

Abstract
Alzheimer’s disease (AD) dementia impacts all facets of higher order cognitive function and is characterized by the presence of distinctive pathological lesions in the gray matter (GM). The profound alterations in GM structure and function have fostered the view that AD impacts are primarily a consequence of GM damage. However, the white matter (WM) represents about 50% of the cerebrum and this area of the brain is substantially atrophied and profoundly abnormal in both sporadic AD (SAD) and familial AD (FAD). We examined the WM biochemistry by ELISA and Western blot analyses of key proteins in 10 FAD cases harboring mutations in the presenilin genes PSEN1 and PSEN2 as well as in 4 non-demented control (NDC) individuals and 4 subjects with SAD. The molecules examined were direct substrates of PSEN1 such as Notch-1 and amyloid precursor protein (APP). In addition, apolipoproteins, axonal transport molecules, cytoskeletal and structural proteins, neurotrophic factors and synaptic proteins were examined. PSEN-FAD subjects had, on average, higher amounts of WM amyloid-beta (Aβ) peptides compared to SAD, which may play a role in the devastating dysfunction of the brain. However, the PSEN-FAD mutations we examined did not produce uniform increases in the relative proportions of Aβ42 and exhibited substantial variability in total Aβ levels. These observations suggest that neurodegeneration and dementia do not depend solely on enhanced Aβ42 levels. Our data revealed additional complexities in PSEN-FAD individuals. Some direct substrates of γ-secretase, such as Notch, N-cadherin, Erb-B4 and APP, deviated substantially from the NDC group baseline for some, but not all, mutation types. Proteins that were not direct γ-secretase substrates, but play key structural and functional roles in the WM, likewise exhibited varied concentrations in the distinct PSEN mutation backgrounds. Detailing the diverse biochemical pathology spectrum of PSEN mutations may offer valuable insights into dementia progression and the design of effective therapeutic interventions for both SAD and FAD.
Keywords: Sporadic Alzheimer’s disease, familial Alzheimer’s disease, presenilin, γ-secretase, white matter, gray matter, amyloid precursor protein, amyloid-beta
Introduction
Alzheimer’s disease (AD) is one of the most devastating disorders affecting the elderly population. The average life expectancy in Western countries has dramatically improved over the last 100 years which resulted in an estimated 35.5 million dementia cases worldwide [1]. From a pathogenic point of view, AD is divided into sporadic (SAD) which represents 99% of cases, and early onset familial (FAD) which accounts for 1% and is caused by autosomal dominant mutations in the amyloid precursor protein (APP), presenilin 1 (PSEN1) and presenilin 2 (PSEN2) genes with loci on chromosomes 21, 14 and 1, respectively [2-4]. There are 40, 197 and 25 currently recorded mutations in the APP, PSEN1 and PSEN2 genes, respectively, that generate an assortment of different AD phenotypes (http://www.molgen.vib-ua.be/ADMutations/). A neuropathological characteristic shared by SAD and FAD is the abundant deposition of amyloid-beta (Aβ) peptides in the extracellular space of the brain parenchyma and the walls of the cerebral vasculature. These peptides are produced by posttranslational cleavage of APP by β-secretase 1 (BACE1) followed by PSEN/γ-secretase and range from 38 to 43 amino acid residues [5]. The γ-secretase is a transmembrane complex composed of 4 different proteins: Nicastrin, anterior pharynx-defective 1 (Aph-1), presenilin enhancer (Pen-2) and PSEN1 or PSEN2 [6].
Two of the major differences between SAD and PSEN-FAD are the relatively early age of onset and the faster clinical course of the latter which may be explained not only by the effects on Aβ, but also by the over 90 substrates on which the PSEN/γ-secretase acts [7]. The peptides derived from the PSEN/γ-secretase cleavage intervene directly in a wide variety of vital functions including: regulation of transcription, cell cycle, cell death, neurite outgrowth and cell differentiation, angiogenesis and tumorigenesis, cell adhesion, targeting of tyrosinase to melanocytes and modulation of voltage gated sodium-and potassium-channels that control the generation and propagation of action potentials [7]. The specific type of mutation and its location along the PSEN molecule will determine explicit secondary and tertiary structures that, in concert with the other 3 components of the γ-secretase complex (Nicastrin, Aph-1 and Pen-2) and the composition of the surrounding membrane, generate a unique quaternary conformation which dictates the final substrate affinity and consequent specific activity. Due to the extraordinarily wide range of pathophysiological effects linked to individual PSEN mutations, a comprehensive description of genotype-phenotype correlation has been difficult to establish. PSEN mutations can result in functional alterations that affect many essential functional pathways. In addition, the PSEN mutations are responsible for a very heterogeneous array of clinical dysfunctions including myoclonus, seizures, extrapyramidal signs, spastic paraparesis, aphasia, agnosia, cerebellar ataxia and psychiatric and behavioral disorders. A diverse spectrum of neuropathologic findings including cotton wool plaques, Lewy bodies, Pick bodies, white matter (WM) changes, ectopic neurons and at times clusters of amyloid core plaques in the WM are evident. For a detailed review of these clinical and pathological features and their putative associations with specific PSEN amino acid substitutions and their structural locations the reader is referred to the excellent reviews of Larner and Doran [8,9], Ryan and Rossor [10] and Mann et al. [11].
Transgenic mouse models harboring PSEN mutations do not produce amyloid plaques; however, these constructs demonstrate abundant pathological alterations in the absence of APP mutations [12]. The mouse PSEN-FAD phenotype includes extensive neurodegeneration with up to 30% or more neuronal loss, widespread synaptic loss, astrogliosis, neurofibrillary-like tangles, defective axonal transport, and decreased PSEN1, APP, synaptophysin, kinesin and acetylcholine as well as increased oxidative stress, cathepsin-D, lysosomal activity, apolipoprotein E (ApoE) and BACE1 [12]. In addition, electrophysiological alterations affecting long-term potentiation, behavioral abnormalities, increased susceptibility to exitotoxic injury, dysregulation of calcium signaling and impaired hippocampal neurogenesis are observed [12]. Furthermore, PSEN1-/- embryos exhibit gross abnormalities of the axial cytoskeleton and spinal ganglia resulting from defective somite segmentation and differentiation that produces marked deformities of the ribcage and death from ensuing respiratory failure [13]. These mice also exhibit defective neurogenesis and loss of neural progenitor cells [13]. Vascular alterations are prominent in the PSEN-FAD transgenic mice with microscopic and ultrastructural changes in the brain microvasculature, such as string and deformed vessels, degenerating capillaries, thickening of the basement membrane and endothelial cell nuclei distortions, all in the absence of cerebral amyloid angiopathy [14]. Moreover, the PSEN1-/- mice develop intracranial hemorrhages of variable severity, suggesting a general failure in Notch signaling pathways which are important in brain vasculature development [15]. Intriguingly, in FAD mutation carriers studied in the Dominantly Inherited Alzheimer Network (DIAN) initiative, brain microhemorrhages were detect ed in 6% of young asymptomatic FAD mutation carriers and in 25% of mildly symptomatic subjects [16].
Alzheimer’s disease has been primarily considered a disorder of the brain gray matter (GM) despite the fact that the WM represents about 50% of the cerebrum and that this area of the brain is substantially atrophied and profoundly abnormal in AD [17-22]. Obvious cerebral WM changes consisting of MRI hyperintensities and anisotropic changes in diffusion tensor MRI together with myelin rarefaction and gliosis at the histological level are present in a substantial fraction of AD cases [23-28]. It is generally believed that the degenerative changes observed in the WM are due to either vascular insufficiency or are second to GM neuronal or axonal loss or a mixture of these [29-31]. However, the lack of consistent correlation between GM and WM damage challenges this contention. Furthermore, in patients with mild cognitive impairment (MCI), WM atrophy precedes GM degeneration, suggesting the provocative argument that axonal and myelin chemical abnormalities may be a primary change. This concept lends support to the ‘retrogenesis hypothesis’ for the etiology of AD which proposes neurodegeneration begins in WM with oligodendrocyte dysfunction and myelin loss, causing defective axonal support and consequential GM neuronal death [32-38]. Our biochemical and morphological data on SAD WM have revealed: 1) increased levels of soluble Aβ40 and Aβ42 peptides [39], 2) a significant decrease in the amounts of myelin-associated proteins and cholesterol [39], 3) ultrastructural changes such as reduction in axonal numbers and diameters, decreased number of oligodendrocytes and astrocytosis [31], 4) dilation of the WM periarterial spaces, suggestive of interstitial fluid stasis that correlates with vascular amyloid burden in the walls of cortical vessels [40], 5) reduction in the density of microvessels [31] and 6) significant quantitative proteomic alterations in molecules related to cytoskeletal maintenance, calcium metabolism and cellular survival compared to non-demented control (NDC) cases [41].
In an earlier publication, we examined the neuropathological and molecular heterogeneity among demented individuals harboring 10 PSEN mutations [42] and focused on the GM APP, Aβ peptides and γ-secretase substrates Notch-1, N-Cadherin and Erb-B4 [42]. In the present investigation, we extended our studies of the same PSEN mutations by quantifying the WM levels of Aβ peptides as well as 21 other WM proteins and 2 GM synaptic molecules. We selected the WM for intensive investigation because this area of the brain is greatly affected in PSEN-FAD [43-47] and SAD. As a comparative frame of reference, we also determined the levels of these proteins in 4 NDC individuals as well as 4 individuals with SAD. The investigated molecules were grouped as direct substrates of PSEN1 (Notch-1, N-cadherin, Erb-B4 and APP) as well as those participating in synaptic function (neurexin and neuroligin in the GM), cytoskeletal functions (neurofilament or NF-light, NF-medium and NF-heavy), axonal transport (α-tubulin, β-tubulin, kinesin, dynein, dynactin and tau), structural functions (α-synuclein, glial fibrillary acidic protein or GFAP and myelin basic protein or MBP), apolipoproteins (ApoE and apolipoprotein A-1 or ApoA-1) and neurotrophic function (vascular endothelial growth factor or VEGF and pigment epithelium derived factor or PEDF). We discuss the inter-PSEN mutant phenotypic variability and quantitative molecular deviations compared to the levels present in SAD and NDC individuals as well as their pathological relevance to ongoing clinical trials.
Material and methods
Human subjects and neuropathological analyses
We utilized the frontal lobe deep WM from a coronal slice at the level of the nucleus accumbens in 10 individuals carrying the following PSEN1 and PSEN2 mutations; (PSEN1: A79V, F105L, Y115C, M146L, A260V, V261F, V261I, P264L and A431E and PSEN2: N141I) as well as 4 SAD cases. These tissues were provided by the Department of Pathology and Laboratory Medicine, Indiana University School of Medicine. For comparison and frame of reference we also included the frontal lobe deep WM from 4 NDC cases. The postmortem interval (PMI) averages for the PSEN1 and PSEN2 mutations, SAD and NDC individuals were 12.3 h, 10.8 h and 3.2 h, respectively. The differences in PMI between the NDC, SAD and PSEN1/2 subjects are due to the different sources of tissue procurement. The NDC cases were obtained from The Brain and Body Donation Program at Banner Sun Health Research Institute (BSHRI). The relatively short average PMI of 3.2 h for the NDC cases is partially due to a rotating team that is on call 24 hours a day and the deaths occurring in the same community as the Institute [48]. On the other hand, the PSEN brains were obtained from different laboratories with different autopsy protocols, operational constraints and postmortem standards. To make comparisons with the PSEN subjects more significant, we included SAD subjects with early disease onset. In the three cohorts, the average ages at death were 53 years for the PSEN-FAD, 64 years for the SAD and 67 years for the NDC group. The mean age at disease onset for the PSEN-FAD mutations and SAD subjects was 45 years and 52 years, respectively. For the PSEN-FAD mutation and SAD cases, APOE genotype, age at death, age at onset, gender, brain weight, and the average number of cotton wool plaques, mature plaques, primitive plaques and diffuse plaques as well as the average number of neurofibrillary tangles (NFT) per mm2 are presented in Table 1. A detailed description of the neuropathological assessments is given in our previous publication [42]. The demographics and neuropathological characteristics for the 4 NDC cases, obtained from BSHRI, such as APOE genotype, age at death, gender, brain weight, total plaque score, plaque density, total NFT score and WM rarefaction (WMR) score are presented in Table 2. In general, these NDC cases were selected for having few or no amyloid deposits and a low Braak NFT stage. The relative lack of pathology is also reflected in the NDC cohort’s average brain weight (1418 g), which contrasted with the low average brain weight of the PSEN-FAD mutations (1073 g) and the SAD cases (1112 g) (Tables 1 and 2). These values excluded one PSEN-FAD and one SAD case which were weighed post-formalin fixation (Table 1).
Table 1.
Presenilin and sporadic Alzheimer’s disease subject demographics and neuropathology scoring
| Case ID | APOE genotype | Age at death(y) | Age of onset (y) | Gender | Brain weight (g) | Avg CWP* | Avg mature plaques* | Avg primitive plaques* | Avg diffuse plaques* | Avg NFT* |
|---|---|---|---|---|---|---|---|---|---|---|
| A79V-PSEN1 | 3/4 | 61 | 50 | F | 1060 | 0.00 | 8.67 | 43.67 | 65.33 | 9.00 |
| F105L-PSEN1 | 2/3 | 68 | 60 | F | 1070 | 0.00 | 1.00 | 0.00 | 78.67 | 17.00 |
| Y115C-PSEN1 | 3/3 | 51 | 41 | M | 1026 | 46.33 | 0.67 | 8.67 | 70.67 | 31.33 |
| N141I-PSEN2 | 3/4 | 57 | 42 | M | 1136 | 49.00 | 2.00 | 1.33 | 67.67 | 65.00 |
| M146L-PSEN1 | 3/3 | 52 | 47 | M | 1150 | 13.67 | 14.00 | 0.00 | 102.00 | 14.33 |
| A260V-PSEN1 | 2/3 | 46 | 40 | M | 1140 | 0.00 | 2.33 | 0.67 | 109.00 | 31.00 |
| V261F-PSEN1 | 3/3 | 47 | 36 | M | 950 | 0.00 | 0.67 | 0.00 | 2.00 | 49.00 |
| V261I-PSEN1 | 3/3 | 55 | 48 | F | 1185 | 0.00 | 1.00 | 0.00 | 1.67 | 0.00 |
| P264L-PSEN1 | 3/3 | 53 | 48 | F | 940 | 0.00 | 8.00 | 0.00 | 12.67 | 14.67 |
| A431E-PSEN1 | 3/3 | 43 | 35 | F | 764 PF | 36.33 | 11.67 | 5.00 | 42.00 | 55.33 |
| SAD-1 | 3/3 | 62 | 47 | M | 910 | 0.00 | 23.33 | 0.00 | 12.00 | 42.33 |
| SAD-2 | 3/4 | 66 | 55 | M | 1325 | 0.00 | 11.67 | 0.00 | 30.67 | 6.33 |
| SAD-3 | 3/4 | 73 | 58 | M | 1236 PF | 0.00 | 5.67 | 0.00 | 15.33 | 12.00 |
| SAD-4 | 3/4 | 55 | 48 | F | 1100 | 0.00 | 36.67 | 0.00 | 21.33 | 55.33 |
All counts are measured in mm2;
PSEN, presenilin; SAD, sporadic Alzheimer’s disease; APOE, apolipoprotein E; y, years; g, grams; Avg, average; CWP, cotton wool plaques; NFT, neurofibrillary tangle; PF, post-fixed; M, male; F, female.
Table 2.
Non-demented control subject demographics and neuropathology scoring
| Case ID | APOE genotype | Age at death (y) | Gender | Brain weight (g) | Total plaque score | Plaque density | Total NFT score | Total WMR score |
|---|---|---|---|---|---|---|---|---|
| NDC-15 | 3/4 | 76 | M | 1375 | 5.5 | sparse | 0 | 1 |
| NDC-16 | 3/3 | 65 | M | 1400 | 0 | zero | 1 | 2 |
| NDC-17 | 3/3 | 53 | M | 1456 | 0 | zero | 1 | 0 |
| NDC-18 | 2/3 | 74 | M | 1440 | 0 | zero | 2 | 2 |
APOE, apolipoprotein E; y, years; g, grams; NFT, neurofibrillary tangle; WMR, white matter rarefaction; M, male. The maximum total plaque score is 15. The maximum total NFT score is 15 and the maximum WMR is 12. See reference number [49] for neuropathology scoring details.
Tissue preparation and biochemical analyses
White matter (1 g) was homogenized in 10 ml of 90% glass-distilled formic acid (GDFA) using the Omni TH tissue grinder (Kennesaw, GA). The homogenates were centrifuged for 1 hr at 4°C at 250,000 x g in a SW41 rotor (Beckman, Brea, CA). The supernatants were collected and placed into 1000 MWCO dialysis tubing. Buffer exchange was performed in deionized H2O (2 changes, 1 h each) then in 0.1 M ammonium bicarbonate (3 changes, 1 h each). The samples were lyophilized and submitted to Western blot or ELISA as described below.
Aβ40 and Aβ42 ELISA
Lyophilized WM proteins were shaken for 4 h at 4°C in 1400 μl 5 M guanidine-hydrochloride, 50 mM Trizma, pH 8.0 and then centrifuged at 100,000 x g for 1 h at 4°C in a Beckman 50.4 Ti rotor. Pierce’s Micro BCA protein assay kit (Rockford, IL) was used to determine the total protein quantity in the supernatant. The quantities of Aβ40 and Aβ42 were measured with kits from Life Technologies Corp (Carlsbad, CA), following the manufacturer’s instructions.
Western blots
Lyophilized WM proteins were reconstituted in 500 μl of 5% SDS, 5 mM EDTA, 20 mM Tris-HCl, pH 7.8 and centrifuged for 20 min in a TLA120.2 (Beckman) rotor at 435,000 x g. Total protein was determined with the Micro BCA protein assay kit (Pierce). For a detailed Western blot protocol, see Maarouf et al [49]. Briefly, equal amounts of total protein from each case were separated on 4-12% Bis-Tris gels (Life Technologies Corp) and transferred onto 0.45 μm nitrocellulose membranes (Life Technologies Corp). All membranes were re-probed with anti-mouse, anti-rabbit actin or GAPDH antibodies to serve as a total protein loading controls. Table 3 provides the descriptions of all antibodies used in this study. Quantity One software (Bio-Rad, Hercules, CA, USA) was used to measure the trace quantity of each band which corresponds to the measured area under each band’s intensity profile curve. Reported units are in optical density (OD) x mm.
Table 3.
Antibodies Used in Western Blots
| Primary antibody | Antigen specificity or immunogen | Secondary antibody | Company/Catalog # |
|---|---|---|---|
| Notch-1 | NICD N-terminal 14 aa | R | Millipore/AB5709 |
| N-Cadherin | aa 802-819 of mouse N-Cadherin | M | BD Transduction Laboratories/610920 |
| Erb-B4 | aa 1258-1308 of human Erb-B4 | R | Santa Cruz/sc-283 |
| CT20APP | Last 20 aa of APP | M | Covance/SIG-39152 |
| Neurexin 1,2,3 | CT tails of α/β neurexins 1,2,3 | R | Synaptic Systems/175003 |
| Neuroligin | aa 718-843 of rat neuroligin 1 | R | Synaptic Systems/129013 |
| α-tubulin | Synthetic peptide of human α-tubulin | R | Cell Signaling/2144S |
| R | |||
| β-tubulin | N-terminal synthetic peptide of human β-tubulin | R | Cell Signaling/2128S |
| Kinesin 5A | Synthetic peptide: aa 1007-1027 | R | Pierce/PA1-642 |
| Dynein | Cytoplasmic dynein from chicken embryo brain | M | Sigma/D5167 |
| Dynactin | aa 1266-1278 of human dynactin-1 | G | Abcam/ab11806 |
| Tau (HT7) | aa 159-163 | M | Pierce/MN1000 |
| NF-Heavy | PO- epitope on heavy NF protein (210 kDa) | M | Abcam/Ab7795 |
| NF-Medium | PO- and non-PO- forms of medium NF protein (160 kDa) | M | Abcam/Ab7794 |
| NF-Light | Recognizes the light NF subunit (~68 kDa) | M | Abcam/Ab7255 |
| α-synuclein | aa 15-123 of Rat synuclein-1 | R | BD Transduction Laboratories/610786 |
| GFAP | Recombinant GFAP | R | Abcam/ab7260 |
| MBP | aa 119-131 | M | Millipore/MAB381 |
| ApoE | Recombinant ApoE | G | Millipore/AB947 |
| ApoA-1 | aa 127-142 of human ApoA-1 | R | Life Technologies/701239 |
| PEDF | Human PEDF | R | BioProducts MD/AB-PEDF1 |
| VEGF165 | Recombinant human VEGF165 | R | Millipore/07-1419 |
| GAPDH | Full-length human GAPDH protein | M | Life Technologies/39-8600 |
| Actin Ab-5 | Clone C4 | M | BD Transduction Laboratories/A65020 |
| Actin | N-terminus of human α-actin | R | Abcam/Ab37063 |
NICD, Notch intracellular domain; aa, amino acid; CT, C-terminal; APP, amyloid precursor protein; NF, neurofilament; GFAP, glial fibrillary acidic protein; MBP, myelin basic protein; Apo, Apolipoprotein; PEDF, Pigment epithelium-derived factor; VEGF, Vascular endothelial growth factor; GAPDH, Glyceraldehyde 3-phosphate dehydrogenase; M, HRP conjugated AffiniPure goat-anti rabbit IgG (catalog # 111-035-144, Jackson Laboratory); R, HRP conjugated AffiniPure goat-anti mouse IgG, (catalog # 115-035-146 Jackson Laboratory); G, HRP conjugated AffiniPure bovine-anti goat IgG (catalog # 805-035-180).
Statistical analyses
One of the challenges in the present study was to determine how direct comparisons on a number of different protein markers could be made using data derived from Western blot densitometry analyses. Since the molecular markers use different scales of measurement, direct comparisons of the raw values were not practical and did not allow for meaningful interpretations of the data. One of our interests was to determine whether the protein levels of the studied PSEN-FAD cases were different from NDC and also how SAD differed from NDC. We were also interested in examining the inter-individual variability of the molecular markers among the different PSEN-FAD cases. We used a method based on the t-distribution for small samples suitable for finding the confidence for small samples as is the case of the NDC group of patients [50]. A linear combination T of Student’s t statistics is proposed as a practical method of obtaining confidence intervals for the common mean, and the distribution function of T is conveniently approximated for the general case [51]. We obtained the confidence interval and used this interval to determine the appropriateness of the FAD and SAD patients. We conducted intervals with 5% type I error rate. The critical region of the test given by the asymptotic theory is modified to try to produce more accurate results in small to moderate sample sizes. Values that fell within the intervals were considered consistent with the NDC cases and those beyond the interval were considered to be inconsistent. We conducted these analyses through SAS 9.2 and PROC IML.
Results
Aβ levels in the GM and WM
The histograms illustrated in Figure 1A and 1B show the levels of GM and WM Aβ peptides, respectively, in 9 PSEN1-FAD and 1 PSEN2-FAD (denoted by asterisk) mutation carriers compared to SAD and NDC cases. The levels of Aβ40 were disproportionately increased relative to Aβ42 in GM in the M146L, V261F, V261I and A431E mutation carriers (Figure 1A) [42], resulting in lower GM Aβ42:40 ratios. Interestingly, 2 of the SAD cases also have relatively low GM Aβ42:40 ratios, although the difference between the 2 Aβ isoforms is much less pronounced than in the 4 PSEN-FAD cases [42]. On the other hand, in the WM, only 2 of the PSEN-FAD cases (N141I and P264L) and 1 of the SAD cases contained more Aβ40 than Aβ42 (Figure 1B). In terms of mean total GM Aβ concentration (Aβ40+Aβ42), this was 2-fold higher in PSEN-FAD cases than in SAD cases (12 μg/g wet weight vs. 6 μg/g wet weight), and, as expected, significantly higher that of the NDC mean (Kruskal-Wallis, p = 0.039), as previously published [42]. The same trends are observed for the WM in which PSEN-FAD cases had a total Aβ mean of 31 ng/mg total protein and the SAD cases had a mean of 4.2 ng/mg total protein while the NDC group had a mean of 0.22 ng/mg total protein (Kruskal Wallis, p = 0.004). In the WM, the differences in total Aβ levels are greater between FAD and SAD (7.4 fold) in comparison to GM.
Figure 1.
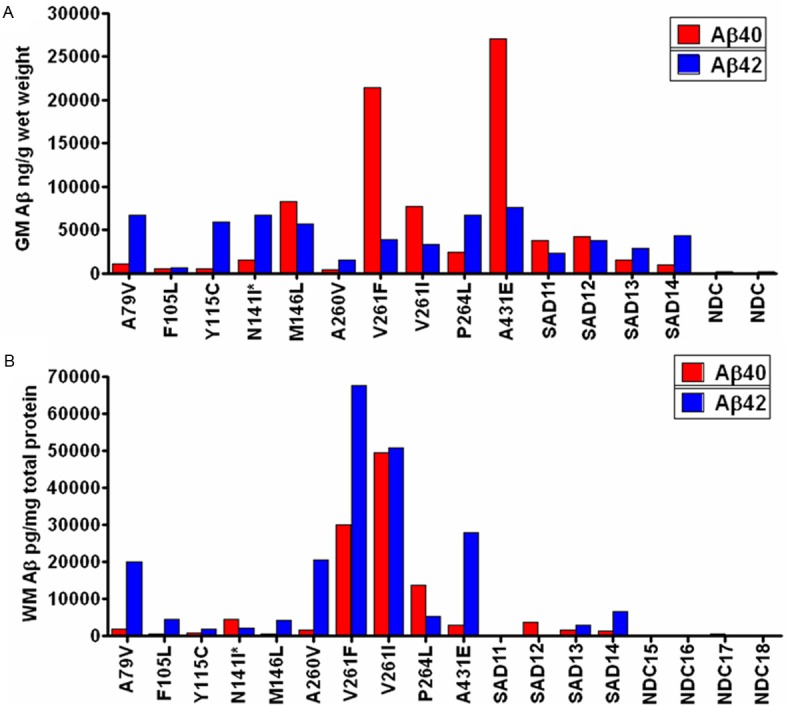
Quantities of Aβ40 (red bars) and Aβ42 (blue bars) as detected by ELISA. A: The levels of Aβ in the gray matter reported in ng/g wet weight of brain tissue [42]. Four of 10 PSEN-FAD subjects had higher amounts of Aβ40 relative to Aβ42. B: Aβ (pg/mg of total protein) was also quantified in the white matter for the same PSEN-FAD individuals. Only 2 PSEN-FAD cases (M141I and P264L) had elevated Aβ40 over Aβ42. GM, gray matter; WM, white matter; SAD = sporadic Alzheimer’s disease; NDC = non-demented control. The asterisk next to N141I denotes that this case has a PSEN2 mutation. All other subjects are PSEN1 carriers. See Tables 1 and 2 for subject key and information.
Western blot results
Figure 2 through Figure 6 display the PSEN-FAD and SAD individual confidence values when compared to the confidence intervals of NDC group. Molecules investigated by Western blot were grouped as follows: 1) direct substrates of PSEN1 in the WM (Notch-1-Figure 2A, N-cadherin-Figure 2B, Erb-B4-Figure 2C, APP-Figure 2D and its C-terminal fragments (CTFs)-Figure 2E), 2) GM synaptic proteins (neurexin-Figure 3A and neuroligin-Figure 3B), 3) proteins performing cytoskeletal functions in the WM (NF-light-Figure 3C, NF-medium-Figure 3D and NF-heavy-Figure 3E), 4) WM axonal transport molecules (α-tubulin-Figure 4A, β-tubulin-Figure 4B, kinesin-Figure 4C, dynein-Figure 4D, dynactin-Figure 4E and tau-Figure 4F), 5) proteins related to structural functions in the WM (α-synuclein-Figure 5A, GFAP-Figure 5B and MBP-Figure 5C), 6) WM apolipoproteins (ApoA-1-Figure 6A and ApoE-Figure 6B), 7) WM neurotrophic proteins (PEDF-Figure 6C and VEGF-Figure 6D).
Figure 2.
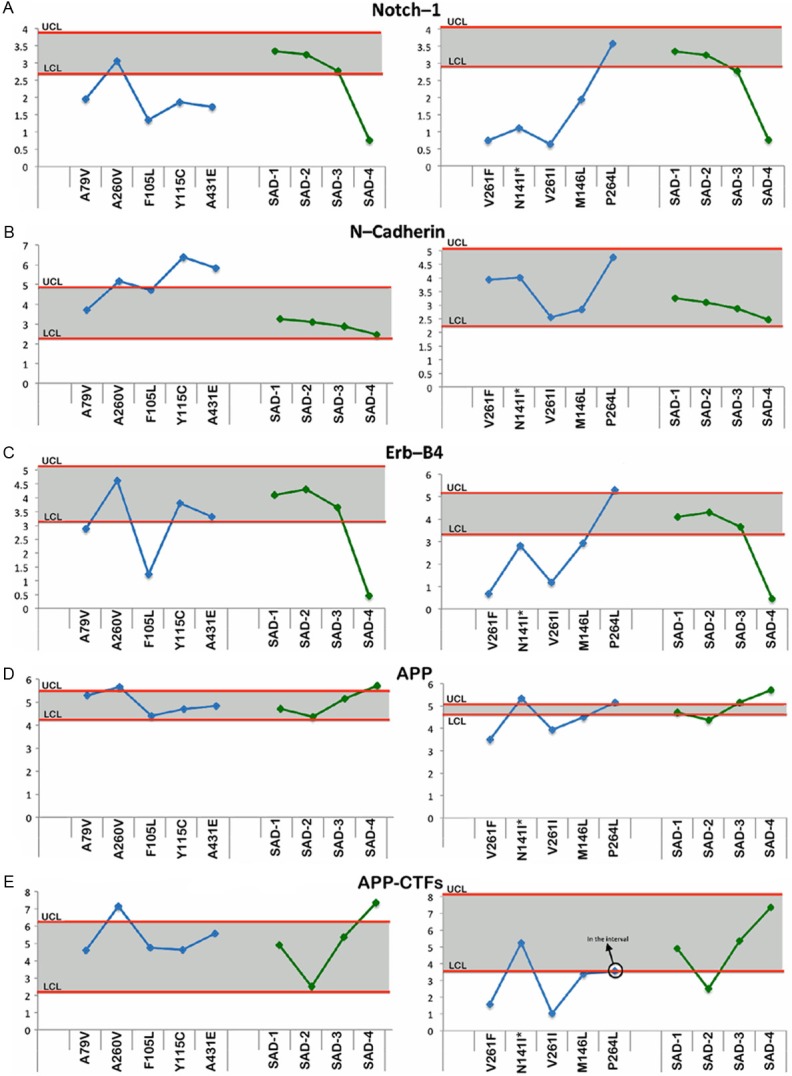
Upper and lower confidence levels (UCL and LCL) derived from Western blot densitometry analyses from non-demented control subjects. The distribution values for each of the interrogated proteins observed in each individual PSEN mutation and SAD cases #1-4 cases are shown along y-axis. A: Notch-1, B: N-cadherin, C: Erb-B4, D: Amyloid precursor protein (APP) and E: Amyloid precursor protein C-terminal fragments (APP-CTFs). All analyses were performed in the white matter.
Figure 6.
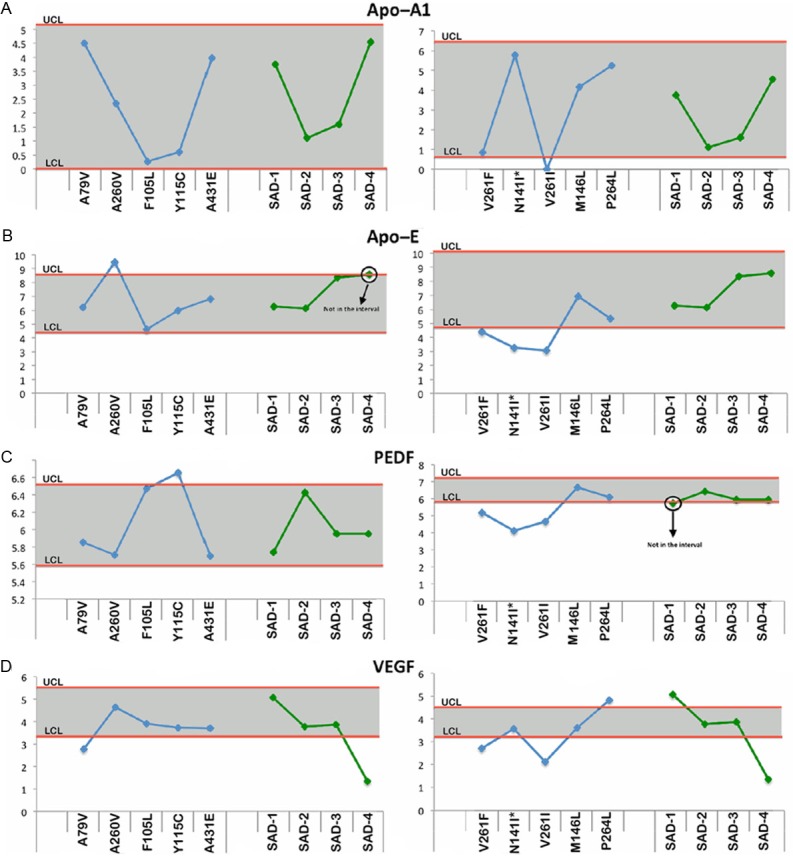
Upper and lower confidence levels (UCL and LCL) derived from Western blot densitometry analyses from non-demented control subjects. The distribution values for each of the interrogated proteins observed in each individual PSEN mutation and SAD cases #1-4 cases are shown along y-axis. A: Apolipoprotein A1 (ApoA1), B: Apolipoprotein E (ApoE), C: Pigment epithelium derived factor (PEDF) and D: Vascular endothelial growth factor (VEGF). All analyses were performed in the white matter.
Figure 3.
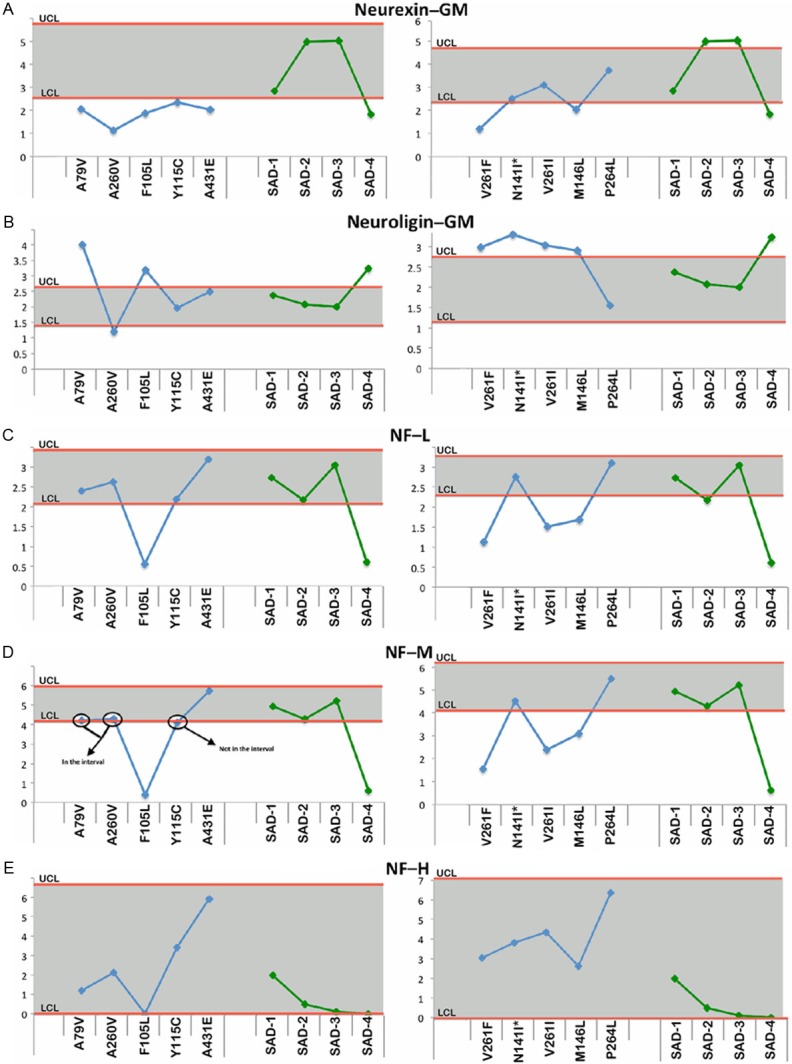
Upper and lower confidence levels (UCL and LCL) derived from Western blot densitometry analyses from non-demented control subjects. The distribution values for each of the interrogated proteins observed in each individual PSEN mutation and SAD cases #1-4 cases are shown along y-axis. A: Neurexin in the gray matter (GM), B: Neuroligin in the gray matter (GM), C: Neurofilament-light (NF-L) chain, D: Neurofilament-medium (NF-M) chain and E: Neurofilament-heavy (NF-H) chain. Unless otherwise noted, all analyses were performed in the white matter.
Figure 4.
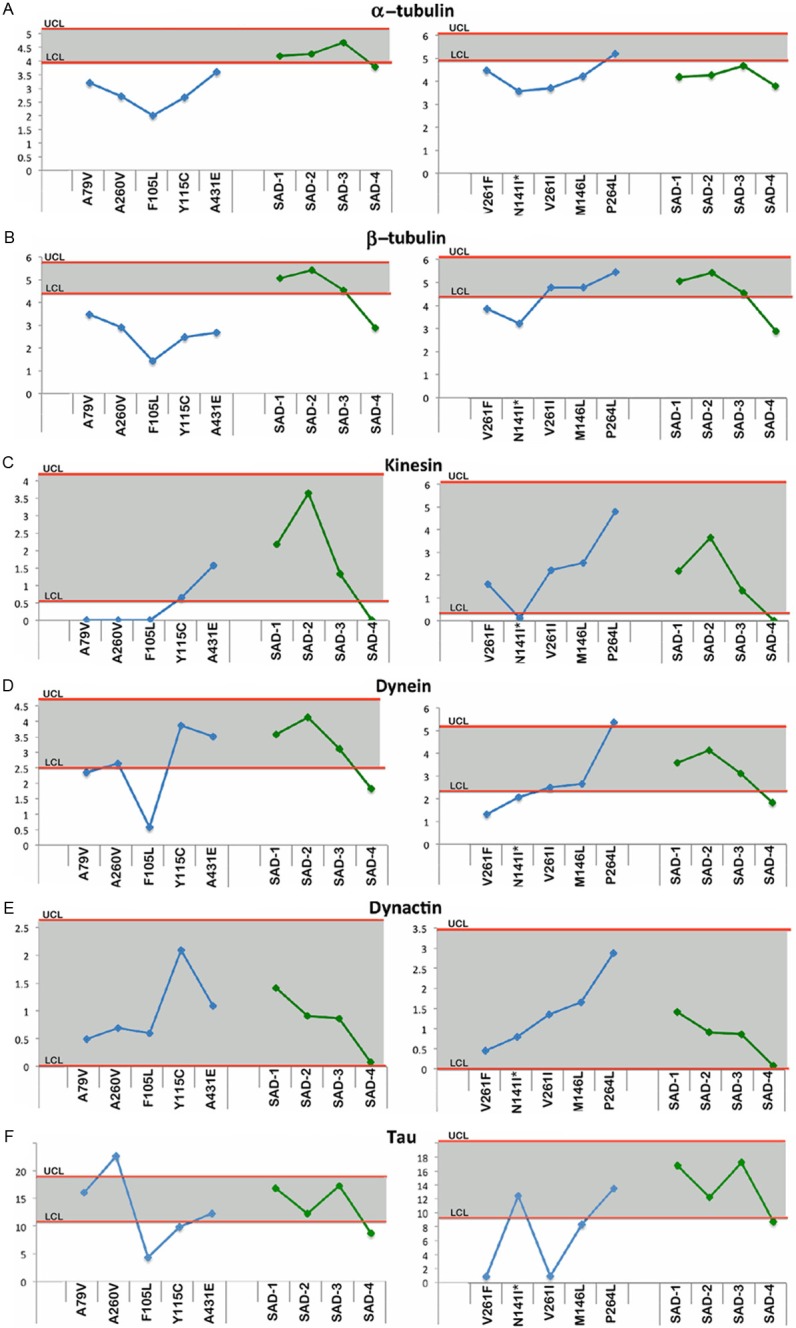
Upper and lower confidence levels (UCL and LCL) derived from Western blot densitometry analyses from non-demented control subjects. The distribution values for each of the interrogated proteins observed in each individual PSEN mutation and SAD cases #1-4 cases are shown along y-axis. A: α-tubulin, B: β-tubulin, C: Kinesin, D: Dynein, E: Dynactin and F: Tau. All analyses were performed in the white matter.
Figure 5.
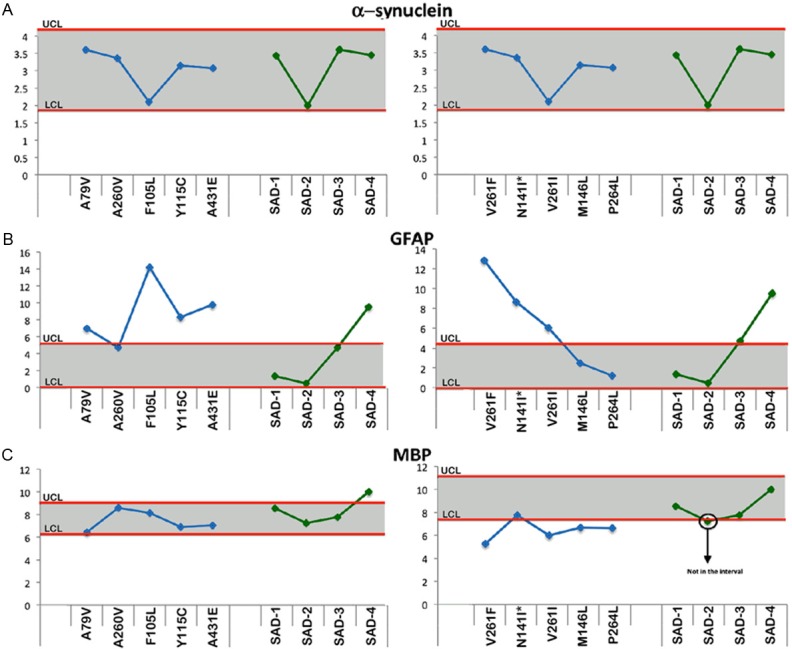
Upper and lower confidence levels (UCL and LCL) derived from Western blot densitometry analyses from non-demented control subjects. The distribution values for each of the interrogated proteins observed in each individual PSEN mutation and SAD cases #1-4 cases are shown along y-axis. A: α-synuclein, B: Glial fibrillary acidic protein (GFAP) and C: Myelin basic protein (MBP). All analyses were performed in the white matter.
Direct substrates of the PSEN
With the exception of the A260V and P264L mutation carriers, the PSEN-FAD subjects had decreased levels of Notch-1 relative to the NDC cohort, while 3 out of 4 SAD individuals had Notch-1 levels within the NDC range (Figure 2A). In general, the amounts of N-cadherin in the PSEN-FAD and SAD groups were consistent with NDC subjects and the SAD cases, with the exception of Y115C and A431E in which the N-cadherin was elevated (Figure 2B). The levels of Erb-B4 fluctuated among the PSEN-FAD subjects with half of the PSEN-FAD mutations having lower levels of this molecule (Figure 2C). Similar to the Notch-1 results (Figure 2A), SAD case #4 also had reduced levels of Erb-B4 when compared to the NDC group (Figure 2C). Full length APP did not demonstrate large differences in the PSEN-FAD and SAD individuals compared to the NDC cases (Figure 2D). In reference to PSEN-FAD CTFs, only A260V showed an increase, whereas V261F and V261I showed a decrease in the levels of these peptides (Figure 2E).
Synaptic molecules
In the GM of PSEN-FAD cases, neurexin was consistently under-represented in 7 (A79V, A260V, F105L, Y115C, A431E, V261F and M146L) of the 10 PSEN-FAD individuals relative to the NDC cohort (Figure 3A). In contrast, neuroligin was elevated in 6 (A79V, F105L, V261F, N141I, V261I and M146L) out of 10 cases with only one case A260V below the lower confidence level in the PSEN-FAD cohort (Figure 3B). In SAD subjects, the levels of GM neurexin and neuroligin, for the most part, were close to the range of the NDC cohort with the exception of SAD case #4 (Figure 3A and 3B).
Cytoskeleton molecules
Six out of 10 PSEN-FAD subjects had levels of NF-light chain similar to the NDC group with the remaining 4 (F105L, V261F, V261I and M146L) PSEN-FAD individuals having lower amounts of this protein (Figure 3C). Neurofilament-medium chain (Figure 3D) followed the same trend as NF-light (Figure 3C) in both the PSEN-FAD and SAD cohort in which only SAD #4 had decreased level vs. the NDC subjects. Relative amounts of NF-heavy chain were all within the NDC range in both the PSEN-FAD and SAD cohorts (Figure 3E).
Axonal transport molecules
With the exception of P264L for α-tubulin and V261I, M146L and P264L for β-tubulin, all the remaining PSEN-FAD had lower amounts of these two proteins relative to the NDC cases (Figure 4A and 4B), suggesting a loss of microtubules when compared to the NDC cohort. The SAD group fell below the lower confidence interval for α-tubulin (Figure 4A). The SAD cases, on the other hand, were mostly within the NDC confidence interval for β-tubulin with the exception of SAD case #4 (Figure 4B). In general, most of the values of the cargo proteins kinesin, dynein and dynactin in the PSEN-FAD and SAD individuals were within the range of the NDC group (Figure 4C-E). Four cases (A79V, A260V, F105L and N141I) in the kinesin graph (Figure 4C) and 3 cases (F105L, V261F and N141I) in the dynein graph (Figure 4D) were underrepresented in the PSEN-FAD group. The cargo proteins kinesin and dynein were decreased relative to NDC in SAD case #4 (Figure 4C and 4D). The relative levels of the microtubule stabilizing tau protein were inconsistent among the PSEN-FAD subjects, with F105L, Y115C, V261F, V261I and M146L having decreased values and the A260V displaying a positive value (Figure 4F), compared to the NDC cases. In the SAD cohort, only case #4 had a value below the NDC confidence limit (Figure 4F).
Structural proteins
The relative levels of α-synuclein in the PSEN-FAD mutations and SAD cases all fell within the range of the NDC group (Figure 5A). On the other hand, the relative amounts of GFAP were increased in 7 of the 10 PSEN-FAD individuals, with the remaining cases falling within the confidence interval of NDC group (Figure 5B). In the SAD versus NDC comparisons, the GFAP values were only increased in SAD case #4 while the other 3 SAD individuals had levels similar to the NDC group (Figure 5B). In reference to MBP, 4 of the 10 PSEN-FAD individuals (V261F, V261L, M146L and P264L) had levels lower than the NDC group (Figure 5C), whereas in the SAD cases MBP levels did not dramatically differ from the controls (Figure 5C).
Apolipoproteins
In general, all the ApoA1 values in the PSEN-FAD mutations fell within the NDC confidence interval with the exception of V261L which had a negative level (Figure 6A). The relative amounts of ApoE in six of the PSEN-FAD cases were within the range of the NDC confidence interval (Figure 6B). Case A79V was increased and cases V2-61F, N141I and V261I were decreased compared to the ND-C cohort (Figure 6B). Both apolipoproteins were within the NDC confidence limits for the SAD cohorts (Figure 6A and 6B).
Neurotrophic factors
The levels of PEDF were increased in PSEN-FAD Y115C and decreased in V261F, N141I and V261I relative to the NDC confidence limits (Figure 6C). In the SAD cases, PEDF levels fell within the confidence interval of NDC (Figure 6C). Lastly, the VEGF analysis only showed 3 PSEN-FAD cases (A-79V, V261F and V261I) with negative levels relative to NDC (Figure 6D). Case #4 was the only SAD individual with a negative value in comparison to the NDC group (Figure 6D).
Discussion
The WM in FAD and SAD is profoundly affected in morphological and volumetric parameters since there is severe atrophy resulting from loss of axons and myelin and concomitant expansion of the lateral ventricles [52-54]. This WM rarefaction is often associated with widespread dilation of the periarterial spaces suggesting chronic retention of interstitial fluid as well as loss of WM tissue [40]. In addition, the presence of ectopic neurons containing NFT and clusters of amyloid plaques in the WM have also been reported in some cases of PSEN-FAD [45,47]. Presently, there is a relative paucity of information regarding the WM biochemical alterations and their contribution to the pathophysiology of SAD and FAD compared to the GM. Therefore, we sought to determine the amount of Aβ peptides as well as analyze 21 different WM structural and functional proteins in 10 different PSEN-FAD mutation carriers. In addition, 2 GM synaptic proteins were assessed. We were concerned about the effect of the wide range of PMI in our PSEN-FAD cohort. Therefore, we performed linear correlations between the PMI and densitometry and immunoassay values observed for the different protein markers, and found that there was no significant correlation between these parameters in any case (p > 0.05).
In reference to the Aβ quantifications, 3 important conclusions can be drawn from our data: 1) The PSEN-FAD mutations do not always produce a uniform increase in the Aβ42:Aβ40 ratios, 2) the PSEN-FAD mutations have, on average, higher total amounts of Aβ peptides, when compared to SAD, which may play a role, besides other factors, in the global devastating dysfunction of the brain in FAD and 3) there is a great deal of individual variability in Aβ amounts among the different PSEN-FAD mutations. Consideration should be given to the fact that Aβ levels in the WM are roughly 1000-fold less than in the GM. This implies that the accumulation of the Aβ peptides mainly occurs in GM extracellular space, neuronal cell bodies, neurites, presynaptic terminals and vascular walls rather than in the long WM axons, associated glial or WM microvasculature. Therefore, the presence of Aβ in the WM of SAD and PSEN-FAD may be due to interstitial fluid diffusion from the GM or alternatively it may represent a small intracellular axonal pool and/or myelin-bound Aβ [55-58] that is elevated in SAD and PSEN-FAD, since these peptides were not increased in the NDC cases.
In a previous publication, we reported a biochemical analysis of the PSEN1 E280A (‘paisa’) mutation in GM samples and observed abundant diffuse amyloid plaques with a peculiar staining pattern distinct to previously described plaques in PSEN cases [59]. In addition, increased amounts of APP-CT99 and Aβ42 were present along with Aβ peptides longer than 40-42 amino acid residues [59]. In more recent studies, examination of young PSEN1 E280A mutation carriers between 18-26 years of age, demonstrated a thinner cerebral cortex [60], greater hippocampal and parahippocampal activation, less precuneus and posterior cingulate deactivation, less GM in parietal regions, and higher CSF and plasma Aβ42 levels compared to non-carriers [61]. It has been recently suggested that CSF levels of Aβ and tau as well as brain amyloid deposition in PSEN-FAD subjects follow a pattern similar to that observed in SAD [62].
It is widely believed that the PSEN-FAD phenotype is a consequence of the increased production of the Aβ42 peptide, thus supporting the amyloid cascade hypothesis as the central factor in the pathogenesis of both SAD and FAD. However, this hypothesis has been challenged since in several PSEN-FAD mutations the prevalent accumulated amyloid form is Aβ40 [42,63-65]. In addition, not all PSEN-FAD mutations cause an increase in the Aβ42:40 ratio [42,64]. Intriguingly, in the PSEN1 insR352 mutation, which expresses a frontotemporal dementia phenotype, there is substantially less Aβ42 accumulation [63], although these experiments were carried out in cultured cells. Furthermore, in the case of the PSEN1 mutation G183V, which is associated with frontotemporal dementia and neuropathologically characterized by Pick-type tauopathy, Aβ deposits and Aβ peptides were not detected [66]. These observations suggest that neurodegeneration and dementia may be phenomena independent of increased Aβ42 production [64].
Our data revealed a great deal of heterogeneity in protein expression among the different PSEN-FAD mutations. For example, 3 of the 10 PSEN1 mutations which deviated the most from the NDC baseline were F105L, V261F and V261I which shared decreased amounts of Notch-1, Erb-B4, NF-light, NF-medium, α-tubulin and tau. In addition, the F105L mutation had also decreased values of neurexin, β-tubulin, kinesin and dynein, while the V261F and V261I mutations had decreased values of APP, APP-CTFs, MBP, ApoE, PEDF and VEGF. The 3 mutations (F105L, V261F and V261I) also shared elevated GFAP. Interestingly, the PSEN mutations V261F and V261I had the most abundant levels of Aβ in the WM. In general, the proteins that revealed the most consistent changes in the PSEN-FAD cohort were Notch-1, neurexin, α-tubulin, β-tubulin and GFAP. Eight of the 10 PSEN-FAD subjects had reduced amounts of Notch-1 relative to the controls, while the 2 remaining cases fell within the range of the NDC cohort. Notch-1 is a receptor that is sequentially cleaved, with the final γ-secretase processing releasing the nuclear transcription factor notch intracellular domain (NICD) which is a key component in tissue development and renewal [67,68]. It is possible that abnormally low levels of NICD may have negative effects on cognition. With the exception of the PSEN2 mutation N141I and the PSEN1 mutations V261I and P264L, all the remaining PSEN mutations carriers had lower neurexin levels than the NDC group. On the other hand, 6 out of the 10 different mutation carriers had increased levels of neuroligin relative to the NDC cases. Presenilins are key proteins in the release of neurotransmitters and regulation of long-term potentiation [69]. The binding partners neurexin and neuroligin are both substrates of the PSEN/γ-secretase and have been described as pivotal proteins in pre-synaptic and post-synaptic junctions [70-72]. These proteins have also been depicted as important regulatory components in the maintenance of vascular functions since they participate in the metabolism of endothelial and vascular smooth muscle cells [73]. In addition, recent experiments suggest that neurexin and neuroligin inhibit and promote angiogenesis, respectively, and influence vascular tone [73].
Some of the direct substrates of the PSEN/γ-secretase, such as Notch-1, N-cadherin, Erb-B4 and APP, appeared to deviate from the confidence intervals established by the NDC protein markers, while in other mutation types these markers were apparently not affected by specific mutations. Proteins that are direct substrates of PSEN could be increased due to substrate accumulation. This may be elicited by conformational changes generated by the specific amino acid location of the different mutations that result in reduced PSEN affinity for the substrate and/or decreased proteolytic function of the active center. In the alternative scenario in which PSEN substrates were decreased, specific mutations may have induced an increased rate of enzymatic activity against specific substrates. Whether the PSEN mutations result in a gain or loss of function is contentious [74,75]. In all likelihood the outcome may not represent a simple, universal dichotomy because some PSEN mutations induce both effects depending on the biophysical properties of the mutated PSEN/γ-secretase molecule in concert with the conformational properties of a specific substrate.
In reference to tau, we observed in only one PSEN-FAD mutation case, the amount of this molecule was increased whereas in 5 other cases tau levels were decreased relative to the NDC cohort. The remaining PSEN-FAD 4 cases fell within the confidence interval established by the NDC cases. Tau integrated within the paired helical filaments (PHF) is a difficult molecule to assess, due to the extreme insolubility of ancillary molecules in most of the detergents and denaturing chaotropic agents that only allow a fractional solubilization of PHF. Detailed chemical analysis of PHF isolated from AD has revealed that their insolubility is mainly due to the high content of glycoproteins and glycolipids present in these structures [76-78] that suggest they are probably derived from stacks of irreversibly denatured cytomembranes, including mitochondria and endoplasmic reticulum, as observed by electron microscopy in human biopsies [79].
Alterations in protein marker molecules which are not known to be direct substrates of PSEN, for example molecules involved in cytoskeletal structural or axonal transport functions were observed to be increased or decreased, depending on the specific PSEN mutation, when compared to the NDC confidence intervals. This may be due to multiple molecular interactions resulting from downstream effects initially caused by direct substrates of the mutated PSEN molecules. Tabulating all confidence interval limits within the PSEN-FAD cohort revealed that 11% fell above, 53% fell within and 36% fell below the relative range of the statistical values established by the NDC group, suggesting a general reduction of proteins in the diseased individuals. Interestingly, the most altered SAD case relative to the NDC confidence limits, was SAD subject #4 who demonstrated decreased Notch-1, Erb-B4, neurexin, NF-light, NF-medium, α-tubulin, β-tubulin, kinesin, dynein, tau and VEGF, but increased values of APP, neuroligin, GFAP and MBP. These alterations may be due to a more severe disease course, as SAD case #4 had the earliest onset and most rapid disease course as well as the highest number of mature amyloid plaques and NFT in this group.
Intriguingly, the relative amount of the 23 protein markers were very heterogeneous among the PSEN-FAD mutations and in many instances their values departed substantially from the NDC cases. It is important to consider that all PSEN-FAD and SAD cases were at the end-stage of an extended disease process and consequently, the biochemical damage and functional disturbances were at their maximum levels of expression. This may account for the gross morphological and biochemical alterations in the integrity of neuronal and glial cells, which severely perturbed the structure and function of vital systems such as axonal transport, synaptic integrity and transmission of action potentials as well as glial and neuronal cytoskeleton scaffolds and neurotrophic factor levels. It could be hypothesized that the individual pathological phenotypic characteristics of the diverse PSEN-FAD mutations are likely to result, not only from Aβ accumulation, but also from alterations of multiple molecular substrates and their additive negative pleiotropic interactions. Thus, the observed early age of onset, severe disease course and neuropathological presentation of the PSEN-FAD mutations may not be solely the result of APP and tau misprocessing, but may be the end-product of cumulative pathological changes exerting deleterious effects on a large number of molecules and cellular functions that could not be substituted or rescued by alternative biochemical pathways.
Although several clinical and radiological studies have examined an assortment of PSEN mutations in cross-sectional studies, very few isolated cases have been examined on a longitudinal basis [80,81]. Two large and long-term immunotherapy clinical trials for populations harboring PSEN-FAD mutations [82-84] are targeting Aβ in younger asymptomatic carriers in the hope that early elimination or diminished production of these peptides by monoclonal antibodies will prevent or modify the relentless course of dementia. The Alzheimer’s Prevention Initiative will utilize a large Colombian cohort with a significant number of individuals carrying the PSEN1 E280A paisa mutation and offers an unparalleled opportunity to test the effects of prophylactic immunotherapeutic in a presymptomatic single PSEN mutation carrier kindred [82]. The DIAN prophylactic immunotherapy project will be implemented in patients harboring several PSEN-FAD mutations [83]. In addition, the Anti-Amyloid Treatment in Asymptomatic Alzheimer’s Disease (A4) prevention clinical trial represents a complementary strategy in which putative dementia-prophylactic Aβ immunotherapy will be provided to asymptomatic individuals recognized by imaging technologies to possess substantial amyloid deposit burdens and hypothesized to be at imminent risk for SAD development [85].
A large number of morphological, functional and chemical studies on PSEN-FAD have been centered on the hypothesis that these neurodegenerative disorders are mainly due to the overexpression of Aβ peptides. However, the cause-effect of the amyloid cascade hypothesis as the only pathogenetic factor in PSEN-FAD remains uncertain in view of the potential pathological effects of altered PSEN/γ-secretase affinity and enzymatic activity on over 90 substrates [7] and their inescapable pleiotropic disturbances on brain homeostasis. This issue is well illustrated by the clinical trials involving the selective inhibition of PSEN/γ-secretase Aβ cleavage by semagacestat, avagacestat, begacestat and ELND006 which were halted because they failed to reach their expected endpoints, and also because they triggered multiple undesirable and sometimes irreversible side-effects [86-90]. The implication of these observations is that the early onset and faster demise associated with PSEN-FAD mutations may not only be due to enhanced APP/Aβ processing or to the lack of their degradation, but rather to the extensive molecular alterations and ripple effects on a multitude of pivotal substrates ultimately caused by a dysfunctional PSEN/γ-secretase. If this hypothesis is correct, a detailed postmortem biochemical assessment of individuals treated with γ-secretase inhibitors will be of enormous value in the interpretation of clinical trial outcomes.
In summary, there is a great need for a better understanding of the clinical, neuropathological and biochemical evolution of PSEN-FAD. The pathology induced by PSEN-FAD mutations involves both the GM and WM in a complex network of potentially devastating direct and indirect impacts on the brain ability to create and transmit action potentials. A comprehensive assessment of the biochemical diversity exhibited by various alleles and their relationship to clinical presentations will be helpful in the interpretation of early interventional trials employing FAD patients. New information on the biochemical pathology of PSEN mutations will offer valuable insights into the etiology and progression of the disease and assist in the design of preventative measures and effective therapeutic interventions for both SAD and FAD.
Acknowledgements
This study was supported by the State of Arizona Alzheimer’s Research Consortium as well as the National Institute on Aging (NIA) grants R01 AG-19795, P30 AG-19610 and P30 AG-10133. The Brain and Body Donation Program at BSHRI is supported by the National Institute of Neurological Disorders and Stroke (U24 NS072026), the Arizona Department of Health Services (contract 211002, Arizona Alzheimer’s Research Center), the Arizona Biomedical Research Commission (contracts 4001, 0011, 05-901 and 1001 to the Arizona Parkinson’s Disease Consortium) and the Michael J. Fox Foundation for Parkinson’s Research. We would like to thank Dean Luehrs, PhD for critical reading of the manuscript and Matthew Grabinski for assistance in Figure preparation. The funders had no role in study design, data collection and analysis, decision to publish or preparation of the manuscript. SAJ has received indirect salary support from Bayer Health Care Pharmaceuticals, Eisai Inc., Elan Pharmaceuticals, Eli Lill & Co., Pfizer, and Genentech and receives royalties from American Psychiatric Publishing. MNS receives grant/contract support from Pfizer, Eisai, Neuronix, Lilly, Avid, Piramal, GE, Avanir, Elan, Functional Neuromodulation and is on the advisory board for Biogen, Lilly, Piramal, Eisai and receives royalties from Wiley and Tenspeed (RandomHouse). TGB receives funding from AVID-Radiopharmaceuticals, Schering-Bayer Pharmaceuticals, GE Healthcare, Piramal Radiopharmaceuticals and Navidea.
Disclosure of conflict of interest
AER, CLM, MM, JW, TAK, IDD, CMW, WMK, MPM and BG have no conflicts of interest.
References
- 1.Wortmann M. Dementia: a global health priority- highlights from an ADI and World Health Organization report. Alzheimers Res Ther. 2012;4:40. doi: 10.1186/alzrt143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Goate A, Chartier-Harlin MC, Mullan M, Brown J, Crawford F, Fidani L, Giuffra L, Haynes A, Irving N, James L, Mant R, Newton P, Rooke K, Roques P, Talbot C, Pericak-Vance M, Roses A, Willson R, Rosser M, Owen M, Hardy J. Segregation of a missense mutation in the amyloid precursor protein gene with familial Alzheimer’s disease. Nature. 1991;349:704–706. doi: 10.1038/349704a0. [DOI] [PubMed] [Google Scholar]
- 3.Sherrington R, Rogaev EI, Liang Y, Rogaeva EA, Levesque G, Ikeda M, Chi H, Lin C, Li G, Holman K, Tsuda T, Mar L, Foncin JF, Bruni AC, Montesi MP, Sorbi S, Rainero I, Pinessi L, Nee L, Chumakov I, Pollen D, Brookes A, Sanseau P, Polinsky RJ, Wasco W, Da Silva HA, Haines JL, Perkicak-Vance MA, Tanzi RE, Roses AD, Fraser PE, Rommens JM, St George-Hyslop PH. Cloning of a gene bearing missense mutations in early-onset familial Alzheimer’s disease. Nature. 1995;375:754–760. doi: 10.1038/375754a0. [DOI] [PubMed] [Google Scholar]
- 4.Levy-Lahad E, Wasco W, Poorkaj P, Romano DM, Oshima J, Pettingell WH, Yu CE, Jondro PD, Schmidt SD, Wang K, Crowley AC, Fu YH, Galas D, Nemens E, Wijsman EM, Bird TD, Schellenberg GD, Tanzi RE. Candidate gene for the chromosome 1 familial Alzheimer’s disease locus. Science. 1995;269:973–977. doi: 10.1126/science.7638622. [DOI] [PubMed] [Google Scholar]
- 5.Zhang H, Ma Q, Zhang YW, Xu H. Proteolytic processing of Alzheimer’s beta-amyloid precursor protein. J Neurochem. 2012;120(Suppl 1):9–21. doi: 10.1111/j.1471-4159.2011.07519.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.De Strooper B, Iwatsubo T, Wolfe MS. Presenilins and gamma-Secretase: Structure, Function, and Role in Alzheimer Disease. Cold Spring Harb Perspect Med. 2012;2:a006304. doi: 10.1101/cshperspect.a006304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haapasalo A, Kovacs DM. The many substrates of presenilin/gamma-secretase. J Alzheimers Dis. 2011;25:3–28. doi: 10.3233/JAD-2011-101065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Larner AJ, Doran M. Clinical phenotypic heterogeneity of Alzheimer’s disease associated with mutations of the presenilin-1 gene. J Neurol. 2006;253:139–158. doi: 10.1007/s00415-005-0019-5. [DOI] [PubMed] [Google Scholar]
- 9.Larner AJ, Doran M. Genotype-phenotype relationships of presenilin-1 mutations in Alzheimer’s disease: an update. J Alzheimers Dis. 2009;17:259–265. doi: 10.3233/JAD-2009-1042. [DOI] [PubMed] [Google Scholar]
- 10.Ryan NS, Rossor MN. Correlating familial Alzheimer’s disease gene mutations with clinical phenotype. Biomark Med. 2010;4:99–112. doi: 10.2217/bmm.09.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mann DM, Pickering-Brown SM, Takeuchi A, Iwatsubo T. Amyloid angiopathy and variability in amyloid beta deposition is determined by mutation position in presenilin-1-linked Alzheimer’s disease. Am J Pathol. 2001;158:2165–2175. doi: 10.1016/s0002-9440(10)64688-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Elder GA, Gama Sosa MA, De Gasperi R, Dickstein DL, Hof PR. Presenilin transgenic mice as models of Alzheimer’s disease. Brain Struct Funct. 2010;214:127–143. doi: 10.1007/s00429-009-0227-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen J, Bronson RT, Chen DF, Xia W, Selkoe DJ, Tonegawa S. Skeletal and CNS defects in Presenilin-1-deficient mice. Cell. 1997;89:629–639. doi: 10.1016/s0092-8674(00)80244-5. [DOI] [PubMed] [Google Scholar]
- 14.Gama Sosa MA, Gasperi RD, Rocher AB, Wang AC, Janssen WG, Flores T, Perez GM, Schmeidler J, Dickstein DL, Hof PR, Elder GA. Age-related vascular pathology in transgenic mice expressing presenilin 1-associated familial Alzheimer’s disease mutations. Am J Pathol. 2010;176:353–368. doi: 10.2353/ajpath.2010.090482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wong PC, Zheng H, Chen H, Becher MW, Sirinathsinghji DJ, Trumbauer ME, Chen HY, Price DL, Van der Ploeg LH, Sisodia SS. Presenilin 1 is required for Notch1 and DII1 expression in the paraxial mesoderm. Nature. 1997;387:288–292. doi: 10.1038/387288a0. [DOI] [PubMed] [Google Scholar]
- 16.Strobel G. Expanding the Network, DIAN Starts Showing Longitudinal Data. (10-11-12). http://www.alzforum.org/new/detail.asp?id=3290.
- 17.Brunetti A, Postiglione A, Tedeschi E, Ciarmiello A, Quarantelli M, Covelli EM, Milan G, Larobina M, Soricelli A, Sodano A, Alfano B. Measurement of global brain atrophy in Alzheimer’s disease with unsupervised segmentation of spin-echo MRI studies. J Magn Reson Imaging. 2000;11:260–266. doi: 10.1002/(sici)1522-2586(200003)11:3<260::aid-jmri4>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 18.Smith CD, Snowdon DA, Wang H, Markesbery WR. White matter volumes and periventricular white matter hyperintensities in aging and dementia. Neurology. 2000;54:838–842. doi: 10.1212/wnl.54.4.838. [DOI] [PubMed] [Google Scholar]
- 19.DeCarli C, Grady CL, Clark CM, Katz DA, Brady DR, Murphy DG, Haxby JV, Salerno JA, Gillette JA, Gonzalez-Aviles A, Rapoport SI. Comparison of positron emission tomography, cognition, and brain volume in Alzheimer’s disease with and without severe abnormalities of white matter. J Neurol Neurosurg Psychiatry. 1996;60:158–167. doi: 10.1136/jnnp.60.2.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kawamura J, Meyer JS, Terayama Y, Weathers S. Leuko-araiosis and cerebral hypoperfusion compared in elderly normals and Alzheimer’s dementia. J Am Geriatr Soc. 1992;40:375–380. doi: 10.1111/j.1532-5415.1992.tb02138.x. [DOI] [PubMed] [Google Scholar]
- 21.Bartzokis G. Alzheimer’s disease as homeostatic responses to age-related myelin breakdown. Neurobiol Aging. 2011;32:1341–1371. doi: 10.1016/j.neurobiolaging.2009.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carmichael O, Schwarz C, Drucker D, Fletcher E, Harvey D, Beckett L, Jack CR Jr, Weiner M, DeCarli C. Longitudinal changes in white matter disease and cognition in the first year of the Alzheimer disease neuroimaging initiative. Arch Neurol. 2010;67:1370–1378. doi: 10.1001/archneurol.2010.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Provenzano FA, Muraskin J, Tosto G, Narkhede A, Wasserman BT, Griffith EY, Guzman VA, Meier IB, Zimmerman ME, Brickman AM. White matter hyperintensities and cerebral amyloidosis: necessary and sufficient for clinical expression of Alzheimer disease? JAMA Neurol. 2013;70:455–461. doi: 10.1001/jamaneurol.2013.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gurol ME, Viswanathan A, Gidicsin C, Hedden T, Martinez-Ramirez S, Dumas A, Vashkevich A, Ayres AM, Auriel E, van Etten E, Becker A, Carmasin J, Schwab K, Rosand J, Johnson KA, Greenberg SM. Cerebral amyloid angiopathy burden associated with leukoaraiosis: A positron emission tomography/magnetic resonance imaging study. Ann Neurol. 2012 doi: 10.1002/ana.23830. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gold BT, Johnson NF, Powell DK, Smith CD. White matter integrity and vulnerability to Alzheimer’s disease: Preliminary findings and future directions. Biochim Biophys Acta. 2012;1822:416–422. doi: 10.1016/j.bbadis.2011.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Back SA, Kroenke CD, Sherman LS, Lawrence G, Gong X, Taber EN, Sonnen JA, Larson EB, Montine TJ. White matter lesions defined by diffusion tensor imaging in older adults. Ann Neurol. 2011;70:465–476. doi: 10.1002/ana.22484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beach TG, Walker R, McGeer EG. Patterns of gliosis in Alzheimer’s disease and aging cerebrum. Glia. 1989;2:420–436. doi: 10.1002/glia.440020605. [DOI] [PubMed] [Google Scholar]
- 28.Hachinski VC, Potter P, Merskey H. Leuko-araiosis. Arch Neurol. 1987;44:21–23. doi: 10.1001/archneur.1987.00520130013009. [DOI] [PubMed] [Google Scholar]
- 29.Di Paola M, Spalletta G, Caltagirone C. In vivo structural neuroanatomy of corpus callosum in Alzheimer’s disease and mild cognitive impairment using different MRI techniques: a review. J Alzheimers Dis. 2010;20:67–95. doi: 10.3233/JAD-2010-1370. [DOI] [PubMed] [Google Scholar]
- 30.Matsusue E, Sugihara S, Fujii S, Ohama E, Kinoshita T, Ogawa T. White matter changes in elderly people: MR-pathologic correlations. Magn Reson Med Sci. 2006;5:99–104. doi: 10.2463/mrms.5.99. [DOI] [PubMed] [Google Scholar]
- 31.Kalback W, Esh C, Castano EM, Rahman A, Kokjohn T, Luehrs DC, Sue L, Cisneros R, Gerber F, Richardson C, Bohrmann B, Walker DG, Beach TG, Roher AE. Atherosclerosis, vascular amyloidosis and brain hypoperfusion in the pathogenesis of sporadic Alzheimer’s disease. Neurol Res. 2004;26:525–539. doi: 10.1179/016164104225017668. [DOI] [PubMed] [Google Scholar]
- 32.Reisberg B, Pattschull-Furlan A, Franssen E, Sclan SG, Kluger A, Dingcong L, Ferris SH. Dementia of the Alzheimer type recapitulates ontogeny inversely on specific ordinal and temporal parameters. In: Kostovic T, Knezevic S, Wisniewski HM, Spillich GJ, editors. Neurodevelopment, Aging and Cognition. Boston: Birkhauser; 1992. pp. 345–369. [Google Scholar]
- 33.Braak H, Braak E. Development of Alzheimer-related neurofibrillary changes in the neocortex inversely recapitulates cortical myelogenesis. Acta Neuropathol. 1996;92:197–201. doi: 10.1007/s004010050508. [DOI] [PubMed] [Google Scholar]
- 34.Bartzokis G. Age-related myelin breakdown: a developmental model of cognitive decline and Alzheimer’s disease. Neurobiol Aging. 2004;25:5–18. doi: 10.1016/j.neurobiolaging.2003.03.001. [DOI] [PubMed] [Google Scholar]
- 35.Braak H, Del Tredici K. Poor and protracted myelination as a contributory factor to neurodegenerative disorders. Neurobiol Aging. 2004;25:19–23. doi: 10.1016/j.neurobiolaging.2003.04.001. [DOI] [PubMed] [Google Scholar]
- 36.Choi SJ, Lim KO, Monteiro I, Reisberg B. Diffusion tensor imaging of frontal white matter microstructure in early Alzheimer’s disease: a preliminary study. J Geriatr Psychiatry Neurol. 2005;18:12–19. doi: 10.1177/0891988704271763. [DOI] [PubMed] [Google Scholar]
- 37.Stricker NH, Schweinsburg BC, Delano-Wood L, Wierenga CE, Bangen KJ, Haaland KY, Frank LR, Salmon DP, Bondi MW. Decreased white matter integrity in late-myelinating fiber pathways in Alzheimer’s disease supports retrogenesis. Neuroimage. 2009;45:10–16. doi: 10.1016/j.neuroimage.2008.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.O’Dwyer L, Lamberton F, Bokde AL, Ewers M, Faluyi YO, Tanner C, Mazoyer B, O’Neill D, Bartley M, Collins DR, Coughlan T, Prvulovic D, Hampel H. Multiple indices of diffusion identifies white matter damage in mild cognitive impairment and Alzheimer’s disease. PLoS One. 2011;6:e21745. doi: 10.1371/journal.pone.0021745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Roher AE, Weiss N, Kokjohn TA, Kuo YM, Kalback W, Anthony J, Watson D, Luehrs DC, Sue L, Walker D, Emmerling M, Goux W, Beach T. Increased A beta peptides and reduced cholesterol and myelin proteins characterize white matter degeneration in Alzheimer’s disease. Biochemistry. 2002;41:11080–11090. doi: 10.1021/bi026173d. [DOI] [PubMed] [Google Scholar]
- 40.Roher AE, Kuo YM, Esh C, Knebel C, Weiss N, Kalback W, Luehrs DC, Childress JL, Beach TG, Weller RO, Kokjohn TA. Cortical and leptomeningeal cerebrovascular amyloid and white matter pathology in Alzheimer’s disease. Mol Med. 2003;9:112–122. [PMC free article] [PubMed] [Google Scholar]
- 41.Castano EM, Maarouf CL, Wu T, Leal MC, Whiteside CM, Lue LF, Kokjohn TA, Sabbagh MN, Beach TG, Roher AE. Alzheimer disease periventricular white matter lesions exhibit specific proteomic profile alterations. Neurochem Int. 2013;62:145–156. doi: 10.1016/j.neuint.2012.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maarouf CL, Daugs ID, Spina S, Vidal R, Kokjohn TA, Patton RL, Kalback WM, Luehrs DC, Walker DG, Castano EM, Beach TG, Ghetti B, Roher AE. Histopathological and molecular heterogeneity among individuals with dementia associated with Presenilin mutations. Mol Neurodegener. 2008;3:20. doi: 10.1186/1750-1326-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fox NC, Kennedy AM, Harvey RJ, Lantos PL, Roques PK, Collinge J, Hardy J, Hutton M, Stevens JM, Warrington EK, Rossor MN. Clinicopathological features of familial Alzheimer’s disease associated with the M139V mutation in the presenilin 1 gene. Pedigree but not mutation specific age at onset provides evidence for a further genetic factor. Brain. 1997;120:491–501. doi: 10.1093/brain/120.3.491. [DOI] [PubMed] [Google Scholar]
- 44.Abe K. Clinical and molecular analysis of neurodegenerative diseases. Tohoku J Exp Med. 1997;181:389–409. doi: 10.1620/tjem.181.389. [DOI] [PubMed] [Google Scholar]
- 45.Takao M, Ghetti B, Murrell JR, Unverzagt FW, Giaccone G, Tagliavini F, Bugiani O, Piccardo P, Hulette CM, Crain BJ, Farlow MR, Heyman A. Ectopic white matter neurons, a developmental abnormality that may be caused by the PSEN1 S169L mutation in a case of familial AD with myoclonus and seizures. J Neuropathol Exp Neurol. 2001;60:1137–1152. doi: 10.1093/jnen/60.12.1137. [DOI] [PubMed] [Google Scholar]
- 46.O’Riordan S, McMonagle P, Janssen JC, Fox NC, Farrell M, Collinge J, Rossor MN, Hutchinson M. Presenilin-1 mutation (E280G), spastic paraparesis, and cranial MRI white-matter abnormalities. Neurology. 2002;59:1108–1110. doi: 10.1212/wnl.59.7.1108. [DOI] [PubMed] [Google Scholar]
- 47.Marcon G, Giaccone G, Cupidi C, Balestrieri M, Beltrami CA, Finato N, Bergonzi P, Sorbi S, Bugiani O, Tagliavini F. Neuropathological and clinical phenotype of an Italian Alzheimer family with M239V mutation of presenilin 2 gene. J Neuropathol Exp Neurol. 2004;63:199–209. doi: 10.1093/jnen/63.3.199. [DOI] [PubMed] [Google Scholar]
- 48.Beach TG, Sue LI, Walker DG, Roher AE, Lue L, Vedders L, Connor DJ, Sabbagh MN, Rogers J. The Sun Health Research Institute Brain Donation Program: description and experience, 1987-2007. Cell Tissue Bank. 2008;9:229–245. doi: 10.1007/s10561-008-9067-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maarouf CL, Daugs ID, Kokjohn TA, Walker DG, Hunter JM, Kruchowsky JC, Woltjer R, Kaye J, Castano EM, Sabbagh MN, Beach TG, Roher AE. Alzheimer’s disease and non-demented high pathology control nonagenarians: comparing and contrasting the biochemistry of cognitively successful aging. PLoS One. 2011;6:e27291. doi: 10.1371/journal.pone.0027291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Berengut D. Some Small-Sample Properties of the Student t Confidence Intervals. The American Statistician. 1981;35:144–147. [Google Scholar]
- 51.Fairweather WR. A Method of Obtaining an Exact Confidence Interval for the Common Mean of Several Normal Populations. Journal of the Royal Statistical Society Series C (Applied Statistics) 1972;21:229–233. [Google Scholar]
- 52.Ringman JM, O’Neill J, Geschwind D, Medina L, Apostolova LG, Rodriguez Y, Schaffer B, Varpetian A, Tseng B, Ortiz F, Fitten J, Cummings JL, Bartzokis G. Diffusion tensor imaging in preclinical and presymptomatic carriers of familial Alzheimer’s disease mutations. Brain. 2007;130:1767–1776. doi: 10.1093/brain/awm102. [DOI] [PubMed] [Google Scholar]
- 53.Bendlin BB, Ries ML, Canu E, Sodhi A, Lazar M, Alexander AL, Carlsson CM, Sager MA, Asthana S, Johnson SC. White matter is altered with parental family history of Alzheimer’s disease. Alzheimers Dement. 2010;6:394–403. doi: 10.1016/j.jalz.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith CD, Chebrolu H, Andersen AH, Powell DA, Lovell MA, Xiong S, Gold BT. White matter diffusion alterations in normal women at risk of Alzheimer’s disease. Neurobiol Aging. 2010;31:1122–1131. doi: 10.1016/j.neurobiolaging.2008.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Li M, Chen L, Lee DH, Yu LC, Zhang Y. The role of intracellular amyloid beta in Alzheimer’s disease. Prog Neurobiol. 2007;83:131–139. doi: 10.1016/j.pneurobio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 56.Decker H, Lo KY, Unger SM, Ferreira ST, Silverman MA. Amyloid-beta peptide oligomers disrupt axonal transport through an NMDA receptor-dependent mechanism that is mediated by glycogen synthase kinase 3beta in primary cultured hippocampal neurons. J Neurosci. 2010;30:9166–9171. doi: 10.1523/JNEUROSCI.1074-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liao MC, Ahmed M, Smith SO, Van Nostrand WE. Degradation of amyloid beta protein by purified myelin basic protein. J Biol Chem. 2009;284:28917–28925. doi: 10.1074/jbc.M109.050856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kotarba AE, Aucoin D, Hoos MD, Smith SO, Van Nostrand WE. Fine mapping of the amyloid beta-protein binding site on myelin basic protein. Biochemistry. 2013;52:2565–2573. doi: 10.1021/bi4001936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Van Vickle GD, Esh CL, Kokjohn TA, Patton RL, Kalback WM, Luehrs DC, Beach TG, Newel AJ, Lopera F, Ghetti B, Vidal R, Castano EM, Roher AE. Presenilin-1 280Glu-->Ala mutation alters C-terminal APP processing yielding longer abeta peptides: implications for Alzheimer’s disease. Mol Med. 2008;14:184–194. doi: 10.2119/2007-00094.VanVickle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Quiroz YT, Stern CE, Reiman EM, Brickhouse M, Ruiz A, Sperling RA, Lopera F, Dickerson BC. Cortical atrophy in presymptomatic Alzheimer’s disease presenilin 1 mutation carriers. J Neurol Neurosurg Psychiatry. 2013;84:556–561. doi: 10.1136/jnnp-2012-303299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reiman EM, Quiroz YT, Fleisher AS, Chen K, Velez-Pardo C, Jimenez-Del-Rio M, Fagan AM, Shah AR, Alvarez S, Arbelaez A, Giraldo M, Acosta-Baena N, Sperling RA, Dickerson B, Stern CE, Tirado V, Munoz C, Reiman RA, Huentelman MJ, Alexander GE, Langbaum JB, Kosik KS, Tariot PN, Lopera F. Brain imaging and fluid biomarker analysis in young adults at genetic risk for autosomal dominant Alzheimer’s disease in the presenilin 1 E280A kindred: a case-control study. Lancet Neurol. 2012;11:1048–1056. doi: 10.1016/S1474-4422(12)70228-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bateman RJ, Xiong C, Benzinger TL, Fagan AM, Goate A, Fox NC, Marcus DS, Cairns NJ, Xie X, Blazey TM, Holtzman DM, Santacruz A, Buckles V, Oliver A, Moulder K, Aisen PS, Ghetti B, Klunk WE, McDade E, Martins RN, Masters CL, Mayeux R, Ringman JM, Rossor MN, Schofield PR, Sperling RA, Salloway S, Morris JC. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 2012;367:795–804. doi: 10.1056/NEJMoa1202753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Amtul Z, Lewis PA, Piper S, Crook R, Baker M, Findlay K, Singleton A, Hogg M, Younkin L, Younkin SG, Hardy J, Hutton M, Boeve BF, Tang-Wai D, Golde TE. A presenilin 1 mutation associated with familial frontotemporal dementia inhibits gamma-secretase cleavage of APP and notch. Neurobiol Dis. 2002;9:269–273. doi: 10.1006/nbdi.2001.0473. [DOI] [PubMed] [Google Scholar]
- 64.Shioi J, Georgakopoulos A, Mehta P, Kouchi Z, Litterst CM, Baki L, Robakis NK. FAD mutants unable to increase neurotoxic Abeta 42 suggest that mutation effects on neurodegeneration may be independent of effects on Abeta. J Neurochem. 2007;101:674–681. doi: 10.1111/j.1471-4159.2006.04391.x. [DOI] [PubMed] [Google Scholar]
- 65.Page RM, Baumann K, Tomioka M, Perez-Revuelta BI, Fukumori A, Jacobsen H, Flohr A, Luebbers T, Ozmen L, Steiner H, Haass C. Generation of Abeta38 and Abeta42 is independently and differentially affected by familial Alzheimer disease-associated presenilin mutations and gamma-secretase modulation. J Biol Chem. 2008;283:677–683. doi: 10.1074/jbc.M708754200. [DOI] [PubMed] [Google Scholar]
- 66.Dermaut B, Kumar-Singh S, Engelborghs S, Theuns J, Rademakers R, Saerens J, Pickut BA, Peeters K, van den Broeck M, Vennekens K, Claes S, Cruts M, Cras P, Martin JJ, Van Broeckhoven C, De Deyn PP. A novel presenilin 1 mutation associated with Pick’s disease but not beta-amyloid plaques. Ann Neurol. 2004;55:617–626. doi: 10.1002/ana.20083. [DOI] [PubMed] [Google Scholar]
- 67.Kopan R, Schroeter EH, Weintraub H, Nye JS. Signal transduction by activated mNotch: importance of proteolytic processing and its regulation by the extracellular domain. Proc Natl Acad Sci U S A. 1996;93:1683–1688. doi: 10.1073/pnas.93.4.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Woo HN, Park JS, Gwon AR, Arumugam TV, Jo DG. Alzheimer’s disease and Notch signaling. Biochem Biophys Res Commun. 2009;390:1093–1097. doi: 10.1016/j.bbrc.2009.10.093. [DOI] [PubMed] [Google Scholar]
- 69.Zhang C, Wu B, Beglopoulos V, Wines-Samuelson M, Zhang D, Dragatsis I, Sudhof TC, Shen J. Presenilins are essential for regulating neurotransmitter release. Nature. 2009;460:632–636. doi: 10.1038/nature08177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Suzuki K, Hayashi Y, Nakahara S, Kumazaki H, Prox J, Horiuchi K, Zeng M, Tanimura S, Nishiyama Y, Osawa S, Sehara-Fujisawa A, Saftig P, Yokoshima S, Fukuyama T, Matsuki N, Koyama R, Tomita T, Iwatsubo T. Activity-dependent proteolytic cleavage of neuroligin-1. Neuron. 2012;76:410–422. doi: 10.1016/j.neuron.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 71.Saura CA, Servian-Morilla E, Scholl FG. Presenilin/gamma-secretase regulates neurexin processing at synapses. PLoS One. 2011;6:e19430. doi: 10.1371/journal.pone.0019430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bot N, Schweizer C, Ben Halima S, Fraering PC. Processing of the synaptic cell adhesion molecule neurexin-3beta by Alzheimer disease alpha- and gamma-secretases. J Biol Chem. 2011;286:2762–2773. doi: 10.1074/jbc.M110.142521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Bottos A, Destro E, Rissone A, Graziano S, Cordara G, Assenzio B, Cera MR, Mascia L, Bussolino F, Arese M. The synaptic proteins neurexins and neuroligins are widely expressed in the vascular system and contribute to its functions. Proc Natl Acad Sci U S A. 2009;106:20782–20787. doi: 10.1073/pnas.0809510106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.De Strooper B. Loss-of-function presenilin mutations in Alzheimer disease. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8:141–146. doi: 10.1038/sj.embor.7400897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wolfe MS. When loss is gain: reduced presenilin proteolytic function leads to increased Abeta42/Abeta40. Talking Point on the role of presenilin mutations in Alzheimer disease. EMBO Rep. 2007;8:136–140. doi: 10.1038/sj.embor.7400896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Goux WJ, Rodriguez S, Sparkman DR. Analysis of the core components of Alzheimer paired helical filaments. A gas chromatography/mass spectrometry characterization of fatty acids, carbohydrates and long-chain bases. FEBS Lett. 1995;366:81–85. doi: 10.1016/0014-5793(95)00486-s. [DOI] [PubMed] [Google Scholar]
- 77.Goux WJ, Rodriguez S, Sparkman DR. Characterization of the glycolipid associated with Alzheimer paired helical filaments. J Neurochem. 1996;67:723–733. doi: 10.1046/j.1471-4159.1996.67020723.x. [DOI] [PubMed] [Google Scholar]
- 78.Goux WJ, Liu B, Shumburo AM, Parikh S, Sparkman DR. A quantitative assessment of glycolipid and protein associated with paired helical filament preparations from Alzheimer’s diseased brain. J Alzheimers Dis. 2001;3:455–466. doi: 10.3233/jad-2001-3504. [DOI] [PubMed] [Google Scholar]
- 79.Gray EG, Paula-Barbosa M, Roher A. Alzheimer’s disease: paired helical filaments and cytomembranes. Neuropathol Appl Neurobiol. 1987;13:91–110. doi: 10.1111/j.1365-2990.1987.tb00174.x. [DOI] [PubMed] [Google Scholar]
- 80.Hull M, Fiebich BL, Dykierek P, Schmidtke K, Nitzsche E, Orszagh M, Deuschl G, Moser E, Schumacher M, Lucking C, Berger M, Bauer J. Early-onset Alzheimer’s disease due to mutations of the presenilin-1 gene on chromosome 14: a 7-year follow-up of a patient with a mutation at codon 139. Eur Arch Psychiatry Clin Neurosci. 1998;248:123–129. doi: 10.1007/s004060050028. [DOI] [PubMed] [Google Scholar]
- 81.Nikisch G, Hertel A, Kiessling B, Wagner T, Krasz D, Hofmann E, Wiedemann G. Three-year follow-up of a patient with early-onset Alzheimer’s disease with presenilin-2 N141I mutation - case report and review of the literature. Eur J Med Res. 2008;13:579–584. [PubMed] [Google Scholar]
- 82.Reiman EM, Langbaum JB, Fleisher AS, Caselli RJ, Chen K, Ayutyanont N, Quiroz YT, Kosik KS, Lopera F, Tariot PN. Alzheimer’s Prevention Initiative: a plan to accelerate the evaluation of presymptomatic treatments. J Alzheimers Dis. 2011;26(Suppl 3):321–329. doi: 10.3233/JAD-2011-0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Morris JC, Aisen PS, Bateman RJ, Benzinger TL, Cairns NJ, Fagan AM, Ghetti B, Goate AM, Holtzman DM, Klunk WE, McDade E, Marcus DS, Martins RN, Masters CL, Mayeux R, Oliver A, Quaid K, Ringman JM, Rossor MN, Salloway S, Schofield PR, Selsor NJ, Sperling RA, Weiner MW, Xiong C, Moulder KL, Buckles VD. Developing an international network for Alzheimer research: The Dominantly Inherited Alzheimer Network. Clin Investig (Lond) 2012;2:975–984. doi: 10.4155/cli.12.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mullard A. Sting of Alzheimer’s failures offset by upcoming prevention trials. Nat Rev Drug Discov. 2012;11:657–660. doi: 10.1038/nrd3842. [DOI] [PubMed] [Google Scholar]
- 85.Sperling R, Aisen P. Anti-amyloid treatment in Asymptomatic* AD A4 Trial. (09-28-2012).http://alzheimer.wustl.edu/education/berg/berg2012/Slides/Sperling.pdf.
- 86.D’Onofrio G, Panza F, Frisardi V, Solfrizzi V, Imbimbo BP, Paroni G, Cascavilla L, Seripa D, Pilotto A. Advances in the identification of gamma-secretase inhibitors for the treatment of Alzheimer’s disease. Expert Opin Drug Discov. 2012;7:19–37. doi: 10.1517/17460441.2012.645534. [DOI] [PubMed] [Google Scholar]
- 87.Wolfe MS. gamma-Secretase as a target for Alzheimer’s disease. Adv Pharmacol. 2012;64:127–153. doi: 10.1016/B978-0-12-394816-8.00004-0. [DOI] [PubMed] [Google Scholar]
- 88.Tomita T. [Development of Alzheimer’s disease treatment based on the molecular mechanism of gamma-secretase activity] . Rinsho Shinkei-gaku. 2012;52:1165–1167. doi: 10.5692/clinicalneurol.52.1165. [DOI] [PubMed] [Google Scholar]
- 89.Lleo A, Saura CA. gamma-secretase substrates and their implications for drug development in Alzheimer’s disease. Curr Top Med Chem. 2011;11:1513–1527. doi: 10.2174/156802611795861004. [DOI] [PubMed] [Google Scholar]
- 90.Hopkins CR. ACS chemical neuroscience molecule spotlight on ELND006: another gamma-secretase inhibitor fails in the clinic. ACS Chem Neurosci. 2011;2:279–280. doi: 10.1021/cn2000469. [DOI] [PMC free article] [PubMed] [Google Scholar]