

Abstract
Viral infections frequently induce acute and chronic inflammatory diseases, yet the contribution of the innate immune response to a detrimental host response remains poorly understood. In virus-infected cells, double-stranded RNA (dsRNA) is generated as an intermediate during viral replication. Cell necrosis (and the release of endogenous dsRNA) is a common event during both sterile and infectious inflammatory processes. The discovery of Toll-like receptor 3 (TLR3) as an interferon-inducing dsRNA sensor led to the assumption that TLR3 was the master sentinel against viral infections. This simplistic view has been challenged by the discovery of at least three members of the DExd/H-box helicase cytosolic sensors of dsRNA that share with TLR3 the Toll–interleukin-1 receptor (TIR) -adapter molecule TIR domain-containing adaptor protein interferon-β (TRIF) for downstream type I interferon signalling. Data are conflicting on the role of TLR3 in protective immunity against viruses in the mouse model. Varying susceptibility to infection and disease outcomes have been reported in TLR3-immunodeficient mice. Surprisingly, the susceptibility to develop herpes simplex virus-1 encephalitis in humans with inborn defects of the TLR3 pathway varies, and TLR3-deficient humans do not show increased susceptibility to other viral infections. Therefore, a current challenge is to understand the protective versus pathogenic contribution of TLR3 in viral infections. We review recent advances in the identification of TLR3-signalling pathways, endogenous and virus-induced negative regulators of the TLR3 cascade, and discuss the protective versus pathogenic role of TLR3 in viral pathogenesis.
Keywords: negative regulation, pathogenesis, Toll-like receptor 3, virus
Introduction
Mammalian cells are able to detect viruses at multiple steps. Therefore, it is likely that cells benefit from redundant mechanisms to detect and limit viral infections. Recognition of viral nucleic acids by intracellular Toll-like receptors (TLRs) entails the early steps of the immune response elicited during viral infections.1 For the past 10 years, efforts have been made to define the role of TLR3 in the antiviral response and to elucidate at the molecular level the TLR3 pathway as a ‘major sentinel’ against viruses. In contrast to TLR7, which recognizes single-strand (ss) RNA (therefore its role could be restricted to certain types of RNA viruses), TLR3 has, at least in principle, the ability to play a crucial role in the antiviral response against most viruses by its ability to sense double-stranded (ds) RNA, a common intermediate of replication among many viruses.1 Intriguingly, dsRNA of non-viral origin and, in particular, endogenous mRNA and RNA released from necrotic cells have been shown to trigger the TLR3 pathway.2,3
Toll-like receptor 3 is broadly expressed and well conserved among vertebrates and its expression has been confirmed in immune and non-immune cells.4 A functional antiviral role of TLR3 against certain viruses has been demonstrated in various human and murine cells as well as in the mouse model (recently reviewed in ref. 5). Interestingly, TLR3 has been implicated in licensing myeloid dendritic cells (DCs) for cross-priming of CD8 T cells in the mouse, and a certain subset of human DCs that highly express TLR3 exhibit a better capacity for cross-presenting apoptotic and necrotic cell antigens after TLR3 stimulation.6 Indeed, at least in part, the strategy of using TLR3 ligands as adjuvants in vaccine therapy is based on the selective high expression of TLR3 in human CD141+ DCs and mouse CD8a+ DC subsets. In contrast to the beneficial roles of TLR3 in antiviral response and cellular immunity, emerging evidence suggests that TLR3 also mediates pathogenesis, which may or may not be directly related to the exacerbation of the antiviral response. This review provides background on the TLR3 pathway, highlights the negative regulation of TLR3 signalling by endogenous and virus-encoded proteins, and discusses emerging evidence on the detrimental contribution of TLR3 in viral pathogenesis.
TLR3 pathway
The TLR3 is located in the endoplasmic reticulum of unstimulated cells, and upon dsRNA stimulation, it traffics to endosomes to encounter its ligand through a pH-dependent mechanism that also requires TLR3 dimerization.7 The interaction between the endoplasmic reticulum membrane protein UNC-93B and TLR3 is crucial for TLR3 trafficking and signalling.8 In addition, recent structural studies have demonstrated that the length (at least 45 bp) and structure of dsRNAs play critical roles in TLR3 recognition and signalling.9 Emerging evidence suggests that TLR proteolysis by asparagine endopeptidase and cathepsins in endolysosomes may represent a general regulatory strategy for all TLRs involved in nucleic acid recognition.10 The TLR3 proteolytic processing regulates stability and endosomal localization, but its role in TLR3 function remains poorly understood.11 Furthermore, TLR3 tyrosine phosphorylation in the endosome is essential for recruiting the adaptor protein Toll–interleukin-1 (IL-1) receptor (TIR) domain–containing adaptor protein interferon-β (IFN-β) (TRIF) to the TIR domain of TLR3. This is in contrast to other endosomal TLRs (TLR7–9) that do not require tyrosine phosphorylation, possibly owing to their MyD88-dependent, TRIF-independent signalling. To recruit TRIF upon recognition and dimerization, Tyr759 and Tyr858 residues in the cytoplasmic domain of TLR3 are sufficient for downstream signalling when phosphorylated.12 Two studies have reported a differential role for the tyrosine kinases c-Src13 and epidermal growth factor receptor14 in mediating TLR3 phosphorylation. These studies support the current model that after dsRNA stimulation, TLR3 binds epidermal growth factor receptor before c-Src does. Interestingly, epidermal growth factor receptor is required for Tyr858 phosphorylation, and Src is required for Tyr759 phosphorylation. In addition, phosphoinositide 3-kinase and Bruton's tyrosine kinase have been implicated in TLR3 phosphorylation. Phosphoinositide 3-kinase is required for the full activation of IFN regulatory transcription factor 3 (IRF3) and nuclear factor-κB (NF-κB),15 but its role in TLR3 signalling remains to be determined. Bruton's tyrosine kinase directly phosphorylates the Tyr759 residue of TLR3.16 In Bruton's tyrosine kinase-deficient macrophages, TLR3-induced phosphoinositide 3-kinase, AKT and mitogen-activated protein kinase (MAPK) signalling and activation of NF-κB, IRF3, and activator protein 1 (AP-1) transcription factors are all defective.16
Triggering TRIF recruitment by TLR3 dimerization and Tyr phosphorylation results in the stimulation of the transcription factors IRF3, NF-κB and AP-1 through two branches. NF-κB activating kinase (NAK) -associated protein 1 mediates TRIF association with the tumour necrosis factor (TNF) receptor-associated factor 3 (TRAF3) together with TRAF family member-associated NF-κB activator (TANK)-binding kinase 1 (TBK1) and IκB kinase ε (IKKε) complex, which subsequently promotes the phosphorylation, dimerization and translocation of IRF3 into the nucleus.17 The second branch drives the activation of NF-κB and AP-1 and is mediated by the protein kinase receptor-interacting protein 1 (RIP1) and the E3 ubiquitin protein ligase TRAF6, which interact with TRIF and recruit TAB2 (TAK1 binding protein 2), TAB3 (TAK1 binding protein 3), and TAK1 (transforming growth factor-β-activated kinase 1). This branch results in the activation of the IKK complex (nuclear factor-κB essential modulator, IKKα, IKKβ), the MAPK pathway, and further activation of NF-κB and AP-1,18 respectively. The RIP1 and TRAF3 branches result in the activation of the antiviral and pro-inflammatory response by the induction of type I IFN and other cytokines.
Intriguingly, RIP1 interaction with TRIF leads to cell death through a Fas-associated protein with death domain/caspase-8-dependent and mitochondrion-independent pathway (RIP1/Fas-associated protein with death domain).19 Indeed, the TLR3–TRIF axis has a cytopathic potential that might be developed during infection.20 Taken together, these data suggest that upon TLR3–TRIF stimulation and activation, three major outcomes can be predicted: (i) the development of the antiviral response mediated by IRF3 activation and further type I IFN production; (ii) a cytopathic effect or cell death through caspase-8 activation by RIP1; and (iii) the generation of a pro-inflammatory environment by the activation of NF-κB and AP-1.
Negative regulators of the TLR3 signalling cascade
Many of the adaptors and molecules involved in TLR3 signalling are shared by other TLR pathways. For example, TRIF is also an adaptor in a branch of the TLR421 and perhaps the TLR2 pathways22 and modulates the TLR5 pathway by inducing proteolytic degradation of TLR5.23 Unexpectedly, evidence is emerging on the role of TRIF in cytosolic sensing of dsRNA. In particular, TRIF is an adaptor of a novel dsRNA sensor complex consisting of the RNA helicases DDX1-DDX21-DHX36 that has been identified in the cytosol of myeloid DCs.24 Hence, the challenge is to understand specificity and redundancy among TLRs and other known and yet to be discovered pathways. Moreover, several positive25 and several negative15,26–30 regulators of the TLR3 pathway have been identified. Here, we focus on the negative regulation of the TLR3 pathway and discuss endogenous and virus-encoded negative regulators of the TLR3 pathway.
Endogenous regulators
The signalling balance must be maintained during the development of any immune response to prevent tissue damage and, potentially, autoimmunity. Emerging evidence demonstrates that endogenous negative regulators play a key role in the fine tuning of innate signalling pathways. Several adaptors and downstream molecules of the TLR3 pathway have been shown to be negatively modulated by various mechanisms and regulators. Among these, we discuss recent advances in the regulation of TRIF, TRAF3, RIP1 and TRAF6 (Table 1).
Table 1.
Endogenous negative regulators of the TLR3-TRIF pathway
| Target | Negative regulators | Proposed mechanism | Function |
|---|---|---|---|
| TRIF | SARM | Binding to TRIF | K48-linked polyubiquitination and proteasomal degradation of TRIF.28 Down-regulation of the signalling cascade.31 |
| TAG | Inhibition by competition | TRIF-specific inhibitory protein; down-regulation of cytokine and chemokine induction; inhibits AP-1 through MAPK pathway.32 | |
| CD11b/Cbl-b | SyK and Cbl-b (E3 ubiquitin ligase) activation | TLR-triggered, active CD11b integrin engages in cross-talk with the MyD88 and TRIF pathways inhibiting TLR signalling.33 | |
| TRIM38 | Ubiquitination | K48-linked polyubiquitination and proteasomal degradation of TRIF; down-regulation of the signalling cascade.35 | |
| Triad3A | TIR domain polyubiquitination | Triad3A interacts and induces degradation of TIR domains of the TRIF and RIP1 proteins.34 | |
| ADAM15 | Proteolytic cleavage of TRIF | ADAM15 serves to curtail TRIF-dependent TLR3 and TLR4 signalling, protecting the host from generating an excessive pro-inflammatory response.38 | |
| TRAF3 | DUBA | Cleavage of ubiquitin linkages | Increasing amounts of DUBA leads to decreased type I IFN response.30 |
| MIP-T3 | Affects TRAF3 ubiquitination only slightly | MIPT-3 prevents TRAF3 from engaging downstream transducers.39 | |
| RIP1 | A20 | Cleavage of ubiquitin linkages | A20 is a de-ubiquitinating enzyme that is induced by LPS in macrophages and that removes ubiquitin chains from its target.41 |
| RIP3 | Inhibition by competition | Presence of RIP3 negatively regulates the TRIF-RIP1-induced NF-κB pathway.40 | |
| Triad3A | TIR domain polyubiquitination | Triad3A interacts and induces degradation of TIR domains of the TRIF and RIP1 proteins.34 | |
| TRAF6 | CYLD | Deubiquitination; autoregulatory loop | Mediates an inhibitory action on the NF-κB pathway by reversing the ubiquitination of TRAF2, TRAF7 and TRAF643,44 |
| A20 | Cleavage of ubiquitin linkages | A20 is a de-ubiquitinating enzyme induced by LPS in macrophages; removes ubiquitin chains from its target.27 | |
| TANK | Inhibits TRAF6 ubiquitination. | TANK in unstimulated cells inhibits TRAF6 activation.45 | |
| Pellino-3 | Ubiquitination | TRIF signalling induces Pellino3 expression, and inhibits the ability of TRAF6 to activate IRF7.46 | |
| USP4 | Cleavage of ubiquitin linkages | Removal of polyubiquitin chains in a DUB activity-dependent manner.47 | |
| SHP | De-ubiquitination | SHP inhibits TLR signalling by regulating the polyubiquitination of TRAF6.48 | |
| NLRX1 | Dissociates of TRAF6 upon TLR stimulation | NLRX1 functions as a negative regulator that inhibits TLR-induced NF-κB activation.49,101 |
A list of definitions for the abbreviations appearing in this Table can be found at the beginning of the article.
A number of molecules have been demonstrated to affect TRIF under sterile and infectious conditions in various studies using in vitro cell approaches. Sterile α- and armadillo-motif-containing protein (SARM) is a TIR domain-containing adaptor that has been shown to inhibit TRIF, resulting in down-regulation of TRIF-dependent cytokine and chemokine induction28 and AP-1 activation through the MAPK pathway.31 TRIF is also involved in the TLR4, MyD88-dependent signalling pathway, and the adaptor translocating chain-associated factor 3 (TRAM) mediates TRIF recruitment. TAG (TRAM adaptor with GOLD domain), a splice variant of TRAM, is an adaptor that negatively modulates TLR4–TRAM signalling from endosomes by displacing TRIF, so inhibiting the signal transduction.32 Interestingly, integrins may play a role in the regulation of some TLRs. The integrin CD11b activates Syk and promotes degradation of TRIF via the E3 ubiquitin ligase Cbl-b.33 Triad3A is an E3 ubiquitin ligase that modulates TLR3 signalling by degradation of the TIR domains of TRIF and RIP1.34 Tripartite motif (TRIM) proteins and, in particular, TRIM38 have been identified as novel negative regulators of the innate immune response. TRIM38 expression is enhanced after TLR3 stimulation and negatively regulates TLR3-mediated type I interferon signalling by targeting TRIF for proteasomal degradation.35 Importantly, TRIM38 negatively regulates TLR3/4- and RIG-I-mediated IFN-β production and antiviral response by targeting NF-κB-activating kinase-associated protein 1 (NAP1)36 and TRAF6.37 Finally, a novel role for ADAM15 (a disintegrin and metalloprotease) as a negative regulator of TLR3 and TLR4 signalling by a mechanism involving TRIF degradation has recently been identified.38
TRAF3 assembles Lys63 linked polyubiquitin chains and forms a protein complex with TBK1 and IKKε, then binds to TRIF and acts as a mediator of the IRF3 activation, which results in the phosphorylation of IRF3. De-ubiquitinating enzymes (DUBs) are proteases that specifically cleave ubiquitin linkages, negating the action of ubiquitin ligases. De-ubiquitinating enzyme A (DUBA) selectively cleaves the polyubiquitin chains on TRAF3, resulting in its dissociation from the downstream signalling complex and the reduction of the type I IFN response.30 Interestingly, MIP-T3 has been suggested to function in the cilia to facilitate infection with viruses that preferentially infect the apical ciliated side of the respiratory and gastrointestinal epithelia by compromising innate immunity,39 although this hypothesis requires further investigation. The negative role of MIP-T3 in the production of type I IFN by inhibition of TRAF3 complex formation is unique as MIP-T3 appears to affect TRAF3 ubiquitination only slightly but is capable of preventing TRAF3 from engaging downstream transducers.39
The RIP1 kinase is negatively regulated by RIP3, Triad3A and A20. Meylan and colleagues have shown that RIP3 inhibits RIP1 by competition, down-regulating the TRIF–RIP1-induced NF-κB pathway.40 Similarly to DUBA, A20 is a de-ubiquitinating enzyme that has been found to act in the cytoplasm as a negative regulator of TLR responses, affecting RIP1 and TRAF6 and thereby terminating TLR-induced NF-κB signalling.41 In addition, A20 interacts with TRIF and inhibits TLR3- and Sendai virus-induced activation of IFN-sensitive response element and IFN-β promoter, suggesting that A20 is a virus-inducible protein that blocks both inflammatory and cellular antiviral responses.42
A variety of negative regulators affect TRAF6, a signal transducer shared by the TRIF and MyD88 axis. TLRs other than TLR3 depend on the adaptor protein MyD88, and its activation from different sources converges in the recruitment of TRAF6. Therefore, it is not surprising to find several molecules that interact with and tightly regulate TRAF6. In addition to A20,27 TRAF6 is negatively regulated by the DUB tumour suppressor cylindromatosis, which negatively regulates the TNF receptor-induced activation of NF-κB and c-Jun N-terminal kinase by reversing ubiquitination of its target and possibly by a transient interaction with nuclear factor-κB essential modulator.43,44 Activation of NF-κB and AP-1 leads to production of pro-inflammatory signals as well as cylindromatosis. Therefore, it generates an autoregulatory loop that controls the immune response through TRAF6. TANK (also known as I-TRAF) has been identified as a TRAF-binding protein. The presence of TANK in unstimulated cells mediates the inhibition of TRAF6 by blocking its ubiquitination. TANK seems to negatively regulate pro-inflammatory cytokine production, rather than the IFN response induced by TLR signalling.45 TRAF6 is also regulated by Pellino-3, a family of proteins that have been shown to be subject to various forms of post-translational modifications. It was demonstrated that TRIF signalling induces its expression, and through its E3 ubiquitin ligase capability, Pellino-3 inhibits TRAF6 and further activation of IRF7, resulting in down-regulation of type I IFN expression.46 A potent negative regulator of TRAF6 is the ubiquitin-specific protease 4 (USP4), which plays an essential role in modulation of the TLR/IL-1R signalling-mediated innate immune response by cleaving ubiquitin linkages in a DUB activity-dependent manner.47 Similarly, the orphan nuclear receptor small heterodimer partner is a transcriptional co-repressor that regulates hepatic metabolic pathways and has been found to inhibit TLR signalling by regulating the polyubiquitination of TRAF6.48 The nucleotide-binding domain leucine-rich repeat containing (NLR) proteins serve as regulators of inflammatory signalling pathways. The mitochondrial NLRX1 protein interacts with TRAF6 and inhibits NF-κB activation. Upon TLR stimulation, NLRX1 dissociates from TRAF6 to allow its function.49 NLRX1 is an important regulator of the antiviral response against RNA viruses,because it also counteracts the RIG-I-MAVS (mitochondrial antiviral signalling protein) cytoplasmic pathway.49
Virus-induced regulators
In the previous section, we described a number of endogenous negative regulators that target TRIF, TRAF3, RIP1 and TRAF6, thereby controlling the signalling cascades mediated by TLR3 stimulation to protect the host from potentially harmful, excessive immune activation. During co-evolution with the host, viruses have evolved mechanisms to counteract cell responses against infection for their own benefit and diminish the immune response at multiple levels. Here we discuss the various strategies that some viruses have developed to negatively regulate the TLR3 pathway by targeting TRIF, TRIM38, TRAF3, TRAF6 and RIP1 (Table 2).
Table 2.
Viral-induced negative regulators of the TLR3-TRIF axis
| Target | Negative regulators | Virus | Function |
|---|---|---|---|
| TRIF | NS3-4A | HCV | Inhibits both NF-κB and IRF3 activation via its interaction with and cleavage of TRIF, leading to IFN-I inhibition.50 |
| 3CD | HAV | Inhibits both NF-κB and IRF3 activation via its interaction with and cleavage of TRIF, leading to IFN-I inhibition.51 | |
| 3C Protease | CVB3 | B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signalling.52 | |
| 3C Protease | EV71 | 3C cleaves TRIF to attenuate the antiviral response mediated by the TLR3 pathway.53 | |
| RTA | KSHV | Replication and transcription Factor (RTA) mediates degradation of TRIF through the ubiquitin-mediated proteosome pathway.54 | |
| TRIM38 | SeV | Induces the expression of cellular TRIM38 upon infection, down-regulating TRIF and further downstream signalLing cascades.35 | |
| M protein | SARS-CoV | M prevents the formation of the TRAF3-TANK-TBK1/IKKε complex inhibiting the activation of IRF3/IRF7.56 | |
| TRAF6 | A52R | VACV | Inhibits TLR3-induced NF-kB activation by sequestering TRAF6 and IRAK2.57 |
A list of definitions for the abbreviations appearing in this Table can be found at the beginning of the article.
One viral strategy to interfere with the TLR3 signalling pathway is to down-regulate the expression of the adaptor TRIF. Some viral non-structural proteins with serine protease activity have been identified to target TRIF for proteolysis. The NS3/4A of hepatitis C virus (HCV),50 the hepatitis A virus (HAV) protease-polymerase processing intermediate (3CD),51 the 3C(pro) cysteine protease of coxsackievirus B3 (CVB3),52 and the enterovirus 71 (EV71) 3C protease53 cleave TRIF and disrupt the TLR3 signalling. Additionally, the replication and transcription activator that is necessary and sufficient for the switch from latent to lytic infection with Kaposi's sarcoma-associated herpesvirus, mediates the degradation of TRIF through the ubiquitin-mediated proteasome pathway.54
Little is known about how viruses may regulate the activity of the endogenous negative regulator TRIM38. As discussed above, the expression of this E3 ligase is up-regulated after TLR3 stimulation. TRIM38 promotes K48-linked polyubiquitination and proteasomal degradation of NAP1 and TRIF and negatively regulates TLR3-mediated type I IFN signalling.35 The picornavirus enterovirus 71 induces TRIM38 degradation,55 suggesting that reduced expression of TRIM38 may be beneficial for the virus by targeting molecules yet to be discovered. The precise role of TRIM38 suppression in picornavirus infection remains unknown. In contrast, TRIM38 expression was induced upon vesicular stomatitis virus infection, an ssRNA virus recognized by RIG-I. Over-expression of TRIM38 decreased IFN-β production and increased vesicular stomatitis virus replication in the presence or absence of polyriboinosinic polyribocytidylic acid [poly(I:C)], suggesting that virus-induced TRIM38 negatively regulates antiviral immune responses.35
Some viral proteins have the ability to interfere with the antiviral pathways by sequestration of cellular proteins such as TRAF3 and TRAF6 that play key roles in the antiviral response. For instance, the M protein of the highly pathogenic severe acute respiratory syndrome coronavirus prevents the formation of the TRAF3-TANK-TBK1/IKKε complex and thereby inhibits TBK1/IKKε-dependent activation of IRF3/IRF7 transcription factors.56 Vaccinia virus inhibits TLR3-induced NF-κB activation by sequestering TRAF6 and IL-1 receptor-associated kinase-like 2 through interaction with the poxvirus protein A52R.57
Taken together, these findings provide evidence of the complex regulatory networks that viruses exploit to counteract the TLR3 signalling pathway. The question that arises is how crucial is TLR3 in antiviral immunity in vivo. Surprisingly, data from the literature suggest that TLR3 may be a double-edged sword that functions in both defence and offence in host immunity to viruses.
TLR3 in human viral pathogenesis
Because TLR3 recognizes dsRNA, which is produced as a replicative intermediate by RNA and DNA viruses, and some viruses contain dsRNA genomes, the TLR3 pathway has the potential to control immunity to most of the clinically relevant viral infections in humans, caused by flaviviruses, hepatitis B and C viruses, herpesvirus, rotavirus, retroviruses, orthomyxoviruses and poxviruses, among others. However, the role of TLR3 in human immunity is still largely unknown, and our understanding is based mostly on studies using human primary or immortalized cells (Table 3, Fig. 1).
Table 3.
TLR3 in human viral pathogenesis
| Virus | Target organ | Disease | Model tested | Role of TLR3 |
|---|---|---|---|---|
| HSV-1 | CNS | Encephalitis, other | Humans with inborn errors of TLR3 | Children with inborn errors in the TLR3 pathway developed HSE; TLR3 is required for protective immunity against HSE by a mechanism that involves production of type I IFN in neurons and oligodendrocytes.59,60 |
| HCV | Liver | Hepatitis, HCC | Hepatoma cells, in vitro | TLR3 mediates pro-inflammatory response.65 |
| Kidney | Glomerulonephritis | Mesangial cells, in vitro | Up-regulation of TLR3 mRNA expression in HCV-positive glomerulonephritis that correlated with enhanced RANTES and MCP-1. | |
| Immune complexes containing viral RNA may activate mesangial TLR3 during HCV infection, thereby contributing to chemokine/cyokine release, effecting proliferation and apoptosis.66 | ||||
| HBV | Liver | Hepatitis, hepatocellular carcinoma | Humans | TLR3 polymorphisms are associated with acute-on-chronic liver failure, and hepatocellular carcinoma.67,68 |
| Influenza, A/H1N1/2009 | Lung | Pneumonia | Humans | A TLR3 polymorphism correlates with increased risk of pneumonia in children.69 |
| Influenza A | CNS, brain | Encephalopathy | Humans | A missense mutation (F3035) in the TLR3 gene correlated with encephalopathy in IAV-infected patients.63 |
| TBEV | CNS | Encephalitis | Humans | The wild-type rs3775291 TLR3 allele was more common among TBE patients than among healthy controls, suggesting that TLR3 may be a risk factor for TBEV infection.64 |
| Rotavirus | Intestine | Gastrointestinal | Humans | Up-regulation of epithelial TLR3 expression during infancy might contribute to the age-dependent susceptibility to rotavirus infection.39 |
| HIV-1 | CD4+ T cells, macrophages, others | Immunodeficiency, encephalitis, other | In vitro and in vivo | Activation of the TLR3 pathway enhances the induction of HIV-specific CD8+ cytotoxic T lymphocytes.72 |
| Activation of the TLR3 pathway with poly(I:C) induces an antiviral state that limits HIV-1 infection in primary human macrophages70 and human fetal astrocytes.71 | ||||
| Potential detrimental contribution of TLR3 in HIV-1-induced myopathies.74 |
A list of definitions for the abbreviations appearing in this Table can be found at the beginning of the article.
Figure 1.
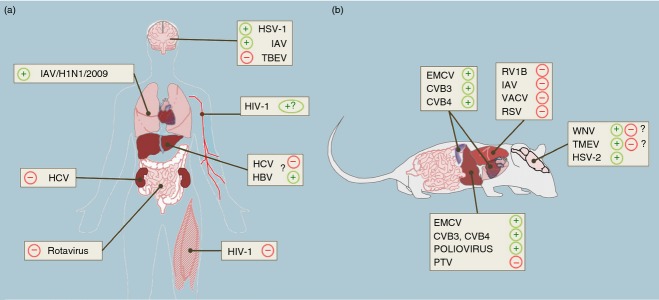
Role of Toll-like receptor 3 (TLR3) in the pathogenesis of viral infections in humans and mice shown by organ type. TLR3 may exhibit protective (+) or detrimental roles (−) in humans (a) and mice (b) depending on virus type. (a) A protective role of TLR3 in the central nervous system (CNS) has been suggested for herpes simplex virus type 1 (HSV-1) and influenza A virus (IAV) -induced encephalitis in humans. In contrast, TLR3 sensing of tick-borne encephalitis virus (TBEV) may contribute to neuropathogenesis in some infected individuals. Although the precise role of TLR3 in influenza infection in the lung remains unknown, children with a TLR3 polymorphism had an increased risk of pneumonia induced by the pandemic IAV/H1N1/2009 strain. Rotavirus is the leading cause of severe diarrhoea in infants and young children worldwide. Up-regulation of TLR3 expression during infancy might contribute to age-dependent susceptibility to rotavirus infection. HIV-1 induces AIDS and it can also cause several neurological disorders. Despite various studies demonstrating an antiviral role of the TLR3 pathway in cell culture, there is little evidence that TLR3 could play a major role in host defence in HIV-1-infected individuals. TLR3 expression is up-regulated in proximity to infiltrating mononuclear cells in biopsy specimens from patients with HIV-1 myopathies. The role of TLR3 in liver diseases induced by human hepatitis C (HCV) and B (HBV) viruses remains to be elucidated. Chronic over-stimulation of the TLR3 pathway may contribute to an unbalanced intrahepatic inflammatory response that is observed in chronic viral hepatitis. Some TLR3 polymorphisms have been associated with hepatocellular carcinoma in patients infected with HBV. (b) The role of TLR3 in the pathogenesis of various viral infections has been studied in TLR3-deficient mice. TLR3 plays a detrimental role in the pathogenesis of rhinovirus type 1B (RV1B), vaccinia virus (VACV), respiratory syncytial virus (RSV) and IAV in the lung. In contrast, TLR3 plays a protective role against the infection with herpes simplex virus type 2 (HSV-2) in the CNS. However, the contribution of TLR3 to West Nile virus (WNV) encephalitis remains controversial. In the liver, TLR3-deficient mice exhibit increased resistance to Punta Toro virus fatal infection, suggesting a detrimental role in phlebovirus pathogenesis. However, TLR3 mediates protection against poliovirus, coxsackievirus B (CVB3 and CVB4), and encephalomyocarditis virus (EMCV) infections in the liver, heart and pancreas of infected mice.
Viral encephalitis
A remarkable exception is the knowledge gained from human patients with inborn errors in the TLR3 pathway who do not display susceptibility to viral infections other than herpes encephalitis.58 Herpes simplex virus (HSV-1) is a highly prevalent neurotropic virus that infects the central nervous system (CNS) and can generate herpes simplex encephalitis in children with inborn errors of TLR3 immunity. It has been shown that impaired TLR3- and UNC-93B-dependent IFN-α/β intrinsic immunity to HSV-1 in the CNS, in neurons and oligodendrocytes in particular, may underlie the pathogenesis of herpes simplex encephalitis in children with TLR3-pathway deficiencies.59,60 Moreover, Li and colleagues have demonstrated that among IFNs, endogenous IFN-λ also participates in TLR3-mediated antiviral activity in primary human astrocytes.61 To date, TLR3 is the only TLR that has been shown to play a non-redundant role for protection against HSV-1 infection in the CNS, although TLR3 may have a redundant role against other viruses in humans.62 Taken together, these findings suggest that the role of human TLR3 in protection against herpes simplex encephalitis is unique.
Moreover, Hidaka and colleagues performed a genetic analysis including TLR genes that could be involved in the recognition of influenza A virus (IAV) on patients with influenza-associated encephalopathy and found a missense mutation (F3035) in the TLR3 gene. This finding correlated the deficiency of TLR3 with the encephalopathy induced by IAV.63 In contrast, a genetic evaluation of TLR3 identified the wild-type TLR3 rs3775291 allele to be a risk factor in tick-borne encephalitis virus infection, a pathogen that can be asymptomatic or can cause severe symptoms in the CNS.64 This study suggests that a functional TLR3 is a risk factor for tick-borne encephalitis virus infection.
Viral hepatitis
In contrast, emerging evidence suggests that TLR3 may exhibit protective as well as detrimental functions in the context of other human viral infections. Hepatitis C virus is an ssRNA virus that induces chronic hepatitis and may lead to hepatocellular carcinoma. TLR3 senses dsRNA of HCV upon infection in cultured hepatoma cells as well as in primary human hepatocytes, leading to NF-κB activation and to secretion of pro-inflammatory cytokines and chemokines. These data suggest that TLR3 may contribute to the intrahepatic and unbalanced pro-inflammatory response that characterizes the pathogenesis of HCV-associated liver diseases.65 Interestingly, a link has been reported between the TLR3 pathway and induction of glomerulonephritis in patients with chronic HCV infection.66 This disease is characterized by inflammation of the blood vessels in the kidneys. In HCV-associated glomerulonephritis, TLR3 mRNA and protein expression are up-regulated in mesangial cells, and upon stimulation with TLR3 ligand, an increment of selected cytokines and chemokines that are characteristic of the disease is observed.66 In addition, some TLR3 polymorphisms have been associated with chronic hepatitis B and hepatitis B-related acute-on-chronic liver failure67 and hepatocellular carcinoma.68
Influenza and rotavirus infections
Furthermore, there is a close relationship between the presence of TLR3 rs5743313/CT polymorphism and an increased risk of pneumonia in children infected by the pandemic A/H1N1/2009 influenza virus.69 Although animal models are being evaluated (discussed below), the precise role of TLR3 against influenza in humans remains to be determined. In the context of rotavirus infection, up-regulation of epithelial TLR3 expression during infancy might contribute to the age-dependent susceptibility to rotavirus infection.39
Human immunodeficiency virus
The contribution of TLR3 in HIV-1 infection is poorly understood. Activation of the TLR3 pathway with poly(I:C) induces an antiviral state that limits HIV-1 infection in primary human macrophages70 and human fetal astrocytes71 by different mechanisms. In addition, when BALB/c mice were immunized with purified recombinant HIV-1 envelope gp120 or influenza haemagglutinin protein together with poly(I:C), epitope-specific CD8+MHC class I molecule-restricted cytotoxic T lymphocytes were primed from naive CD8+ T cells in vivo, suggesting that activation of the TLR3 pathway enhances class I processing of exogenous protein and induction of HIV-specific CD8+ cytotoxic T lymphocytes.72 Interestingly, a common TLR3 allele confers immunologically mediated protection from HIV-1.73 Taken together, these findings suggest that triggering of TLR3 with specific ligands could have therapeutic potential against HIV-1 infection in humans. However, extreme caution is advised because TLR3 may also make a detrimental contribution of TLR3 in HIV-1-induced pathogenesis. For example, TLR3 is elevated and it is expressed in proximity to infiltrating mononuclear cells in biopsy specimens from patients with HIV myopathies.74
Immunity to viral infection in TLR3-deficient mice
Viral infections in mice with TLR3 deficiency are useful models to better understand the complexity of TLR3-mediated immune responses in vivo. The role of TLR3 has been tested in at least 17 different viral infections in mice (Table 4, Fig. 1). Most of the viruses studied so far have neurotropic, respiratory, liver or cardiac tropisms.
Table 4.
Viral pathogenesis in TLR3-deficient mice
| Virus | Target organ | Disease | Route of inoculation; model tested | Phenotype in TLR3−/− mice |
|---|---|---|---|---|
| WNV | CNS (neurons) | Encephalitis | Subcutaneous, intraperitoneal C57BL/6J | Detrimental role: higher viral burden in the periphery but lower load in the brain; diminished inflammatory response and neuropathology.77 |
| Protective role: absence of TLR3 enhances WNV mortality in mice and increases viral burden in the brain but modest changes in peripheral viral loads; TLR3 serves a protective role against WNV by restricting replication in neurons.78 | ||||
| HSV-1 | CNS (neurons, astrocytes, oligodendrocytes) | Encephalitis | Skin flank scarification, C57BL/6J | Protective role: higher viral load at infection site; lower number of HSV-1 specific CD8+ T cells.83 |
| HSV-2 | Female genital tract; CNS (astrocytes) | Genital herpes; meningitis, myelitis. encephalitis | Intravaginal, subcutaneous C57BL/6J | Protective role: higher viral load in cerebellum and medulla spinalis; Aggravation of CNS disease score.82 |
| T3 reovirus | CNS | Encephalitis | i.cb. or i.c. C57BL/6 × B129 | No differences in CNS injury; survival and viral load were not determined.84 |
| PV | CNS (brain, spinal cord) | Poliomyelitis | Transgenic mice expressing human PVR C57BL/6-PVRTg21 | Protective role: lower survival rate and increased viral burden in the liver, spleen, and kidney; serum IFN-α levels were blunted.79 |
| TEMV strains BeAn and GDVII | CNS | Encephalitis, Demyelination | Hemisphere; SLJ and C57BL/6J | TLR3-deficient susceptible SJL mice accelerated the development of demyelinating disease; TLR3-deficient resistant B6 mice remained disease free; Protective role: higher viral load in brain and spinal cord; severe demyelination.80 |
| Detrimental role: activation of TLR3 with poly IC prior to viral infection exacerbated disease development, whereas such activation after viral infection restrained disease development.80 | ||||
| IAV | Lung (airways) | Pneumonia | intranasal, C57BL/6 | Detrimental role: increased survival rate and viral burden in the lung; lower pro-inflammatory response in bronchoalveolar air space.86 |
| RSV | Lung | Bronchiolitis, pneumonia, asthma exacerbations | Intratracheal C57BL/6J, BALB/c | RSV sensitizes the airway epithelium to subsequent viral and bacterial exposures by up-regulating TLRs and increasing their membrane localization.87 |
| TLR3 contributes to formation of lung oedema through nucleotide/P2Y Purinergic receptor-mediated impairment of alveolar fluid clearance.88 | ||||
| RV1B | Lung (airways) | Chronic inflammatory disease of the airways (asthma) | intranasal C57BL/6J | Detrimental role: reduced lung inflammatory responses and airways responsiveness; TLR3 initiates pro-inflammatory signalling pathways leading to airways inflammation and hyperresponsiveness.85 |
| Vaccinia | Lung, multiorgan | Respiratory, other | intranasal C57BL/6 | Detrimental role: increased survival rate and lower viral load in the respiratory tract; less pro-inflammatory response in serum, lung, and bronchoalveolar lavage fluid.89 |
| MCMV | Multiorgan | Systemic | intraperitoneal C57BL/6J, C57BL/6J × B129 | Protective role: lower survival rate; increased viral load in spleen; and decreased IFN production in serum. No difference in CD8 T or CD4 T cells.84 |
| LCMV | CNS, multiorgan | Encephalo-myelitis, other | intraperitoneal, foot pad C57BL/6J × B129 | No difference in CD4 and CD8 T cells; Survival rate and viral load, not tested.84 |
| VSV | CNS, multiorgan | Encephalo-myelitis, other | intravenous C57BL/6J × B129 | No difference in CD4 and CD8 T cells; Survival rate and viral load, not tested.84 |
| CVB3 | Heart | Chronic myocarditis | intraperitoneal C57BL/6J | Protective role: increased viral burden in the heart and serum, but lower IFN-γ and pro-inflammatory responses.92 |
| CVB4 | Heart, liver | Chronic myocarditis, hepatitis | intraperitoneal C57BL/6 × B129 NOD | Protective role: lower survival rate; increased viral burden and inflammation in the heart.93 |
| Protective role: lower survival rate; increased viral burden and inflammation in the heart; phenotype rescued by WT NOD macrophages.94 | ||||
| EMCV, myocarditic variant | Heart | Myocarditis | intraperitoneal C57BL/6J | Protective role: lower survival rate; increased viral burden in the heart and liver; inhibited inflammatory response in heart.90 |
| PTV | Liver, multiorgan | Hepatitis, other | subcutaneous C57BL/6J | Detrimental role: increased survival rate, lower viral load in serum, and less pro-inflammatory response in liver and serum.96 |
icb, intracerebal; ic, intracranial.
A list of definitions for the abbreviations appearing in this Table can be found at the beginning of the article.
Viral infections of the CNS
The role of TLR3 has been studied in TLR3-deficient mice with various genetic backgrounds with the following neurotropic viruses: West Nile virus (WNV) (Flaviviridae, positive-sense ssRNA), poliovirus and Theiler's murine encephalomyelitis virus (TMEV) (Picornaviridae, positive-sense ssRNA), HSV-1 and HSV-2 (Herpesviridae, dsDNA) and T3 reovirus (Reoviridae, segmented dsRNA). The CNS is separated from the rest of the body by the blood–brain barrier; the cerebrospinal fluid is also separated from the periphery by the blood–cerebrospinal fluid barrier of the choroid-plexus epithelium.75 This structure makes the brain an immune-privileged tissue where, during viral infection, the tight balance between a controlled antiviral response and excessive immune activation determines the pathological outcome.75 West Nile virus infection induces encephalitis in humans and mice.76 The role of TLR3 in WNV encephalitis remains controversial, and both protective and pathogenic roles have been reported in the mouse model. Perhaps these conflicting data reflect the use of different routes of virus inoculation and strains. For instance, TLR3 deficiency protected mice that were infected intraperitoneally from WNV lethal infection, reduced blood–brain barrier permeability, and decreased systemic TNF-α and IL-6 production suggesting a detrimental role of TLR3.77 In contrast, increased mortality was observed in TLR3-deficient mice in association with elevated virus burden in neurons in the brain, suggesting that TLR3 has a protective role against WNV in the CNS.78 In addition, contrasting results on the role of pattern recognition receptors in vitro and in vivo have been reported for some viruses. For example, type I IFN production in response to poliovirus infection is mediated by MDA5 in primary cultured kidney cells in vitro.79 However, the same group found that serum IFN-α levels, viral load in non-neural tissues, and mortality rates did not differ significantly between MDA5-deficient mice and wild-type mice. In contrast, serum IFN production was abrogated and the viral load in non-neural tissues and mortality rates were both markedly higher in TRIF-deficient and TLR3-deficient mice than in wild-type mice. These results suggest that multiple pathways are involved in the antiviral response in mice and that the TLR3–TRIF-mediated signalling pathway plays an essential role in the antiviral response against poliovirus infection in the mouse model.79
The role of TLR3-mediated signalling in the protection and pathogenesis of TMEV-induced demyelinating disease is intriguing and warrants further investigation. In this case, activation of TLR3 signal transduction before TMEV infection leads to pathogenesis via over-activation of the pathogenic immune response. In contrast, TLR3-mediated signalling during viral infection protects against TMEV demyelinating disease by reducing the viral load and modulating immune responses.80
Herpes simplex virus 1 is a major cause of encephalitis, whereas HSV-2 can give rise to meningitis as well as encephalitis, sacral radiculitis and myelitis in humans.81 The role of TLR3 in herpes simplex encephalitis has been recently studied in the mouse model using the HSV-2 strain.82 Interestingly, TLR3-deficient mice were hyper-susceptible to HSV-2 infection in the CNS after vaginal inoculation. Although TLR3-deficient mice did not exhibit a global defect in innate immune responses to HSV, astrocytes were defective in HSV-induced type I IFN production. Overall, these data suggest that TLR3 acts in astrocytes to sense HSV-2 infection immediately after entry into the CNS, possibly preventing HSV from spreading beyond the neurons mediating entry into the CNS.82 Although HSV-1 strain has been studied in TLR3-deficient mice,83 the role of TLR3 in HSV-1 infection of the CNS has not been fully investigated. However, the study above has revealed a key function of TLR3 in CD8 T-cell priming to HSV-1.83 Finally, the role of TLR3 in reovirus-induced neuropathogenesis has been addressed using the T3 reovirus strain Dearing (T3D).84 No differences were observed in CNS injury, viral loads, and mortality in TLR3-deficient compared with wild-type mice, suggesting that at least for the T3D strain, TLR3 does not play a significant biological role in reovirus neuropathogenesis.84
Viral respiratory infections
The contribution of TLR3 in respiratory viral infections has been tested for rhinovirus, influenza A virus, vaccinia, and respiratory syncitial virus in the mouse. Rhinovirus is the most frequent cause of asthma exacerbations and can be studied in vivo by using the rhinovirus type 1 RV1B strain, a minor group virus that replicates in mouse lungs. By using TLR3-deficient mice, Wang and colleagues showed that lack of TLR3 was beneficial for the infected mice. TLR3-deficient mice showed reduced lung inflammatory responses and reduced airway responsiveness, suggesting that in the context of rhinovirus infection, binding of viral dsRNA to TLR3 initiates pro-inflammatory signalling pathways leading to airway inflammation and hyper-responsiveness.85 Similarly, in the absence of TLR3, IAV-infected mice had increased survival rates even though the viral burden in lungs was elevated, and it correlated with significantly reduced pro-inflammatory mediators as well as a lower number of CD8+ T lymphocytes in the bronchoalveolar airspace, suggesting a detrimental pro-inflammatory role of TLR3 in the lungs of IAV-infected mice.86
Furthermore, respiratory syncytial virus preferentially infects airway epithelial cells, causing upper respiratory infections, asthma exacerbations and pneumonia. Respiratory syncytial virus induces TLR3 up-regulation, leading to a priming of lung epithelial cells for subsequent exposure to extracellular dsRNA. Upon TLR3 stimulation, activation of NF-κB and increased levels of IL-8 amplify epithelial cell responsiveness, resulting in an altered inflammatory response.87 Additionally, Aeffner and colleagues reported that dsRNA induces similar pulmonary dysfunctions to respiratory syncytial virus infection in mice. Respiratory syncytial virus induces nucleotide/P2Y purinergic receptor-mediated impairment of alveolar fluid clearance, which results in the formation of lung oedema.88
The role of TLR3 in the pathogenesis of vaccinia virus, a prototype poxvirus, has been investigated using a recombinant strain Western Reserve vaccinia virus that expresses firefly luciferase in the mouse. TLR3-deficient mice had substantially lower viral replication in the respiratory tract and diminished dissemination of virus to abdominal organs. Mice lacking TLR3 had reduced disease morbidity and lung inflammation that correlated with reduced recruitment of leucocytes to the lung. Interestingly, mice lacking TLR3 also had lower levels of inflammatory cytokines, including IL-6, monocyte chemoattractant protein-1 and TNF-α in serum and/or bronchoalveolar lavage fluid, but levels of IFN-β did not differ between genotypes of mice.89 Taken together, these results show that, in the respiratory tract, TLR3 might have a detrimental role and its stimulation leads to potential over-activation of the pro-inflammatory response in the airways that is detrimental to the host.
Viral infections of the heart and the pancreas
Mice that are TLR3-deficient have been shown to be highly susceptible to encephalomyocarditis virus (EMCV)90,91 and coxsackieviruses B3 and B492–94 infections, resulting in significant mortality. These viruses induce cardiac injury or diabetes depending on the virus strain, and are considered a model of virus-induced autoimmune inflammation in the heart and pancreas. Lack of TLR3 leads to an impaired expression of pro-inflammatory response in the heart and the liver of mice infected with EMCV, and in the pancreas of mice infected with the EMCV variant of pancreas β-cell tropism, facilitating the increment of viral burden in these tissues.108,109 Greater cardiac damage and increased viral load in heart, serum and liver are also observed in TLR3-deficient mice compared with immunocompetent mice infected with coxsackieviruses B3 and B4.92–94 Remarkably, TLR3 and its adaptor TRIF may have differential roles in viral myocarditis resulting in different T helper type 2 (Th2) responses that uniquely influence the progression to chronic myocarditis. In particular, TLR3-deficient mice developed an IL-4-dominant Th2 response during acute coxsackievirus B3 myocarditis with elevated markers of alternative activation, whereas TRIF-deficient mice had elevated the Th2-associated cytokine IL-33. Although TLR3- or TRIF-deficient mice developed similarly worse acute coxsackievirus B3 myocarditis and viral replication compared with control mice, disease was significantly worse in TRIF- than in TLR3-deficient mice. This is the first report in the literature to demonstrate a differential role of TRIF and TLR3 in a mouse model of viral pathogenesis. These findings, although intriguing, suggest that TLR3 may also signal independently of TRIF under at least some circumstances. Indeed, emerging evidence demonstrates the existence of a new branch of TLR3 signalling that does not lead to gene induction but affects many cellular properties, such as cell migration, adhesion and proliferation.95 Surprisingly, for this effect of TLR3 signalling, the adaptor proteins TRIF and MyD88 were not required. The effects of the new pathway were mediated by the proto-oncoprotein c-Src, which bound to TLR3 after dsRNA stimulation of cells.95 This is the first example in the literature of TLR-mediated cellular effects of an adaptor-independent effect of any TLR.
Viral infections of the liver
Finally, the role of TLR3 in viral hepatitis using TLR3-deficient mice remains poorly understood. A caveat in the literature is that the viruses studied so far (EMCV, coxsackievirus B4, poliovirus and the phlevovirus Punta Toro virus) are not strictly hepatotropic and induce multi-organ pathogenesis. Nevertheless, mice lacking TLR3 expression developed higher viral load in the liver and increased mortality compared with their controls after intraperitoneal infections with EMCV90 and coxsackievirus B494 and intravenous infection with poliovirus.79 In contrast, TLR3-deficient mice demonstrate increased resistance to lethal infection and have reduced liver disease associated with hepatotropic Punta Toro virus infection.96 Although infectious challenge produced comparable liver and serum viral loads, mice lacking TLR3 were able to clear systemic virus at a slightly faster rate, and exhibited fewer inflammatory mediators. In particular, TLR3-deficient mice had lower levels of IL-6, monocyte chemoattractant protein-1, IFN-γ, and RANTES than wild-type animals. Therefore, the TLR3-mediated response to Punta Toro virus infection is detrimental to disease outcome. Although IL-6 is critical to establish antiviral defence in this model, it also contributes to pathogenesis when released in excess through a TLR3-mediated mechanism.96 In addition, non-specific activation of innate immunity can drastically enhance susceptibility to immune destruction of the liver through activation of TLR3.97 Immune destruction of the liver requires first the priming of liver-specific T cells. These activated T cells then have to migrate into the target organ, where autoimmunity finally occurs. Usually, the liver does not attract liver-specific T cells because chemokines are expressed at low levels in the liver. However, pro-inflammatory signals derived from ligation of TLR3 can lead to homing of CD8+ T cells to the liver with subsequent enhancement of liver disease by a mechanism mediated by IFN-α- and TNF-α-dependent up-regulation of genes and their products, such as the chemokine CXCL9, involved in T-cell homing and migration.97 This pathogenic mechanism may explain observations suggesting that autoimmunity, including hepatitis, can be promoted by viral infections.
TLR3: lessons learned from mice
Overall, data from the mouse model suggest that the protective versus pathogenic contribution of TLR3 in viral infections is multifactorial. However, some conclusions can be drawn. First, there are differences among type of viruses, but the role of TLR3 in viral pathogenesis does not exhibit a differential trend among ssRNA, dsRNA and DNA viruses. Second, TLR3 may exhibit differential roles that are tissue specific. In this regard, the detrimental contribution of the TLR3 pathway in autoimmune hepatitis through activation of certain cytokines and chemokines involved in T-cell migration to the liver is remarkable. However, TLR3 is protective in three and detrimental in only one of the four hepatotropic viruses tested to date in the mouse (the phlebovirus Punta Toro virus). Perhaps among these viruses, the pathogenesis of Punta Toro virus represents a better example of virus-induced hepatitis in the absence of other major organ involvement. In the CNS, TLR3 seems to be protective rather than detrimental against infection with certain viruses, and at least in the case of the flavivirus WNV, controversial results remain to be further evaluated. In contrast, studies suggest that TLR3 may have a pathogenic role in the lung at least in the context of the four respiratory viruses that have been evaluated. Third, experimental factors (e.g. route of virus inoculation and mouse strain) may affect the interpretation of the results obtained in certain studies.
Concluding remarks and perspectives
Although the role of TLR3 in protection against viral infections may vary among viruses, a detrimental contribution of TLR3 in viral pathogenesis is emerging from studies in the mouse and in certain viral infections in humans (Fig. 1). TLR3 participates in both defence and offence in host immunity to viruses. A working model of the role of TLR3 in antiviral (protective) and immunoregulatory (detrimental) responses based on data from the literature is shown in Fig. 2. We have much to learn about the mechanisms that drive TLR3-mediated disease in the context of viral infections and, importantly, in autoimmunity and in cancers that occur after certain infections.
Figure 2.
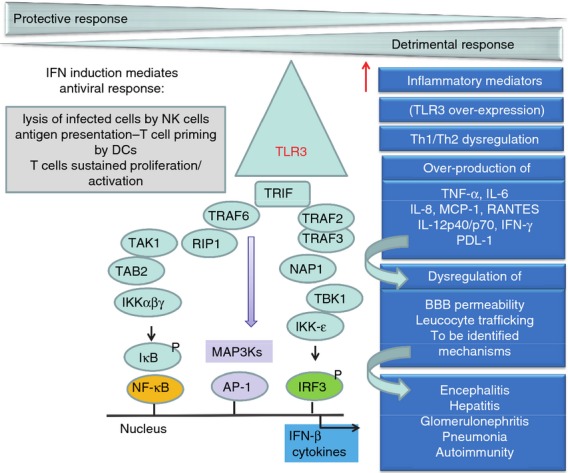
Toll-like receptor 3 (TLR3) participates in both defence and offence in host immunity to viruses. TLR3 recognizes dsRNA, a common intermediate of replication among many viruses. TLR3 dimerization and Tyr phosphorylation trigger the recruitment of the adaptor protein Toll–interleukin-1 receptor domain-containing adaptor protein interferon-β (TRIF), and induce the activation of the transcription factors interferon (IFN) regulatory transcription factor 3 (IRF3), nuclear factor-κB (NF-κB) and activator protein-1 (AP-1) through two branches. TLR3 activation leads to (i) the development of an antiviral response mediated by IRF3 activation and further type I IFN production; (ii) cell death through a Fas-associated protein with death domain /caspase-8-dependent and mitochondrion-independent pathway (RIP1/ Fas-associated protein with death domain); and (iii) the generation of a pro-inflammatory environment by the activation of NF-κB and AP-1, and the mitogen-activated protein kinases (MAPK) the extracellular signal-regulated protein kinase 1/2 (ERK), the p38 MAP kinases (p38), and the c-Jun N-terminal kinase (JNK), which differentially regulate many cellular functions including inflammation-mediated inflammatory processes. Despite the detrimental role of TLR3 in viral pathogenesis being poorly understood, three major mechanisms have been identified: (i) over-expression of TLR3 (only observed in some viral infections but not others); (ii) over-production of cytokines [tumour necrosis factor-α (TNF-α), interleukin-6 (IL-6), IL-12p40/p70, and IFN-γ], chemokines [regulated on activation, normal T-celll expressed and secreted (RANTES), IL-8, and monocyte chemoattractant protein-1 (MCP-1)], and the immune suppressive molecule programmed death ligand-1 (PDL-1); (iii) dysregulation of T helper type 1 and 2 (Th1/Th2) polarization. However, the precise molecular mechanisms that lead to TLR3 hyper-responsiveness are unknown. Over-production of inflammatory mediators leads to dysregulation of leucocyte trafficking, blood–brain barrier (BBB) permeability, and mechanisms to be identified. A detrimental role of TLR3 has been identified in mice infected with West Nile virus (WNV) and Theiler's murine encephalomyelitis (TMEV) (encephalitis), Punta Toro virus (PTV) (hepatitis), and in respiratory infections caused by respiratory syncytial virus (RSV), vaccinia virus, influenza A virus and rhinovirus RV1B (pneumonia). In humans, TLR3 may contribute to glomerulonephritis in patients with chronic hepatitis C virus infection. Finally, TLR3 hyper-responsiveness may play a detrimental role in virus-triggered autoimmunity.
The role of TLR3 in the fine-tune regulation of T-cell polarization may have a major pathogenic impact in the context of some viral infections. Virally infected cells generate signals that act on DCs to favour cross-priming. Upon dsRNA recognition, TLR3 induces type-I IFN secretion known to enhance cross-presentation by DCs and to mediate an antiviral immunity against a number of viral infections.98 This event ultimately results in Th1 responses and cytotoxic T lymphocyte activation.98 The beneficial or detrimental role of TLR3 balancing the response towards Th1 immunity upon tissue-specific viral infections would greatly diverge depending on the ability of the cellular environment to modulate and balance it by generating a Th2 counterpart response. Moreover, a single viral pathogen conventionally defined as Th1-inducing bears a variety of ligands capable of engaging multiple TLRs that can also stimulate a Th2 response, although to a different degree. Therefore, a better understanding of the Th1/Th2 response modulation through TLR3 stimulation in virus-driven tissue-specific pathogenesis is essential. In the context of chronic RNA viral infections that result in sustained IFN-α/β signalling, without viral control, the TLR3–TRIF axis may play important roles in determining how the balance between antiviral and immunoregulatory pathways affect protective versus detrimental responses.
It will be interesting to determine whether TLR3 has a non-redundant, protective role against viral infections other than in herpes simplex encephalitis in humans. A current challenge is to fully understand this remarkable and perhaps unique role. Another intriguing challenge is to identify TLR3-dependent, TRIF-independent pathways, and their contributions not only during infections but also in autoimmunity and cancer. Our understanding of the potential differential roles of TLR3 in certain organs is in its infancy, and at least in liver97 and kidney,99 activation of TLR3 may induce autoimmune hepatitis and glomerulonephritis, respectively. Triggering of TLR3 with specific ligands such as poly(I:C) is being evaluated to enhance the antiviral response.5,70,100 Based on the emerging detrimental role of TLR3 in certain diseases, the question remains how therapeutic activation of TLR3 for antiviral purposes may aggravate pathogenesis in some individuals. We can probably expect the development of specific therapeutic approaches to diminish TLR3-driven disease in the future.
Acknowledgments
RPL and SNM wrote the manuscript. Figs 1 and 2 were created by RPL and SNM respectively. We apologize to any investigators whose work we have omitted due to space limitations. We acknowledge funding to the authors' Laboratory by Public Health Service Grants to SNM (NS061179, AI088423 and DK089314 from the National Institute of Neurological Disorders and Stroke, the National Institute of Allergy and Infectious Disease, and the National Institute of Diabetes and Digestive and Kidney Diseases, respectively). The founders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. Diana Winters (Drexel University College of Medicine Academic Publishing Services) is acknowledged for manuscript editing.
Glossary
- AP-1
activator protein 1
- BTK
Bruton's tyrosine kinase
- CNS
central nervous system
- CVB
coxsackievirus B
- CYLD
cylindromatosis
- DC
dendritic cell
- dsRNA
double-stranded RNA
- DUB
de-ubiquitinating enzyme
- ECMV
encephalomyocarditis virus
- EGFR
epidermal growth factor receptor
- EV71
enterovirus 71
- FADD
Fas-associated protein with death domain
- HBV
hepatitis B virus
- HCV
hepatitis C virus
- HSE
herpes simplex encephalitis
- IAV
influenza A virus
- IFN
interferon
- IKKε
IκB kinase ε
- IRAK2
interleukin-1 receptor-associated kinase-like 2
- IRF3
interferon regulatory transcription factor 3
- ISRE
interferon-sensitive response element
- JNK
c-Jun N-terminal kinase
- LCMV
lymphocytic choriomeningitis virus
- MAPK
mitogen-activated protein kinase
- MAVS
mitochondrial antiviral signalling protein
- MCMV
mouse cytomegalovirus
- MCP-1
monocyte chemoattractant protein-1
- NAP1
NAK-associated protein 1
- NEMO
nuclear factor-κB essential modulator
- NF-κB
nuclear factor-κB
- NLR
nucleotide-binding domain leucine-rich repeat containing
- PI3K
phosphoinositide 3-kinase
- poly(I:C)
polyriboinosinic polyribocytidylic acid
- PTV
Punta Toro virus
- PV
poliovirus
- RANTES
regulated on activation, normal T-cell expressed and secreted
- RIP1
receptor-interacting protein 1
- RSV
respiratory syncytial virus
- RTA
replication and transcription activator
- RV1B
rhinovirus type 1B
- SARM
sterile α- and armadillo-motif-containing
- SHP
small heterodimer partner
- SOCS1
suppressor of cytokine signalling 1
- ssRNA
single-stranded RNA
- STAT1
signal transducer and activator of transcription 1
- TAG
TRAM adaptor with GOLD domain
- TAK1
transforming growth factor-β-activated kinase 1
- TBEV
tick-borne encephalitis virus
- TBK1
TANK-binding kinase 1
- TIR
Toll–interleukin-1 receptor
- TLR
Toll-like receptor 3
- TMEV
Theiler's murine encephalomyelitis
- TNF
tumour necrosis factor
- TRAF3
tumour necrosis factor receptor-associated factor 3
- TRAM
translocating chain-associating membrane
- TRIF
TIR domain-containing adaptor protein interferon-β
- TRIM
tripartite motif
- USP4
ubiquitin-specific protease 4
- VSV
vesicular stomatitis virus
- WNV
West Nile virus
Conflict of interest
None.
References
- 1.Thompson MR, Kaminski JJ, Kurt-Jones EA, Fitzgerald KA. Pattern recognition receptors and the innate immune response to viral infection. Viruses. 2011;3:920–40. doi: 10.3390/v3060920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kariko K, Ni H, Capodici J, Lamphier M, Weissman D. mRNA is an endogenous ligand for Toll-like receptor 3. J Biol Chem. 2004;279:12542–50. doi: 10.1074/jbc.M310175200. [DOI] [PubMed] [Google Scholar]
- 3.Cavassani KA, Ishii M, Wen H, Schaller MA, Lincoln PM, Lukacs NW, et al. TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events. J Exp Med. 2008;205:2609–21. doi: 10.1084/jem.20081370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mikami T, Miyashita H, Takatsuka S, Kuroki Y, Matsushima N. Molecular evolution of vertebrate Toll-like receptors: evolutionary rate difference between their leucine-rich repeats and their TIR domains. Gene. 2012;503:235–43. doi: 10.1016/j.gene.2012.04.007. [DOI] [PubMed] [Google Scholar]
- 5.Matsumoto M, Oshiumi H, Seya T. Antiviral responses induced by the TLR3 pathway. Rev Med Virol. 2011;21:67–77. doi: 10.1002/rmv.680. [DOI] [PubMed] [Google Scholar]
- 6.Jongbloed SL, Kassianos AJ, McDonald KJ, et al. Human CD141+ (BDCA-3)+ dendritic cells (DCs) represent a unique myeloid DC subset that cross-presents necrotic cell antigens. J Exp Med. 2010;207:1247–60. doi: 10.1084/jem.20092140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang Y, Liu L, Davies DR, Segal DM. Dimerization of Toll-like receptor 3 (TLR3) is required for ligand binding. J Biol Chem. 2010;285:36836–41. doi: 10.1074/jbc.M110.167973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 2008;452:234–8. doi: 10.1038/nature06726. [DOI] [PubMed] [Google Scholar]
- 9.Liu L, Botos I, Wang Y, Leonard JN, Shiloach J, Segal DM, Davies DR. Structural basis of toll-like receptor 3 signaling with double-stranded RNA. Science. 2008;320:379–81. doi: 10.1126/science.1155406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ewald SE, Engel A, Lee J, Wang M, Bogyo M, Barton GM. Nucleic acid recognition by Toll-like receptors is coupled to stepwise processing by cathepsins and asparagine endopeptidase. J Exp Med. 2011;208:643–51. doi: 10.1084/jem.20100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garcia-Cattaneo A, Gobert FX, Muller M, Toscano F, Flores M, Lescure A, Del Nery E, Benaroch P. Cleavage of Toll-like receptor 3 by cathepsins B and H is essential for signaling. Proc Natl Acad Sci USA. 2012;109:9053–8. doi: 10.1073/pnas.1115091109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sarkar SN, Elco CP, Peters KL, Chattopadhyay S, Sen GC. Two tyrosine residues of Toll-like receptor 3 trigger different steps of NF-κB activation. J Biol Chem. 2007;282:3423–7. doi: 10.1074/jbc.C600226200. [DOI] [PubMed] [Google Scholar]
- 13.Johnsen IB, Nguyen TT, Ringdal M, Tryggestad AM, Bakke O, Lien E, Espevik T, Anthonsen MW. Toll-like receptor 3 associates with c-Src tyrosine kinase on endosomes to initiate antiviral signaling. EMBO J. 2006;25:3335–46. doi: 10.1038/sj.emboj.7601222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Yamashita M, Chattopadhyay S, Fensterl V, Saikia P, Wetzel JL, Sen GC. Epidermal growth factor receptor is essential for Toll-like receptor 3 signaling. Sci Signal. 2012;5:ra50. doi: 10.1126/scisignal.2002581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun H, Zhuang G, Chai L, Wang Z, Johnson D, Ma Y, et al. TIPE2 controls innate immunity to RNA by targeting the phosphatidylinositol 3-kinase-Rac pathway. J Immunol. 2012;189:2768–73. doi: 10.4049/jimmunol.1103477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee KG, Xu S, Kang ZH, et al. Bruton's tyrosine kinase phosphorylates Toll-like receptor 3 to initiate antiviral response. Proc Natl Acad Sci USA. 2012;109:5791–6. doi: 10.1073/pnas.1119238109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sato S, Sugiyama M, Yamamoto M, Watanabe Y, Kawai T, Takeda K, Akira S. Toll/IL-1 receptor domain-containing adaptor inducing IFN-β (TRIF) associates with TNF receptor-associated factor 6 and TANK-binding kinase 1, and activates two distinct transcription factors, NF- κB and IFN-regulatory factor-3, in the Toll-like receptor signaling. J Immunol. 2003;171:4304–10. doi: 10.4049/jimmunol.171.8.4304. [DOI] [PubMed] [Google Scholar]
- 18.Alexopoulou L, Holt AC, Medzhitov R, Flavell RA. Recognition of double-stranded RNA and activation of NF-κB by Toll-like receptor 3. Nature. 2001;413:732–8. doi: 10.1038/35099560. [DOI] [PubMed] [Google Scholar]
- 19.Han KJ, Su X, Xu LG, Bin LH, Zhang J, Shu HB. Mechanisms of the TRIF-induced interferon-stimulated response element and NF-κB activation and apoptosis pathways. J Biol Chem. 2004;279:15652–61. doi: 10.1074/jbc.M311629200. [DOI] [PubMed] [Google Scholar]
- 20.Ruckdeschel K, Pfaffinger G, Haase R, Sing A, Weighardt H, Hacker G, Holzmann B, Heesemann J. Signaling of apoptosis through TLRs critically involves toll/IL-1 receptor domain-containing adapter inducing IFN-β, but not MyD88, in bacteria-infected murine macrophages. J Immunol. 2004;173:3320–8. doi: 10.4049/jimmunol.173.5.3320. [DOI] [PubMed] [Google Scholar]
- 21.Yamamoto M, Sato S, Hemmi H, et al. Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway. Science. 2003;301:640–3. doi: 10.1126/science.1087262. [DOI] [PubMed] [Google Scholar]
- 22.Petnicki-Ocwieja T, Chung E, Acosta DI, et al. TRIF mediates Toll-like receptor 2-dependent inflammatory responses to Borrelia burgdorferi. Infect Immun. 2013;81:402–10. doi: 10.1128/IAI.00890-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi YJ, Im E, Pothoulakis C, Rhee SH. TRIF modulates TLR5-dependent responses by inducing proteolytic degradation of TLR5. J Biol Chem. 2010;285:21382–90. doi: 10.1074/jbc.M110.115022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Z, Kim T, Bao M, et al. DDX1, DDX21, and DHX36 helicases form a complex with the adaptor molecule TRIF to sense dsRNA in dendritic cells. Immunity. 2011;34:866–78. doi: 10.1016/j.immuni.2011.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coll RC, O'Neill LA. New insights into the regulation of signalling by toll-like receptors and nod-like receptors. J Innate Immun. 2010;2:406–21. doi: 10.1159/000315469. [DOI] [PubMed] [Google Scholar]
- 26.An H, Zhao W, Hou J, et al. SHP-2 phosphatase negatively regulates the TRIF adaptor protein-dependent type I interferon and proinflammatory cytokine production. Immunity. 2006;25:919–28. doi: 10.1016/j.immuni.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 27.Boone DL, Turer EE, Lee EG, et al. The ubiquitin-modifying enzyme A20 is required for termination of Toll-like receptor responses. Nat Immunol. 2004;5:1052–60. doi: 10.1038/ni1110. [DOI] [PubMed] [Google Scholar]
- 28.Carty M, Goodbody R, Schroder M, Stack J, Moynagh PN, Bowie AG. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat Immunol. 2006;7:1074–81. doi: 10.1038/ni1382. [DOI] [PubMed] [Google Scholar]
- 29.Zhang M, Wang L, Zhao X, Zhao K, Meng H, Zhao W, Gao C. TRAF-interacting protein (TRIP) negatively regulates IFN-β production and antiviral response by promoting proteasomal degradation of TANK-binding kinase 1. J Exp Med. 2012;209:1703–11. doi: 10.1084/jem.20120024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kayagaki N, Phung Q, Chan S, et al. DUBA: a deubiquitinase that regulates type I interferon production. Science. 2007;318:1628–32. doi: 10.1126/science.1145918. [DOI] [PubMed] [Google Scholar]
- 31.Peng J, Yuan Q, Lin B, et al. SARM inhibits both TRIF- and MyD88-mediated AP-1 activation. Eur J Immunol. 2010;40:1738–47. doi: 10.1002/eji.200940034. [DOI] [PubMed] [Google Scholar]
- 32.Palsson-McDermott EM, Doyle SL, McGettrick AF, et al. TAG, a splice variant of the adaptor TRAM, negatively regulates the adaptor MyD88-independent TLR4 pathway. Nat Immunol. 2009;10:579–86. doi: 10.1038/ni.1727. [DOI] [PubMed] [Google Scholar]
- 33.Han C, Jin J, Xu S, Liu H, Li N, Cao X. Integrin CD11b negatively regulates TLR-triggered inflammatory responses by activating Syk and promoting degradation of MyD88 and TRIF via Cbl-b. Nat Immunol. 2010;11:734–42. doi: 10.1038/ni.1908. [DOI] [PubMed] [Google Scholar]
- 34.Fearns C, Pan Q, Mathison JC, Chuang TH. Triad3A regulates ubiquitination and proteasomal degradation of RIP1 following disruption of Hsp90 binding. J Biol Chem. 2006;281:34592–600. doi: 10.1074/jbc.M604019200. [DOI] [PubMed] [Google Scholar]
- 35.Xue Q, Zhou Z, Lei X, Liu X, He B, Wang J, Hung T. TRIM38 negatively regulates TLR3-MEdiated IFN-β signaling by targeting TRIF for degradation. PLoS ONE. 2012;7:e46825. doi: 10.1371/journal.pone.0046825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhao W, Wang L, Zhang M, Wang P, Yuan C, Qi J, Meng H, Goa C. Tripartite motif-containing protein 38 negatively regulates TLR3/4- and RIG-I-mediated IFN-β production and antiviral response by targeting NAP1. J Immunol. 2012;188:5311–8. doi: 10.4049/jimmunol.1103506. [DOI] [PubMed] [Google Scholar]
- 37.Zhao W, Wang L, Zhang M, Yuan C, Gao C. E3 ubiquitin ligase tripartite motif 38 negatively regulates TLR-mediated immune responses by proteasomal degradation of TNF receptor-associated factor 6 in macrophages. J Immunol. 2012;188:2567–74. doi: 10.4049/jimmunol.1103255. [DOI] [PubMed] [Google Scholar]
- 38.Ahmed S, Maratha A, Butt AQ, Shevlin E, Miggin SM. TRIF-mediated TLR3 and TLR4 signaling is negatively regulated by ADAM15. J Immunol. 2013;190:2217–28. doi: 10.4049/jimmunol.1201630. [DOI] [PubMed] [Google Scholar]
- 39.Pott J, Stockinger S, Torow N, et al. Age-dependent TLR3 expression of the intestinal epithelium contributes to rotavirus susceptibility. PLoS Pathog. 2012;8:e1002670. doi: 10.1371/journal.ppat.1002670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meylan E, Burns K, Hofmann K, Blancheteau V, Martinon F, Kelliher M, Tschopp J. RIP1 is an essential mediator of Toll-like receptor 3-induced NF-κB activation. Nat Immunol. 2004;5:503–7. doi: 10.1038/ni1061. [DOI] [PubMed] [Google Scholar]
- 41.Wertz IE, O'Rourke KM, Zhou H, et al. De-ubiquitination and ubiquitin ligase domains of A20 downregulate NF-κB signalling. Nature. 2004;430:694–9. doi: 10.1038/nature02794. [DOI] [PubMed] [Google Scholar]
- 42.Wang YY, Li L, Han KJ, Zhai Z, Shu HB. A20 is a potent inhibitor of TLR3- and Sendai virus-induced activation of NF-κB and ISRE and IFN-β promoter. FEBS Lett. 2004;576:86–90. doi: 10.1016/j.febslet.2004.08.071. [DOI] [PubMed] [Google Scholar]
- 43.Yoshida H, Jono H, Kai H, Li JD. The tumor suppressor cylindromatosis (CYLD) acts as a negative regulator for toll-like receptor 2 signaling via negative cross-talk with TRAF6 AND TRAF7. J Biol Chem. 2005;280:41111–21. doi: 10.1074/jbc.M509526200. [DOI] [PubMed] [Google Scholar]
- 44.Trompouki E, Hatzivassiliou E, Tsichritzis T, Farmer H, Ashworth A, Mosialos G. CYLD is a deubiquitinating enzyme that negatively regulates NF-κB activation by TNFR family members. Nature. 2003;424:793–6. doi: 10.1038/nature01803. [DOI] [PubMed] [Google Scholar]
- 45.Kawagoe T, Takeuchi O, Takabatake Y, Kato H, Isaka Y, Tsujimura T, Akira S. TANK is a negative regulator of Toll-like receptor signaling and is critical for the prevention of autoimmune nephritis. Nat Immunol. 2009;10:965–72. doi: 10.1038/ni.1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Siednienko J, Jackson R, Mellett M, et al. Pellino3 targets the IRF7 pathway and facilitates autoregulation of TLR3- and viral-induced expression of type I interferons. Nat Immunol. 2012;13:1055–62. doi: 10.1038/ni.2429. [DOI] [PubMed] [Google Scholar]
- 47.Zhou F, Zhang X, van Dam H, Ten Dijke P, Huang H, Zhang L. Ubiquitin-specific protease 4 mitigates Toll-like/interleukin-1 receptor signaling and regulates innate immune activation. J Biol Chem. 2012;287:11002–10. doi: 10.1074/jbc.M111.328187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yuk JM, Shin DM, Lee HM, et al. The orphan nuclear receptor SHP acts as a negative regulator in inflammatory signaling triggered by Toll-like receptors. Nat Immunol. 2011;12:742–51. doi: 10.1038/ni.2064. [DOI] [PubMed] [Google Scholar]
- 49.Allen IC, Moore CB, Schneider M, et al. NLRX1 protein attenuates inflammatory responses to infection by interfering with the RIG-I-MAVS and TRAF6-NF-κB signaling pathways. Immunity. 2011;34:854–65. doi: 10.1016/j.immuni.2011.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li K, Foy E, Ferreon JC, et al. Immune evasion by hepatitis C virus NS3/4A protease-mediated cleavage of the Toll-like receptor 3 adaptor protein TRIF. Proc Natl Acad Sci USA. 2005;102:2992–7. doi: 10.1073/pnas.0408824102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qu L, Feng Z, Yamane D, Liang Y, Lanford RE, Li K, Lemon SM. Disruption of TLR3 signaling due to cleavage of TRIF by the hepatitis A virus protease-polymerase processing intermediate, 3CD. PLoS Pathog. 2011;7:e1002169. doi: 10.1371/journal.ppat.1002169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mukherjee A, Morosky SA, Delorme-Axford E, Dybdahl-Sissoko N, Oberste MS, Wang T, Coyne CB. The coxsackievirus B 3C protease cleaves MAVS and TRIF to attenuate host type I interferon and apoptotic signaling. PLoS Pathog. 2011;7:e1001311. doi: 10.1371/journal.ppat.1001311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lei X, Sun Z, Liu X, Jin Q, He B, Wang J. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J Virol. 2011;85:8811–8. doi: 10.1128/JVI.00447-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahmad H, Gubbels R, Ehlers E, Meyer F, Waterbury T, Lin R, Zhang L. Kaposi sarcoma-associated herpesvirus degrades cellular Toll-interleukin-1 receptor domain-containing adaptor-inducing β-interferon (TRIF) J Biol Chem. 2011;286:7865–72. doi: 10.1074/jbc.M110.191452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Liu X, Lei X, Zhou Z, Sun Z, Xue Q, Wang J, Hung T. Enterovirus 71 induces degradation of TRIM38, a potential E3 ubiquitin ligase. Virol J. 2011;8:61. doi: 10.1186/1743-422X-8-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Siu KL, Kok KH, Ng MH, Poon VK, Yuen KY, Zheng BJ, Jin DY. Severe acute respiratory syndrome coronavirus M protein inhibits type I interferon production by impeding the formation of TRAF3.TANK.TBK1/IKKε complex. J Biol Chem. 2009;284:16202–9. doi: 10.1074/jbc.M109.008227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Harte MT, Haga IR, Maloney G, et al. The poxvirus protein A52R targets Toll-like receptor signaling complexes to suppress host defense. J Exp Med. 2003;197:343–51. doi: 10.1084/jem.20021652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhang SY, Jouanguy E, Ugolini S, et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science. 2007;317:1522–7. doi: 10.1126/science.1139522. [DOI] [PubMed] [Google Scholar]
- 59.Lafaille FG, Pessach IM, Zhang SY, et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature. 2012;491:769–73. doi: 10.1038/nature11583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Guo Y, Audry M, Ciancanelli M, et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J Exp Med. 2011;208:2083–98. doi: 10.1084/jem.20101568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Li J, Ye L, Wang X, Hu S, Ho W. Induction of interferon-γ contributes to Toll-like receptor 3-mediated herpes simplex virus type 1 inhibition in astrocytes. J Neurosci Res. 2012;90:399–406. doi: 10.1002/jnr.22758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sancho-Shimizu V, Perez de Diego R, Jouanguy E, Zhang SY, Casanova JL. Inborn errors of anti-viral interferon immunity in humans. Curr Opin Virol. 2011;1:487–96. doi: 10.1016/j.coviro.2011.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hidaka F, Matsuo S, Muta T, Takeshige K, Mizukami T, Nunoi H. A missense mutation of the Toll-like receptor 3 gene in a patient with influenza-associated encephalopathy. Clin Immunol. 2006;119:188–94. doi: 10.1016/j.clim.2006.01.005. [DOI] [PubMed] [Google Scholar]
- 64.Kindberg E, Vene S, Mickiene A, Lundkvist A, Lindquist L, Svensson L. A functional Toll-like receptor 3 gene (TLR3) may be a risk factor for tick-borne encephalitis virus (TBEV) infection. J Infect Dis. 2011;203:523–8. doi: 10.1093/infdis/jiq082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Li K, Li NL, Wei D, Pfeffer SR, Fan M, Pfeffer LM. Activation of chemokine and inflammatory cytokine response in hepatitis C virus-infected hepatocytes depends on Toll-like receptor 3 sensing of hepatitis C virus double-stranded RNA intermediates. Hepatology. 2012;55:666–75. doi: 10.1002/hep.24763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wornle M, Schmid H, Banas B, et al. Novel role of toll-like receptor 3 in hepatitis C-associated glomerulonephritis. Am J Pathol. 2006;168:370–85. doi: 10.2353/ajpath.2006.050491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rong Y, Song H, You S, et al. Association of Toll-like receptor 3 polymorphisms with chronic hepatitis B and hepatitis B-related acute-on-chronic liver failure. Inflammation. 2013;36:413–8. doi: 10.1007/s10753-012-9560-4. [DOI] [PubMed] [Google Scholar]
- 68.Li G, Zheng Z. Toll-like receptor 3 genetic variants and susceptibility to hepatocellular carcinoma and HBV-related hepatocellular carcinoma. Tumour Biol. 2013;34:1589–94. doi: 10.1007/s13277-013-0689-z. [DOI] [PubMed] [Google Scholar]
- 69.Esposito S, Molteni CG, Giliani S, et al. Toll-like receptor 3 gene polymorphisms and severity of pandemic A/H1N1/2009 influenza in otherwise healthy children. Virol J. 2012;9:270. doi: 10.1186/1743-422X-9-270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Swaminathan G, Rossi F, Sierra LJ, Gupta A, Navas-Martin S, Martin-Garcia J. A role for microRNA-155 modulation in the anti-HIV-1 effects of Toll-like receptor 3 stimulation in macrophages. PLoS Pathog. 2012;8:e1002937. doi: 10.1371/journal.ppat.1002937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Rivieccio MA, Suh HS, Zhao Y, Zhao ML, Chin KC, Lee SC, Brosnan C. TLR3 ligation activates an antiviral response in human fetal astrocytes: a role for viperin/cig5. J Immunol. 2006;177:4735–41. doi: 10.4049/jimmunol.177.7.4735. [DOI] [PubMed] [Google Scholar]
- 72.Fujimoto C, Nakagawa Y, Ohara K, Takahashi H. Polyriboinosinic polyribocytidylic acid [poly(I:C)]/TLR3 signaling allows class I processing of exogenous protein and induction of HIV-specific CD8+ cytotoxic T lymphocytes. Int Immunol. 2004;16:55–63. doi: 10.1093/intimm/dxh025. [DOI] [PubMed] [Google Scholar]
- 73.Sironi M, Biasin M, Cagliani R, et al. A common polymorphism in TLR3 confers natural resistance to HIV-1 infection. J Immunol. 2012;188:818–23. doi: 10.4049/jimmunol.1102179. [DOI] [PubMed] [Google Scholar]
- 74.Schreiner B, Voss J, Wischhusen J, et al. Expression of toll-like receptors by human muscle cells in vitro and in vivo: TLR3 is highly expressed in inflammatory and HIV myopathies, mediates IL-8 release and up-regulation of NKG2D-ligands. FASEB J. 2006;20:118–20. doi: 10.1096/fj.05-4342fje. [DOI] [PubMed] [Google Scholar]
- 75.de Vries HE, Kooij G, Frenkel D, Georgopoulos S, Monsonego A, Janigro D. Inflammatory events at blood–brain barrier in neuroinflammatory and neurodegenerative disorders: implications for clinical disease. Epilepsia. 2012;53(Suppl 6):45–52. doi: 10.1111/j.1528-1167.2012.03702.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Suthar MS, Diamond MS, Gale M., Jr West Nile virus infection and immunity. Nat Rev Microbiol. 2013;11:115–28. doi: 10.1038/nrmicro2950. [DOI] [PubMed] [Google Scholar]
- 77.Wang T, Town T, Alexopoulou L, Anderson JF, Fikrig E, Flavell RA. Toll-like receptor 3 mediates West Nile virus entry into the brain causing lethal encephalitis. Nat Med. 2004;10:1366–73. doi: 10.1038/nm1140. [DOI] [PubMed] [Google Scholar]
- 78.Daffis S, Samuel MA, Suthar MS, Gale M, Jr, Diamond MS. Toll-like receptor 3 has a protective role against West Nile virus infection. J Virol. 2008;82:10349–58. doi: 10.1128/JVI.00935-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Abe Y, Fujii K, Nagata N, et al. The toll-like receptor 3-mediated antiviral response is important for protection against poliovirus infection in poliovirus receptor transgenic mice. J Virol. 2012;86:185–94. doi: 10.1128/JVI.05245-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jin YH, Kaneyama T, Kang MH, Kang HS, Koh CS, Kim BS. TLR3 signaling is either protective or pathogenic for the development of Theiler's virus-induced demyelinating disease depending on the time of viral infection. J Neuroinflammation. 2011;8:178. doi: 10.1186/1742-2094-8-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tyler KL. Herpes simplex virus infections of the central nervous system: encephalitis and meningitis, including Mollaret's. Herpes. 2004;11(Suppl 2):57A–64A. [PubMed] [Google Scholar]
- 82.Reinert LS, Harder L, Holm CK, et al. TLR3 deficiency renders astrocytes permissive to herpes simplex virus infection and facilitates establishment of CNS infection in mice. J Clin Invest. 2012;122:1368–76. doi: 10.1172/JCI60893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davey GM, Wojtasiak M, Proietto AI, Carbone FR, Heath WR, Bedoui S. Cutting edge: priming of CD8 T cell immunity to herpes simplex virus type 1 requires cognate TLR3 expression in vivo. J Immunol. 2010;184:2243–6. doi: 10.4049/jimmunol.0903013. [DOI] [PubMed] [Google Scholar]
- 84.Edelmann KH, Richardson-Burns S, Alexopoulou L, Tyler KL, Flavell RA, Oldstone MB. Does Toll-like receptor 3 play a biological role in virus infections? Virology. 2004;322:231–8. doi: 10.1016/j.virol.2004.01.033. [DOI] [PubMed] [Google Scholar]
- 85.Wang Q, Miller DJ, Bowman ER, et al. MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness. PLoS Pathog. 2011;7:e1002070. doi: 10.1371/journal.ppat.1002070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Le Goffic R, Balloy V, Lagranderie M, Alexopoulou L, Escriou N, Flavell R, Chignard M, Si-Tahar M. Detrimental contribution of the Toll-like receptor (TLR)3 to influenza A virus-induced acute pneumonia. PLoS Pathog. 2006;2:e53. doi: 10.1371/journal.ppat.0020053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Groskreutz DJ, Monick MM, Powers LS, Yarovinsky TO, Look DC, Hunninghake GW. Respiratory syncytial virus induces TLR3 protein and protein kinase R, leading to increased double-stranded RNA responsiveness in airway epithelial cells. J Immunol. 2006;176:1733–40. doi: 10.4049/jimmunol.176.3.1733. [DOI] [PubMed] [Google Scholar]
- 88.Aeffner F, Traylor ZP, Yu EN, Davis IC. Double-stranded RNA induces similar pulmonary dysfunction to respiratory syncytial virus in BALB/c mice. Am J Physiol Lung Cell Mol Physiol. 2011;301:L99–109. doi: 10.1152/ajplung.00398.2010. [DOI] [PubMed] [Google Scholar]
- 89.Hutchens M, Luker KE, Sottile P, Sonstein J, Lukacs NW, Nunez G, Curtis J, Luker G. TLR3 increases disease morbidity and mortality from vaccinia infection. J Immunol. 2008;180:483–91. doi: 10.4049/jimmunol.180.1.483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Hardarson HS, Baker JS, Yang Z, et al. Toll-like receptor 3 is an essential component of the innate stress response in virus-induced cardiac injury. Am J Physiol Heart Circ Physiol. 2007;292:H251–8. doi: 10.1152/ajpheart.00398.2006. [DOI] [PubMed] [Google Scholar]
- 91.McCartney SA, Vermi W, Lonardi S, et al. RNA sensor-induced type I IFN prevents diabetes caused by a β cell-tropic virus in mice. J Clin Invest. 2011;121:1497–507. doi: 10.1172/JCI44005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Negishi H, Osawa T, Ogami K, et al. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc Natl Acad Sci USA. 2008;105:20446–51. doi: 10.1073/pnas.0810372105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Abston ED, Coronado MJ, Bucek A, et al. Th2 regulation of viral myocarditis in mice: different roles for TLR3 versus TRIF in progression to chronic disease. Clin Dev Immunol. 2012;2012:129486. doi: 10.1155/2012/129486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Richer MJ, Lavallee DJ, Shanina I, Horwitz MS. Toll-like receptor 3 signaling on macrophages is required for survival following coxsackievirus B4 infection. PLoS ONE. 2009;4:e4127. doi: 10.1371/journal.pone.0004127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yamashita M, Chattopadhyay S, Fensterl V, Zhang Y, Sen GC. A TRIF-independent branch of TLR3 signaling. J Immunol. 2012;188:2825–33. doi: 10.4049/jimmunol.1103220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Gowen BB, Hoopes JD, Wong MH, Jung KH, Isakson KC, Alexopoulou L, Flavell R, Sidwell R. TLR3 deletion limits mortality and disease severity due to Phlebovirus infection. J Immunol. 2006;177:6301–7. doi: 10.4049/jimmunol.177.9.6301. [DOI] [PubMed] [Google Scholar]
- 97.Lang KS, Georgiev P, Recher M, et al. Immunoprivileged status of the liver is controlled by Toll-like receptor 3 signaling. J Clin Invest. 2006;116:2456–63. doi: 10.1172/JCI28349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schulz O, Diebold SS, Chen M, et al. Toll-like receptor 3 promotes cross-priming to virus-infected cells. Nature. 2005;433:887–92. doi: 10.1038/nature03326. [DOI] [PubMed] [Google Scholar]
- 99.Yamashita M, Millward CA, Inoshita H, Saikia P, Chattopadhyay S, Sen GC, Emancipator SN. Antiviral innate immunity disturbs podocyte cell function. J Innate Immun. 2012;5:231–41. doi: 10.1159/000345255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mazaleuskaya L, Veltrop R, Ikpeze N, Martin-Garcia J, Navas-Martin S. Protective role of Toll-like receptor 3-induced type I interferon in murine coronavirus infection of macrophages. Viruses. 2012;4:901–23. doi: 10.3390/v4050901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Xia X, Cui J, Wang HY, et al. NLRX1 negatively regulates TLR-induced NF-κB signaling by targeting TRAF6 and IKK. Immunity. 2011;34:843–53. doi: 10.1016/j.immuni.2011.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]