW L-NAME and 8 mg/kg BW GSNO in 0.15 mL saline (n = 6); (iv) 50 mg/kg BW L-NAME in 0.15 mL saline and 8 mg/kg BW SNAP in 0.15 mL DMSO (n = 9); and (v) 0.15 mL DMSO and 0.15 mL saline (SNAP control, n = 7). Blood pressures were measured on day 14 through day 20, a 4-h urine sample was taken on day 20, and animals were sacrificed on day 21. Pups were counted and weighed individually.
Results:
SNAP and GSNO significantly decreased systolic, diastolic, and mean arterial pressures in PE-induced rats from day 14 through day 20 (P < 0.05). Pup weights in SNAP and GSNO groups were higher than in L-NAME group but lower than in controls (P ≤ 0.001). SNAP and GSNO partially reversed growth retardation.
Conclusion:
Elevated blood pressure, proteinuria, and intrauterine growth restriction associated with PE were induced in Sprague-Dawley rats using L-NAME. These were partially reversed with the use of GSNO and SNAP. The mechanism of action of these S-nitrosothiols (RSNOs) should be further explored.
Keywords: Blood pressure, N-nitro-l-arginine methyl ester, pre-eclampsia, S-nitrosoglutathione, S-nitroso-N-acetylpenicillamine
INTRODUCTION
Hypertensive disorders such as chronic hypertension, gestational hypertension, and pre-eclampsia (PE) complicate approximately 5-7% of all pregnancies,[1] with PE being dominant, affecting 3-5% of pregnancies.[2] The diagnosis of PE is made based on the presence of both hypertension and proteinuria after 20 weeks of gestation. Changes that are observed in PE are usually pregnancy-induced and regress after delivery.[3]
The symptoms of PE not only reflect damage to the uterus, fetus, and placenta, but to the kidneys as well.[4] Though PE is of unknown etiology, there are risk factors that are associated with the disorder, such as gestational and type 1 diabetes mellitus, obesity, and chronic hypertension.[5] It has been reported that PE originates in the placenta.[6] Placental vascular abnormalities associated with PE contribute to fetal intrauterine growth restriction (IUGR). PE is among the leading causes of IUGR and results from the impairment of materno-fetal exchanges. These impairments in materno-fetal exchanges coincide with the onset of PE in the third trimester of pregnancy.[7]
Nitric oxide (NO), which is generated by the endothelial cells, has been implicated in the pathogenesis of PE. Its production plays an integral role in homeostatic vasodilation and is believed to contribute to the vasodilation of normal pregnancy.[8] The biosynthesis of NO and cGMP increases during pregnancy in rats.[9] The plasma level and urinary excretion of cGMP are also increased in human gestation, but the status of the biosynthesis of NO in normal human pregnancy and PE is controversial.[10]
Choi et al. reported that NO production increased during normal pregnancy and decreased in PE.[11] Seligman et al. also showed that there was a decrease in NO production in pre-eclamptic individuals.[12] In an earlier study, a decrease in amniotic cGMP was found in PE.[13] cGMP is believed to reflect the overall production of NO, therefore a decrease in cGMP reflects a decrease in NO. However, other investigators reported an increase in NO production in PE.[14,15,16] It is believed that the increase may be part of a compensatory mechanism to offset the pathological effects of PE.[15,16]
To get a better understanding of PE, several animal models of the disease have been developed using various compounds to manipulate certain mechanisms. The inhibition of NO synthesis using N-nitro-L-arginine methyl ester (L-NAME) is one such mechanism and is used to induce PE in rat models.[17] The hallmark features of PE, proteinuria, and hypertension are seen in these models on administration of nitric oxide synthase (NOS) inhibitors. It is believed that these models can greatly assist in understanding the pathophysiology of this hypertensive disease. This study investigates the effects of S-nitrosothiols (RSNOs) such as S-nitroso-N-acetylpenicillamine and S-nitrosoglutathione (GSNO) on rats with the classical features of PE induced using L-NAME.
MATERIALS AND METHODS
Animal care
Approval for the use of rats in all experiments was obtained from the UHWI/UWIFMS Ethics Committee (AN5, 2006/2007). The animals were housed in the Department of Basic Medical Sciences at the University of the West Indies, Mona. They were maintained there under the supervision of the attendants. They were fed tap water and LabDiet 5008 Formula Diet ad libitum. Each experimental group had 6-10 rats to allow for statistical analysis.
Female Sprague-Dawley rats weighing 200-300 g were housed in the departmental animal house and entrained by a 12 h: 12 h light: Dark cycle. They were acclimatized to laboratory conditions for 2 weeks before being studied. Following acclimatization, they were housed overnight with male Sprague-Dawley rats to facilitate mating. The day prior to co-housing was noted as day 1 of gestation; from then on, the animals were checked weekly for weight gain to confirm pregnancy and their blood pressures monitored as well.
On day 14 (after being mated), the animals were separated into five groups and treated as follows for 7 days: (i) 0.3 mL 0.9% saline (control, n = 10), (ii) 50 mg/kg Body Weight (BW) L-NAME in 0.3 mL saline (n = 10), (iii) 50 mg/kg BW L-NAME and 8 mg/kg BW GSNO in 0.15 mL saline (n = 6), (iv) 50 mg/kg BW L-NAME in 0.15 mL saline and 8 mg/kg BW SNAP in 0.15 mL dimethyl sulfoxide (DMSO; n = 9); (v) 0.15 mL DMSO and 0.15 mL saline (SNAP control, n = 7).
The animals were treated for 7 days and their blood pressures measured throughout the 7 days of treatment. Animals were kept in metabolic cages for 4 h for urine collection. These samples were taken for each animal on day 20 of gestation for protein analysis. They were sacrificed on day 21 by cardiac puncture. The pups were removed, numbered, and immediately weighed individually.
Blood pressure measurements
The blood pressures of the animals were obtained by applying a non-invasive method using the CODA 6 Blood Pressure (BP) System from Total Protein Kit Sigma-Aldrich (Missouri, USA). Blood pressure readings were taken in one set consisting of 25 cycles including 5 acclimation cycles 10 min after the animal was treated. The system automatically eliminated the acclimation cycles as well as any erroneous readings.
The systolic blood pressure (SBP), diastolic blood pressure (DBP), and mean arterial pressure (MAP) of all the animals in each of the five groups (control, L-NAME, GSNO, SNAP, and SNAPC) were measured on days 1, 7, 14, 16, 18, and 20 using the CODA 6 BP system.
Urine and blood analyses
Each animal was placed separately in metabolic cages for 4 h for urine collection on day 20. This was done in the absence of food or water to avoid contamination by fallen food particles. Also, as blood pressure was measured in the same animals, it was necessary to avoid stress, hence the limited time for urine collection. Urine was collected, dispensed in 2 mL microcentrifuge tubes, centrifuged at 3000 rpm for 10 min, and stored at −20°C until ready for use.
The protein concentration of each urine sample was determined with the Total Protein Kit (Sigma) which uses the micro pyrogallol red method. The assays were done according to the directions given in the kit and the absorbance was measured at 600 nm. The urine samples were analyzed for creatinine concentration at the Chemical Pathology Laboratory at the University Hospital of the West Indies (UHWI). Samples were assayed using the alkaline picrate method which relies on the Jaffe reaction, in which the samples form a yellow/orange color when treated with alkaline picrate.[18,19] The color intensity (which is proportional to the creatinine concentration of the sample) was measured using the Architect c8000 spectrophotometer at a wavelength of 500 nm. The level of proteinuria was determined by calculating mg protein/mg of creatinine.
Statistical analyses
Data are presented as the means ± SEM. Blood pressure parameters values were analyzed using one-way analysis of variance, while urine and pup weight values were evaluated using Student's t-test. P < 0.05 was considered statistically significant in all cases.
RESULTS
Animals treated with L-NAME showed sustained elevated SBP from day 14 (168.6 ± 1.32 mmHg) through to day 20 (163.2 ± 1.85 mmHg), which was significantly higher than the other groups (P < 0.05) [Figure 1]. Both control groups (animals treated with saline only (control) and animals treated with saline plus DMSO (SNAPC)) had relatively consistent SBP throughout the entire gestation period. Animals that were treated with SNAP and GSNO had significantly lower (P < 0.05) SBP than the other groups from day 14 and day 16 onward (respectively).
Figure 1.
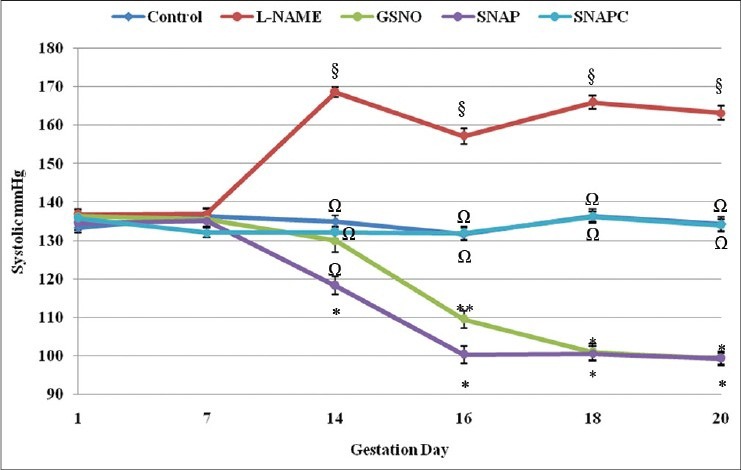
Systolic pressure of the different groups from day 1 through to day 20 of gestation. Significant differences (P < 0.05) are indicated by different symbols on the different gestation days
The DBP of the animals treated with L-NAME showed marked increases post-treatment, which remained elevated through to day 20 [Figure 2]. DBP in the L-NAME treated group was significantly (P < 0.05) higher than in the other groups. The controls had fairly consistent DBP throughout the gestation period with slight changes that were not significant. The DBP of the SNAP and GSNO groups were lowered post-treatment and remained low through to day 20. Though the GSNO group showed similarity to both the control groups and the SNAP group on day 14, the latter (80.8 ± 1.96 mmHg) was significantly different (P < 0.05) from both controls (SNAPC: 93.2 ± 1.74 mmHg and control: 90.0 ± 1.38 mmHg) and the L-NAME group (124.0 ± 1.41 mmHg), but similar to the GSNO (87.7 ± 3.01 mmHg) group.
Figure 2.
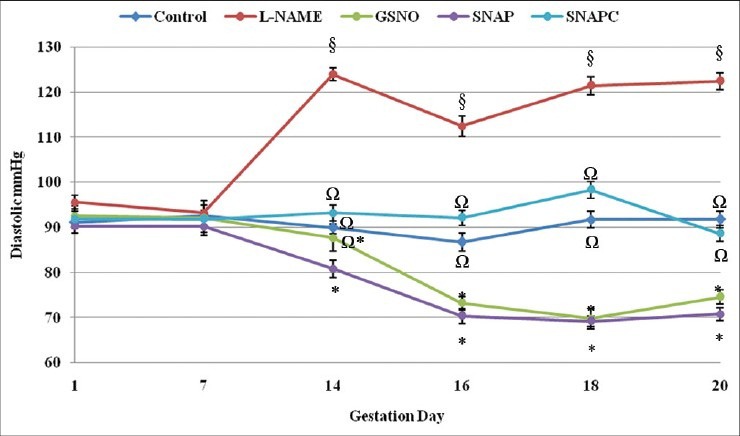
Diastolic pressure of the different groups from day 1 through to day 20 of gestation. Significant differences (P < 0.05) are indicated by different symbols on the different gestation days
The results observed for MAP of the different animal groups were similar to those of the SBP. The L-NAME group had significant (P < 0.05) sustained elevated pressure from day 14 through to day 20 of gestation (post-treatment) compared with the other groups [Figure 3]. The MAP in the control groups remained constant and that of the SNAP and GSNO groups decreased and remained significantly (P < 0.05) low through to day 20.
Figure 3.
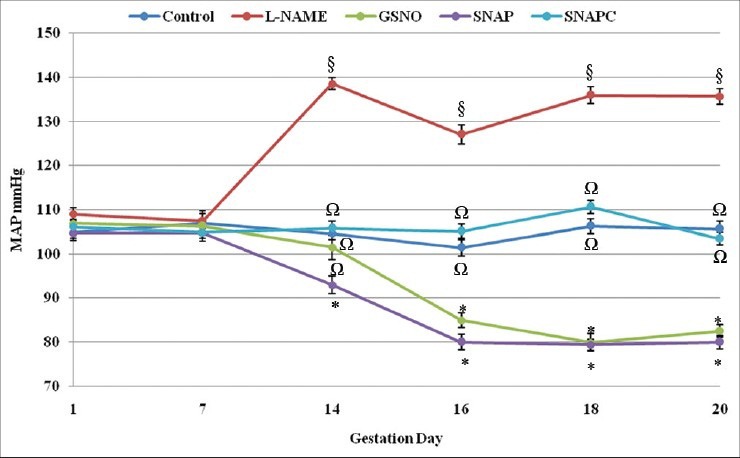
Mean arterial pressure (MAP) of the different groups from day 1 through to day 20 of gestation. Significant differences (P < 0.05) are indicated by different symbols on the different gestation days
The L-NAME treated animals showed a significant increase (P < 0.05) in the level of proteinuria (4.9 ± 1.16 mg protein/mg creatinine) compared with the control group (2.3 ± 0.38 mg protein/mg creatinine) [Figure 4]. However, both SNAP and GSNO showed an unexpected increase rather than a decrease in proteinuria compared with the controls. The level of proteinuria in the SNAP (8.5 ± 1.79 mg protein/mg creatinine), GSNO (6.5 ± 2.46 mg protein/mg creatinine), and L-NAME (4.9 ± 1.16 mg protein/mg creatinine) groups was all significantly higher (P < 0.05) than that of the control groups (2.3 ± 0.38 mg protein/mg creatinine and SNAPC, 3.3 ± 0.41 mg protein/mg creatinine).
Figure 4.
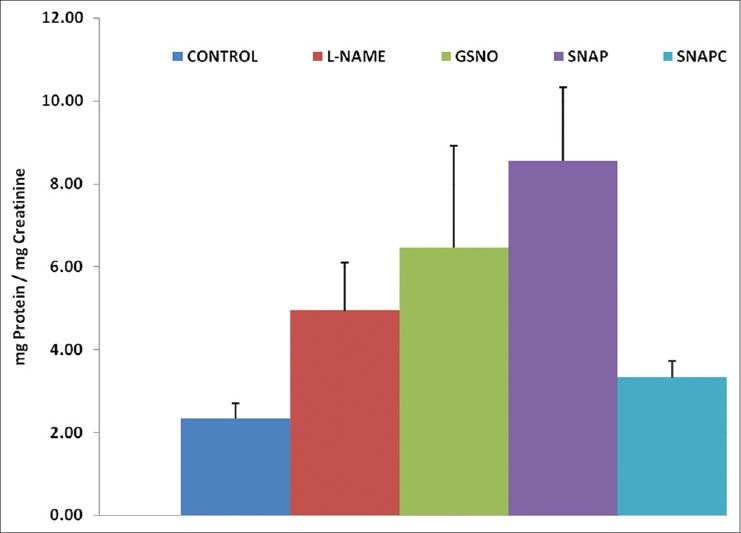
Urinary protein levels (mg protein/mg creatinine) of the different groups. Significant differences (P < 0.05) are indicated by different letters
Pup weight is a measure of growth retardation which is associated with PE. The reversal of growth retardation was observed in both the SNAP and GSNO groups [Figure 5]. Both of these groups (GSNO, 4.1 ± 0.08 g and SNAP, 3.9 ± 0.08 g) weighed significantly lower (P < 0.05) compared with the control group (4.9 ± 0.07 g), but were significantly higher in weight than the L-NAME group (3.4 ± 0.10 g). The SNAP group did not show any significant difference compared with the SNAPC group (4.2 ± 0.08 g).
Figure 5.
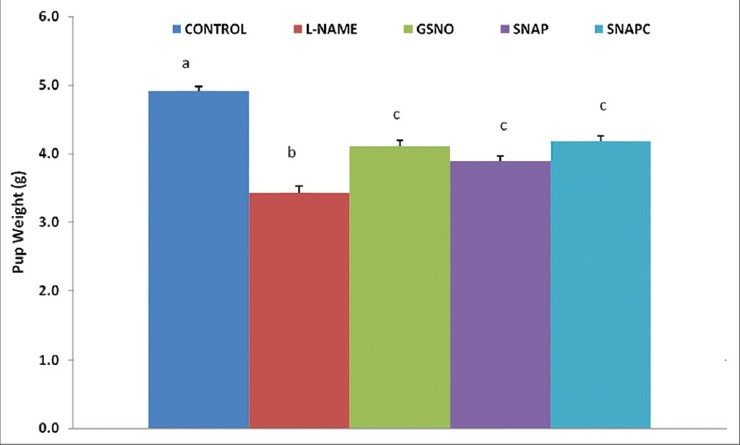
Average pup weight (g) of each rat group. Significant differences (P < 0.05) are indicated by different letters
DISCUSSION
In this study, the blood pressure measurements remained fairly constant throughout gestation in the control groups, but increased in the L-NAME group and decreased in the GSNO and SNAP groups in the third week. Since the gestation period for Sprague-Dawley rats on average is 3 weeks (21 days), the experiment was designed to mirror the condition in humans. PE occurs in the third trimester or after 20 weeks of gestation in humans, hence the time of induction in the rat model was day 14, which is the beginning of the third week. Hypertension (one of the hallmark characteristics of PE) was successfully induced by L-NAME and reversed using SNAP and GSNO. In Sprague-Dawley rats, BPs greater than 129/91 mmHg are considered high. Proteinuria and the associated IUGR were also observed in the induced models.
L-NAME has been used with much success to induce PE-like symptoms in pregnant rats. Whether administered orally or intravenously, the effects are similar: Elevated blood pressure, proteinuria, and growth retardation.[20,21] L-NAME is an analog of l-arginine which competes at the active site of NOS, resulting in a reduction in the synthesis of NO, which subsequently leads to elevated blood pressures.[22] The increase in SBP, DBP, and MAP on the first day of administration reflects the potent inhibitory action of L-NAME in reducing the production of NO. Though the values fluctuated over the course of treatment, elevated blood pressure measurements were sustained over the 7-day period (days 14-20). On the contrary, both SNAP and GSNO lowered blood pressures through the release of the potent vasodilator, NO. The process of lowering blood pressure first involves the activation of the enzyme guanylate cyclase by the released NO. This subsequently leads to the generation of cGMP. Increased levels of cGMP lead to vascular smooth muscle relaxation via a decrease in calcium release from intracellular stores. The blood pressure is lowered as a result of decreased peripheral arterial vascular resistance.[23]
Both SNAP and GSNO ultimately lowered blood pressure to a similar extent; however, the blood pressure measurements (SBP, DBP, and MAP) of the animals that were treated with SNAP were lower than those treated with GSNO, at the time of initial administration of both drugs (day 14). When compared with controls, blood pressure measurements of the animals treated with SNAP were significantly lower (P ≤ 0.05) from the initial point of administration (day 14) onward. Significant lowering in blood pressure measurements in the GSNO group was not observed at this time, but rather from day 16 onward (P ≤ 0.05), implying that SNAP has a more rapid onset of action than GSNO. The rapid onset of action of SNAP has been confirmed in previous studies done by Mathews and Kerr in 1993, which showed that GSNO was more stable and had a longer half-life (159 h) than SNAP (1.15 h).[24] NO is released faster from SNAP as it is less stable than GSNO; therefore, SNAP would act faster than GSNO in lowering the blood pressure.
The potency of SNAP and GSNO in lowering BP was demonstrated even in the presence of L-NAME. At a dose of 8 mg/kg BW, both SNAP and GSNO were very efficient in lowering blood pressures with L-NAME at a higher dosage of 50 mg/kg BW being co-administered. L-NAME on its own increased SBP, DBP, and MAP significantly, but when co-administered with each of the RSNOs, its effects were countered significantly in all three blood pressure measurements. These findings support previously reported data of the hypotensive nature of GSNO and SNAP.[25]
Elevated levels of urinary protein indicate kidney dysfunction. Treatment with GSNO and SNAP caused a further increase in proteinuria compared with the L-NAME group, but this was not significant. Studies have shown that L-NAME causes kidney damage,[26,27] hence the high levels in the L-NAME group, but there is no report saying that RSNOs cause kidney damage. As both GSNO and SNAP were co- administered with L-NAME separately (SNAP + L-NAME and GSNO + L-NAME), it is not clear which compound could have caused possible irreversible damage to the kidneys, and therefore the high levels of protein. It has been reported that DMSO can cause kidney damage.[28] Therefore, the use of the solvent DMSO in the SNAP group also introduces another uncertainty as to the source of possible renal insult. It is, however, not certain how much damage the DMSO could have caused as the difference in urinary protein in the SNAPC group (3.3 ± 0.41 mg protein/mg creatinine) compared with the control group (2.3 ± 0.38 mg protein/mg creatinine) was not significant.
Partial reversal of growth retardation was observed in the GSNO group where the weight of the pups was higher than in the L-NAME group but lower than in the control group. On the other hand, reversal was observed in the SNAP group, where the weight of the pups was higher than in the L-NAME group and similar to that of the SNAPC group. However, when the SNAPC group was compared with the control group, it was observed that DMSO might have caused some interference with the weight of the pups as these groups were significantly different (P < 0.05). There have been no documented reports of DMSO affecting the pup weight. However, as with the results observed for the level of proteinuria, it was observed that DMSO might indeed have an effect which contributed to growth retardation. This, however, is not for certain, so further investigations need to be done to confirm.
It has already been established that L-NAME causes growth retardation in rats.[21,27] This was observed in the L-NAME group which had significantly lower (P < 0.05) pup weights compared with the weights of the pups in the control group. There has been no report, however, of the effect of RSNOs on pup weight in PE-induced rats. As observed in the blood pressure results of the RSNO groups where L-NAME was co-administered, the countering action of GSNO and SNAP was observed. It is suspected that the growth retarding effect of L-NAME is countered by NO which is released from GSNO and SNAP. Further investigations are required to determine the mechanism of action of these compounds in countering the effects of L-NAME in the reversal of growth retardation. Another important observation is the concentration of GSNO and SNAP compared with L-NAME (8 mg/kg BW to 50 mg/kg BW, respectively) and how effective the latter was in reversing the restricted growth at such a low dose. The effect of co-administering L-NAME and GSNO or SNAP at different concentrations could also be investigated.
In addition to the observed reduction in pup weight in the L-NAME treated animals, there were other observations, such as hind limb deformation, that were made. This is consistent with previously published reports.[29,30] Another important observation was that the pups from mothers treated with L-NAME died post-delivery, while the pups from mothers of the control and RSNO groups survived. The death of the pups of the L-NAME group could be as a result of poor lung development resulting from IUGR.[31] Insufficient pulmonary surfactant levels could also explain why these pups did not survive. Pulmonary surfactant is a lipoprotein which is produced by type 2 cells in the lung. It is essential for normal breathing as it reduces alveolar surface tension, and its production is initiated in the third trimester of gestation in fetal lung. Insufficient surfactant production by the lungs can result in respiratory distress syndrome which is observed in premature and low birth weight infants.[32,33]
CONCLUSION
It has been shown that the major symptoms associated with PE were induced in Sprague-Dawley rats by intravenous administration of L-NAME (50 mg/kg BW daily). These hallmark features – elevated blood pressure, proteinuria, and associated IUGR – were successfully induced. The classic symptoms of PE were partially reversed with the use of GSNO and SNAP; blood pressure was lowered and IUGR reversed. The high levels of proteinuria observed in these groups indicate possible impaired glomerular permeability which could result from glomerular capillary hypertension due to the presence of L-NAME. The failure of SNAP and GSNO to lower the level of urinary proteins requires further investigation. Based on the results obtained, the use of SNAP and GSNO in treating PE can be further explored.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Shah DM. Role of the rennin-angiotensin system in the pathogenesis of preeclampsia. Am J Physiol Renal Physiol. 2005;288:F614–25. doi: 10.1152/ajprenal.00410.2003. [DOI] [PubMed] [Google Scholar]
- 2.Maynard SE, Venkatesha S, Thadhani R, Karumanchi SA. Soluble FMS-like tyrosine kinase 1 and endothelial dysfunction in the pathogenesis of preeclampsia. Pediatr Res. 2005;57:1R–7R. doi: 10.1203/01.PDR.0000159567.85157.B7. [DOI] [PubMed] [Google Scholar]
- 3.Roberts JM, Redman CW. Pre-eclampsia: More than pregnancy-induced hypertension. Lancet. 1993;341:1447–51. doi: 10.1016/0140-6736(93)90889-o. [DOI] [PubMed] [Google Scholar]
- 4.Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc Natl Acad Sci U S A. 1987;84:9265–9. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wagner LK. Diagnosis and management of preeclampsia. Am Fam Physician. 2004;70:2317–24. [PubMed] [Google Scholar]
- 6.Redman CW. Preeclampsia and the placenta. Placenta. 1991;12:301–8. doi: 10.1016/0143-4004(91)90339-h. [DOI] [PubMed] [Google Scholar]
- 7.Beauséjour A, Bibeau K, Lavoie JC, St-Louis J, Brochu M. Placental oxidative stress in a rat model of preeclampsia. Placenta. 2007;28:52–8. doi: 10.1016/j.placenta.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 8.Vallance P, Chan N. Endothelial function and nitric oxide: Clinical relevance. Heart. 2001;85:342–50. doi: 10.1136/heart.85.3.342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Conrad KP, Joffe GM, Kruszyna H, Kruzyna R, Rochelle LG, Smith RP, et al. Identification of increased nitric oxide biosynthesis during pregnancy in rats. FASEB J. 1993;7:566–71. [PubMed] [Google Scholar]
- 10.Sladek SM, Magness RR, Conrad KP. Nitric oxide and pregnancy. Am J Physiol. 1997;272:R441–63. doi: 10.1152/ajpregu.1997.272.2.R441. [DOI] [PubMed] [Google Scholar]
- 11.Choi JW, Im MW, Pai SH. Nitric oxide production increases during normal pregnancy and decreases in preeclampsia. Ann Clin Lab Sci. 2002;32:257–63. [PubMed] [Google Scholar]
- 12.Seligman SP, Buyon JP, Clancy RM, Young BK, Ambramson SB. The role of nitric oxide in the pathogenesis of preeclampsia. Am J Obstet Gynecol. 1994;171:944–8. doi: 10.1016/s0002-9378(94)70064-8. [DOI] [PubMed] [Google Scholar]
- 13.Rizzo G, Capponi A, Rinaldo D, Arduini D, Romanini C. Low cyclic guanosine monophosphate levels in the amniotic fluid of pre-eclamptic pregnancies. Br J Obstet Gynaecol. 1996;103:834–7. doi: 10.1111/j.1471-0528.1996.tb09884.x. [DOI] [PubMed] [Google Scholar]
- 14.Baker PN, Davidge ST, Roberts JM. Plasma from women with pre-eclampsia increases endothelial cell nitric oxide production. Hypertension. 1995;26:244–8. doi: 10.1161/01.hyp.26.2.244. [DOI] [PubMed] [Google Scholar]
- 15.Norris LA, Higgins JR, Darling MR, Walshe JJ, Bonnar J. Nitric oxide in the uteroplacental, and peripheral circulations in preeclampsia. Obstet Gynecol. 1999;93:958–63. doi: 10.1016/s0029-7844(99)00007-1. [DOI] [PubMed] [Google Scholar]
- 16.Ranta V, Viinikka L, Halmesmaki E, Ylikorkala O. Nitric oxide production with preeclampsia. Obstet Gynecol. 1999;93:442–5. doi: 10.1016/s0029-7844(98)00465-7. [DOI] [PubMed] [Google Scholar]
- 17.Pandhi P, Saha L, Malhotra S. Prolonged blockade of nitric oxide synthesis in pregnant rats as a model of preeclampsia. Indian J Pharmacol. 2001;33:92–5. [Google Scholar]
- 18.Cook JG. Factors influencing the assay of creatinine. Ann Clin Biochem. 1975;12:219–32. doi: 10.1177/000456327501200162. [DOI] [PubMed] [Google Scholar]
- 19.Slot C. Plasmacreatininedetermination: A new and specific Jaffé reaction method. Scand J Clin Lab Invest. 1965;17:381–7. doi: 10.3109/00365516509077065. [DOI] [PubMed] [Google Scholar]
- 20.Molnár M, Hertelendy F. Nomega-nitro-L-arginine, an inhibitor of nitric oxide synthesis, increases blood pressure in rats and reverses the pregnancy-induced refracteriness to vasopressor agents. Am J Obstet Gynecol. 1992;166:1560–7. doi: 10.1016/0002-9378(92)91634-m. [DOI] [PubMed] [Google Scholar]
- 21.Yallampalli C, Garfield RE. Inhibition of nitric oxide synthesis in rats during pregnancy produces signs similar to those of preeclampsia. Am J Obstet Gynecol. 1993;169:1316–20. doi: 10.1016/0002-9378(93)90299-x. [DOI] [PubMed] [Google Scholar]
- 22.Änggård E. Nitric oxide: Mediator, murderer and medicine. Lancet. 1994;343:1199–206. doi: 10.1016/s0140-6736(94)92405-8. [DOI] [PubMed] [Google Scholar]
- 23.McGrowder D, Ragoobirsingh D, Dasgupta T. Effects of S-nitroso-N-acetyl-penicillamine administration on glucose tolerance and plasma levels of insulin and glucagon in the dog. Nitric Oxide. 2001;5:402–12. doi: 10.1006/niox.2001.0360. [DOI] [PubMed] [Google Scholar]
- 24.Mathews WR, Kerr SW. Biological activity of s-nitrosothiols: The role of nitric oxide. J Pharmacol Exp Ther. 1993;267:1529–37. [PubMed] [Google Scholar]
- 25.McGrowder D, Ragoobirsingh D, Dasgupta T. The hyperglycemic effect of S-nitrosoglutathione in the dog. Nitric Oxide. 1999;3:481–91. doi: 10.1006/niox.1999.0254. [DOI] [PubMed] [Google Scholar]
- 26.Baylis C, Mituka B, Deng A. Chronic blockade of nitric oxide sysnthesis in the rat produces systemic hypertension and glomerular damage. J Clin Invest. 1992;90:278–81. doi: 10.1172/JCI115849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Molnár M, Sütö T, Tóth T, Hertelendy F. Prolonged blockade of nitric oxide synthesis in gravid rats produces sustained hypertension, proteinuria, thrombocytopenie and intrauterine growth retardation. Am J Obstet Gynecol. 1994;170:1458–66. doi: 10.1016/s0002-9378(94)70179-2. [DOI] [PubMed] [Google Scholar]
- 28.Kedar I, Jacob ET, Bar-Natan N, Ravid M. Dimethyl sulfoxide in acute ischemia of the kidney. Ann N Y Acad Sci. 2006;411:131–4. doi: 10.1111/j.1749-6632.1983.tb47294.x. [DOI] [PubMed] [Google Scholar]
- 29.Tiboni GM, Giampietro F, Di Giulio C. The nitric oxide synthesis inhibitor N-Nitro-L-Arginine Methyl Ester (L-Name) causes limb defects in mouse fetuses: Protective effect of acute hyperoxia. Pediatr Res. 2003;54:69–76. doi: 10.1203/01.PDR.0000069840.78984.76. [DOI] [PubMed] [Google Scholar]
- 30.Ravishankar V, Buhimschi CS, Booth CJ, Bhandari V, Norwitz E, Copel J, et al. Fetal nucleated red blood cells in a rat model of intrauterine growth restriction induced by hypoxia and nitric oxide synthase inhibition. Am J Obstet Gynecol. 2007;196:482.e1–8. doi: 10.1016/j.ajog.2006.12.020. [DOI] [PubMed] [Google Scholar]
- 31.Diaz V, Lebras-Isabet M, Denjean A. Effect of Nomega-nitro-L-arginine methyl ester-induced intrauterine growth restriction on postnatal lung growth in rats. Pediatr Res. 2005;58:557–61. doi: 10.1203/01.PDR.0000179398.62365.43. [DOI] [PubMed] [Google Scholar]
- 32.Condon JC, Jeyasuria P, Faust JM, Mendelson CR. Surfactant protein secreted by the maturing mouse fetal lung acts as a hormone that signals the initiation of parturition. Proc Natl Acad Sci U S A. 2004;101:4978–83. doi: 10.1073/pnas.0401124101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sakai K, Kohri T, Kweon MN, Kishino Y. A non-glycosylated form of pulmonary surfactant protein A appears in Rat amniotic fluid. Eur Respir J. 1994;7:88–93. doi: 10.1183/09031936.94.07010088. [DOI] [PubMed] [Google Scholar]